Live Genomics for Pathogen Monitoring in Public Health



Abstract
:1. Introduction
1.1. Following Microbes in Public Health Microbiology
1.2. Comparative Genomics in Public Health
1.3. Approaching Species Definition in the Genomics Era
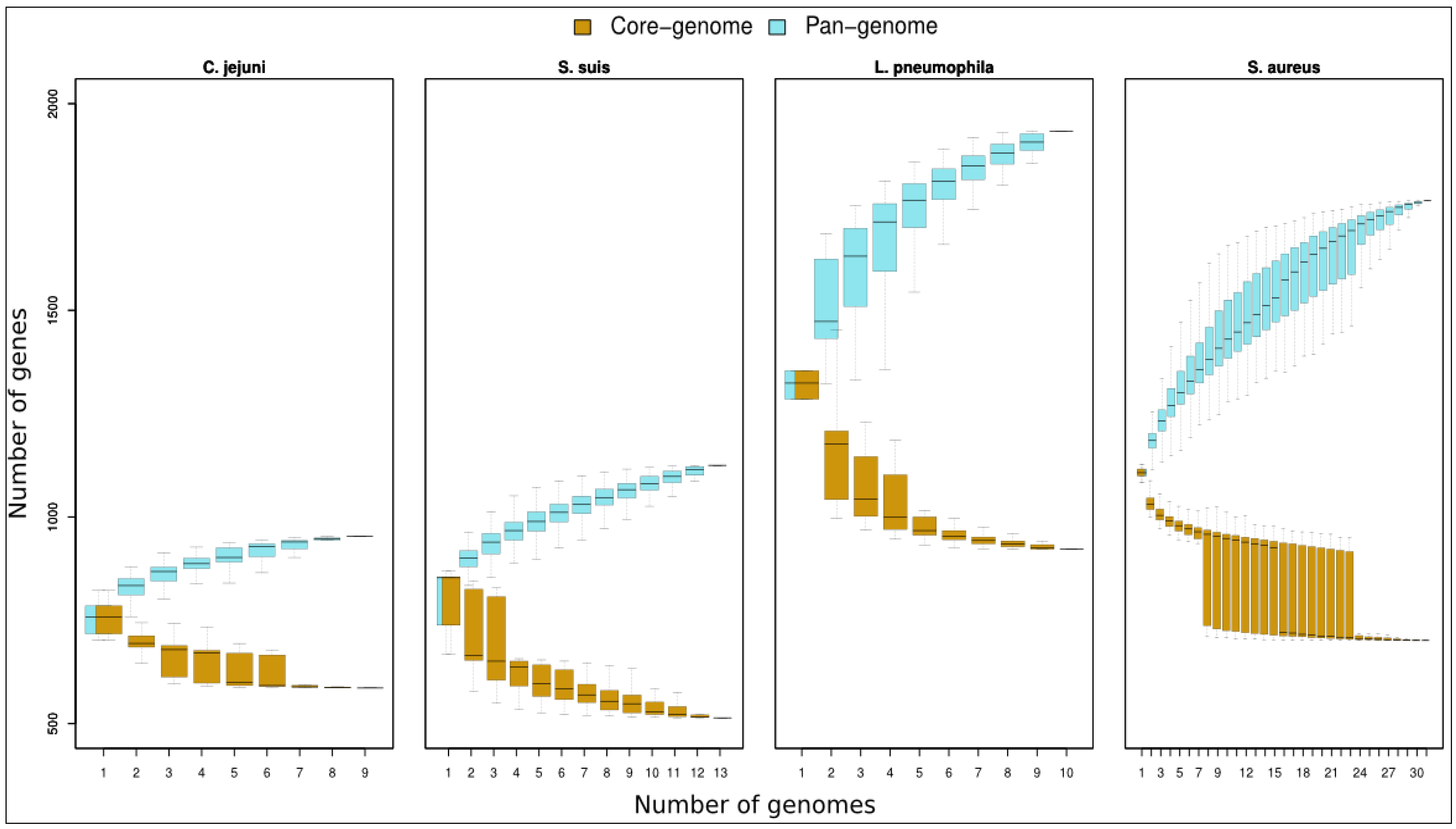
2. Pan-Genome

2.1. Core Genome
2.2. Dispensable-Genome
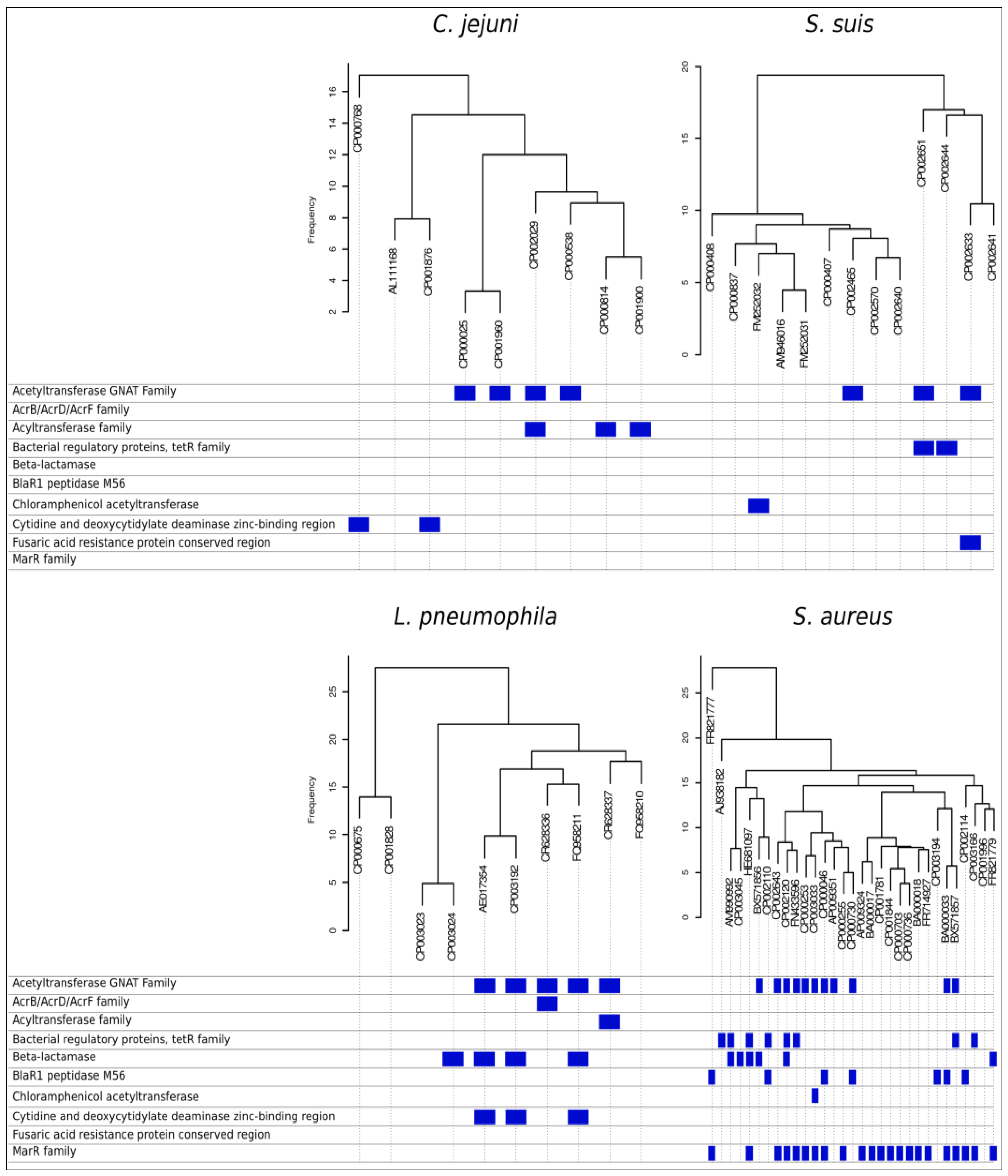
3. SNPs/InDels Profiles
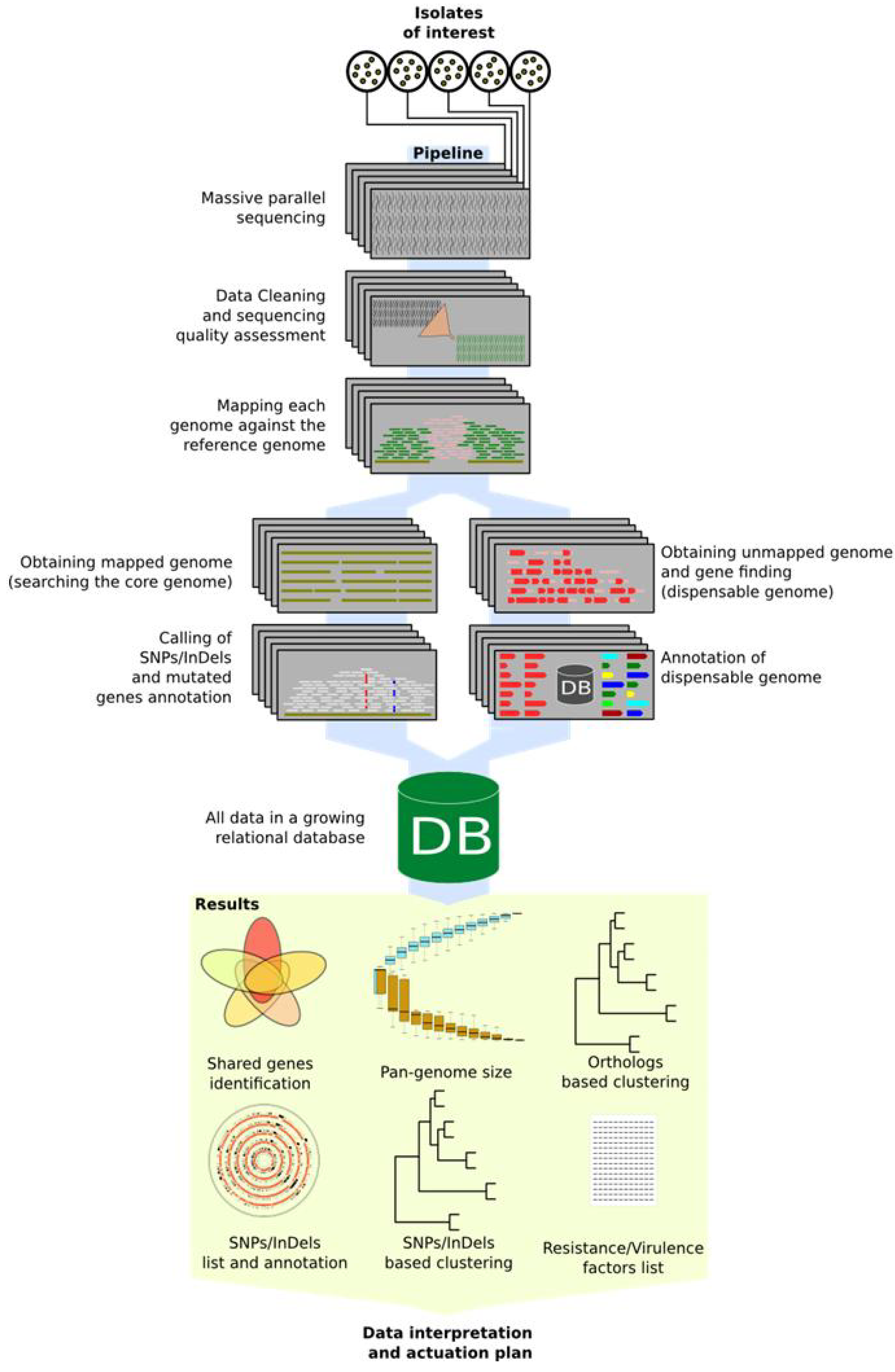
4. Automatic Pipelines


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C. jejuni | S. suis | L. pneumophila | S. aureus | |
|---|---|---|---|---|
| Number of genomes | 9 | 13 | 10 | 31 |
| Average genome length | 1,678,553.8 bp | 2,090,478.5 bp | 3,302,389.7 bp | 2,894,586.7 bp |
| Pan-genome size | 953 | 1,125 | 1,933 | 1,765 |
4.1. Pipeline—A Little Bioinformatics
4.2. Pan-genome Data Mining Pipeline

5. Conclusions

Supplementary Files
Acknowledgments
Conflicts of Interest
References and Notes
- Freifeld, A.G.; Bow, E.J.; Sepkowitz, K.A.; Boeckh, M.J.; Ito, J.I.; Mullen, C.A.; Raad, I.I.; Rolston, K.V; Young, J.-A.H.; Wingard, J.R. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2011, 52, e56–e93. [Google Scholar] [CrossRef]
- Björkman, J.; Andersson, D.I. The cost of antibiotic resistance from a bacterial perspective. Drug Resist. Updates 2000, 3, 237–245. [Google Scholar] [CrossRef]
- McCormick, J.B. Epidemiology of emerging/re-emerging antimicrobial-resistant bacterial pathogens. Curr. Opin. Microbiol. 1998, 1, 125–129. [Google Scholar] [CrossRef]
- Fraimow, H.S.; Tsigrelis, C. Antimicrobial resistance in the intensive care unit: Mechanisms, epidemiology, and management of specific resistant pathogens. Crit. Care Clin. 2011, 27, 163–205. [Google Scholar] [CrossRef]
- The European Committee on Antimicrobial Susceptibility Testing. Available online: http://www.eucast.org/ (accessed on 26 September 2013).
- Blot, S.I.; Vandewoude, K.H.; Hoste, E.A.; Colardyn, F.A. Outcome and attributable mortality in critically ill patients with bacteremia involving methicillin-susceptible and methicillin-resistant Staphylococcus aureus. Arch. Intern. Med. 2002, 162, 2229–2235. [Google Scholar] [CrossRef]
- Cosgrove, S.E.; Qi, Y.; Kaye, K.S.; Harbarth, S.; Karchmer, A.W.; Carmeli, Y. The impact of methicillin resistance in Staphylococcus aureus bacteremia on patient outcomes: mortality, length of stay, and hospital charges. Infect. Contr. Hosp. Epidemiol. 2005, 26, 166–174. [Google Scholar]
- Barenfanger, J.; Drake, C.; Kacich, G. Clinical and financial benefits of rapid bacterial identification and antimicrobial susceptibility testing. J. Clin. Microbiol. 1999, 37, 1415–1418. [Google Scholar]
- Osih, R.B.; McGregor, J.C.; Rich, S.E.; Moore, A.C.; Furuno, J.P.; Perencevich, E.N.; Harris, A.D. Impact of empiric antibiotic therapy on outcomes in patients with Pseudomonas aeruginosa bacteremia. Antimicrob. Agents Chemother. 2007, 51, 839–844. [Google Scholar] [CrossRef]
- Thom, K.A.; Schweizer, M.L.; Osih, R.B.; McGregor, J.C.; Furuno, J.P.; Perencevich, E.N.; Harris, A.D. Impact of empiric antimicrobial therapy on outcomes in patients with Escherichia coli and Klebsiella pneumoniae bacteremia: A cohort study. BMC Infect. Dis. 2008, 8, 116. [Google Scholar] [CrossRef]
- De Paoli, P. Bio-banking in microbiology: from sample collection to epidemiology, diagnosis and research. FEMS Microbial. Rev. 2005, 29, 897–910. [Google Scholar] [CrossRef]
- Mencacci, A.; Leli, C.; Cardaccia, A.; Meucci, M.; Moretti, A.; D’Alò, F.; Farinelli, S.; Pagliochini, R.; Barcaccia, M.; Bistoni, F. Procalcitonin predicts real-time PCR results in blood samples from patients with suspected sepsis. PloS One 2012, 7, e53279. [Google Scholar]
- Lehmann, L.E.; Hunfeld, K.P.; Emrich, T.; Haberhausen, G.; Wissing, H.; Hoeft, A.; Stüber, F. A multiplex real-time PCR assay for rapid detection and differentiation of 25 bacterial and fungal pathogens from whole blood samples. Med. Microbial. Immunol. 2008, 197, 313–324. [Google Scholar] [CrossRef]
- Endimiani, A.; Hujer, K.M.; Hujer, A.M.; Kurz, S.; Jacobs, M.R.; Perlin, D.S.; Bonomo, R.A. Are we ready for novel detection methods to treat respiratory pathogens in hospital-acquired pneumonia? Clin. Infect. Dis. 2011, 52 Suppl 4, S373–S383. [Google Scholar]
- Siširak, M.; Hukić, M. An outbreak of multidrug-resistant Serratia marcescens: The importance of continuous monitoring of nosocomial infections. Acta medica academica 2013, 42, 25–31. [Google Scholar] [CrossRef]
- Fernández, J.A.; López, P.; Orozco, D.; Merino, J. Clinical study of an outbreak of Legionnaire’s disease in Alcoy, Southeastern Spain. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 21, 729–735. [Google Scholar] [CrossRef]
- Warny, M.; Pepin, J.; Fang, A.; Killgore, G.; Thompson, A.; Brazier, J.; Frost, E.; McDonald, L.C. Toxin production by an emerging strain of Clostridium difficile associated with outbreaks of severe disease in North America and Europe. Lancet 2005, 366, 1079–1084. [Google Scholar] [CrossRef]
- Reuter, S.; Harrison, T.G.; Köser, C.U.; Ellington, M.J.; Smith, G.P.; Parkhill, J.; Peacock, S.J.; Bentley, S.D.; Török, M.E. A pilot study of rapid whole-genome sequencing for the investigation of a Legionella outbreak. BMJ Open 2013, 3, e002175. [Google Scholar]
- Gardy, J.L.; Johnston, J.C.; Ho Sui, S.J.; Cook, V.J.; Shah, L.; Brodkin, E.; Rempel, S.; Moore, R.; Zhao, Y.; Holt, R.; et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. New Engl. J. Med. 2011, 364, 730–739. [Google Scholar] [CrossRef]
- Sabat, A.J.; Budimir, A.; Nashev, D.; Sá-Leão, R.; van Dijl, J.M; Laurent, F.; Grundmann, H.; Friedrich, A.W. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Eurosurveillance 2013, 18, 20380. [Google Scholar]
- Rohde, H.; Qin, J.; Cui, Y.; Li, D.; Loman, N.J.; Hentschke, M.; Chen, W.; Pu, F.; Peng, Y.; Li, J.; et al. Open-source genomic analysis of Shiga-toxin-producing E. coli O104:H4. New Engl. J. Med. 2011, 365, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Global Microbial Identifier. Available online: www.globalmicrobialidentifier.org (accessed on 26 September 2013).
- Johnson, J.L. Use of nucleic-acid homologies in the taxonomy of anaerobic bacteria. Int. J. Syst. Bacteriol 1973, 23, 308–315. [Google Scholar] [CrossRef]
- Staley, J.T. Biodiversity: are microbial species threatened? Curr. Opin. Biotechnol. 1997, 8, 340–345. [Google Scholar] [CrossRef]
- Spratt, B.G. Multilocus sequence typing: molecular typing of bacterial pathogens in an era of rapid DNA sequencing and the internet. Curr. Opin. Microbiol. 1999, 2, 312–316. [Google Scholar] [CrossRef]
- Maiden, M.C.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Kisand, V.; Lettieri, T. Genome sequencing of bacteria: sequencing, de novo assembly and rapid analysis using open source tools. BMC Genomics 2013, 14, 211. [Google Scholar] [CrossRef]
- Perkins, T.T.; Tay, C.Y.; Thirriot, F.; Marshall, B. Choosing a benchtop sequencing machine to characterise Helicobacter pylori genomes. PloS One 2013, 8, e67539. [Google Scholar]
- Palm, D.; Johansson, K.; Ozin, A.; Friedrich, A.W.; Grundmann, H.; Larsson, J.T.; Struelens, M.J. Molecular epidemiology of human pathogens: how to translate breakthroughs into public health practice, Stockholm, November 2011. Eurosurveillance 2012, 17, 20054. [Google Scholar]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, S. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Blount, Z.D.; Barrick, J.E.; Davidson, C.J.; Lenski, R.E. Genomic analysis of a key innovation in an experimental Escherichia coli population. Nature 2012, 489, 513–518. [Google Scholar] [CrossRef]
- Cuadros-Orellana, S.; Martin-Cuadrado, A.-B.; Legault, B.; D’Auria, G.; Zhaxybayeva, O.; Papke, R.T.; Rodriguez-Valera, F. Genomic plasticity in prokaryotes: The case of the square haloarchaeon. ISME J 2007, 1, 235–245. [Google Scholar] [CrossRef]
- Morowitz, M.J.; Denef, V.J.; Costello, E.K.; Thomas, B.C.; Poroyko, V.; Relman, D.A.; Banfield, J.F. Strain-resolved community genomic analysis of gut microbial colonization in a premature infant. Proc. Natl. Acad. Sci. USA 2010, 108, 1128–1133. [Google Scholar]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Novais, A.; Comas, I.; Baquero, F.; Cantón, R.; Coque, T.M.; Moya, A.; González-Candelas, F.; Galán, J.-C. Evolutionary trajectories of beta-lactamase CTX-M-1 cluster enzymes: predicting antibiotic resistance. PLoS Pathogens 2010, 6, e1000735. [Google Scholar] [CrossRef]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR—a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008, 6, 181–186. [Google Scholar] [CrossRef]
- Geric, B.; Johnson, S.; Gerding, D.N.; Grabnar, M.; Rupnik, M. Frequency of binary toxin genes among Clostridium difficile strains that do not produce large clostridial toxins. J. Clin. Microbiol. 2003, 41, 5227–5232. [Google Scholar] [CrossRef]
- D’Auria, G.; Jiménez-Hernández, N.; Peris-Bondia, F.; Moya, A.; Latorre, A. Legionella pneumophila pangenome reveals strain-specific virulence factors. BMC Genom. 2010, 11, 181. [Google Scholar] [CrossRef]
- Mira, A.; Martín-Cuadrado, A.B.; D’Auria, G.; Rodríguez-Valera, F. The bacterial pan-genome:a new paradigm in microbiology. Int. Microbiol. 2010, 13, 45–57. [Google Scholar]
- Liang, W.; Zhao, Y.; Chen, C.; Cui, X.; Yu, J.; Xiao, J.; Kan, B. Pan-genomic analysis provides insights into the genomic variation and evolution of Salmonella Paratyphi A. PloS One 2012, 7, e45346. [Google Scholar]
- Den Bakker, H.C.; Switt, A.M.; Govoni, G.; Cummings, C.A.; Ranieri, M.L.; Degoricija, L.; Hoelzer, K.; Rodriguez-Rivera, L.D.; Brown, S.; Bolchacova, E.; et al. Genome sequencing reveals diversification of virulence factor content and possible host adaptation in distinct subpopulations of Salmonella enterica. BMC Genom. 2011, 12, 425. [Google Scholar] [CrossRef]
- Verma, M.; Lal, D.; Kaur, J.; Saxena, A.; Kaur, J.; Anand, S.; Lal, R. Phylogenetic analyses of phylum Actinobacteria based on whole genome sequences. Res. Microbiol. 2013, 164, 718–728. [Google Scholar] [CrossRef]
- Rannala, B.; Yang, Z. Phylogenetic inference using whole genomes. Annu Rev Genom Hum Genet 2008, 9, 217–231. [Google Scholar] [CrossRef]
- Parkhill, J.; Wren, B.W. Bacterial epidemiology and biology—lessons from genome sequencing. Genome Boil. 2011, 12, 230. [Google Scholar] [CrossRef]
- Morelli, G.; Song, Y.; Mazzoni, C.J.; Eppinger, M.; Roumagnac, P.; Wagner, D.M.; Feldkamp, M.; Kusecek, B.; Vogler, A.J.; Li, Y.; et al. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Genet. 2010, 42, 1140–1143. [Google Scholar] [CrossRef]
- Hudson, C.R.; Quist, C.; Lee, M.D.; Keyes, K.; Dodson, S.V.; Morales, C.; Sanchez, S.; White, D.G.; Maurer, J.J. Genetic relatedness of Salmonella isolates from nondomestic birds in Southeastern United States. J. Clin. Microbiol. 2000, 38, 1860–1865. [Google Scholar]
- Lamoth, F.; Jaton, K.; Prod’hom, G.; Senn, L.; Bille, J.; Calandra, T.; Marchetti, O. Multiplex blood PCR in combination with blood cultures for improvement of microbiological documentation of infection in febrile neutropenia. J. Clin. Microbiol. 2010, 48, 3510–3516. [Google Scholar] [CrossRef]
- Gordon, D.M. Geographical structure and host specificity in bacteria and the implications for tracing the source of coliform contamination. Microbiology 2001, 147, 1079–1085. [Google Scholar]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef]
- Altmann, A.; Weber, P.; Bader, D.; Preuss, M.; Binder, E.B.; Müller-Myhsok, B. A beginners guide to SNP calling from high-throughput DNA-sequencing data. Hum. Genet. 2012, 131, 1541–1554. [Google Scholar] [CrossRef]
- Kislyuk, A.O.; Katz, L.S.; Agrawal, S.; Hagen, M.S.; Conley, A.B.; Jayaraman, P.; Nelakuditi, V.; Humphrey, J.C.; Sammons, S.A.; Govil, D.; et al. A computational genomics pipeline for prokaryotic sequencing projects. Bioinformatics 2010, 26, 1819–1826. [Google Scholar] [CrossRef]
- Sherman—A tool to simulate FastQ files for high-throughput sequencing experiments. Available online: http://www.bioinformatics.babraham.ac.uk/projects/sherman/ (accessed on 26 September 2013).
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Martin, J.; Sykes, S.; Young, S.; Kota, K.; Sanka, R.; Sheth, N.; Orvis, J.; Sodergren, E.; Wang, Z.; Weinstock, G.M.; et al. Optimizing read mapping to reference genomes to determine composition and species prevalence in microbial communities. PLoS One 2012, 7, e36427. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Chevreux, B.; Wetter, T.; Suhai, S. Genome sequence assembly using trace signals and additional sequence information. In Proceedings of German Conference on Bioinformatics; Hannover: Germany, 1999; pp. 45–56. [Google Scholar]
- Delcher, A.L.; Harmon, D.; Kasif, S.; White, O.; Salzberg, S.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y. HMM-FRAME: Accurate protein domain classification for metagenomic sequences containing frameshift errors. BMC Bioinformatics 2011, 12, 198. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; Eddy, S.R.; Birney, E.; Bateman, A.; Durbin, R. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998, 26, 320–322. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Chen, K.; Wylie, T.; Larson, D.E.; McLellan, M.D.; Mardis, E.R.; Weinstock, G.M.; Wilson, R.K.; Ding, L. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 2009, 25, 2283–2285. [Google Scholar] [CrossRef]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria, 2012; ISBN 3-900051-07-0. [Google Scholar]
- Morgan, M.; Anders, S.; Lawrence, M.; Aboyoun, P.; Pagès, H.; Gentleman, R. ShortRead: A bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 2009, 25, 2607–2608. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
D'Auria, G.; Schneider, M.V.; Moya, A. Live Genomics for Pathogen Monitoring in Public Health. Pathogens 2014, 3, 93-108. https://doi.org/10.3390/pathogens3010093
D'Auria G, Schneider MV, Moya A. Live Genomics for Pathogen Monitoring in Public Health. Pathogens. 2014; 3(1):93-108. https://doi.org/10.3390/pathogens3010093
Chicago/Turabian StyleD'Auria, Giuseppe, Maria Victoria Schneider, and Andrés Moya. 2014. "Live Genomics for Pathogen Monitoring in Public Health" Pathogens 3, no. 1: 93-108. https://doi.org/10.3390/pathogens3010093