Pseudomonas aeruginosa Genome Evolution in Patients and under the Hospital Environment

Abstract
:1. Introduction
2. Results and Discussion
2.1. Strains
2.1.1. Description of KK Related-Strains
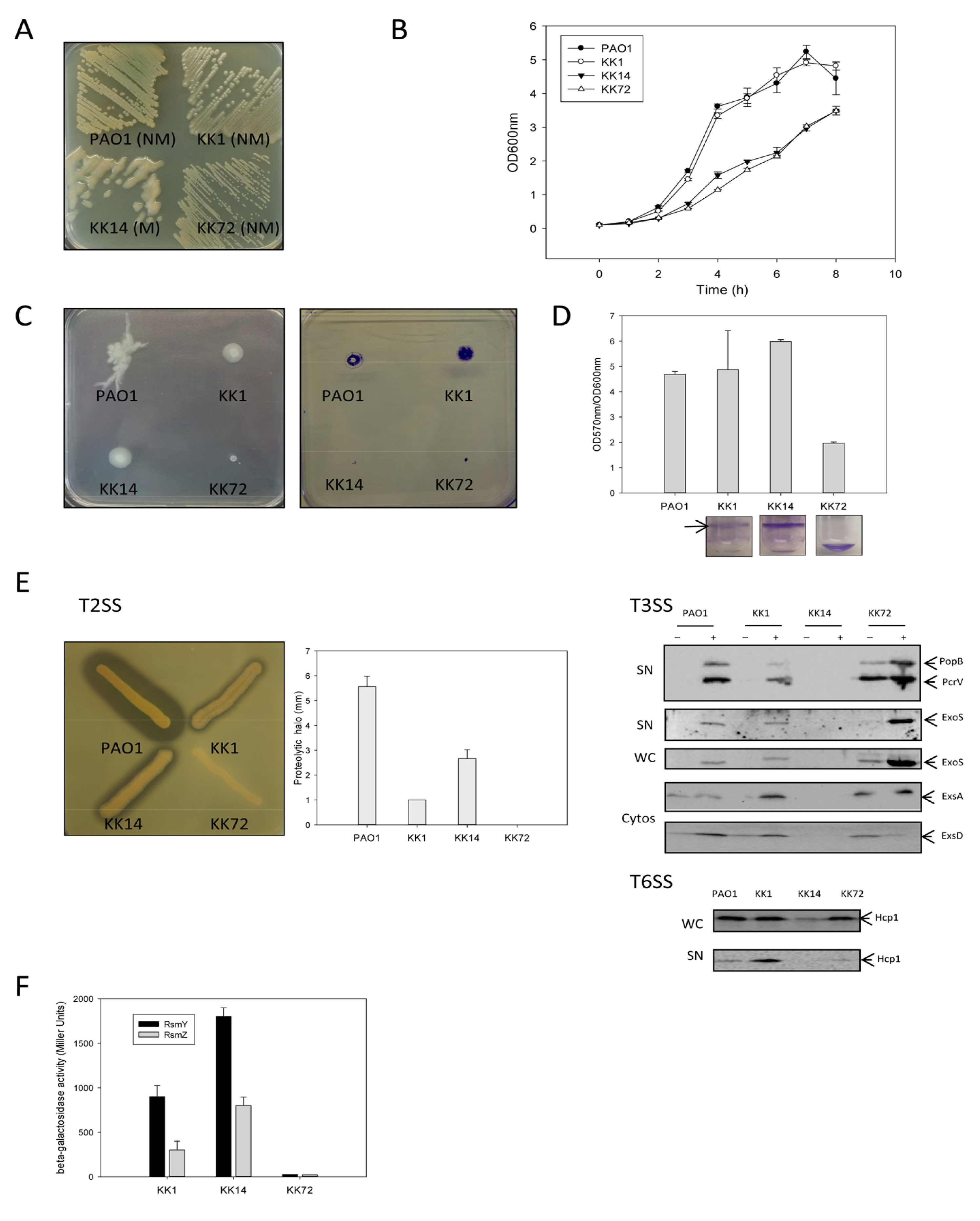
2.1.1.1. Growth Characteristics
2.1.1.2. Motility and Biofilm Capacities
2.1.1.3. Proteolytic Activity, T3SS and T6SS

2.1.1.4. sRNA Rsm
2.1.2. Description of ST395 Related-Strains
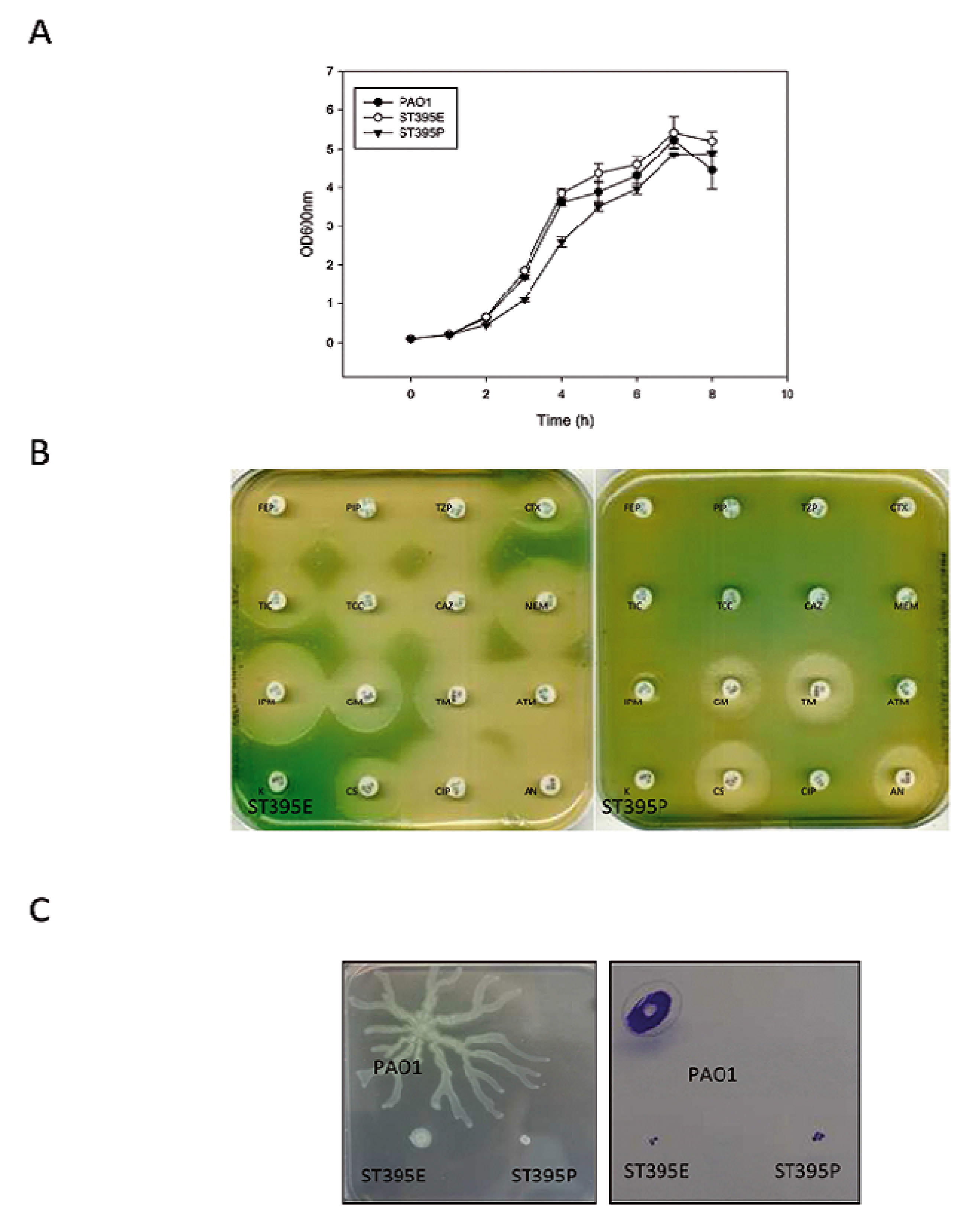
2.1.2.1. Growth Characteristics
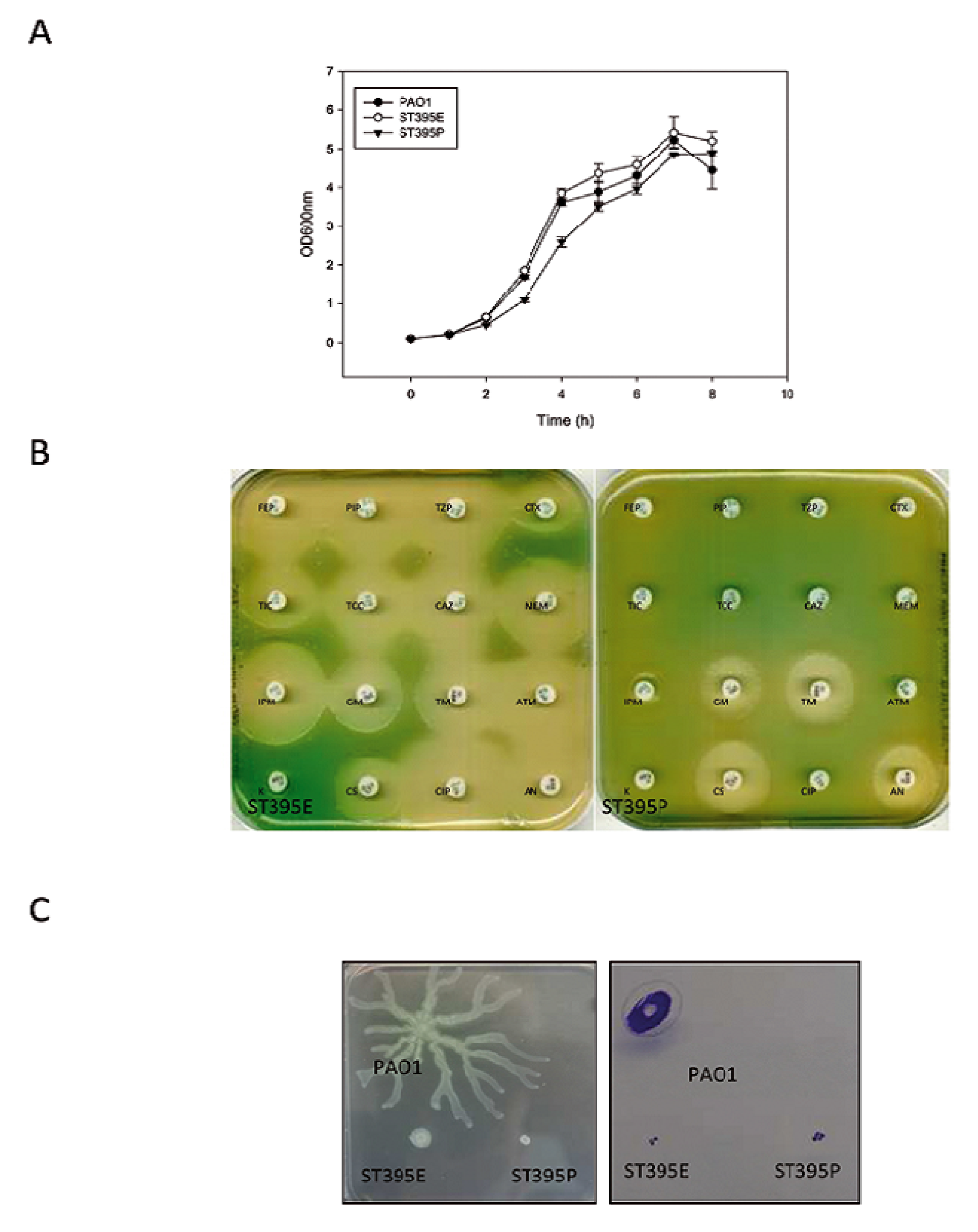
2.1.2.2. Antibiotic Resistance
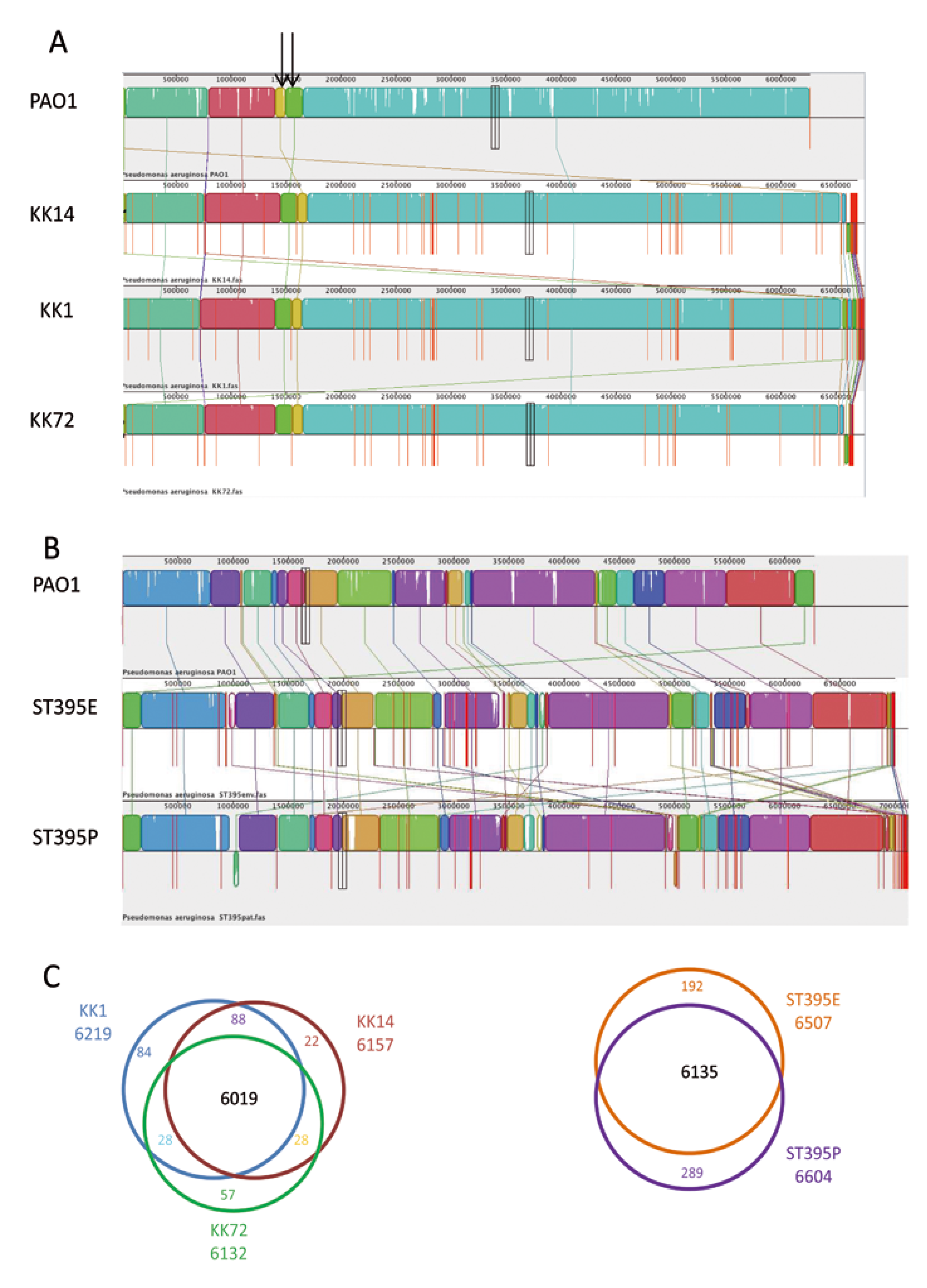
2.2. Genome Examination
2.2.1. KK Strain Microevolution and Phenotypic Consequences


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | PAO1 | KK1 | KK14 | KK72 |
|---|---|---|---|---|
| Numbers of reads | NA | 10,357,778 | 10,357,778 | 10,357,778 |
| Average read length (bp) | NA | 90 | 90 | 90 |
| Sequence coverage | NA | 138 | 140 | 140 |
| DNA scaffolds | NA | 95 | 78 | 125 |
| DNA total number bases | 6,264,404 | 6,759,575 | 6,690,898 | 6,657,327 |
| ORF | 5571 | 6219 | 6157 | 6132 |
| Genes with EC number (enzymes) | NA | 978 | 978 | 974 |
| Strains | PAO1 | ST395E | ST395P |
|---|---|---|---|
| Numbers of reads | NA | 9,422,224 | 9,880,000 |
| Average read length | NA | 90 | 90 |
| Sequence coverage | NA | 121 | 125 |
| DNA scaffolds | NA | 55 | 86 |
| DNA total number bases | 6,264,404 | 6,993,173 | 7,133,660 |
| ORF | 5571 | 6507 | 6604 |
| Genes with EC number (enzymes) | NA | 982 | 982 |
| Location in PAO1 genome | Phagic origin | KK1 | KK14 | KK72 | |
|---|---|---|---|---|---|
| Region KK_1 | PA3357-PA3391 (24 kb) | no | + (33.119 kb) | + (33.119 kb) | - |
| Region KK_2 | PA0632-PA0649 | yes | - | - | - |
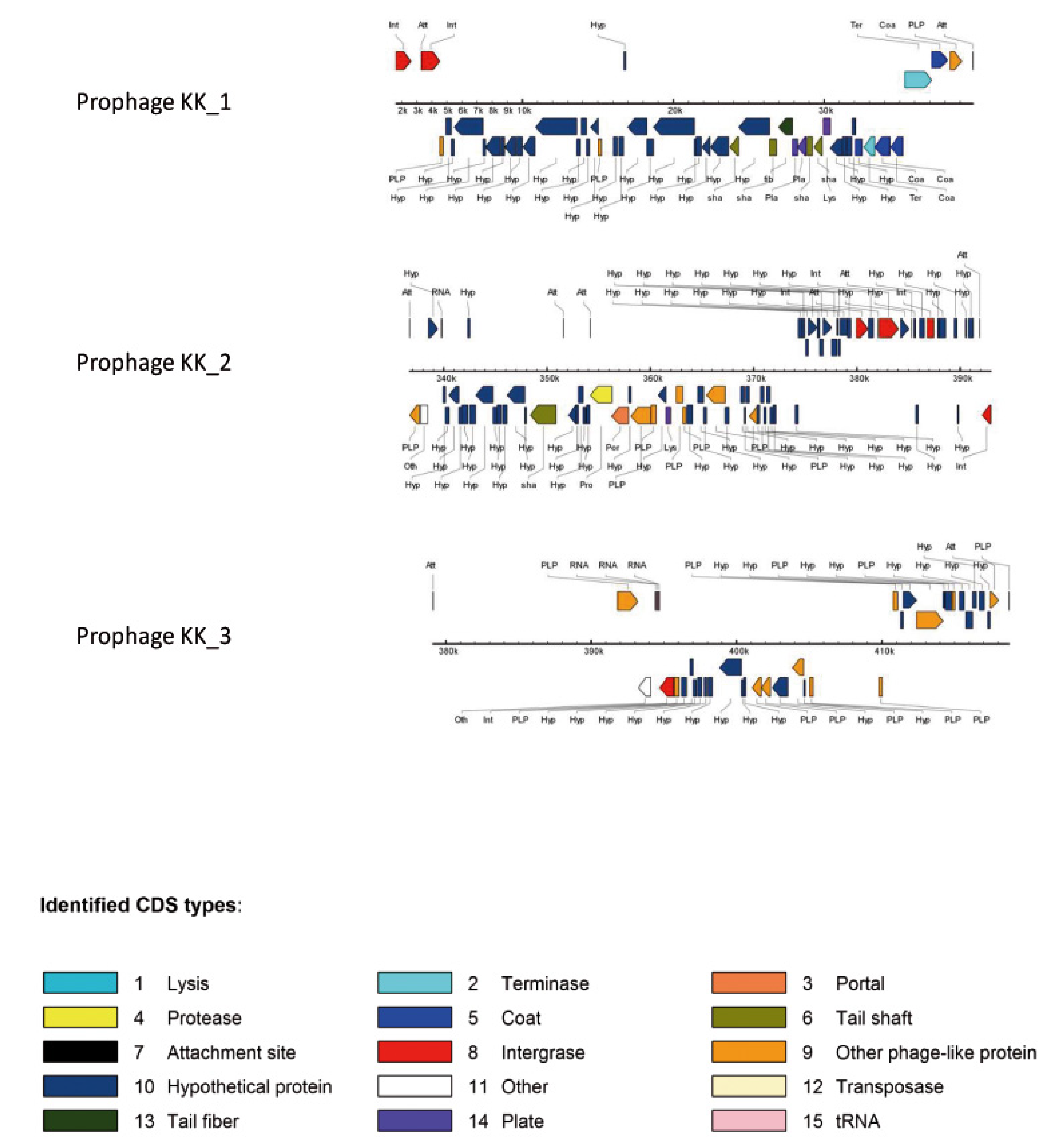
| Region KK_3 Prophage KK_1 | PA0729.1 | yes ΦCTX | + (38.859 kb) | + (38.859 kb) | - |
| Region KK_4 Prophage KK_2 | PA0820-PA0826.1 | yes F10 | + (>54.093 kb) | + (>54.074 kb) | + (>54.093 kb) |
| Region KK_5 | PA1087-PA1094 | no | + (12.703 kb) Novel locus | + (12.703 kb) Novel locus | + (12.703 kb) Novel locus |
| Region KK_6 Prophage KK_3 | PA1796.1-PA1796.4 | yes Φ297 | + (51.617 kb) | - | + (51.625 kb) |
| Region KK_7 Prophage-like_KK_4 | PA4673.1 | ?? | + (>24.424 kb) | + (22.616 kb) | + (24.664 kb) |
| Region KK_8 | PA2593-PA2594 | no | + (31.824 kb) | + (34.971 kb) | + (31.814 kb) |
| Region KK_9 | PA2819.1-PA2819.3 | no | + (123.358 kb) | + (122.554 kb) | + (122.566 kb) |
| Region KK_10 | PA2583.1 | no | + (102.482 kb) | + (>83.975 kb) | + (102.482 kb) |
| Region KK_11 | PA3768-3769 | no | + (6.955 kb) | + (6.955 kb) | + (6.955 kb) |
| Region KK_12 | Upstream PA2077 | no | + (57.2 kb) | + (57.2 kb) | + (57.2 kb) |
2.2.1.1. Major Genomic Changes between KK Strains

2.2.1.2. Minor Genomic Changes between KK Strains
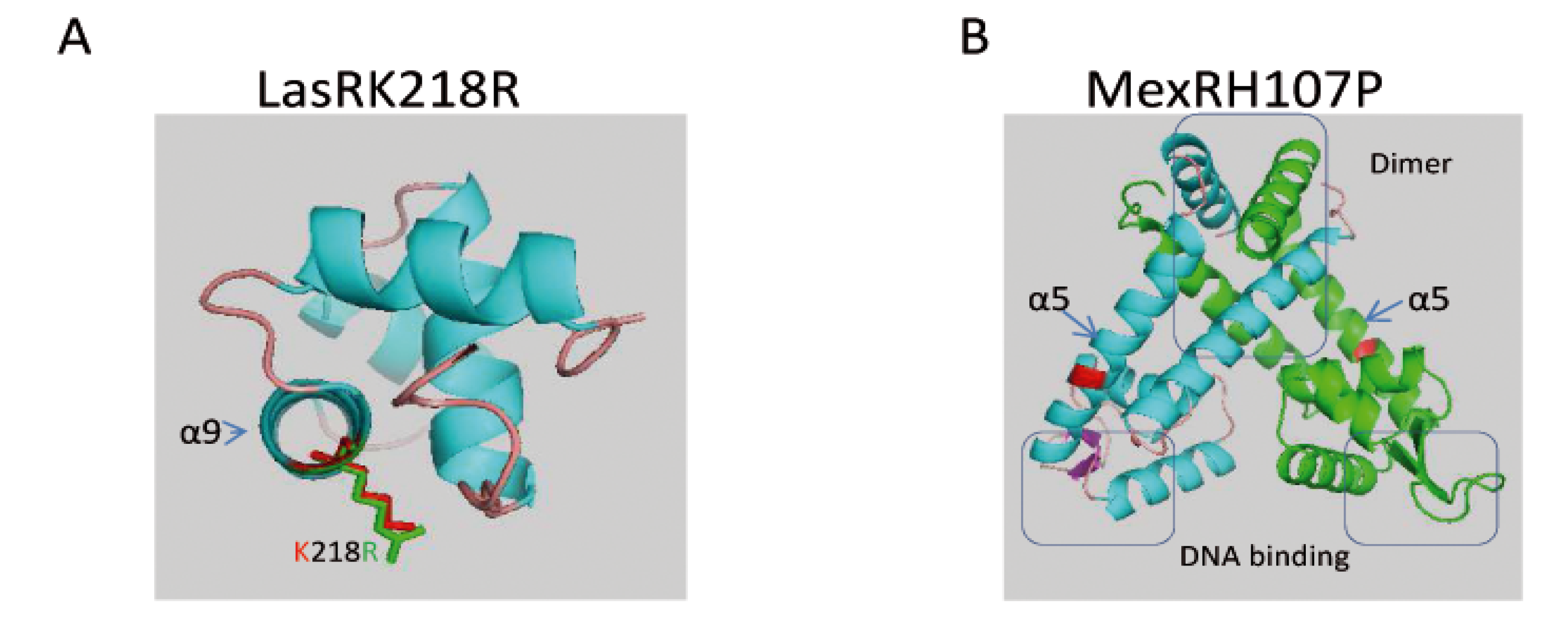
2.2.1.3. Genetic Changes Related to Evolution of Phenotypic Traits of KK Strains over Time in CF Patient


2.2.2. ST395 Strain Microevolution and Phenotypic Consequences
| Location in PAO1 genome | Phagic origin | ST395E | ST395P | |
|---|---|---|---|---|
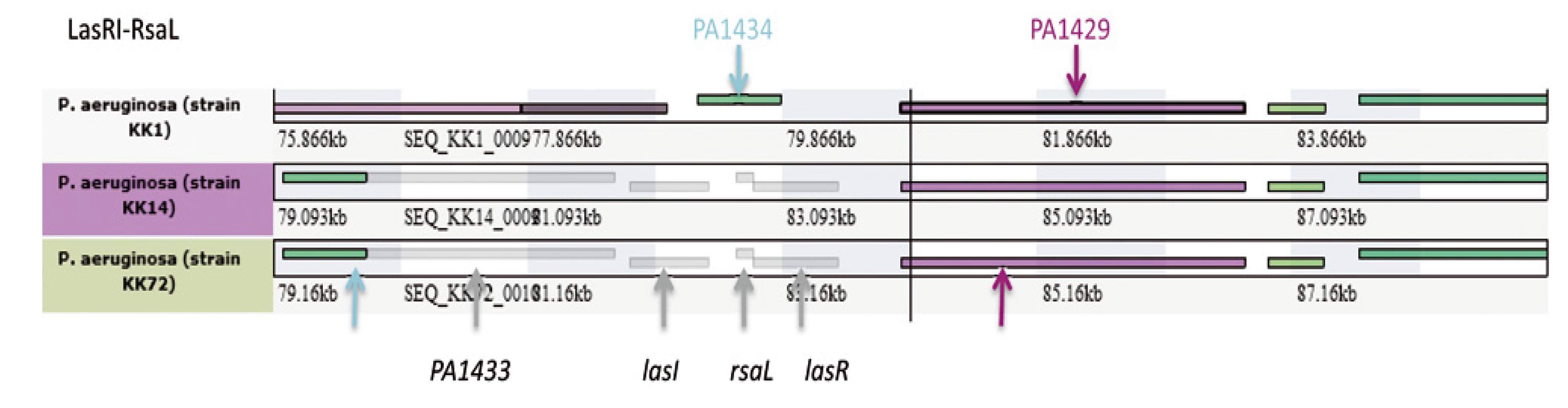
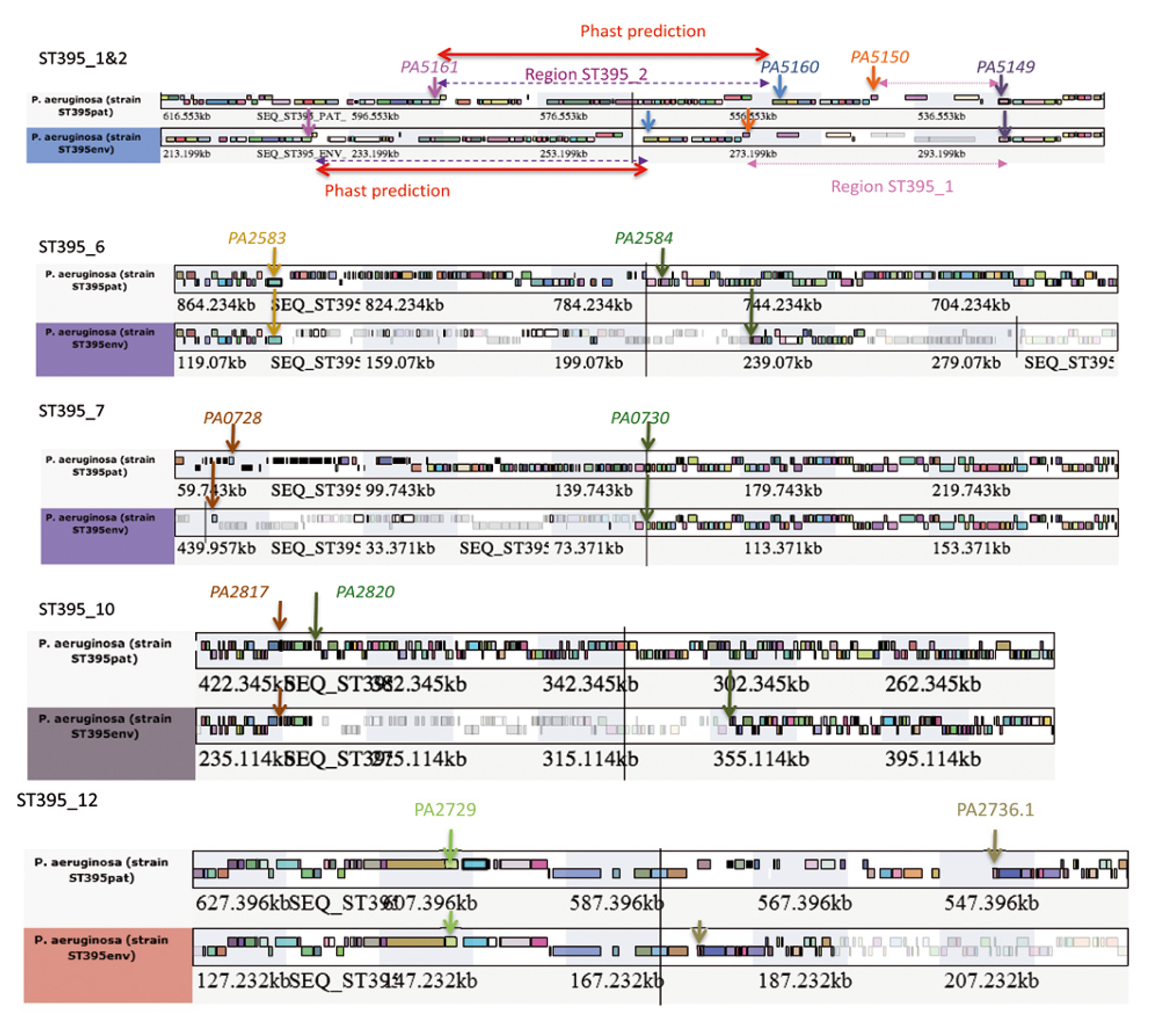
| Region ST395_1&2 | PA5149.1 | no | + (26.483 kb) | + (12.871 kb) |
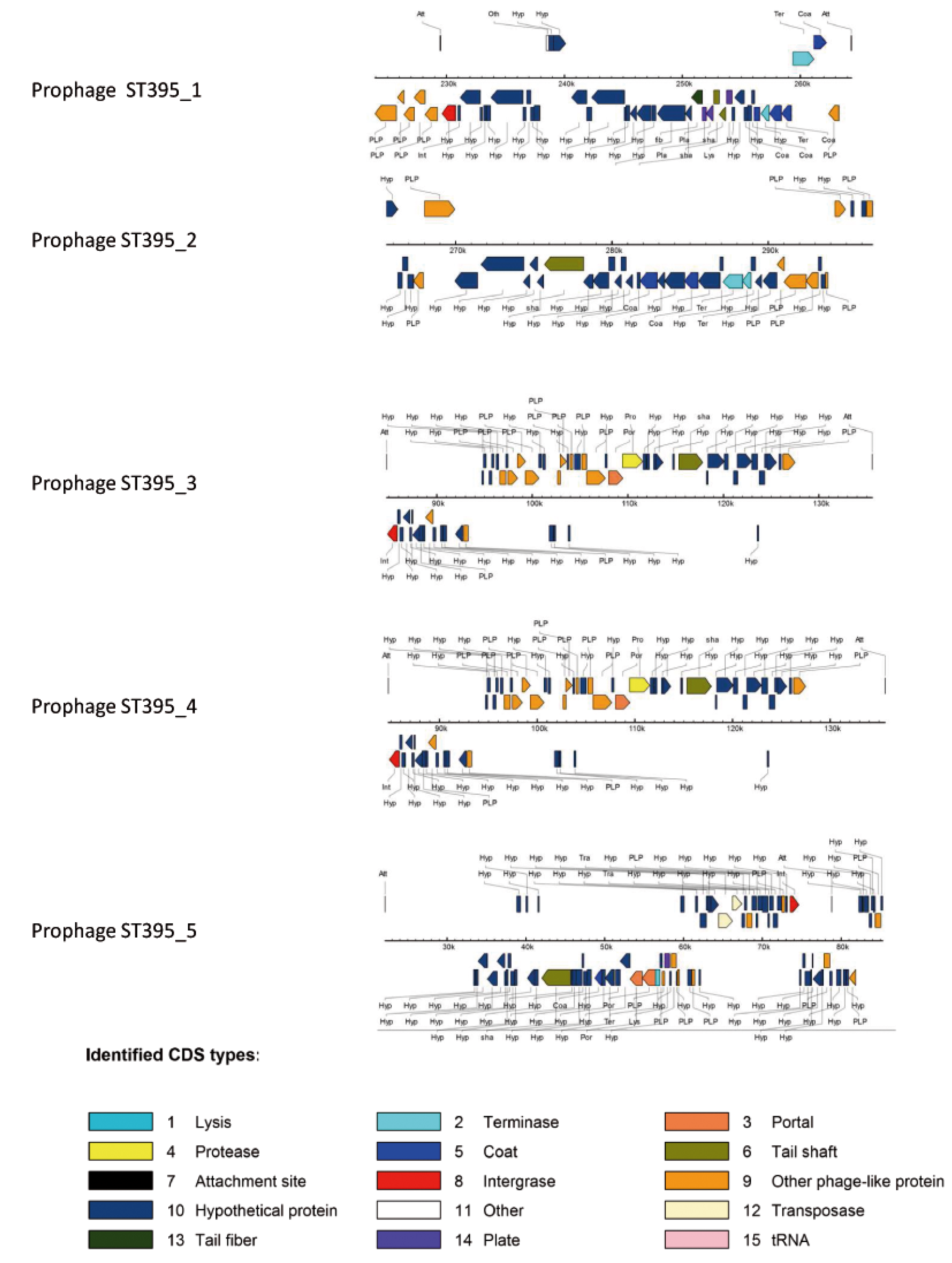
| Region ST395_1&2 Prophage ST395_1 | PA5160.1 | yes ΦCTX | + (40.4 kb) | + (22.7 kb) |
| Region ST395_3 Prophage ST395_2 | PA2603.1 | yes Φ297 | + (31 kb) | - |
| Region ST395_4 Prophage ST395_3 | PA4138-PA4139 | yes F10 | + (42,196kb) | ? |
| Region ST395_5 Prophage ST395_4 | PA2794-PA2795 | yes F116 | - | + (70.9 kb) |
| Region ST395_6 | PA2583.1 | no | + (99.342 kb) | + (79.699 kb) |
| Region ST395_7 | PA0728 -PA0730 | no | + (91 kb) | + (104.3 kb) |
| Region ST395_8 | phnA -phnB genes | no | - | + (74 kb) |
| Region ST395_9 Prophage ST395_5 | PA3824.1 | yes B3 | - | + (63.1 kb) |
| Region ST395_10 | PA2817-PA2820 | no | + (104.5 kb) | + (7.46 kb) |
| Region ST395_11 | PA0976.1 | no | + PAPI-1 like | + PAPI-1 like |
| Region ST395_12 | PA2730-PA2736.1 | no | + (25.6 kb) | + (57.25 kb) |
| Region ST395_13 | PA4231-PA4232 | no | + (19.6 kb) | + (19.6 kb) |
| Region ST395_14 | PA3835-PA3836 | no | + (PAGI-9) (7.192 kb) | + (PAGI-9) (7.192 kb) |
2.2.2.1. Major Genomic Changes between ST395 Strains

2.2.2.2. Minor Genomic Changes between ST395 Strains
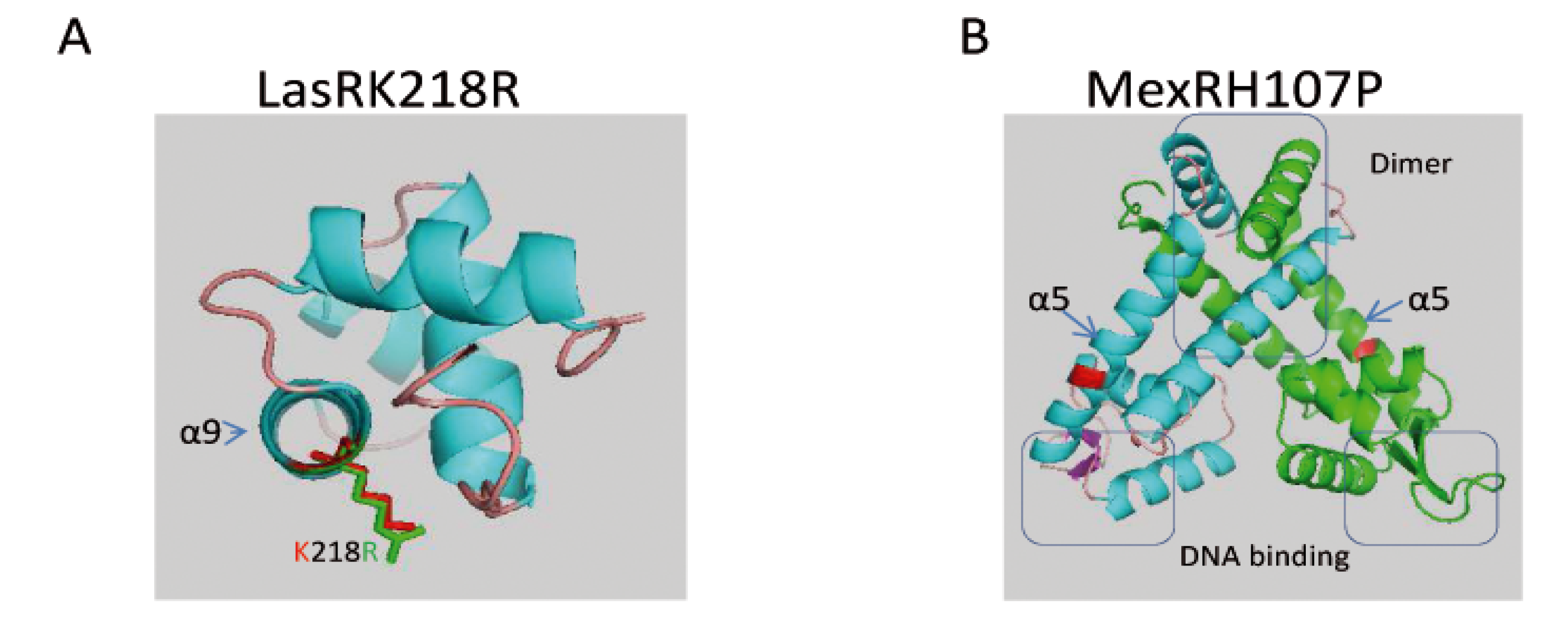
2.2.2.3. Genetic Changes Related to ST395P Multi-Drug Resistance towards Antibiotics as Compared to ST395E
| ST395E | ST395P | |
|---|---|---|
| Resistance to ß lactams | ||
| AmpC derepression | ||
| AmpD, AmpDh2, AmpDh3 | A134V, wt, R66C | T139M and A134V, wt, R66C |
| OprD | wt | c703t SNP introducing a premature stop codon |
| MexAB-OprM overproduction | ||
| MexR | wt | H107P |
| NalC | G71E | G71E |
| NalD | wt | wt |
| ArmR | wt | wt |
| MexXY-OprM overproduction | See Resistance to aminoglycosides | See Resistance to aminoglycosides |
| MexCD-OprJ overproduction | ||
| NfxB | wt | wt |
| Resistance to aminoglycosides | ||
| Aminoglycoside modifying enzymes | ||
| APH(3')-IIb | wt | wt |
| MexXY-OprM overproduction | ||
| MexZ (agrZ mutant) | wt | del nt452-459 of mexZ gene |
| RplA (agrW1 mutant) | wt | wt |
| Fmt (agrW1 mutant) | wt | wt |
| FolD (agrW1 mutant) | wt | wt |
| ArmZ (agrW1 mutant) | wt | wt |
| rplU-rpmA promoter (agrW1 mutant) | wt | Insertion of 2g at -186nt before the start codon of the rplU gene |
| ParRS (agrW2 mutant) | ParR wt, ParS wt | ParR wt, ParS V216A |
| PA2572-PA2573 | wt,R206G Q210K S217A N236D | wt,R206G Q210K S217A N236D |
| Resistance to fluoroquinolones | ||
| DNA gyrase and topoisomerase | ||
| GyrA | 925 aa (PA01 923 aa) | 925 aa T83I |
| ParC | wt | S87L |
| GyrB | wt | wt |
| ParE | V200M | V200M |
| MexAB-OprM overproduction | See Resistance to ß lactams | See Resistance to ß lactams |
| MexXY-OprM overproduction | See Resistance to aminoglycosides | See Resistance to aminoglycosides |
| MexCD-OprJ overproduction | See Resistance to ß lactams | See Resistance to ß lactams |
| MexEF-OprN overproduction | MexT wt, MexS wt | MexT R48C, MexS T19P |
3. Experimental Section
3.1. Origin of Strains
3.2. Genome Sequencing and Bioinformatics
3.3. Phenotypic Studies
3.3.1. Antibiograms
3.3.2. Motilities
3.3.3. Biofilm
3.3.4. Transcriptional Activities
3.3.5. T2SS
3.3.6. T3SS
3.3.7. T6SS
3.3.8. Modelling
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dark, M.J. Whole-genome sequencing in bacteriology: State of the art. Infec. Drug Resist. 2013, 6, 115–123. [Google Scholar] [CrossRef]
- Marvig, R.L.; Jochumsen, N.; Johansen, H.K.; Hoiby, N.; Molin, S.; Sommer, M.O.; Jelsbak, L.; Folkesson, A. Draft genome sequences of Pseudomonas aeruginosa B3 strains isolated from a cystic fibrosis patient undergoing antibiotic chemotherapy. Genome Announc. 2013, 1, e00804-13. [Google Scholar]
- Marvig, R.L.; Johansen, H.K.; Molin, S.; Jelsbak, L. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 2013, 9, e1003741. [Google Scholar] [CrossRef] [Green Version]
- Jeukens, J.; Boyle, B.; Bianconi, I.; Kukavica-Ibrulj, I.; Tummler, B.; Bragonzi, A.; Levesque, R.C. Complete genome sequence of persistent cystic fibrosis isolate Pseudomonas aeruginosa strain rp73. Genome Announc. 2013, 1, e00568-13. [Google Scholar]
- Bezuidt, O.K.; Klockgether, J.; Elsen, S.; Attree, I.; Davenport, C.F.; Tummler, B. Intraclonal genome diversity of Pseudomonas aeruginosa clones CHA and TB. BMC Genom. 2013, 14, 416. [Google Scholar] [CrossRef] [Green Version]
- Klockgether, J.; Miethke, N.; Kubesch, P.; Bohn, Y.S.; Brockhausen, I.; Cramer, N.; Eberl, L.; Greipel, J.; Herrmann, C.; Herrmann, S.; et al. Intraclonal diversity of the pseudomonas aeruginosa cystic fibrosis airway isolates TBCF10839 and TBCF121838: Distinct signatures of transcriptome, proteome, metabolome, adherence and pathogenicity despite an almost identical genome sequence. Environ. Microbiol. 2013, 15, 191–210. [Google Scholar] [CrossRef]
- Naughton, S.; Parker, D.; Seemann, T.; Thomas, T.; Turnbull, L.; Rose, B.; Bye, P.; Cordwell, S.; Whitchurch, C.; Manos, J. Pseudomonas aeruginosa AES-1 exhibits increased virulence gene expression during chronic infection of cystic fibrosis lung. PloS One 2011, 6, e24526. [Google Scholar] [CrossRef]
- Cramer, N.; Klockgether, J.; Wrasman, K.; Schmidt, M.; Davenport, C.F.; Tummler, B. Microevolution of the major common pseudomonas aeruginosa clones C and PA14 in cystic fibrosis lungs. Environ. Microbiol. 2011, 13, 1690–1704. [Google Scholar] [CrossRef]
- Chung, J.C.; Becq, J.; Fraser, L.; Schulz-Trieglaff, O.; Bond, N.J.; Foweraker, J.; Bruce, K.D.; Smith, G.P.; Welch, M. Genomic variation among contemporary Pseudomonas aeruginosa isolates from chronically infected cystic fibrosis patients. J. Bacteriol. 2012, 194, 4857–4866. [Google Scholar] [CrossRef]
- Xiong, J.; Alexander, D.C.; Ma, J.H.; Deraspe, M.; Low, D.E.; Jamieson, F.B.; Roy, P.H. Complete sequence of pOZ176, a 500-kilobase IncP-2 plasmid encoding IMP-9-mediated carbapenem resistance, from outbreak isolate Pseudomonas aeruginosa 96. Antimicrob. Agents. Ch. 2013, 57, 3775–3782. [Google Scholar] [CrossRef]
- Ciornei, C.D.; Novikov, A.; Beloin, C.; Fitting, C.; Caroff, M.; Ghigo, J.M.; Cavaillon, J.M.; Adib-Conquy, M. Biofilm-forming Pseudomonas aeruginosa bacteria undergo lipopolysaccharide structural modifications and induce enhanced inflammatory cytokine response in human monocytes. Innate Immun. 2010, 16, 288–301. [Google Scholar] [CrossRef]
- Bastonero, S.; Le Priol, Y.; Armand, M.; Bernard, C.S.; Reynaud-Gaubert, M.; Olive, D.; Parzy, D.; de Bentzmann, S.; Capo, C.; Mege, J.L. New microbicidal functions of tracheal glands: Defective anti-infectious response to Pseudomonas aeruginosa in cystic fibrosis. PloS One 2009, 4, e5357. [Google Scholar] [CrossRef]
- Mougous, J.D.; Cuff, M.E.; Raunser, S.; Shen, A.; Zhou, M.; Gifford, C.A.; Goodman, A.L.; Joachimiak, G.; Ordonez, C.L.; Lory, S.; et al. A virulence locus of Pseudomonas aeruginosa encodes a protein secretion apparatus. Science 2006, 312, 1526–1530. [Google Scholar] [CrossRef]
- Yahr, T.L.; Frank, D.W. Transcriptional organization of the trans-regulatory locus which controls exoenzyme s synthesis in Pseudomonas aeruginosa. J. Bacteriol. 1994, 176, 3832–3838. [Google Scholar]
- McCaw, M.L.; Lykken, G.L.; Singh, P.K.; Yahr, T.L. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol. Microbiol. 2002, 46, 1123–1133. [Google Scholar] [CrossRef]
- Chatterjee, S.; Chaudhury, S.; McShan, A.C.; Kaur, K.; De Guzman, R.N. Structure and biophysics of type III secretion in bacteria. Biochemistry 2013, 52, 2508–2517. [Google Scholar] [CrossRef]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nature reviews. Microbiology 2009, 7, 654–665. [Google Scholar] [CrossRef]
- Engel, J.; Balachandran, P. Role of Pseudomonas aeruginosa type III effectors in disease. Curr. Opin. Microbiol. 2009, 12, 61–66. [Google Scholar] [CrossRef]
- Cisz, M.; Lee, P.C.; Rietsch, A. ExoS controls the cell contact-mediated switch to effector secretion in Pseudomonas aeruginosa. J. Bacteriol. 2008, 190, 2726–2738. [Google Scholar] [CrossRef]
- Lapouge, K.; Schubert, M.; Allain, F.H.; Haas, D. Gac/Rsm signal transduction pathway of gamma-proteobacteria: From RNA recognition to regulation of social behaviour. Mol. Microbiol. 2008, 67, 241–253. [Google Scholar]
- Brencic, A.; McFarland, K.A.; McManus, H.R.; Castang, S.; Mogno, I.; Dove, S.L.; Lory, S. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol. Microbiol. 2009, 73, 434–445. [Google Scholar] [CrossRef]
- Cholley, P.; Thouverez, M.; Hocquet, D.; van der Mee-Marquet, N.; Talon, D.; Bertrand, X. Most Multidrug-Resistant Pseudomonas aeruginosa Isolates from Hospitals in Eastern France Belong to a Few Clonal Types. J. Clin. Microbiol. 2011, 49, 2578–2583. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infec. 2012, 18, 268–281. [Google Scholar] [CrossRef]
- Wallgene. Available online: https://www.wallgene.com/WallGene/papers (accessed on 4 April 2014).
- Mathee, K.; Narasimhan, G.; Valdes, C.; Qiu, X.; Matewish, J.M.; Koehrsen, M.; Rokas, A.; Yandava, C.N.; Engels, R.; Zeng, E.; et al. Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 3100–3105. [Google Scholar] [CrossRef]
- Nowak-Thompson, B.; Chaney, N.; Wing, J.S.; Gould, S.J.; Loper, J.E. Characterization of the pyoluteorin biosynthetic gene cluster of Pseudomonas fluorescens Pf-5. J. Bacteriol. 1999, 181, 2166–2174. [Google Scholar]
- Hoffman, L.R.; D'Argenio, D.A.; MacCoss, M.J.; Zhang, Z.; Jones, R.A.; Miller, S.I. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 2005, 436, 1171–1175. [Google Scholar] [CrossRef]
- Brown, N.L.; Shih, Y.C.; Leang, C.; Glendinning, K.J.; Hobman, J.L.; Wilson, J.R. Mercury transport and resistance. Biochem. Soc. Trans. 2002, 30, 715–718. [Google Scholar]
- Deretic, V.; Konyecsni, W.M. A procaryotic regulatory factor with a histone H1-like carboxy-terminal domain: Clonal variation of repeats within algP, a gene involved in regulation of mucoidy in Pseudomonas aeruginosa. J. Bacteriol. 1990, 172, 5544–5554. [Google Scholar]
- Wilderman, P.J.; Vasil, A.I.; Johnson, Z.; Vasil, M.L. Genetic and biochemical analyses of a eukaryotic-like phospholipase D of Pseudomonas aeruginosa suggest horizontal acquisition and a role for persistence in a chronic pulmonary infection model. Mol. Microbiol. 2001, 39, 291–303. [Google Scholar] [CrossRef]
- Passador, L.; Cook, J.M.; Gambello, M.J.; Rust, L.; Iglewski, B.H. Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 1993, 260, 1127–1130. [Google Scholar]
- de Kievit, T.; Seed, P.C.; Nezezon, J.; Passador, L.; Iglewski, B.H. RsaL, a novel repressor of virulence gene expression in pseudomonas aeruginosa. J. Bacteriol. 1999, 181, 2175–2184. [Google Scholar]
- Fukushima, J.; Ishiwata, T.; Kurata, M.; You, Z.; Okuda, K. Intracellular receptor-type transcription factor, lasr, contains a highly conserved amphipathic region which precedes the putative helix-turn-helix DNA binding motif. Nucleic Acids Res. 1994, 22, 3706–3707. [Google Scholar] [CrossRef]
- Vannini, A.; Volpari, C.; Gargioli, C.; Muraglia, E.; Cortese, R.; De Francesco, R.; Neddermann, P.; Marco, S.D. The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. EMBO J. 2002, 21, 4393–4401. [Google Scholar] [CrossRef]
- Martin, D.W.; Schurr, M.J.; Mudd, M.H.; Govan, J.R.; Holloway, B.W.; Deretic, V. Mechanism of conversion to mucoidy in Pseudomonas aeruginosa infecting cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 1993, 90, 8377–8381. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Wang, Y.; Moradali, M.F.; Hay, I.D.; Rehm, B.H. Insights into the assembly of the alginate biosynthesis machinery in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2013, 79, 3264–3272. [Google Scholar] [CrossRef]
- Sall, K.M.; Casabona, G.; Bordi, C.; Huber, P.; de Bentzmann, S.; Attrée, I.; Elsen, S. A gacS deletion in Pseudomonas aeruginosa cystic fibrosis isolate CHA shapes its virulence. PloS One 2014, in press. [Google Scholar]
- Mikkelsen, H.; McMullan, R.; Filloux, A. The Pseudomonas aeruginosa reference strain PA14 displays increased virulence due to a mutation in LadS. PloS One 2011, 6, e29113. [Google Scholar] [CrossRef]
- Bordi, C.; Lamy, M.C.; Ventre, I.; Termine, E.; Hachani, A.; Fillet, S.; Roche, B.; Bleves, S.; Mejean, V.; Lazdunski, A.; et al. Regulatory RNAs and the HptB/RetS signalling pathways fine-tune Pseudomonas aeruginosa pathogenesis. Mol. Microbiol. 2010, 76, 1427–1443. [Google Scholar] [CrossRef]
- Goodman, A.L.; Merighi, M.; Hyodo, M.; Ventre, I.; Filloux, A.; Lory, S. Direct interaction between sensor kinase proteins mediates acute and chronic disease phenotypes in a bacterial pathogen. Genes Dev. 2009, 23, 249–259. [Google Scholar] [CrossRef]
- Roux, L.; Filloux, A.; Sivaneson, M.; de Bentzmann, S.; Bordi, C. The LadS hybrid histidine kinase triggers Pseudomonas aeruginosa chronic infection by forming a multicomponent signal transduction system with the GacS/GacA two component system. Env. Microbiol. 2014, in press. [Google Scholar]
- Ventre, I.; Goodman, A.L.; Vallet-Gely, I.; Vasseur, P.; Soscia, C.; Molin, S.; Bleves, S.; Lazdunski, A.; Lory, S.; Filloux, A. Multiple sensors control reciprocal expression of Pseudomonas aeruginosa regulatory RNA and virulence genes. Proc. Natl. Acad. Sci. USA 2006, 103, 171–176. [Google Scholar] [CrossRef]
- Kong, W.; Chen, L.; Zhao, J.; Shen, T.; Surette, M.G.; Shen, L.; Duan, K. Hybrid sensor kinase PA1611 in Pseudomonas aeruginosa regulates transitions between acute and chronic infection through direct interaction with rets. Mol. Microbiol. 2013, 88, 784–797. [Google Scholar] [CrossRef]
- Petrova, O.E.; Sauer, K. SagS contributes to the motile-sessile switch and acts in concert with BfiSR to enable Pseudomonas aeruginosa biofilm formation. J. Bacteriol. 2011, 193, 6614–6628. [Google Scholar] [CrossRef]
- Petrova, O.E.; Sauer, K. The novel two-component regulatory system BfiSR regulates biofilm development by controlling the small RNA Rsmz through CafA. J. Bacteriol. 2010, 192, 5275–5288. [Google Scholar] [CrossRef]
- Moscoso, J.A.; Mikkelsen, H.; Heeb, S.; Williams, P.; Filloux, A. The Pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ. Microbiol. 2011, 13, 3128–3138. [Google Scholar] [CrossRef]
- Intile, P.J.; Diaz, M.R.; Urbanowski, M.L.; Wolfgang, M.C.; Yahr, T.L. The AlgZR Two-Component System Recalibrates the RsmAYZ Posttranscriptional Regulatory System To Inhibit Expression of the Pseudomonas aeruginosa Type III Secretion System. J. Bacteriol. 2014, 196, 357–366. [Google Scholar] [CrossRef]
- Yahr, T.L.; Wolfgang, M.C. Transcriptional regulation of the Pseudomonas aeruginosa type III secretion system. Mol. Microbiol. 2006, 62, 631–640. [Google Scholar] [CrossRef]
- Coggan, K.A.; Wolfgang, M.C. Global regulatory pathways and cross-talk control Pseudomonas aeruginosa environmental lifestyle and virulence phenotype. Curr. Issues Mol. Biol. 2012, 14, 47–70. [Google Scholar]
- Jones, A.K.; Fulcher, N.B.; Balzer, G.J.; Urbanowski, M.L.; Pritchett, C.L.; Schurr, M.J.; Yahr, T.L.; Wolfgang, M.C. Activation of the pseudomonas aeruginosa algu regulon through muca mutation inhibits cyclic amp/vfr signaling. J. Bacteriol. 2010, 192, 5709–5717. [Google Scholar] [CrossRef]
- Lykken, G.L.; Chen, G.; Brutinel, E.D.; Chen, L.; Yahr, T.L. Characterization of ExsC and ExsD self-association and heterocomplex formation. J. Bacteriol. 2006, 188, 6832–6840. [Google Scholar] [CrossRef]
- Winstanley, C.; Langille, M.G.; Fothergill, J.L.; Kukavica-Ibrulj, I.; Paradis-Bleau, C.; Sanschagrin, F.; Thomson, N.R.; Winsor, G.L.; Quail, M.A.; Lennard, N.; et al. Newly introduced genomic prophage islands are critical determinants of in vivo competitiveness in the liverpool epidemic strain of Pseudomonas aeruginosa. Genome Res. 2009, 19, 12–23. [Google Scholar]
- Chen, M.; Yan, Y.; Zhang, W.; Lu, W.; Wang, J.; Ping, S.; Lin, M. Complete genome sequence of the type strain pseudomonas stutzeri cgmcc 1.1803. J. Bacteriol. 2011, 193, 6095. [Google Scholar] [CrossRef]
- He, J.; Baldini, R.L.; Deziel, E.; Saucier, M.; Zhang, Q.; Liberati, N.T.; Lee, D.; Urbach, J.; Goodman, H.M.; Rahme, L.G. The broad host range pathogen Pseudomonas aeruginosa strain pa14 carries two pathogenicity islands harboring plant and animal virulence genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2530–2535. [Google Scholar] [CrossRef]
- Wilson, M.A.; Ringe, D.; Petsko, G.A. The atomic resolution crystal structure of the YajL (ThiJ) protein from Escherichia coli: A close prokaryotic homologue of the parkinsonism-associated protein DJ-1. J. Mol. Biol. 2005, 353, 678–691. [Google Scholar] [CrossRef]
- Rocchetta, H.L.; Burrows, L.L.; Pacan, J.C.; Lam, J.S. Three rhamnosyltransferases responsible for assembly of the A-band D-rhamnan polysaccharide in Pseudomonas aeruginosa: A fourth transferase, WbpL, is required for the initiation of both A-band and B-band lipopolysaccharide synthesis. Mol. Microbiol. 1998, 28, 1103–1119. [Google Scholar] [CrossRef]
- McManus, H.R.; Dove, S.L. The CgrA and CgrC proteins form a complex that positively regulates cupA fimbrial gene expression in Pseudomonas aeruginosa. J. Bacteriol. 2011, 193, 6152–6161. [Google Scholar] [CrossRef]
- Vallet-Gely, I.; Sharp, J.S.; Dove, S.L. Local and global regulators linking anaerobiosis to cupA fimbrial gene expression in Pseudomonas aeruginosa. J. Bacteriol. 2007, 189, 8667–8676. [Google Scholar] [CrossRef]
- Tsusaki, K.; Nishimoto, T.; Nakada, T.; Kubota, M.; Chaen, H.; Sugimoto, T.; Kurimoto, M. Cloning and sequencing of trehalose synthase gene from Pimelobacter sp. R48. BBA 1996, 1290, 1–3. [Google Scholar]
- Valvano, M.A.; Marolda, C.L.; Bittner, M.; Glaskin-Clay, M.; Simon, T.L.; Klena, J.D. The rfaE gene from Escherichia coli encodes a bifunctional protein involved in biosynthesis of the lipopolysaccharide core precursor ADP-L-glycero-D-manno-heptose. J. Bacteriol. 2000, 182, 488–497. [Google Scholar] [CrossRef]
- Battle, S.E.; Rello, J.; Hauser, A.R. Genomic islands of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 2009, 290, 70–78. [Google Scholar]
- Kung, V.L.; Khare, S.; Stehlik, C.; Bacon, E.M.; Hughes, A.J.; Hauser, A.R. An rhs gene of Pseudomonas aeruginosa encodes a virulence protein that activates the inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 1275–1280. [Google Scholar]
- Mérens, A.; Delacour, H.; Plésiat, P.; Cavallo, J.D.; Jeannot, K. Pseudomonas aeruginosa et résistance aux antibiotiques. Revue Francophone Des Laboratoires 2011, 29–62. [Google Scholar]
- Poole, K. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J. Mol. Microbiol. biotechnol. 2001, 3, 255–264. [Google Scholar]
- Li, H.; Luo, Y.F.; Williams, B.J.; Blackwell, T.S.; Xie, C.M. Structure and function of oprd protein in Pseudomonas aeruginosa: From antibiotic resistance to novel therapies. IJMM 2012, 302, 63–68. [Google Scholar]
- Lister, P.D.; Wolter, D.J.; Hanson, N.D. Antibacterial-resistant Pseudomonas aeruginosa: Clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 2009, 22, 582–610. [Google Scholar] [CrossRef]
- Juan, C.; Macia, M.D.; Gutierrez, O.; Vidal, C.; Perez, J.L.; Oliver, A. Molecular mechanisms of beta-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 2005, 49, 4733–4738. [Google Scholar] [CrossRef]
- Fukuoka, T.; Ohya, S.; Narita, T.; Katsuta, M.; Iijima, M.; Masuda, N.; Yasuda, H.; Trias, J.; Nikaido, H. Activity of the carbapenem panipenem and role of the OprD (D2) protein in its diffusion through the Pseudomonas aeruginosa outer membrane. Antimicrob. Agents Chemother. 1993, 37, 322–327. [Google Scholar] [CrossRef]
- Lim, D.; Poole, K.; Strynadka, N.C. Crystal structure of the MexR repressor of the mexRAB-oprM multidrug efflux operon of pseudomonas aeruginosa. J. Biol. Chem. 2002, 277, 29253–29259. [Google Scholar] [CrossRef]
- Andresen, C.; Jalal, S.; Aili, D.; Wang, Y.; Islam, S.; Jarl, A.; Liedberg, B.; Wretlind, B.; Martensson, L.G.; Sunnerhagen, M. Critical biophysical properties in the pseudomonas aeruginosa efflux gene regulator mexr are targeted by mutations conferring multidrug resistance. Protein Sci. 2010, 19, 680–692. [Google Scholar] [CrossRef]
- Matsuo, Y.; Eda, S.; Gotoh, N.; Yoshihara, E.; Nakae, T. MexZ-mediated regulation of mexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol. Lett. 2004, 238, 23–28. [Google Scholar]
- Guenard, S.; Muller, C.; Monlezun, L.; Benas, P.; Broutin, I.; Jeannot, K.; Plesiat, P. Multiple mutations lead to MexXY-OprM-dependent aminoglycoside resistance in clinical strains of pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2014, 58, 221–228. [Google Scholar] [CrossRef]
- Piddock, L.J. Mechanisms of fluoroquinolone resistance: An update 1994–1998. Drugs 1999, 58 (Suppl. 2), 11–18. [Google Scholar] [CrossRef]
- Claudel-Renard, C.; Chevalet, C.; Faraut, T.; Kahn, D. Enzyme-specific profiles for genome annotation: PRIAM. Nucleic Acids Res. 2003, 31, 6633–6639. [Google Scholar] [CrossRef]
- Lechat, P.; Souche, E.; Moszer, I. Syntview - an interactive multi-view genome browser for next-generation comparative microorganism genomics. BMC bioinformatics 2013, 14, 277. [Google Scholar] [CrossRef]
- Phast. Available online: http://phast.wishartlab.com (accessed on 4 April 2014).
- Rashid, M.H.; Rao, N.N.; Kornberg, A. Inorganic polyphosphate is required for motility of bacterial pathogens. J. Bacteriol. 2000, 182, 225–227. [Google Scholar] [CrossRef]
- Michel, G.P.; Durand, E.; Filloux, A. XphA/XqhA, a novel GspCD subunit for type II secretion in Pseudomonas aeruginosa. J. Bacteriol. 2007, 189, 3776–3783. [Google Scholar] [CrossRef]
- Goure, J.; Pastor, A.; Faudry, E.; Chabert, J.; Dessen, A.; Attree, I. The V antigen of Pseudomonas aeruginosa is required for assembly of the functional PopB/PopD translocation pore in host cell membranes. Infect. Immun. 2004, 72, 4741–4750. [Google Scholar] [CrossRef]
- Thibault, J.; Faudry, E.; Ebel, C.; Attree, I.; Elsen, S. Anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J. Biol. Chem. 2009, 284, 15762–15770. [Google Scholar] [CrossRef]
- Casabona, M.G.; Silverman, J.M.; Sall, K.M.; Boyer, F.; Coute, Y.; Poirel, J.; Grunwald, D.; Mougous, J.D.; Elsen, S.; Attree, I. An ABC transporter and an outer membrane lipoprotein participate in posttranslational activation of type VI secretion in Pseudomonas aeruginosa. Environ. Microbiol. 2013, 15, 471–486. [Google Scholar] [CrossRef]
- Ashish, A.; Paterson, S.; Mowat, E.; Fothergill, J.L.; Walshaw, M.J.; Winstanley, C. Extensive diversification is a common feature of Pseudomonas aeruginosa populations during respiratory infections in cystic fibrosis. J. Cystic Fibrosis 2013, 12, 790–793. [Google Scholar] [CrossRef]
- Wilder, C.N.; Allada, G.; Schuster, M. Instantaneous within-patient diversity of Pseudomonas aeruginosa quorum-sensing populations from cystic fibrosis lung infections. Infect. immun. 2009, 77, 5631–5639. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lucchetti-Miganeh, C.; Redelberger, D.; Chambonnier, G.; Rechenmann, F.; Elsen, S.; Bordi, C.; Jeannot, K.; Attrée, I.; Plésiat, P.; De Bentzmann, S. Pseudomonas aeruginosa Genome Evolution in Patients and under the Hospital Environment. Pathogens 2014, 3, 309-340. https://doi.org/10.3390/pathogens3020309
Lucchetti-Miganeh C, Redelberger D, Chambonnier G, Rechenmann F, Elsen S, Bordi C, Jeannot K, Attrée I, Plésiat P, De Bentzmann S. Pseudomonas aeruginosa Genome Evolution in Patients and under the Hospital Environment. Pathogens. 2014; 3(2):309-340. https://doi.org/10.3390/pathogens3020309
Chicago/Turabian StyleLucchetti-Miganeh, Céline, David Redelberger, Gaël Chambonnier, François Rechenmann, Sylvie Elsen, Christophe Bordi, Katy Jeannot, Ina Attrée, Patrick Plésiat, and Sophie De Bentzmann. 2014. "Pseudomonas aeruginosa Genome Evolution in Patients and under the Hospital Environment" Pathogens 3, no. 2: 309-340. https://doi.org/10.3390/pathogens3020309