
Distribution of Type I Restriction–Modification Systems in Streptococcus suis: An Outlook

Abstract
:1. Introduction
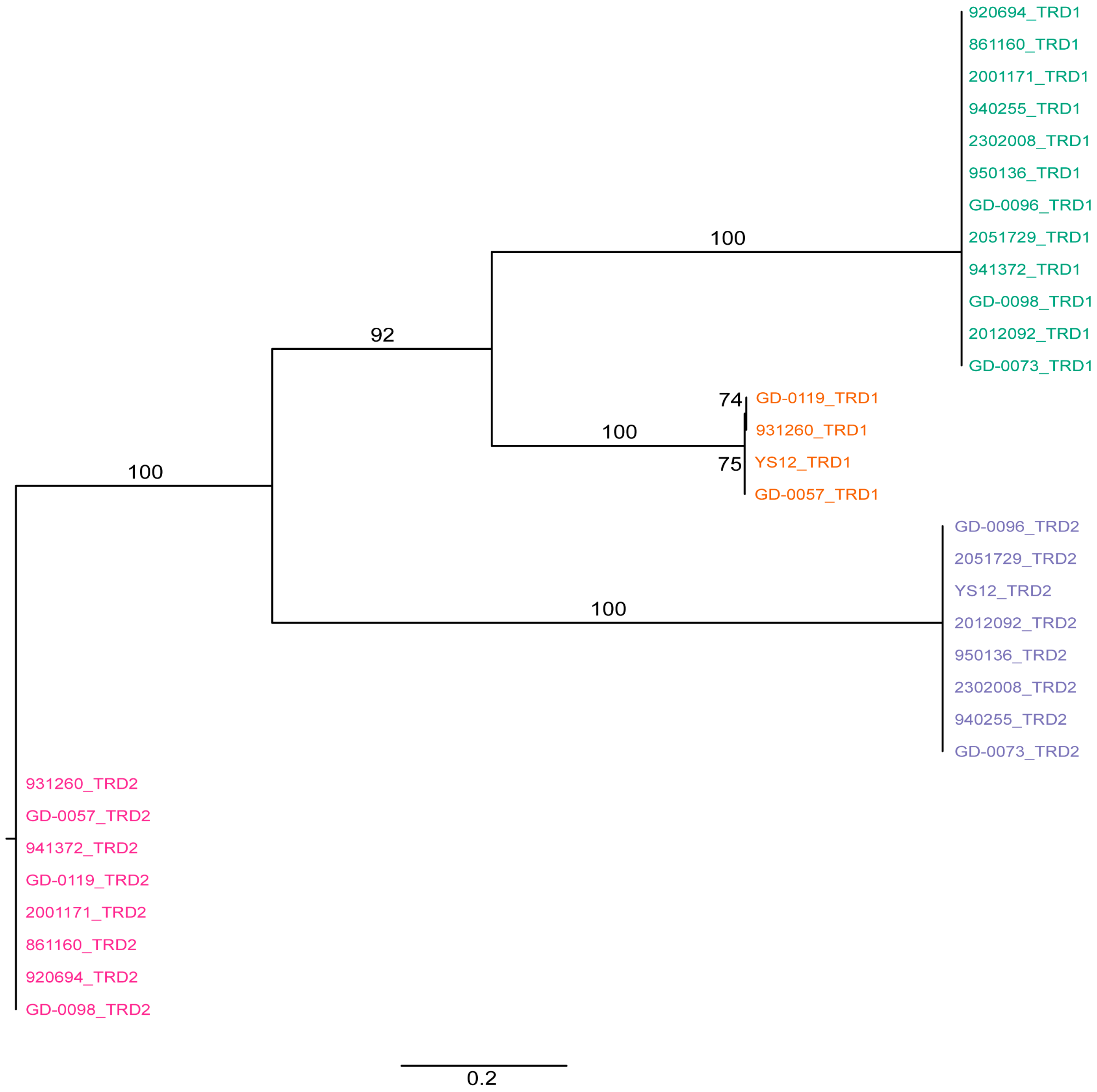
2. Results and Discussion
3. Materials and Methods
3.1. Bacterial Isolates
3.2. Whole Genome Sequencing and Characterization of Isolates
3.3. Restriction–Modification System Search
3.4. Phylogenetic Analysis of Target Recognition Domains
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G.; Murray, N.E. Highlights of the DNA cutters: A short history of the restriction enzymes. Nucleic Acids Res. 2014, 42, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The epigenomic landscape of prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed]
- Vitoriano, I.; Vitor, J.M.; Oleastro, M.; Roxo-Rosa, M.; Vale, F.F. Proteome variability among Helicobacter pylori isolates clustered according to genomic methylation. J. Appl. Microbiol. 2013, 114, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Manso, A.S.; Chai, M.H.; Atack, J.M.; Furi, L.; De Ste Croix, M.; Haigh, R.; Trappetti, C.; Ogunniyi, A.D.; Shewell, L.K.; Boitano, M.; et al. A random six-phase switch regulates pneumococcal virulence via global epigenetic changes. Nat. Commun. 2014, 5, 5055. [Google Scholar] [CrossRef] [PubMed]
- Loenen, W.A.; Dryden, D.T.; Raleigh, E.A.; Wilson, G.G. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014, 42, 20–44. [Google Scholar] [CrossRef] [PubMed]
- Goyette-Desjardins, G.; Auger, J.P.; Xu, J.; Segura, M.; Gottschalk, M. Streptococcus suis, an important pig pathogen and emerging zoonotic agent-an update on the worldwide distribution based on serotyping and sequence typing. Emerg. Microbes Infect. 2014, 3, e45. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, H.F.; Nghia, H.D.; Taylor, W.; Schultsz, C. Streptococcus suis: An emerging human pathogen. Clin. Infect. Dis. 2009, 48, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Willemse, N.; Howell, K.J.; Weinert, L.A.; Heuvelink, A.; Pannekoek, Y.; Wagenaar, J.A.; Smith, H.E.; van der Ende, A.; Schultsz, C. An emerging zoonotic clone in the netherlands provides clues to virulence and zoonotic potential of Streptococcus suis. Sci. Rep. 2016, 6, 28984. [Google Scholar] [CrossRef] [PubMed]
- Sekizaki, T.; Osaki, M.; Takamatsu, D.; Shimoji, Y. Distribution of the SsuDAT1I restriction-modification system among different serotypes of Streptococcus suis. J. Bacteriol. 2001, 183, 5436–5440. [Google Scholar] [CrossRef] [PubMed]
- Sekizaki, T.; Otani, Y.; Osaki, M.; Takamatsu, D.; Shimoji, Y. Evidence for horizontal transfer of SsuDAT1I restriction-modification genes to the Streptococcus suis genome. J. Bacteriol. 2001, 183, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Sekizaki, T.; Takamatsu, D.; Osaki, M.; Shimoji, Y. Different foreign genes incidentally integrated into the same locus of the Streptococcus suis genome. J. Bacteriol. 2005, 187, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—A database for DNA restriction and modification: Enzymes, genes and genomes. Nucleic Acids Res. 2015, 43, D298–D299. [Google Scholar] [CrossRef] [PubMed]
- Schultsz, C.; Jansen, E.; Keijzers, W.; Rothkamp, A.; Duim, B.; Wagenaar, J.A.; van der Ende, A. Differences in the population structure of invasive Streptococcus suis strains isolated from pigs and from humans in the Netherlands. PLoS ONE 2012, 7, e33854. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Coupland, P.G.; Stevenson, A.E.; Callendrello, A.; Bentley, S.D.; Hanage, W.P. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat. Commun. 2014, 5, 5471. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, P.H.; Touchon, M.; Rocha, E.P. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014, 42, 10618–10631. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhang, W.; Zheng, H.; Lan, R.; Wang, H.; Du, P.; Bai, X.; Ji, S.; Meng, Q.; Jin, D.; et al. Minimum core genome sequence typing of bacterial pathogens: A unified approach for clinical and public health microbiology. J. Clin. Microbiol. 2013, 51, 2582–2591. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis comparison tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Kawai, M.; Uchiyama, I.; Kobayashi, I. Domain movement within a gene: A novel evolutionary mechanism for protein diversification. PLoS ONE 2011, 6, e18819. [Google Scholar] [CrossRef] [PubMed]
- Holden, M.T.; Hauser, H.; Sanders, M.; Ngo, T.H.; Cherevach, I.; Cronin, A.; Goodhead, I.; Mungall, K.; Quail, M.A.; Price, C.; et al. Rapid evolution of virulence and drug resistance in the emerging zoonotic pathogen Streptococcus suis. PLoS ONE 2009, 4, e6072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Leigh, J.A.; Heath, P.J.; Luque, I.; Tarradas, C.; Dowson, C.G.; Whatmore, A.M. Development of a multilocus sequence typing scheme for the pig pathogen Streptococcus suis: Identification of virulent clones and potential capsular serotype exchange. J. Clin. Microbiol. 2002, 40, 3671–3680. [Google Scholar] [CrossRef] [PubMed]
- Feil, E.J.; Li, B.C.; Aanensen, D.M.; Hanage, W.P.; Spratt, B.G. eBURST: Inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 2004, 186, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated profile hmm searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
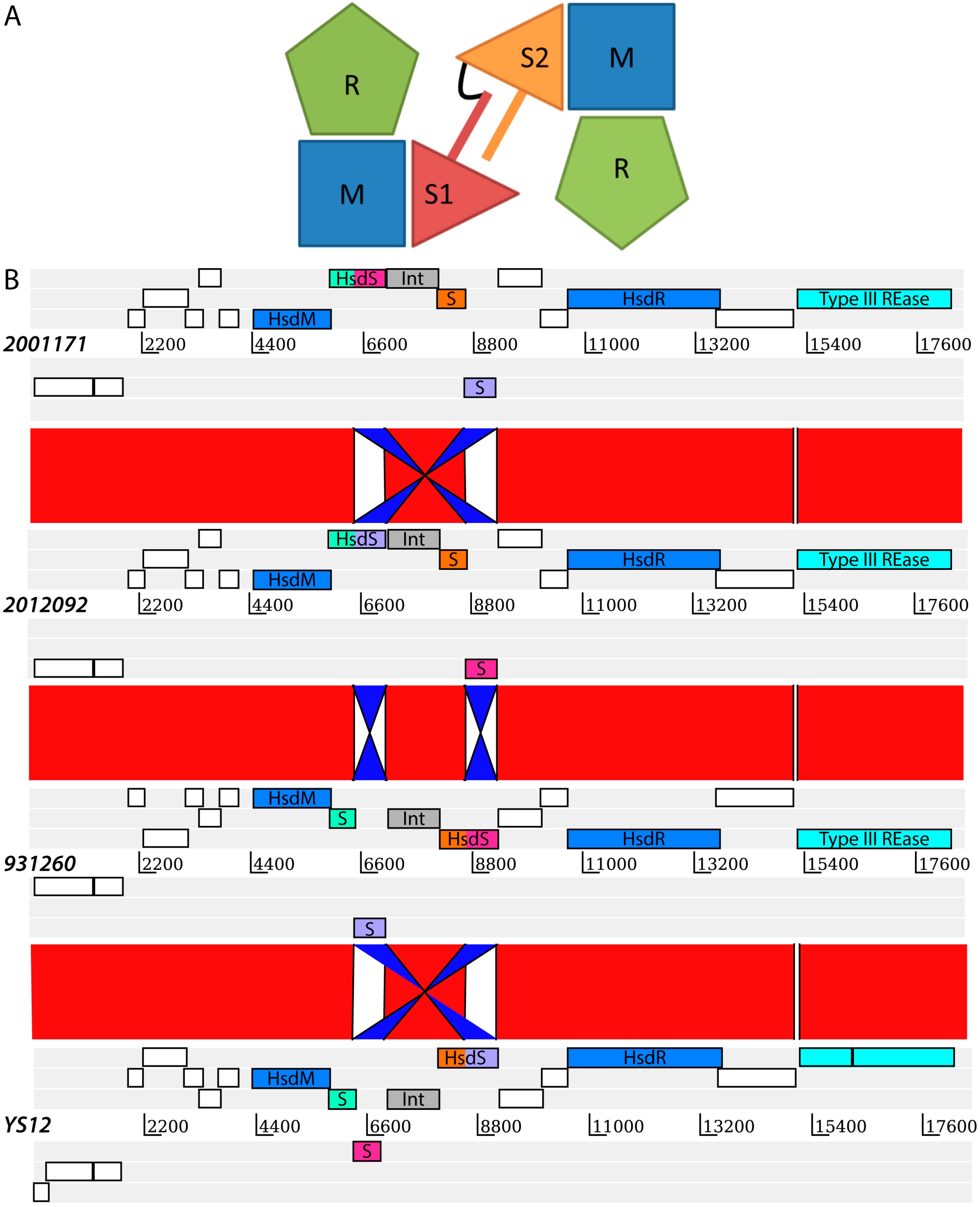

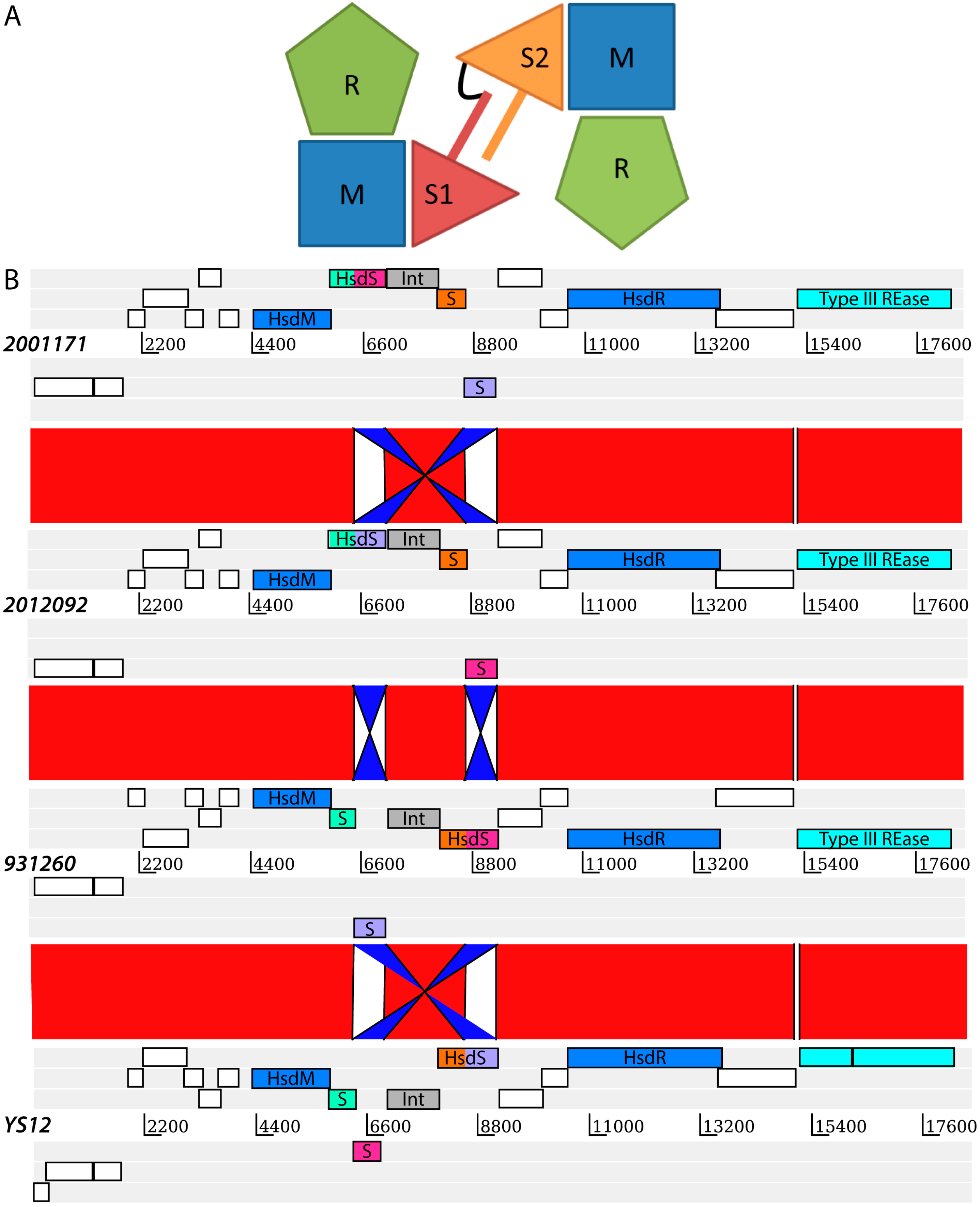{kind=link}
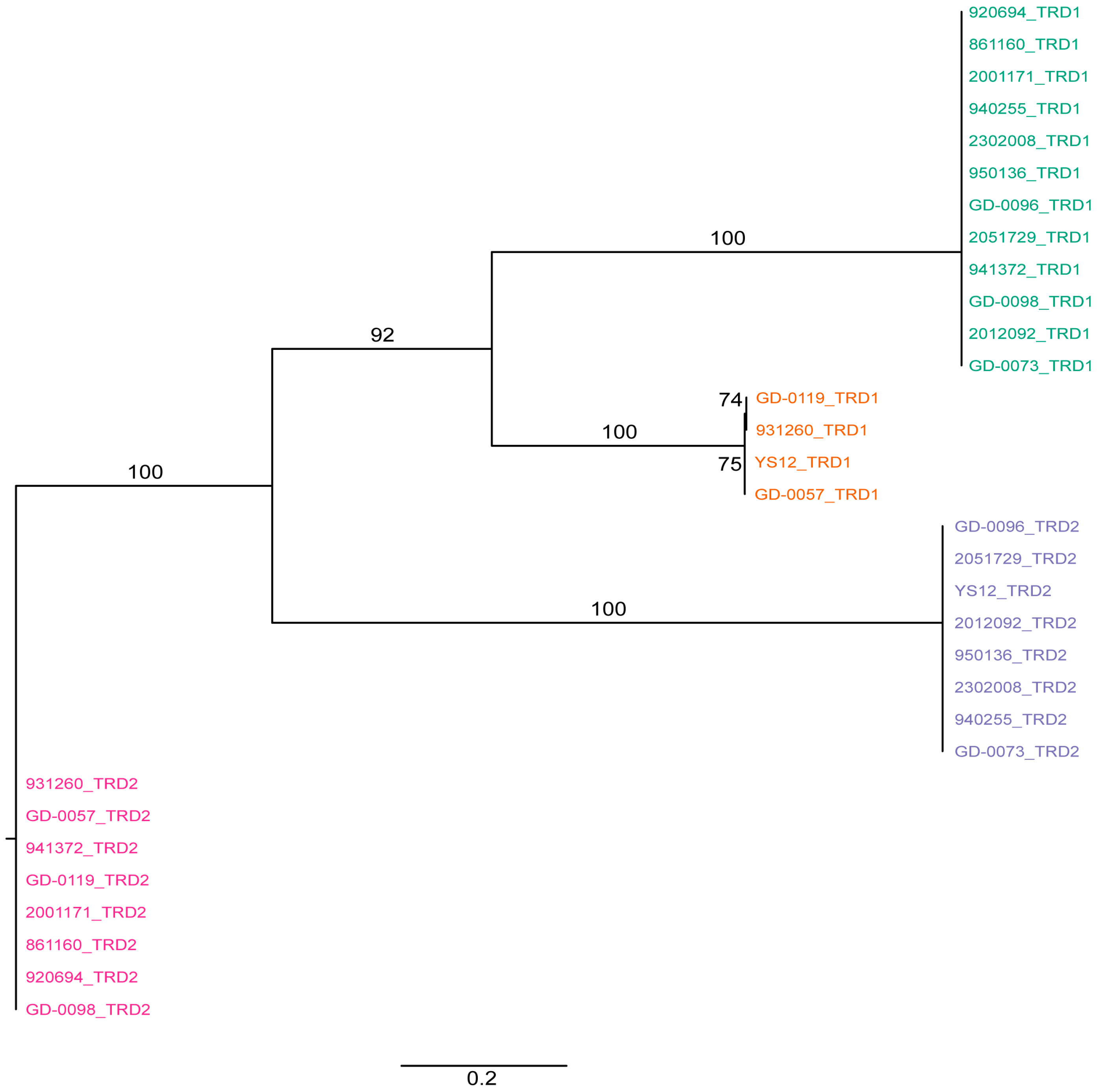{kind=link}
| Clonal Complex (Nr. of isolates) | SsuPORF1588P † Nr. of isolates | SsuPORF1273P † Nr. of isolates | SsuPORF652P † Nr. of isolates | SsuCC20P Nr. of isolates | Ssu13ORF242P † Nr. of isolates |
|---|---|---|---|---|---|
| CC1 (44) | 44 | 44 | 42 a | 0 | 0 |
| CC13 (7) | 6 b | 0 | 0 | 0 | 0 |
| CC16 (31) | 31 | 29 c | 3 d | 0 | 0 |
| CC20 (16) | 16 | 16 | 3 e | 15 f | 0 |
| CC27/CC29 (9) | 9 | 0 | 0 | 0 | 0 |
| Singleton (9) | 4 | 2 | 1 | 0 | 2 |
| Total (116) | 110 | 91 | 49 | 15 | 2 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Willemse, N.; Schultsz, C. Distribution of Type I Restriction–Modification Systems in Streptococcus suis: An Outlook. Pathogens 2016, 5, 62. https://doi.org/10.3390/pathogens5040062
Willemse N, Schultsz C. Distribution of Type I Restriction–Modification Systems in Streptococcus suis: An Outlook. Pathogens. 2016; 5(4):62. https://doi.org/10.3390/pathogens5040062
Chicago/Turabian StyleWillemse, Niels, and Constance Schultsz. 2016. "Distribution of Type I Restriction–Modification Systems in Streptococcus suis: An Outlook" Pathogens 5, no. 4: 62. https://doi.org/10.3390/pathogens5040062