Genetic Diversity of Tick-Borne Rickettsial Pathogens; Insights Gained from Distant Strains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
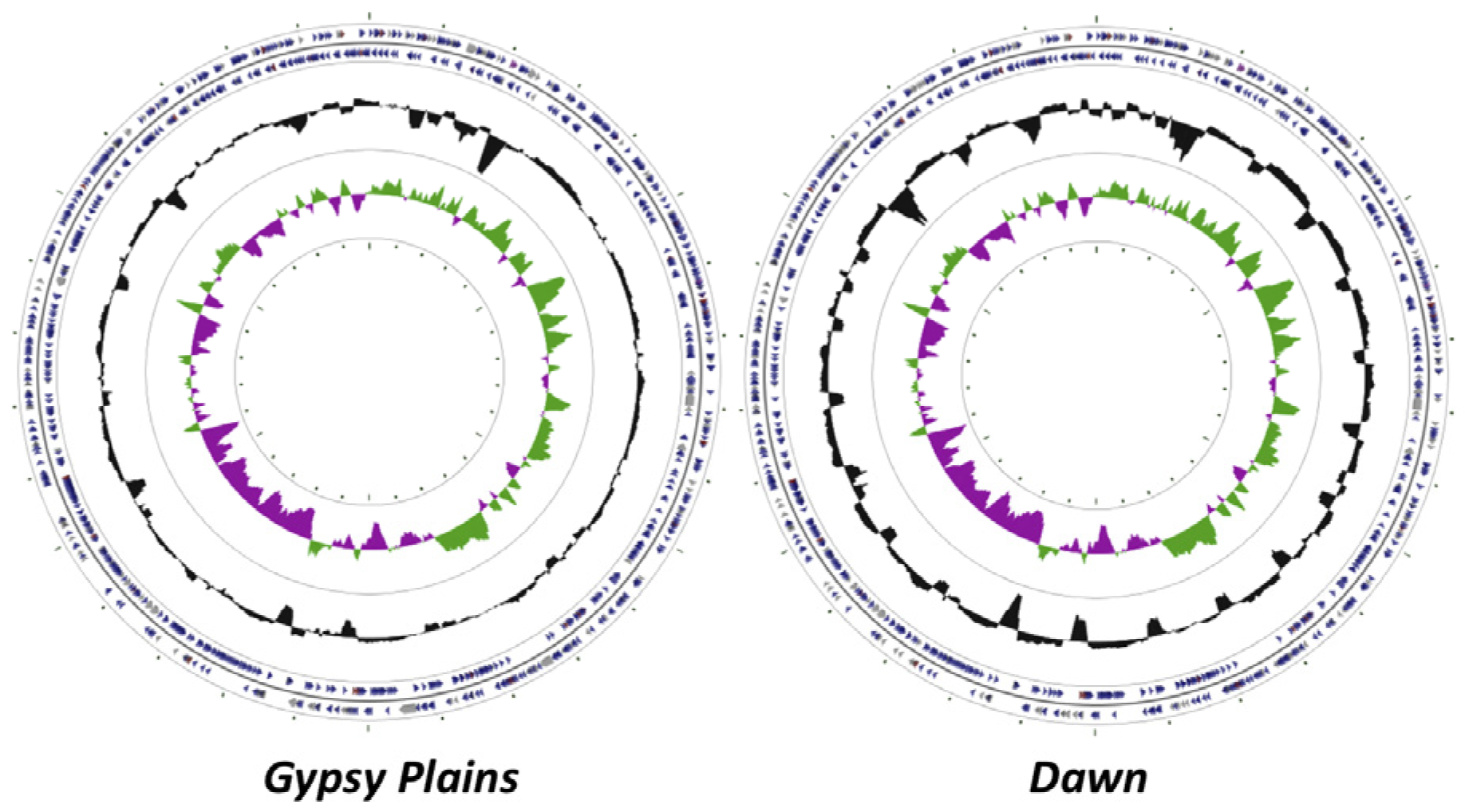
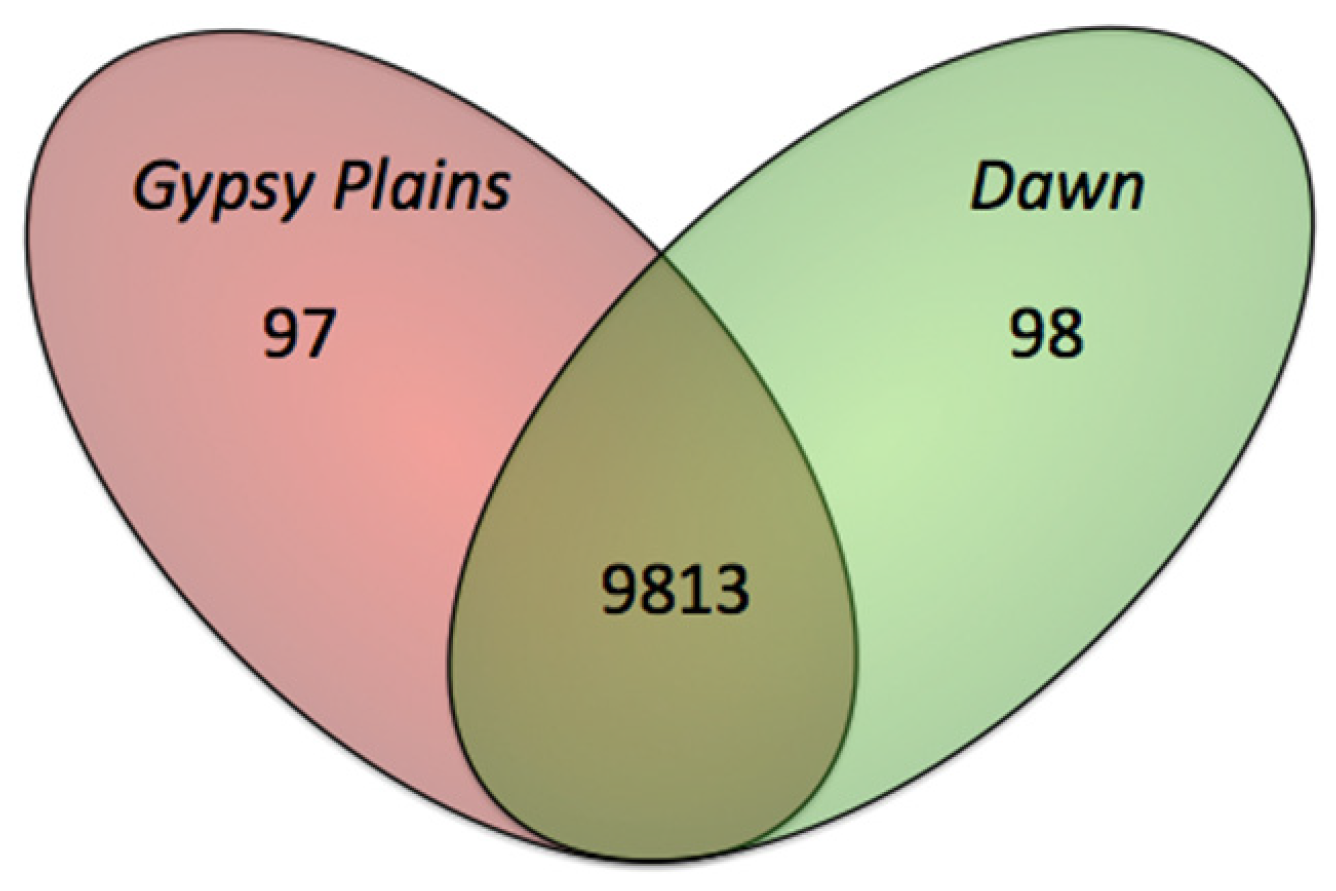
2.1. Genome Sequences

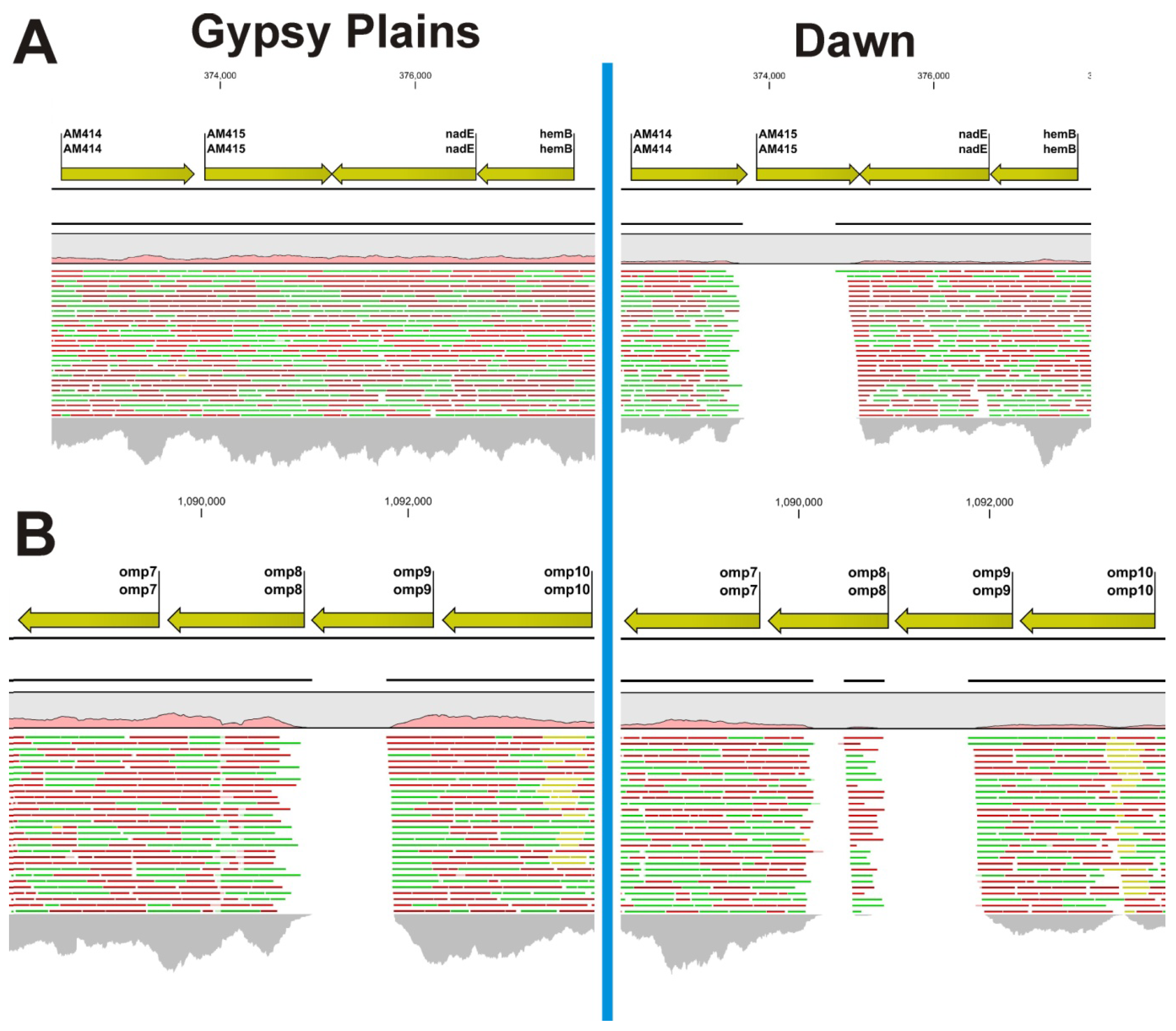
2.2. Identification of INDELs as Genetic Markers between the Attenuated and Virulent Strains


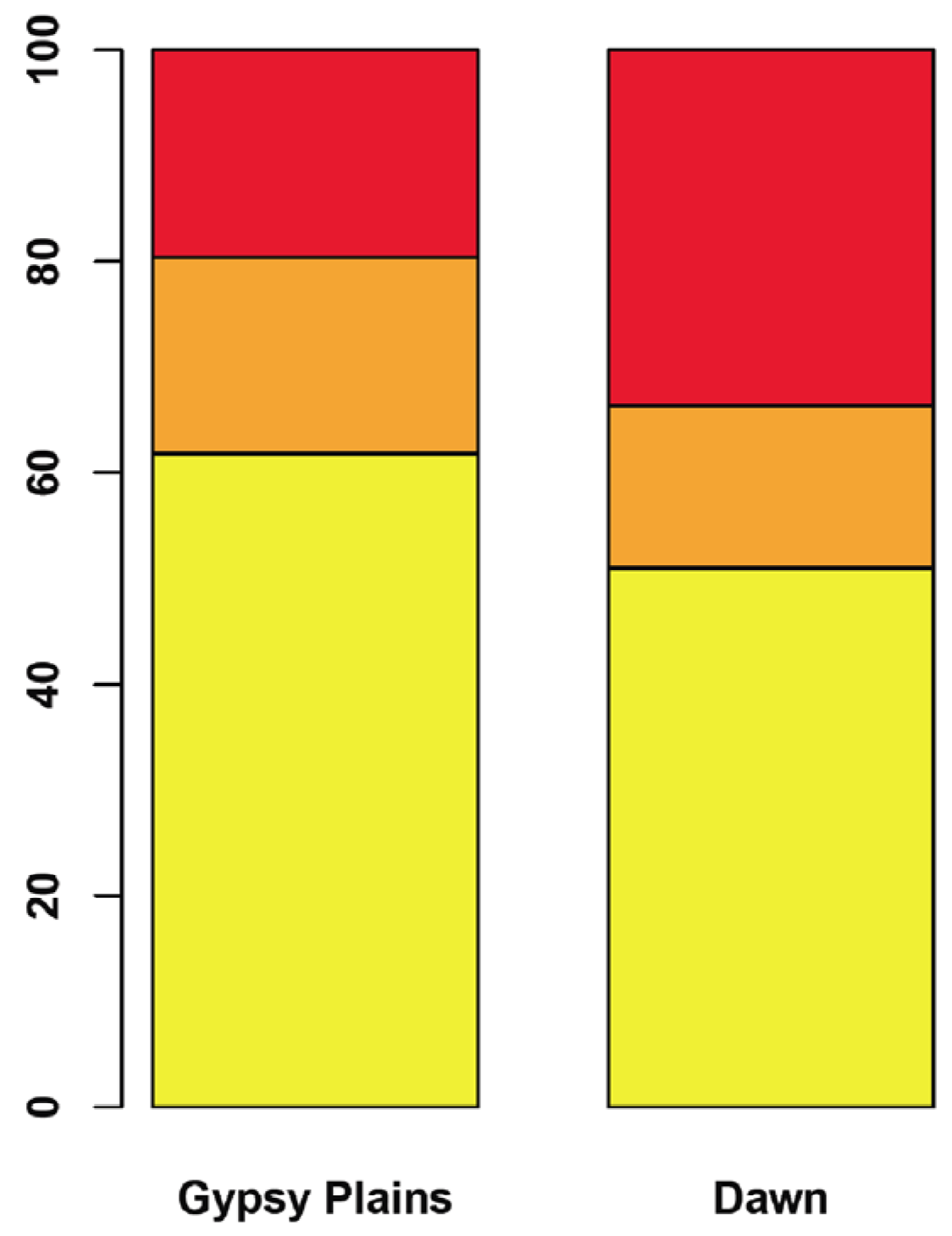
2.3. SNPs that Segregate with Virulence and Transmission Phenotypes


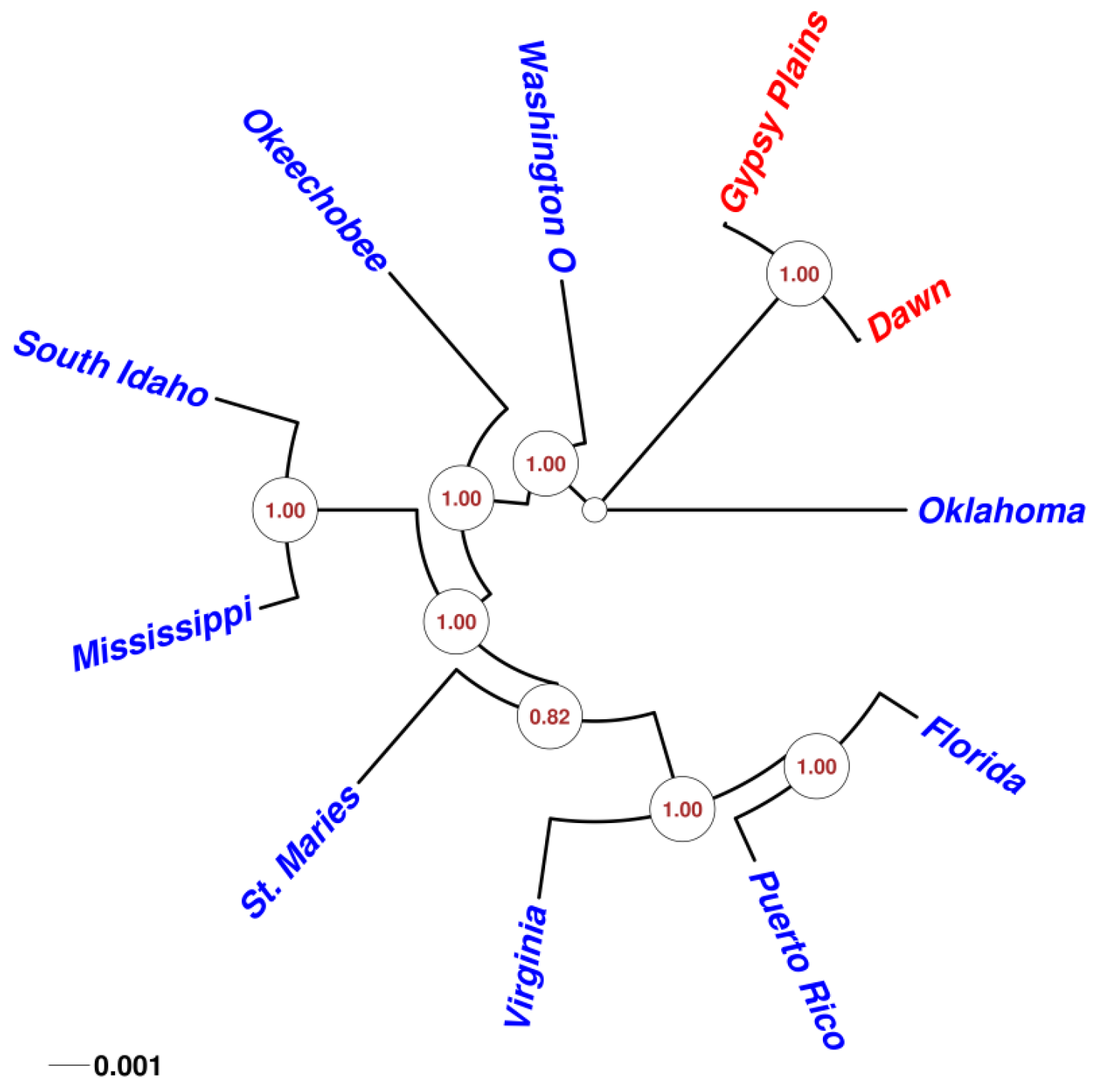
2.4. Phylogenetic Analysis in 11 A. marginale Genomes

3. Discussion
4. Experimental Section
4.1. Genome Sequencing/de novo Assembly/Annotation
4.2. SNP Calling and Annotation
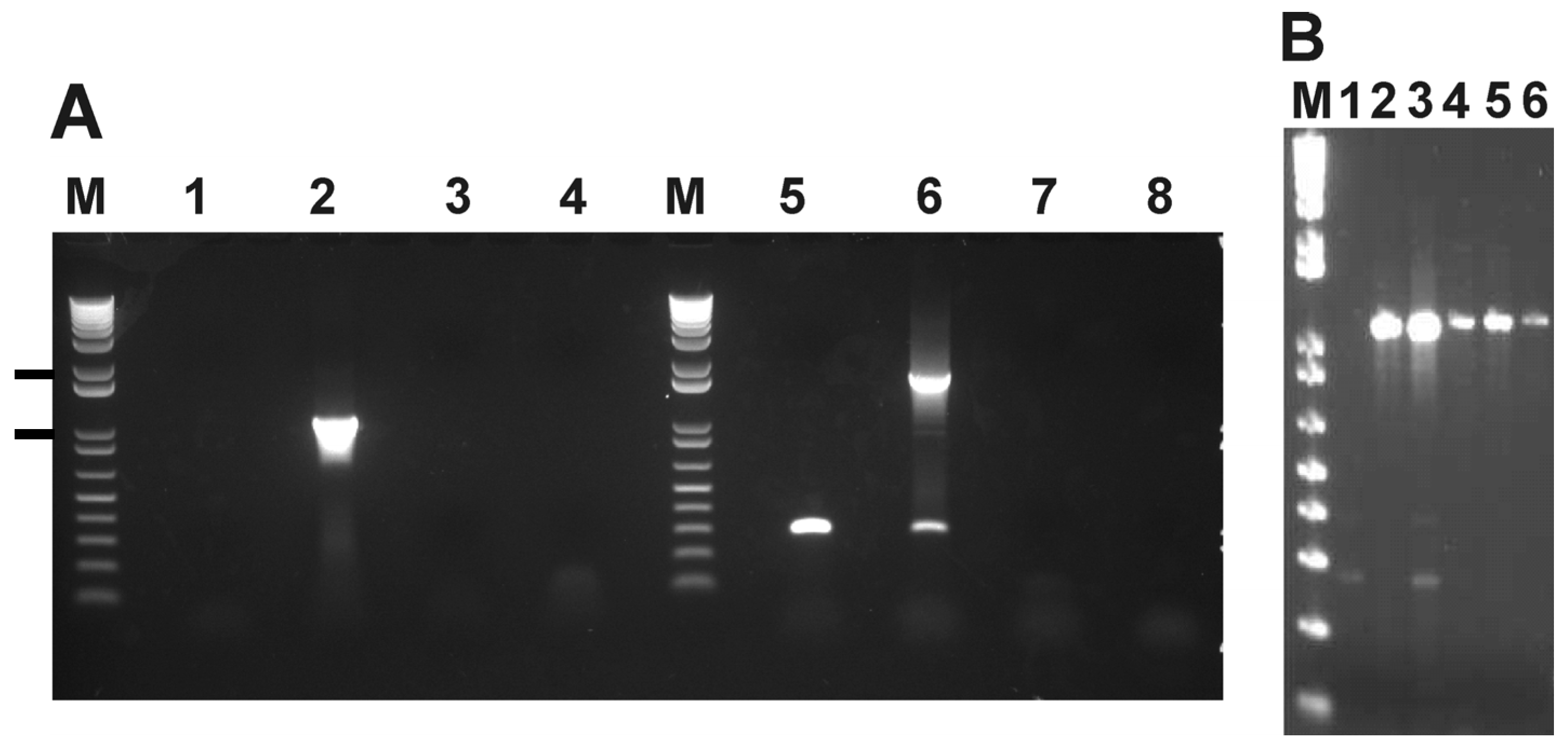
4.3. Identification of a Genetic Marker
4.4. Phylogenetic Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bryant, J.; Chewapreecha, C.; Bentley, S.D. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol. 2012, 7, 1283–1296. [Google Scholar] [CrossRef]
- Rokas, A.; Williams, B.L.; King, N.; Carroll, S.B. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 2003, 425, 798–804. [Google Scholar] [CrossRef]
- Palmer, G.H. Anaplasma Vaccines. In Veterinary Protozoan and Hemoparasite Vaccines; Wright, I.G., Ed.; CRC Press: Boca Raton, FL, USA, 1989; pp. 2–29. [Google Scholar]
- Brayton, K.A.; Kappmeyer, L.S.; Herndon, D.R.; Dark, M.J.; Tibbals, D.L.; Palmer, G.H.; McGuire, T.C.; Knowles, D.P., Jr. Complete genome sequencing of Anaplasma marginale reveals that the surface is skewed to two superfamilies of outer membrane proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 844–849. [Google Scholar] [CrossRef]
- Dark, M.J.; Al-Khedery, B.; Barbet, A.F. Multistrain genome analysis identifies candidate vaccine antigens of Anaplasma marginale. Vaccine 2011, 29, 4923–4932. [Google Scholar] [CrossRef]
- Dark, M.J.; Herndon, D.R.; Kappmeyer, L.S.; Gonzales, M.P.; Nordeen, E.; Palmer, G.H.; Knowles, D.P., Jr.; Brayton, K.A. Conservation in the face of diversity: Multistrain analysis of an intracellular bacterium. BMC Genomics 2009, 10. [Google Scholar] [CrossRef]
- Bock, R.E.; de Vos, A.J. Immunity following use of Australian tick fever vaccine: A review of the evidence. Aust. Vet. J. 2001, 79, 832–839. [Google Scholar] [CrossRef]
- Shkap, V.; Leibovitz, B.; Krigel, Y.; Molad, T.; Fish, L.; Mazuz, M.; Fleiderovitz, L.; Savitsky, I. Concomitant infection of cattle with the vaccine strain Anaplasma marginale ss centrale and field strains of A. marginale. Vet. Microbiol. 2008, 130, 277–284. [Google Scholar] [CrossRef]
- Herndon, D.R.; Palmer, G.H.; Shkap, V.; Knowles, D.P., Jr.; Brayton, K.A. Complete genome sequence of Anaplasma marginale subsp. centrale. J. Bacteriol. 2010, 192, 379–380. [Google Scholar] [CrossRef]
- Bock, R.E.; deVos, A.J.; Kingston, T.G.; Carter, P.D. Assessment of a low virulence Australian isolate of Anaplasma marginale for pathogenicity, immunogenicity and transmissibility by Boophilus microplus. Vet. Parasitol. 2003, 118, 121–131. [Google Scholar] [CrossRef]
- Bock, R.E.; de Vos, A.J.; Kingston, T.G.; McLellan, D.J. Effect of breed of cattle on innate resistance to infection with Babesia bovis, B bigemina and Anaplasma marginale. Aust. Vet. J. 1997, 75, 337–340. [Google Scholar] [CrossRef]
- Macmillan, H.; Norimine, J.; Brayton, K.A.; Palmer, G.H.; Brown, W.C. Physical linkage of naturally complexed bacterial outer membrane proteins enhances immunogenicity. Infect. Immun. 2008, 76, 1223–1229. [Google Scholar] [CrossRef]
- Pierle, S.A.; Dark, M.J.; Dahmen, D.; Palmer, G.H.; Brayton, K.A. Comparative genomics and transcriptomics of trait-gene association. BMC Genomics 2012, 13, 669. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Davies, J.E. Origins, acquisition and dissemination of antibiotic resistance determinants. Ciba Found. Symp. 1997, 207, 15–27, discussion 27–35. [Google Scholar]
- Ochman, H.; Lawrence, J.G.; Groisman, E.A. Lateral gene transfer and the nature of bacterial innovation. Nature 2000, 405, 299–304. [Google Scholar] [CrossRef]
- Dumler, J.S. Fitness and freezing: Vector biology and human health. J. Clin. Investig. 2010, 120, 3087–3090. [Google Scholar] [CrossRef]
- Stermann, M.; Sedlacek, L.; Maass, S.; Bange, F.C. A promoter mutation causes differential nitrate reductase activity of Mycobacterium tuberculosis and Mycobacterium bovis. J. Bacteriol. 2004, 186, 2856–2861. [Google Scholar] [CrossRef]
- Read, T.D.; Salzberg, S.L.; Pop, M.; Shumway, M.; Umayam, L.; Jiang, L.; Holtzapple, E.; Busch, J.D.; Smith, K.L.; Schupp, J.M.; et al. Comparative genome sequencing for discovery of novel polymorphisms in Bacillus anthracis. Science 2002, 296, 2028–2033. [Google Scholar] [CrossRef]
- Noh, S.M.; Brayton, K.A.; Knowles, D.P.; Agnes, J.T.; Dark, M.J.; Brown, W.C.; Baszler, T.V.; Palmer, G.H. Differential expression and sequence conservation of the Anaplasma marginale msp2 gene superfamily outer membrane proteins. Infect. Immun. 2006, 74, 3471–3479. [Google Scholar] [CrossRef]
- Ballouz, S.; Francis, A.R.; Lan, R.; Tanaka, M.M. Conditions for the evolution of gene clusters in bacterial genomes. PLoS Comput. Biol. 2010, 6, e1000672. [Google Scholar] [CrossRef]
- De la Fuente, J.; Garcia-Garcia, J.C.; Blouin, E.F.; Kocan, K.M. Differential adhesion of major surface proteins 1a and 1b of the ehrlichial cattle pathogen Anaplasma marginale to bovine erythrocytes and tick cells. Int. J. Parasitol. 2001, 31, 145–153. [Google Scholar] [CrossRef]
- Felsheim, R.F.; Chavez, A.S.; Palmer, G.H.; Crosby, L.; Barbet, A.F.; Kurtti, T.J.; Munderloh, U.G. Transformation of Anaplasma marginale. Vet. Parasitol. 2010, 167, 167–174. [Google Scholar] [CrossRef]
- Angus, B.M. The history of the cattle tick Boophilus microplus in Australia and achievements in its control. Int. J. Parasitol. 1996, 26, 1341–1355. [Google Scholar] [CrossRef]
- De la Fuente, J.; Naranjo, V.; Ruiz-Fons, F.; Hofle, U.; Fernandez De Mera, I.G.; Villanua, D.; Almazan, C.; Torina, A.; Caracappa, S.; Kocan, K.M.; et al. potential vertebrate reservoir hosts and invertebrate vectors of Anaplasma marginale and A. phagocytophilum in central Spain. Vector Borne Zoonotic Dis. 2005, 5, 390–401. [Google Scholar] [CrossRef]
- Kuttler, K.L. Anaplasma infections in wild and domestic ruminants: A review. J. Wildl. Dis. 1984, 20, 12–20. [Google Scholar] [CrossRef]
- Letts, G.A. Feral animals in the northern territory. Aust. Vet. J. 1964, 40, 84–88. [Google Scholar] [CrossRef]
- Anisimov, A.P.; Lindler, L.E.; Pier, G.B. Intraspecific diversity of Yersinia pestis. Clin. Microbiol. Rev. 2004, 17, 434–464. [Google Scholar] [CrossRef]
- Shapiro, B.J.; Friedman, J.; Cordero, O.X.; Preheim, S.P.; Timberlake, S.C.; Szabo, G.; Polz, M.F.; Alm, E.J. Population genomics of early events in the ecological differentiation of bacteria. Science 2012, 336, 48–51. [Google Scholar] [CrossRef]
- Papke, R.T.; Gogarten, J.P. Ecology. How bacterial lineages emerge. Science 2012, 336, 45–46. [Google Scholar] [CrossRef]
- Neafsey, D.E.; Lawniczak, M.K.; Park, D.J.; Redmond, S.N.; Coulibaly, M.B.; Traore, S.F.; Sagnon, N.; Costantini, C.; Johnson, C.; Wiegand, R.C.; et al. SNP genotyping defines complex gene-flow boundaries among African malaria vector mosquitoes. Science 2010, 330, 514–517. [Google Scholar] [CrossRef]
- Turner, T.L.; Hahn, M.W.; Nuzhdin, S.V. Genomic islands of speciation in Anopheles gambiae. PLoS Biol. 2005, 3, e285. [Google Scholar] [CrossRef] [Green Version]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Muller, W.E.; Wetter, T.; Suhai, S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef]
- Bonfield, J.K.; Whitwham, A. Gap5--editing the billion fragment sequence assembly. Bioinformatics 2010, 26, 1699–1703. [Google Scholar] [CrossRef]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Angiuoli, S.V.; Gussman, A.; Klimke, W.; Cochrane, G.; Field, D.; Garrity, G.; Kodira, C.D.; Kyrpides, N.; Madupu, R.; Markowitz, V.; et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. Omics 2008, 12, 137–141. [Google Scholar] [CrossRef]
- Salzberg, S.L.; Delcher, A.L.; Kasif, S.; White, O. Microbial gene identification using interpolated Markov models. Nucleic Acids Res. 1998, 26, 544–548. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Picard tools. Available online: http://picard.sourceforge.net (accessed on June 2013).
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Altshuler, D.; Pollara, V.J.; Cowles, C.R.; Van Etten, W.J.; Baldwin, J.; Linton, L.; Lander, E.S. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 2000, 407, 513–516. [Google Scholar] [CrossRef]
- Brockman, W.; Alvarez, P.; Young, S.; Garber, M.; Giannoukos, G.; Lee, W.L.; Russ, C.; Lander, E.S.; Nusbaum, C.; Jaffe, D.B. Quality scores and SNP detection in sequencing-by-synthesis systems. Genome Res. 2008, 18, 763–770. [Google Scholar]
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef]
- Angiuoli, S.V.; Salzberg, S.L. Mugsy: Fast multiple alignment of closely related whole genomes. Bioinformatics 2011, 27, 334–342. [Google Scholar]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Lanave, C.; Preparata, G.; Saccone, C.; Serio, G. A new method for calculating evolutionary substitution rates. J. Mol. Evol. 1984, 20, 86–93. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pierlé, S.A.; Imaz-Rosshandler, I.; Kerudin, A.A.; Sambono, J.; Lew-Tabor, A.; Rolls, P.; Rangel-Escareño, C.; Brayton, K.A. Genetic Diversity of Tick-Borne Rickettsial Pathogens; Insights Gained from Distant Strains. Pathogens 2014, 3, 57-72. https://doi.org/10.3390/pathogens3010057
Pierlé SA, Imaz-Rosshandler I, Kerudin AA, Sambono J, Lew-Tabor A, Rolls P, Rangel-Escareño C, Brayton KA. Genetic Diversity of Tick-Borne Rickettsial Pathogens; Insights Gained from Distant Strains. Pathogens. 2014; 3(1):57-72. https://doi.org/10.3390/pathogens3010057
Chicago/Turabian StylePierlé, Sebastián Aguilar, Ivan Imaz-Rosshandler, Ammielle Akim Kerudin, Jacqueline Sambono, Ala Lew-Tabor, Peter Rolls, Claudia Rangel-Escareño, and Kelly A. Brayton. 2014. "Genetic Diversity of Tick-Borne Rickettsial Pathogens; Insights Gained from Distant Strains" Pathogens 3, no. 1: 57-72. https://doi.org/10.3390/pathogens3010057