
Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis

College of Veterinary Medicine, Department of Infectious Diseases, The University of Georgia, 501 D.W. Brooks Drive, Athens, GA 30602, USA
Pathogens 2017, 6(1), 10; https://doi.org/10.3390/pathogens6010010
Submission received: 23 December 2016
/
Accepted: 3 March 2017
/
Published: 9 March 2017
(This article belongs to the Special Issue Host Defense Against Bacteria)
{kind=link}
Abstract
:Cystic fibrosis (CF) affects 70,000 patients worldwide. Morbidity and mortality in CF is largely caused by lung complications due to the triad of impaired mucociliary clearance, microbial infections and chronic inflammation. Cystic fibrosis airway inflammation is mediated by robust infiltration of polymorphonuclear neutrophil granulocytes (PMNs, neutrophils). Neutrophils are not capable of clearing lung infections and contribute to tissue damage by releasing their dangerous cargo. Pseudomonas aeruginosa is an opportunistic pathogen causing infections in immunocompromised individuals. P. aeruginosa is a main respiratory pathogen in CF infecting most patients. Although PMNs are key to attack and clear P. aeruginosa in immunocompetent individuals, PMNs fail to do so in CF. Understanding why neutrophils cannot clear P. aeruginosa in CF is essential to design novel therapies. This review provides an overview of the antimicrobial mechanisms by which PMNs attack and eliminate P. aeruginosa. It also summarizes current advances in our understanding of why PMNs are incapable of clearing P. aeruginosa and how this bacterium adapts to and resists PMN-mediated killing in the airways of CF patients chronically infected with P. aeruginosa.
1. Antimicrobial Mechanisms of Neutrophils against Pseudomonas aeruginosa
1.1. P. aeruginosa, an Opportunistic Pathogen
Pseudomonas aeruginosa has a widespread occurrence in aqueous environments in nature [1,2]. This ubiquitous, Gram-negative pathogen has a large genome that enables its adaptation to diverse growth conditions and infections in many species, ranging from nematodes to vertebrates, including humans [3,4]. P. aeruginosa uses several virulence factors to establish infection [3]. P. aeruginosa can be found in the environment in two main, different growth forms, as planktonic bacteria and biofilms [5]. Planktonic bacteria harbor several surface appendages that mediate motility (flagellum, pilus) and virulence (secretion systems) [6,7]. P. aeruginosa residing in biofilms becomes metabolically less active and more resistant to attacks of the immune system or any medical treatment [8,9,10]. The medical relevance of P. aeruginosa biofilms is so high that this bacterium became the model organism for microbial biofilm research. While this review focuses on interactions between P. aeruginosa and neutrophils, several excellent reviews provide deeper insights into different aspects of P. aeruginosa biology.
Healthy humans are protected against P. aeruginosa and do not typically suffer from infections caused by this bacterium. Immunocompromised individuals or patients with deficient clearance mechanisms are the ones mainly infected by P. aeruginosa. P. aeruginosa is an opportunistic pathogen posing a significant medical problem to society. P. aeruginosa is one of the main leading causes (18%–20%) of nosocomial lung infections (also called hospital-acquired or health care-associated pneumonia) [11,12,13,14]. This is due to the high prevalence of immunocompromised patients, biofilm growth of P. aeruginosa on plastic surfaces of medical devices, increased occurrence of multidrug-resistant strains in hospital wards and healthcare personnel carrying this bacterium [15,16,17,18]. Neutropenic patients are immunocompromised and also frequently infected with P. aeruginosa [19]. Chemotherapy of cancer patients often results in neutropenia and P. aeruginosa lung infections [20,21]. Susceptibility of human immunodeficiency virus (HIV) patients to P. aeruginosa is also high; 8%–25% of HIV patients with pneumonia are infected with this bacterium [14]. Chronic obstructive pulmonary disease (COPD) patients are also frequently colonized in their lungs with P. aeruginosa (4%–15% of adult COPD patients) and present diverse symptoms ranging from mild bronchitis to pneumonia with sepsis [22,23,24].
In addition to the transient infections mentioned above, P. aeruginosa can also establish persistent lung infections, such as in patients with cystic fibrosis (CF), non-CF bronchiectasis and primary ciliary dyskinesia (PCD). Although more prevalent than CF, less is known about the diverse etiologies of non-CF bronchiectasis. Non-CF bronchiectasis patients typically develop permanent damage and dilation of the lower airways due to prior pneumonia and become infected with organisms, like P. aeruginosa, despite any obvious abnormalities in their immune system [24]. PCD is characterized by impaired mucociliary clearance of the airways that prevents mucus transport and enables persistent infections with bacteria, including P. aeruginosa [25]. Although P. aeruginosa infects the lungs of human patients with a variety of conditions, the disease most tightly associated with this bacterium is CF.
1.2. Cystic Fibrosis
Cystic fibrosis is a common genetic disease affecting 70,000 people worldwide [26]. Mutations in the cystic fibrosis transmembrane regulator (CFTR) anion channel alter normal ion and fluid transport across the airway epithelium, lead to thickened mucus formation, impaired mucociliary clearance, bacterial adherence and inflammation [26,27]. The vicious cycle of impaired clearance-infection-inflammation drives long-term lung damage, bronchiectasis, airflow obstruction and death in CF [27]. In the lungs, CFTR is primarily expressed in epithelial cells, and this deficiency is the primary cause of lung disease. It is still debated whether mucociliary clearance enables microbial colonization first that drives subsequent inflammation or the epithelial CFTR defect triggers a hyperinflammatory phenotype prior the appearance of any microbes. The recently-developed pig animal model of CF lung disease started to clarify some of these unanswered questions [28]. Although CF lungs host polymicrobial infections, P. aeruginosa is one of the major pathogens infecting a large part of CF patients [26,27,29]. According to the 2015 Cystic Fibrosis Foundation Patient Registry Annual Data Report, while P. aeruginosa used to be the most prevalent microorganism infecting CF patients, its prevalence continues to decrease. On the other hand, Staphylococcus aureus prevalence is increasing. These trends could be partially due to aggressive, early strategies to eradicate P. aeruginosa and more sensitive methods to detect Gram-positive microorganisms. Chronic P. aeruginosa infection has been linked to more rapid progression of lung disease and mortality in CF [30,31,32]. P. aeruginosa infects CF patients early in life and becomes a persistent pathogen in subsequent years [33]. Its persistence in the CF lung is thought to be due largely to its ability to form biofilms [34,35,36]. In biofilms, P. aeruginosa bacteria are embedded in a self-produced polymer matrix mainly consisting of the polysaccharide alginate [36]. P. aeruginosa biofilms also contain self-produced or foreign DNA in CF airways [36]. Biofilms protect bacteria against attacks of the immune system or antibiotic treatments and provide the advantage to survive in the environment of the CF lung.
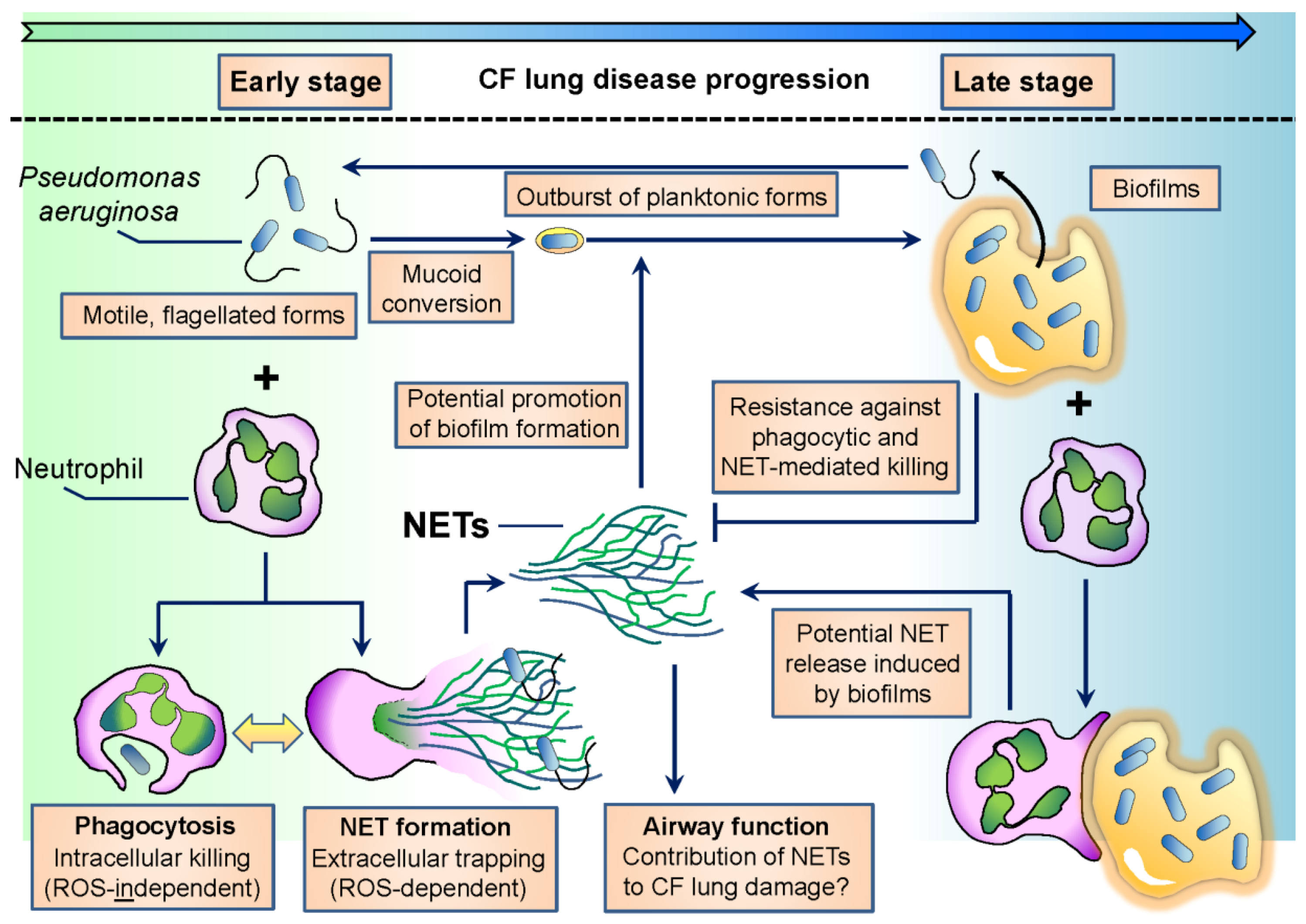
Early isolates of P. aeruginosa in CF are planktonic, characterized by high motility and flagellum expression [37] (Figure 1). The flagellum is crucial for P. aeruginosa to establish infection in several hosts since flagellum-deficient strains are severely reduced in virulence [38,39]. On the other hand, over the course of CF infection, one of the most characteristic changes in P. aeruginosa phenotype is the loss of flagellar motility [33,37] (Figure 1). Adaptation of P. aeruginosa to airways in chronic CF also involves mucoid conversion, alginate production, a decrease in virulence factor expression and biofilm formation [37]. Culturing P. aeruginosa in CF sputum results in similar changes [40]. The flagellum is also needed to initiate biofilm formation by P. aeruginosa [41,42]. In chronic stages of CF airway disease, P. aeruginosa is dominantly found in the form of biofilms. Biofilm cultures are resistant against the attacks of the immune system and medical treatments, including antibiotics, and ensure long-term survival of the bacterium in the host. Several excellent reviews deal with the details of the formation, regulation and structure of P. aeruginosa biofilms [8,43,44,45,46].
It is of high clinical relevance to understand how P. aeruginosa can persist in CF lungs for years and why it is not efficiently eliminated by the immune system, particularly neutrophils that are present in large numbers and drive inflammation [31].
1.3. Neutrophils Are Key to Eliminate P. aeruginosa
P. aeruginosa is a frequent pathogen in neutropenic patients irrespective of the cause of low PMN numbers (chemotherapy, HIV) [14,20,21]. Humans deficient in key neutrophil-mediated antimicrobial mechanisms, such as specific granule deficiency or leukocyte adhesion deficiency, are prone to P. aeruginosa infection [47]. PMN depletion in mice leads to enhanced mortality in acute P. aeruginosa lung infection models [48,49]. In neutropenic mice, a 105-times smaller dose of P. aeruginosa is enough to lead to the same lethal effect in a lung infection model than in mice with normal PMN numbers [48]. Similar effects were shown in rabbits, as well [50]. Airway epithelial control of PMN infiltration has been shown to be crucial to fight P. aeruginosa in murine airways [51,52]. The antimicrobial peptide LL-37, highly expressed in PMNs, also augments P. aeruginosa clearance [53,54]. Overall, PMNs play a critical role in fighting P. aeruginosa.
1.4. PMN Recruitment to the Airways
The appearance of P. aeruginosa in mammalian airways is always accompanied by robust PMN infiltration that is driven by chemoattractant molecules primarily released by airway epithelial cells or already recruited leukocytes, including PMNs. Interleukin 8 (IL-8) is one of the most potent PMN-recruiting chemokines that can be released by epithelial cells, PMNs and macrophages and is recognized by two chemokine receptors, CXCR1 and CXCR2 (C-X-C motif chemokine receptor 1 and 2) [55]. Concentrations of IL-8 in CF airways is elevated and correlated with lung damage [56,57,58,59]. Elevated sputum IL-8 levels also correlated with P. aeruginosa infection in a cohort of CF patients [60]. IL-8 is also detected in the exhaled breath condensate of CF patients, and it is elevated in patients infected with P. aeruginosa [61]. PMNs and macrophages can be major sources of IL-8 in CF since both phagocytes release IL-8 in response to both microbial lipopolysaccharide (LPS) and host stimuli (IL-1β, TNF-α) [62]. Airway epithelial cells release IL-8 in response to P. aeruginosa, LPS or its extracellular pigment pyocyanin [63,64,65,66]. PMN elastase present in CF airway fluids has also been shown to induce IL-8 release in airway epithelial cells [67]. In addition to IL-8 release stimulated by exogenous microbial or host stimuli, enhanced endogenous IL-8 production has also been proposed to drive early inflammation in CF. CFTR-deficient airway epithelial cells secrete larger amounts of IL-8 than their wild-type counterparts, indicating an epithelium-derived proinflammatory effect of IL-8 in CF [68,69,70]. Endoplasmic reticulum stress and exaggerated NF-κB activation induced by misfolded CFTR is the likely reason for enhanced chemokine production in CF epithelial cells [71,72,73]. Among the many potential PMN-recruiting molecules present in the CF sputum [74], the complement activation product C5a has been shown to be important to optimal mucosal defense against P. aeruginosa in the murine lung [75]. Formation of C5a has been demonstrated in CF airway fluids [76]. Whether C5a is a main PMN recruiter in CF airways remains to be elucidated as PMN serine proteases were suggested to degrade and inactivate the C5a receptor on PMNs [77]. Although not as potent as the previous molecules, the proinflammatory cytokine IL-1β can also direct PMN migration. Elevated levels of IL-1β, a proinflammatory cytokine produced by inflammasomes, have been detected in CF airways and correlated with lung damage [59,78]. Although CF macrophages harbor an intact inflammasome capable of releasing IL-1β in response to P. aeruginosa, it remains to be better understood whether enhanced IL-1β levels seen in CF are due to intrinsic increase in NF-kB activation [79]. Although on a per cell basis, PMNs release far less IL-1β than macrophages, considering the total amount of PMNs in CF airways, PMNs could be a pathologically relevant source of IL-1β in CF [80]. The lipid leukotriene B4 is a product of the arachidonic acid metabolic pathway, released by leukocytes, including PMNs, and one of the most potent molecules driving PMN chemotaxis. Leukotrienes are present in CF sputum [81,82,83,84]. CF PMNs were shown to have decreased chemotaxis in response to LTB4 compared to non-CF PMNs, while their chemotactic response to formyl peptides was the same [85]. Leukotriene B4 is also detectable in exhaled breath condensates of CF patients and is elevated upon P. aeruginosa infection [61]. IL-17, a potent PMN-recruiting cytokine produced by Th17 cells, is essential to prevent chronic P. aeruginosa infection in mice [86]. CF airways have elevated levels of IL-17 [61,87]. IL-23, a cytokine closely related to IL-17, is essential to protect mice from P. aeruginosa lung infection [88]. IL-17 immunolocalizes to PMNs and monocytes in CF airways [89,90]. Based on elevated levels of IL-17 in CF and the essential role of the IL-17/IL-23 axis in regulating PMN recruitment, it has also been proposed that CF is a Th17 disease [91,92].
In addition to host molecules, pathogen-associated molecular patterns (PAMPs) can also be strongly chemotactic for PMNs, but their presence in CF airway fluids and contribution to PMN recruitment are less clear. Formyl peptides shedding from bacteria are sensed by PMNs in lower nanomolar concentrations by two different receptors, FPR1 and FPR2 [93]. Formyl peptides have been found in CF sputum [74]. P. aeruginosa flagellin is detectable in the airways of chronic CF patients [94] that is known to stimulate IL-1β release in epithelial cells and macrophages [95]. The Pseudomonas quinolone signal (PQS) that is a quorum sensing molecule of P. aeruginosa has been shown to recruit PMNs via the MAPkinase p38, while it did not affect bactericidal function or induce apoptosis [96].
Thus, PMNs are recruited to CF airways by several mechanisms [74]. It remains to be elucidated whether any one of the several PMN chemoattractants plays a major role and could be targeted therapeutically.
Overall, PMNs provide the most efficient branch of the immune system to fight P. aeruginosa lung infections.
1.5. Intracellular Killing Following Phagocytosis
What mechanisms are used by PMNs to eliminate P. aeruginosa in healthy hosts? The most efficient way of eliminating P. aeruginosa by PMNs is classical phagocytosis and subsequent intracellular killing. PMNs are highly phagocytic leukocytes capable of rapidly engulfing up to 10–20 bacteria per cell in a short period of time [97]. Bacteria first have to be recognized by PMNs. PMNs are equipped to bind to several PAMPs expressed on the surface of bacteria via their pattern-recognition receptors (PRRs). C-type lectins, among them Dectin-1 especially, are important phagocytic PRRs expressed on PMNs essential for uptake and elimination of fungal pathogens [98]. Another PRR, TREM-1, recognizes several microbes and mediates their phagocytosis in PMNs [99]. NOD1 and NOD2 are nonphagocytic, intracellular PRRs binding cytosolic microbial ligands [97,100]. NOD1 participates in the immune response against P. aeruginosa [101]. The nonphagocytic Toll-like receptors (TLRs) recognize a wide range of microbial PAMPs (DNA, RNA, peptides, lipids and carbohydrates) and prime PMNs for enhanced effector responses, including phagocytosis [100]. Microbial uptake of PMNs is several times accelerated when bacteria have been labelled by the immune system. This process called opsonization ensures faster recognition and killing of microbes and is especially important for fighting pathogens that are not recognized directly by PMNs. Classical opsonins include immunoglobulin G (IgG) and the complement activation product C3b. PMNs express two complement receptors prior to activation, CR3 and CR4, both binding to C3bi [100]. Upon stimulation, PMNs mobilize another opsonin receptor, CR1 to their surface recognizing C3b/C4b complexes further enhancing their capacity to engulf microbes [100]. Mannan-binding lectin is a soluble PRR that binds C1q on the microbial surface and promotes phagocytosis in PMNs by binding to the C1q receptor [100]. IgGs are recognized by PMNs via low and high affinity Fc receptors, FcRs [102]. The low affinity FcRs, FcγRIIA and FcγRIIIB, are expressed in PMNs prior to activation, while the surface expression of the high affinity FcγRI on PMNs is increased upon microbial stimulation [100]. Pentraxins form a phylogenetically old group of pentameric plasma proteins that recognize bacterial or fungal PAMPs and serve as the third major type of opsonin receptors [102]. P. aeruginosa is sensitive against the action of pentraxin PTX3 [103], and the risk of P. aeruginosa airway colonization in CF patients is affected by PTX3 genetic variations [104].
P. aeruginosa can be phagocytosed by PMNs similarly to several other microbes [102]. Both FcRs and CR3 have been implicated to participate in opsonic phagocytosis of P. aeruginosa (for details, see [103]). An early report indicated the involvement of CD14 in P. aeruginosa nonopsonic phagocytosis [104]. TLRs likely do not directly mediate nonopsonic uptake since deficiency in MyD88 does not impair mouse macrophage phagocytosis of P. aeruginosa [105]. Very likely, several receptors and their activation pathways are responsible for optimal phagocytosis of P. aeruginosa [103].
Phagosomes containing P. aeruginosa fuse intracellularly with PMN granules storing an arsenal of antimicrobial molecules (detailed in [106]). The phagolysosome fusion creates a special, confined niche in which bacteria can be eliminated efficiently in a precise surgical way without significant leaking of the dangerous PMN cargo into the environment [100]. Four types of PMN granules have been categorized and referred to as primary (or azurophil), secondary (or specific), tertiary (or gelatinase) granules and secretory vesicles [106]. Secretory vesicles are membrane vesicles derived from the plasma membrane primarily containing plasma membrane proteins and extracellular milieu [106]. Primary, secondary and tertiary granules have specific contents and fuse with the phagosome in a reverse order, tertiary granules first and primary granules last [106]. Granule content can also be released into the extracellular milieu when the granule membrane fuses with the plasma membrane via a process called degranulation [106]. Extracellular mobilization of secretory vesicles and gelatinase granules can be achieved relatively easily in PMNs upon weak stimulation, while primary and secondary granules are only released in the extracellular medium when they leak during phagocytosis [106].
PMNs possess several antimicrobial mechanisms that contribute to microbial killing in the phagolysosome. The production of reactive oxygen species (ROS) is a hallmark of PMN activation. ROS comprise short-lived, reactive molecules, ions and radicals that are all derivatives of molecular oxygen [107]. The phagocytic NADPH oxidase expressed in PMNs produces superoxide anions from molecular oxygen as the primary type of ROS [108]. Superoxide can dismutate to hydrogen peroxide (H2O2) spontaneously or with the help of the superoxide dismutase enzyme [108]. Formed H2O2 will be used by myeloperoxidase (MPO), a peroxidase present in large quantities in primary granules of PMNs, to produce highly reactive hypochloric acid (HOCl) [108]. Further, ROS that can be produced by PMNs involve singlet oxygen, hydroxyl radical, chloramines and peroxynitrites [107,108]. ROS are thought to damage microbes primarily by direct chemical attack, but an alternative hypothesis has also been proposed that ROS-driven ion fluxes are actually necessary for optimal phagosomal digestion [97,109,110]. PMNs harbor several antimicrobial products that attack microbes in an ROS-independent manner. Primary granules contain bacterial permeability increasing protein (BPI), neutrophil elastase (NE), proteinase 3, cathepsins, defensins and lysozyme, just to name a few [106]. BPI is a pluripotent protein that neutralizes bacterial LPS, promotes the delivery of bacterial outer membrane antigens to dendritic cells and has antibacterial activity against P. aeruginosa [111,112]. Neutrophil elastase, cathepsins and proteinase 3 are serine proteinases whose functions are to degrade the extracellular matrix and to attack microbial proteins [113]. NE-deficient mice are more susceptible to P. aeruginosa lung infection than their wild-type counterparts [114], while cathepsin G-deficient animals have lower bacterial lung burden [115]. Defensins are antimicrobial peptides released from PMNs in large amounts that kill P. aeruginosa efficiently [116]. Lysozyme treatment ameliorates P. aeruginosa pneumonia [117,118]. LL-37, an antimicrobial peptide belonging to the cathelicidin family, is highly expressed in PMNs, stored in secondary granules and efficient in attacking P. aeruginosa [53]. Thus, PMNs are well-equipped with redundant antimicrobial mechanisms to fight diverse pathogens.
1.6. Neutrophil Extracellular Traps
In addition to intracellular killing [119], PMNs also trap and kill extracellular microbes via another mechanism referred to as neutrophil extracellular trap (NET) formation [120]. NETs are composed of a DNA scaffold associated with histones and neutrophil granule components, such as MPO and NE [120,121,122]. NET formation has been originally proposed to be an active way of cell death involving cytoskeletal changes, plasma and nuclear membrane disintegration and DNA extrusion [120,123]. Later on, alternative mechanisms of NET release were discovered that do not require cell death, occur much faster and include mitochondrial DNA [124]. Several microorganisms including P. aeruginosa were shown to trigger NET release in PMNs. Only NET-forming PMNs release protein-DNA complexes (myeloperoxidase-DNA or neutrophil elastase-DNA); not apoptotic or necrotic PMNs [120,125,126,127,128]. Despite the vast amount of literature published in the last 12 years on NETs, signaling pathways leading to NET formation are still largely unknown, and only the involvement of a few molecules is known. The NADPH oxidase has been shown to be required for NET release early on since PMNs isolated from chronic granulomatous disease (CGD) patients with nonfunctional NADPH oxidase did not form NETs in response to different microbes [123,129]. Residual NADPH oxidase activity in CGD has been indicated to determine PMNs’ ability to release NETs [130]. Similarly, MPO, NE and the citrullination of histones mediated by peptidylarginine deiminase 4 (PAD4) were also shown to be required for NET formation [131]. Citrullinated histones are only present in NETs, not in resting PMNs [132]. PAD4-deficient murine PMNs do not form NETs in vitro [133,134], and PAD4-deficient mice have impaired NET-mediated antibacterial defenses [135]. The fact that the purpose of NET formation is extracellular trapping of microbes by PMNs to help localize the infection is well accepted. It remains though controversial whether NETs are also efficient in killing trapped microorganisms [136]. Extracellular DNA (ecDNA) has been shown to be antimicrobial against P. aeruginosa in in vitro studies [137,138]. EcDNA itself is thought to scavenge large amounts of cations and thereby limit microbial growth [137,138]. NET-associated antimicrobial proteins and peptides were suggested to harm trapped microorganisms [120]. This has been challenged by questioning whether the extracellular concentration of NET-related enzymes and proteins is sufficient to support such a killing effect [139]. NET-linked myeloperoxidase was shown to kill microorganisms in vitro in the presence of hydrogen peroxide [140]. The exact microbicidal potential of NETs, especially in vivo, remains to be elucidated.
In addition to the beneficial antimicrobial role of NETs, several studies suggested their contribution to the pathology of numerous conditions, including CF lung disease. NETs were shown to drive or contribute to the pathology of numerous autoimmune diseases, such as systemic lupus erythematosus, rheumatoid arthritis and small-vessel vasculitis [141]. In gout, a dual pro- and anti-inflammatory role has been proposed for NETs in disease pathogenesis [142]. NETs have also been linked to cancer, diabetes and viral infections [143,144,145]. Although NETs are currently the most studied field of PMN biology, several questions need to be answered with regard to their roles in antimicrobial defenses and clinical manifestation of several diseases.
1.7. PMN Microvesicles
The most recently-described mechanism by which PMNs attack microbes is the release of microvesicles [146]. Vesicle (diameter 100–1000 nm) shedding from live cells obtained significant attention is the past few years as a novel tool of cell-to-cell communication [147]. They are involved in antigen presentation, the transfer of receptor proteins or RNA and host defense. PMNs also release microvesicles that have in vitro antimicrobial activity against S. aureus [146]. The amount of PMN-derived microvesicles increases robustly during inflammatory conditions [148]. A diverse set of host and microbial stimuli was shown to induce microvesicle release from PMNs [148]. In addition to direct microbial killing, PMN-derived microvesicles were also shown to modulate the function of other immune cells [148]. Although a new area of research, PMN-derived microvesicles represent an exciting field with potential future implication to host defense, autoimmunity and immunomodulation.
1.8. Oxidative and Non-Oxidative PMN Mechanisms against P. aeruginosa
The PMN antimicrobial mechanisms highlighted above can also traditionally be classified as oxidative and non-oxidative, based on their dependence on NADPH oxidase-produced ROS. P. aeruginosa is fairy resistant against the oxidative mechanisms and is mainly susceptible to non-oxidative actions of PMNs. This is well-supported by human clinical data showing that patients with impaired or completely absent PMN respiratory burst (CGD, myeloperoxidase-deficiency) do not suffer from P. aeruginosa infections [149,150,151]. This indicates that the NADPH oxidase or myeloperoxidase are not required to eliminate P. aeruginosa from healthy hosts, no matter what their contribution to bacterial killing is. CGD PMNs kill P. aeruginosa efficiently in vitro, further supporting the previous conclusion [152]. P. aeruginosa catalase has been proposed to be responsible for its resistance against oxidative PMN killing mechanisms [152]. P. aeruginosa-induced ROS production in PMNs could in fact support long-term survival of the bacterium since hydrogen peroxide was shown to promote mucoid conversion of P. aeruginosa [153]. On the other hand, several molecules of the non-oxidative PMN antimicrobial repertoire are efficient tools to kill P. aeruginosa: NE, lysozyme, defensins and cathelicidins [53,114,116,117,118]. Thus, reducing oxidative and promoting non-oxidative PMN antimicrobial mechanisms holds the best promise to achieve significant improvements in P. aeruginosa-infected patients by targeting PMNs. Whether this could work in CF remains to be determined since CF patients are infected with other microbes. S. aureus, another main pathogen in CF airways [154], is mainly killed by oxidative mechanisms of PMNs [97] and is also a main pathogen in CGD [155].
In summary, PMNs possess several antimicrobial tools whose optimal coordination in time and space is required for efficient elimination of microbes involving P. aeruginosa. If mistakes occur, PMN activation causes host damage with potential, serious, clinical consequences. Therefore, it is essential to understand which mechanisms of PMNs are the most efficient to attack pathogens and how to avoid PMN-mediated pathology occurring in several diseases, including CF.
2. PMN Dysfunction in CF
Airway inflammation in CF is the product of a complex set of innate immune interactions. The PMN is a pivotal cellular player influencing the outcome of these interactions. CF airways contain large numbers of PMNs that fail to successfully eliminate bacteria and cause lung damage. The main points of PMN dysfunction in CF are listed in this section with cited literature that provides more detailed insights into each direction.
2.1. PMN Components Correlate with CF Lung Disease Severity
PMN density in CF sputum correlates with CF lung disease severity (measured as forced expiratory volume in one second, FEV1) [57,78]. Sputum and blood concentrations of NE and MPO in CF patients are associated with declines in lung function [57,58,78,156,157]. NE is a major risk factor for bronchiectasis in CF children [158,159]. Sputum NE levels have been shown to be the best predictors of CF lung function decline so far [158,160]. Improper diminishment of inflammation during an exacerbation is linked to failure to recover respiratory function and increased risk of subsequent re-exacerbation in patients with CF [161]. IL-8, a major PMN chemoattractant that both airway epithelial cells and PMN themselves produce, has also been associated with CF lung function decline [57,58]. Interlekuin-1β (IL-1β), a proinflammatory cytokine recruiting PMNs mainly produced by macrophages, but also secreted by activated PMNs, has also been linked to CF lung damage [78]. These data obtained from clinical samples of CF patients clearly show that PMNs are the clinically most important leukocyte in CF airways, and PMN-mediated inflammation contributes to lung disease.
2.2. CFTR Deficiency in PMNs
CF is a genetic disorder potentially affecting all cells expressing CFTR. PMN functions could be also primarily affected by CFTR deficiency. Since the first detection of CFTR in PMNs [162], reports described impaired intracellular killing of P. aeruginosa, diminished chloride transport, reduced cytosolic calcium changes, altered response to N-formyl-methionyl-leucyl-phenylalanine and diminished degranulation in human or murine CFTR-deficient PMNs [163,164,165,166,167,168,169]. On the other hand, NET formation of CF PMNs and their intrinsic superoxide production was not different from that of non-CF PMNs [170,171]. Concerns about the in vivo relevance of primary CFTR deficiency in PMNs in CF are raised by indirect clinical observations. In CF, PMN dysfunction has been reported and widely accepted only in one organ, the lung. If CFTR were to affect essential antimicrobial functions of PMNs in vivo, the involvement of several other organs would be expected, as is seen in PMN-specific genetic disorders, such as CGD [150,172]. Another possibility is that CFTR deficiency in PMNs can be overcome by parallel mechanisms in each organ, except for the lung. An important argument for PMN-mediated lung damage in CF is the fact that CFTR-deficient airway epithelial cells are major drivers of PMN recruitment. Although exciting results are emerging with regard to the direct or indirect mechanisms by which CFTR affects PMN functions in CF, future research using novel tools like a PMN-specific conditional CFTR knockout mouse strain could help better answer this question. Several excellent reviews provide a deeper insight into this field [173,174,175].
2.3. CF Airway PMNs
A neglected, but clinically very important area of research is the investigation of PMNs found in the airways of CF patients. Studies by a few groups described that CF airway PMNs are different from those found in the blood of the same CF patient [176,177,178,179,180,181,182]. PMNs in CF airways have been shown to have enhanced expression of certain metabolic and stress pathways including CD39, CXCR4, CD114 and RAGE [177]. It has been suggested that a shift in the expression of their nutrient transporters affecting glucose and inorganic phosphate transports is part of their adaptation to the CF airway environment [176]. CF airway PMNs have a reduced respiratory burst and altered expressions of TLRs [179,182,183]. Although these cells are present at the site of infection and drive CF airway inflammation, our knowledge about them is limited at this point. Future studies will have to determine whether their potentially altered metabolic and antimicrobial phenotypes are due to CFTR deficiency, factors found in CF airways, in the tissues surrounding them during migration, or a combination of all of them.
2.4. NETs in CF
The presence of large amounts of PMNs in CF airways has been detected early on, but the mechanism by which PMNs discharge their DNA and granule content still remains uncertain even today. PMNs were thought to die in CF by apoptosis followed by necrosis due to neglect (lack of removal of apoptotic cells, secondary necrosis). PMNs did not form the focus of CF research for a long time partially because successful pharmaceutical targeting of a necrotic PMN death pathway seemed very unlikely. In addition, PMN-driven inflammation might have had dual roles in CF airways: PMNs likely fulfil antimicrobial functions while they also contribute to lung damage. Before clearly understanding the exact role of PMNs in CF airway disease, targeting them in CF is problematic.
With the discovery of NETs, a new, alternative explanation for PMN dysfunction in CF emerged [120]. By releasing NETs, CF airway PMNs would respond to the presence of bacteria, but at the same time would also release their DNA and granule content. EcDNA is present in large amounts in CF airways, and DNAse therapy has been used since the 1980s. Although DNAse therapy improves lung function of CF patients [184,185,186,187] and has a remarkable safety profile over several decades, its effect is not universal [188], does not completely degrade ecDNA [189] and does not improve lung inflammation [186]. ecDNA in CF airways is derived from the host [190], mainly PMNs [78,156,190]. NETs are abundantly present in CF airways [191,192]. Negative correlations were found between CF sputum ecDNA concentrations and lung function measures [58,193]. Neutrophils undergoing NET formation were detected in CF sputum samples [191,192,194]. Histone citrullination, a histone modification characteristic for NET formation, but not for neutrophil apoptosis or necrosis, was also detected in CF sputum [191]. Despite the abundance of NETs in CF airways, NETs have not yet been quantitatively correlated with CF airway disease, it remains unknown what are the main stimuli of NET formation in CF, and what is the molecular-cellular mechanism of NET release by CF airway PMNs.
2.5. Anti-Inflammatory Strategies in CF
Although we do not completely understand the details of PMN-mediated inflammation in CF, anti-inflammatory strategies have been tested and are being used to improve CF lung disease. While several excellent reviews provide deeper insight into this field, here, only the main approaches will be mentioned [195,196,197,198]. Inhibition of NE represents a rather classical approach to diminish PMN-mediated lung damage in CF [199,200,201]. Phosphodiesterase (PDE4) inhibitors reduce cAMP synthesis and thereby have direct inhibitory action on inflammatory cell signaling and PMN recruitment [195]. Attractive lipid mediator targets are lipoxins, mainly LXA4, that are generated from arachidonic acid and have a general suppressive role on inflammation, including PMN effector mechanisms [195]. Resolvins are other lipid mediators of interest because they also have general anti-inflammatory, pro-resolution effects, including inhibition of PMN respiratory burst, chemotaxis and attachment [195]. The cannabinoid receptor CB2 is highly expressed on immune cells, and its activation promotes anti-inflammatory effects, including reduced proinflammatory cytokine production and leukocyte migration [195]. Leukotriene modulators target two enzymes, 5-lipoxygenase and leukotriene A4 hydrolase, in the hope of significantly reducing LTB4 levels and preventing PMN recruitment [195]. As mentioned above, Th17 cells play an important role in orchestrating PMN recruitment to the airways. Targeting IL-17 with therapeutic monoclonal antibodies works in asthma and autoimmune diseases and represents a novel, exciting approach to inhibit PMN recruitment and inflammation in CF [202,203].
In summary, PMNs in CF fail to eliminate respiratory pathogens, including P. aeruginosa. PMNs could provide a promising and most likely powerful target for anti-inflammatory pharmaceutical intervention. PMN targeting in CF remains infeasible until we clearly understand their contribution to antimicrobial defense and lung damage.
3. Adaptation of P. aeruginosa to Neutrophil-Mediated Attacks in CF
P. aeruginosa is a major pathogen infecting the airways of CF patients [37]. P. aeruginosa is not cleared from CF airways in spite of being surrounded by several PMNs. This is likely due to its successful adaptation to the environment found in the CF lung including PMNs. In this section, the mechanisms are discussed that are potential contributors to P. aeruginosa adaptation against PMN-mediated effector mechanisms in CF.
3.1. Loss of Flagellar Motility
As mentioned previously, early isolates of P. aeruginosa are motile. Motile bacteria are easy to recognize by the immune system, and retaining a motile flagellum would lead to rapid microbial clearance. Phagocytes (macrophages) much more likely recognize and phagocytose flagellated P. aeruginosa than its aflagellated counterpart [103,204] (Figure 1). PMNs also phagocytose P. aeruginosa in a flagellum-dependent manner [205]. Impaired phagocytosis of flagellum-deficient P. aeruginosa is due to the loss of its motility, not flagellum expression [105]. Chronic CF isolates of P. aeruginosa are typically nonmotile and resist phagocytic clearance of macrophages [206]. It has been presumed that host-selective pressure inspires P. aeruginosa to lose its motility in chronic CF, especially in patients with poor clinical conditions [207,208]. The flagellum is also required for P. aeruginosa-induced superoxide formation and NET release in human PMNs [205]. Lack of flagellar motility, not flagellum expression per se, is responsible for impaired NET release induced by flagellated P. aeruginosa [205]. This is consistent with previous data showing that early CF isolates of P. aeruginosa induce larger amounts of NETs and are more resistant against NET-mediated attacks than late isolates obtained from the same CF patients [128,170] (Figure 1). Whether loss of swimming motility alone is responsible for reduction in P. aeruginosa-initiated NET release or loss of other NET-inducing or appearance of NET-inhibitory factors also contribute, remains to be studied. PMN products have been shown to attack P. aeruginosa flagellin. NE degrades bacterial flagellin and represses its transcription [209,210]. Lactoferrin, a PMN antibacterial product, attacks flagellin-dependent biofilm formation of P. aeruginosa [211]. Extrusion of NETs induced by motile P. aeruginosa must primarily occur in early disease stages characterized by flagellated bacterial forms (Figure 1). Its occurrence in chronic CF is, however, also likely since P. aeruginosa flagellin has been detected in sputa of CF patients chronically infected with P. aeruginosa, independently of the presence of mucoid bacterial strains [94]. Planktonic, flagellated forms of P. aeruginosa break out from biofilms in the environment, and this could also happen in chronic CF airways (Figure 1).
Overall, flagellum-provided motility is essential to establish infection in CF airways, but its downregulation is required to avoid immune recognition and to enable long-term bacterial adaptation to the host in chronic infection. PMNs likely represent one of the main factors of the CF airway environment responsible for the described changes in P. aeruginosa motility (Figure 1).
3.2. Characteristic Image of Chronic CF Airways: Suspension Biofilms of P. aeruginosa Surrounded by PMNs
Laboratory biofilms of P. aeruginosa have been studied for a long time. In chronic CF airways, P. aeruginosa does not form; however, these flat, thick biofilms covering the airway surface but reside instead in three-dimensional biofilms also called suspension biofilms or non-attached aggregates [212,213,214,215,216]. These bacterial aggregates accompanied by large amounts of PMNs represent the typical clinical image found in the airways of CF patients chronically infected with P. aeruginosa (Figure 1) [212,213,214,215,216].
This mode of biofilm growth is likely not unique to CF airways as P. aeruginosa has been reported to form similar aggregates under different stress conditions, in the absence of any mammalian cells. The P. aeruginosa PAO1 strain was grown in the presence of the detergent sodium dodecyl sulfate, and macroscopic aggregates consisting of respiring bacterial cells embedded in an extracellular matrix composed of DNA and acidic polysaccharides formed [217]. Aggregated bacteria showed a significantly increased rate of survival over planktonic cells in the presence of the detergent [217]. Starvation of P. aeruginosa results in similar aggregate cultures in liquid medium that also contains extracellular DNA originating from bacteria [218,219,220]. Phenazines induce DNA release from P. aeruginosa via H2O2 generation [221,222]. The structure of the suspension biofilms is similar to those of the laboratory biofilms [220]. Important roles of calcium and c-di-GMP signaling in supporting suspension biofilm growth of P. aeruginosa have just been revealed [223,224]. Bacterial growth rates inside of P. aeruginosa suspension aggregates negatively correlated with the size of these aggregates in the CF lung [213]. Quorum sensing molecules, PQS and AHL-12, produced at early stages of biofilm formation of P. aeruginosa stimulate PMN chemotaxis [96,225]. Similarly, N-(3-oxododecanoyl)-l-homoserine lactone (3OC12-HSL), that controls virulence factor and biofilm formation in P. aeruginosa, has also been shown to induce PMN chemotaxis [226]. Interactions involving PMNs and such P. aeruginosa suspension aggregates are clinically highly relevant to CF airway disease and should be studied in great detail (Figure 1).
3.3. P. aeruginosa Resists NET-Mediated Attacks
The number of PMNs surrounding suspension aggregates in CF airways negatively correlates with bacterial growth rates in these aggregates [213]. These data suggest that PMNs limit P. aeruginosa growth in chronic CF, but instead of completely eliminating this bacterium, PMNs force P. aeruginosa to hide in the specialized environment of suspension aggregate cultures. The situation between PMNs and non-attached aggregates of P. aeruginosa can be considered as a compromise between the pathogen and the immune system. This view is somewhat new since the current opinion considers chronic CF airways as a hyperinflammatory environment. The presence of P. aeruginosa aggregates argues against this since bacteria found inside of these biofilms are nonmotile, nonvirulent showing a very slow metabolism. Several lines of evidence suggest that mucoid conversion and biofilm formation make P. aeruginosa resistant against most of the PMN antimicrobial effector mechanisms, including NET formation. Mucoid conversion of P. aeruginosa is a characteristic change over the course of CF lung infections and has been shown to prevent bacterial phagocytosis by macrophages and NET release from PMNs [227,228,229]. P. aeruginosa growing in biofilms activates the complement system less than planktonic forms [230]. P. aeruginosa LPS was suggested to be the main inducer of the classical pathway of complement activation by biofilms [230]. Biofilms trigger a strongly reduced respiratory burst in PMNs compared to P. aeruginosa planktonic forms [231]. P. aeruginosa bacteria residing in biofilms are able to respond to the presence of PMNs by inducing production of quorum sensing-controlled virulence factors, including rhamnolipids [232]. Rhamnolipids efficiently induce PMN lysis [233,234]. Induction of protective bacterial mechanism in response to PMNs supported a “launch a shield” model by which P. aeruginosa biofilms are surrounded by rhamnolipids that will eliminate immune cells including PMNs [43,232]. In support of this, inhibition of rhamnolipid synthesis in P. aeruginosa by inactivation of the rhamnolipid rhlA gene disabled bacterial protection against PMNs [235]. PMNs exhibiting some phagocytic activity have been observed on the surface of laboratory P. aeruginosa biofilms indicating that PMNs are capable of “chewing off” microbes from the biofilm surface [236,237]. Mucoid P. aeruginosa has also been reported to be fairly resistant against NET-mediated killing [170]. A main question remains how PMNs respond to suspension biofilms of P. aeruginosa (Figure 1).
3.4. Neutrophil Components Promote Biofilm Formation of P. aeruginosa
In addition to previous data indicating that P. aeruginosa is resistant in chronic CF airways against PMN effector mechanisms, several lines of evidence suggest that PMNs actually help biofilm growth of the bacterium. ecDNA is a main component of P. aeruginosa aggregates, and ecDNA obtained from necrotic PMNs has been shown to support aggregate growth of P. aeruginosa [214]. NET formation can likely be the source of PMN DNA in CF airways, since P. aeruginosa elicits robust NET release in PMNs [122,128,170,205]. Pyocyanin, a redox pigment and toxin of P. aeruginosa, is present in chronic CF airways and promotes the biofilm-forming ability of ecDNA [64,221]. Pyocyanin itself has also been shown to enhance NET release from human PMNs [130]. Whether PMNs respond to P. aeruginosa aggregates also with NET release remains an open question (Figure 1). If PMNs attempt to engulf bacterial suspension aggregates, NET extrusion could be their answer since according to a recent theory, PMNs preferentially release NETs in response to large particles that are impossible to phagocytose, including fungal hyphae and microcrystals [142,238,239] (Figure 1). PMNs have been shown to promote laboratory biofilm growth of P. aeruginosa in vitro [240,241,242]. Thus, P. aeruginosa not only becomes resistant against PMN effector mechanisms, but it uses PMNs to promote its biofilm growth in CF airways.
4. Conclusions
PMNs are well-equipped to eliminate P. aeruginosa. PMNs kill P. aeruginosa mainly intracellularly following phagocytosis. Neutrophils can also kill bacteria by releasing NETs or microvesicles. The proportion between intra- and extracellular killing mechanisms is crucial in determining the extents of PMN-mediated bacterial killing and collateral tissue damage in any disease. In immunocompromised individuals, such as CF patients, PMNs cannot clear P. aeruginosa. The interaction between neutrophils and P. aeruginosa is one of the most important features in CF airways. P. aeruginosa adapts to PMN-mediated attacks in CF by avoiding phagocytosis and forming resistant suspension microcolonies. PMNs are not capable of removing these suspension biofilms and release their dangerous antimicrobial cargo into the airway lumen to contribute to tissue damage. In summary, P. aeruginosa and PMNs engage in a complex, two-sided interaction in CF airways (Figure 1). It is essential to study its details to better understand CF airway inflammation and to design future neutrophil-based anti-inflammatory therapies in CF.
Conflicts of Interest
The author declares no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; nor in the decision to publish the results.
References
- Vasil, M.L. Pseudomonas aeruginosa: Biology, mechanisms of virulence, epidemiology. J. Pediatr. 1986, 108, 800–805. [Google Scholar] [CrossRef]
- Pollack, M. The virulence of Pseudomonas aeruginosa. Rev. Infect. Dis. 1984, 6 (Suppl. 3), S617–S626. [Google Scholar] [CrossRef] [PubMed]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.; Levy, S.B.; Jackson, R.W. Pseudomonas genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed]
- Rahme, L.G.; Stevens, E.J.; Wolfort, S.F.; Shao, J.; Tompkins, R.G.; Ausubel, F.M. Common virulence factors for bacterial pathogenicity in plants and animals. Science 1995, 268, 1899–1902. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.R.; Isabella, V.M.; Lewis, K. Pseudomonas aeruginosa biofilms in disease. Microb. Ecol. 2014, 68, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Huber, P.; Basso, P.; Reboud, E.; Attree, I. Pseudomonas aeruginosa renews its virulence factors. Environ. Microbiol. Rep. 2016, 8, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Rybtke, M.; Hultqvist, L.D.; Givskov, M.; Tolker-Nielsen, T. Pseudomonas aeruginosa biofilm infections: Community structure, antimicrobial tolerance and immune response. J. Mol. Biol. 2015, 427, 3628–3645. [Google Scholar] [CrossRef] [PubMed]
- Rasamiravaka, T.; Labtani, Q.; Duez, P.; El Jaziri, M. The formation of biofilms by Pseudomonas aeruginosa: A review of the natural and synthetic compounds interfering with control mechanisms. BioMed Res. Int. 2015, 2015, 759348. [Google Scholar] [CrossRef] [PubMed]
- Tolker-Nielsen, T. Pseudomonas aeruginosa biofilm infections: From molecular biofilm biology to new treatment possibilities. APMIS Suppl. 2014, 122, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Spencer, R.C. Predominant pathogens found in the European prevalence of infection in intensive care study. Eur. J. Clin. Microbiol. Infect. Dis. 1996, 15, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Koulenti, D.; Lisboa, T.; Brun-Buisson, C.; Krueger, W.; Macor, A.; Sole-Violan, J.; Diaz, E.; Topeli, A.; DeWaele, J.; Carneiro, A.; et al. Spectrum of practice in the diagnosis of nosocomial pneumonia in patients requiring mechanical ventilation in european intensive care units. Crit. Care Med. 2009, 37, 2360–2368. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.; Moore, G. Pseudomonas aeruginosa in hospital water systems: Biofilms, guidelines, and practicalities. J. Hosp. Infect. 2015, 89, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, S.; Sun, H.Y.; Yu, V.L.; Weingarten, J.A. Pneumonia due to Pseudomonas aeruginosa: Part I: Epidemiology, clinical diagnosis, and source. Chest 2011, 139, 909–919. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, M.; Lepper, P.M.; Haller, M. Ecology of Pseudomonas aeruginosa in the intensive care unit and the evolving role of water outlets as a reservoir of the organism. Am. J. Infect. Control 2005, 33, S41–S49. [Google Scholar] [CrossRef] [PubMed]
- Beer, D.; Vandermeer, B.; Brosnikoff, C.; Shokoples, S.; Rennie, R.; Forgie, S. Bacterial contamination of health care workers’ pagers and the efficacy of various disinfecting agents. Pediatr. Infect. Dis. J. 2006, 25, 1074–1075. [Google Scholar] [CrossRef] [PubMed]
- Adair, C.G.; Gorman, S.P.; Feron, B.M.; Byers, L.M.; Jones, D.S.; Goldsmith, C.E.; Moore, J.E.; Kerr, J.R.; Curran, M.D.; Hogg, G.; et al. Implications of endotracheal tube biofilm for ventilator-associated pneumonia. Intensive Care Med. 1999, 25, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.J.; Edwards, J.R.; Culver, D.H.; Gaynes, R.P. Nosocomial infections in combined medical-surgical intensive care units in the united states. Infect. Control Hosp. Epidemiol. 2000, 21, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Rolston, K.V.; Bodey, G.P. Pseudomonas aeruginosa infection in cancer patients. Cancer Investig. 1992, 10, 43–59. [Google Scholar] [CrossRef]
- Carratala, J.; Roson, B.; Fernandez-Sevilla, A.; Alcaide, F.; Gudiol, F. Bacteremic pneumonia in neutropenic patients with cancer: Causes, empirical antibiotic therapy, and outcome. Arch. Intern. Med. 1998, 158, 868–872. [Google Scholar] [CrossRef] [PubMed]
- Chatzinikolaou, I.; Abi-Said, D.; Bodey, G.P.; Rolston, K.V.; Tarrand, J.J.; Samonis, G. Recent experience with Pseudomonas aeruginosa bacteremia in patients with cancer: Retrospective analysis of 245 episodes. Arch. Intern. Med. 2000, 160, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Bellettato, C.M.; Braccioni, F.; Romagnoli, M.; Casolari, P.; Caramori, G.; Fabbri, L.M.; Johnston, S.L. Infections and airway inflammation in chronic obstructive pulmonary disease severe exacerbations. Am. J. Respir. Crit. Care Med. 2006, 173, 1114–1121. [Google Scholar] [CrossRef] [PubMed]
- Patel, I.S.; Seemungal, T.A.; Wilks, M.; Lloyd-Owen, S.J.; Donaldson, G.C.; Wedzicha, J.A. Relationship between bacterial colonisation and the frequency, character, and severity of COPD exacerbations. Thorax 2002, 57, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.J.; Dehnbostel, J.; Blackwell, T.S. Pseudomonas aeruginosa: Host defence in lung diseases. Respirology 2010, 15, 1037–1056. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.; Chodhari, R.; Collins, N.; Copeland, F.; Hall, P.; Harcourt, J.; Hariri, M.; Hogg, C.; Lucas, J.; Mitchison, H.M.; et al. Primary ciliary dyskinesia: Current state of the art. Arch. Dis. Child. 2007, 92, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, B.Z.; Haaf, J.B.; Leal, T.; Noel, S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: Current perspectives. Clin. Pharmacol. 2016, 8, 127–140. [Google Scholar] [PubMed]
- Cohen, T.S.; Prince, A. Cystic fibrosis: A mucosal immunodeficiency syndrome. Nat. Med. 2012, 18, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.Y.; Ji, X.B.; Lu, H.W.; Yang, J.W.; Xu, J.F. Distribution of major pathogens from sputum and bronchoalveolar lavage fluid in patients with noncystic fibrosis bronchiectasis: A systematic review. Chin. Med. J. (Engl.) 2015, 128, 2792–2797. [Google Scholar] [PubMed]
- Jacques, I.; Derelle, J.; Weber, M.; Vidailhet, M. Pulmonary evolution of cystic fibrosis patients colonized by Pseudomonas aeruginosa and/or burkholderia cepacia. Eur. J. Pediatr. 1998, 157, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Miller, S.; Sorscher, E.J. Cystic fibrosis. N. Engl. J. Med. 2005, 352, 1992–2001. [Google Scholar] [CrossRef] [PubMed]
- Talwalkar, J.S.; Murray, T.S. The approach to Pseudomonas aeruginosa in cystic fibrosis. Clin. Chest Med. 2016, 37, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.L.; Gibson, R.L.; McNamara, S.; Yim, D.; Emerson, J.; Rosenfeld, M.; Hiatt, P.; McCoy, K.; Castile, R.; Smith, A.L.; et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Dis. 2001, 183, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 2001, 9, 50–52. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Hoiby, N.; Ciofu, O.; Bjarnsholt, T. Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol. 2010, 5, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Hoiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Microbiol. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Haiko, J.; Westerlund-Wikstrom, B. The role of the bacterial flagellum in adhesion and virulence. Biology 2013, 2, 1242–1267. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Verma, A.; Kuravi, S.; Si-Tahar, M.; Chignard, M.; Ramphal, R. The role of flagellin versus motility in acute lung disease caused by Pseudomonas aeruginosa. J. Infect. Dis. 2007, 196, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.C.; Jyot, J.; Goodman, A.L.; Ramphal, R.; Lory, S. Pseudomonas aeruginosa regulates flagellin expression as part of a global response to airway fluid from cystic fibrosis patients. Proc. Natl. Acad. Sci. USA 2004, 101, 6664–6668. [Google Scholar] [CrossRef] [PubMed]
- Picioreanu, C.; Kreft, J.U.; Klausen, M.; Haagensen, J.A.; Tolker-Nielsen, T.; Molin, S. Microbial motility involvement in biofilm structure formation—A 3D modelling study. Water Sci. Technol. 2007, 55, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.; Aaes-Jorgensen, A.; Molin, S.; Tolker-Nielsen, T. Involvement of bacterial migration in the development of complex multicellular structures in Pseudomonas aeruginosa biofilms. Mol. Microbiol. 2003, 50, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Alhede, M.; Bjarnsholt, T.; Givskov, M.; Alhede, M. Pseudomonas aeruginosa biofilms: Mechanisms of immune evasion. Adv. Appl. Microbiol. 2014, 86, 1–40. [Google Scholar] [PubMed]
- Jensen, P.O.; Givskov, M.; Bjarnsholt, T.; Moser, C. The immune system vs. Pseudomonas aeruginosa biofilms. FEMS Immunol. Med. Microbiol. 2010, 59, 292–305. [Google Scholar] [CrossRef]
- Wagner, V.E.; Iglewski, B.H.P. Aeruginosa biofilms in cf infection. Clin. Rev. Allergy Immunol. 2008, 35, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.M.; Pereira, M.O. Pseudomonas aeruginosa diversification during infection development in cystic fibrosis lungs—A review. Pathogens 2014, 3, 680–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, T.; Sullivan, K.E. Infections in patients with inherited defects in phagocytic function. Clin. Microbiol. Rev. 2003, 16, 597–621. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.Y.; Priebe, G.P.; Ray, C.; Van Rooijen, N.; Pier, G.B. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect. Immun. 2009, 77, 5300–5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.C.; Strieter, R.M.; Mehrad, B.; Newstead, M.W.; Zeng, X.; Standiford, T.J. CXC chemokine receptor CXCR2 is essential for protective innate host response in murine Pseudomonas aeruginosa pneumonia. Infect. Immun. 2000, 68, 4289–4296. [Google Scholar] [CrossRef] [PubMed]
- Kurahashi, K.; Sawa, T.; Ota, M.; Kajikawa, O.; Hong, K.; Martin, T.R.; Wiener-Kronish, J.P. Depletion of phagocytes in the reticuloendothelial system causes increased inflammation and mortality in rabbits with Pseudomonas aeruginosa pneumonia. Am. J. Physiol Lung Cell. Mol. Physiol. 2009, 296, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Mijares, L.A.; Wangdi, T.; Sokol, C.; Homer, R.; Medzhitov, R.; Kazmierczak, B.I. Airway epithelial MyD88 restores control of Pseudomonas aeruginosa murine infection via an IL-1-dependent pathway. J. Immunol. 2011, 186, 7080–7088. [Google Scholar] [CrossRef] [PubMed]
- Anas, A.A.; van Lieshout, M.H.; Claushuis, T.A.; de Vos, A.F.; Florquin, S.; de Boer, O.J.; Hou, B.; Van’t Veer, C.; van der Poll, T. Lung epithelial MyD88 drives early pulmonary clearance of Pseudomonas aeruginosa by a flagellin dependent mechanism. Am. J. Physiol Lung Cell. Mol. Physiol. 2016, 311, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, P.E.; McHugh, B.; Gwyer Findlay, E.; Mackellar, A.; Mackenzie, K.J.; Gallo, R.L.; Govan, J.R.; Simpson, A.J.; Davidson, D.J. Cathelicidin host defence peptide augments clearance of pulmonary Pseudomonas aeruginosa infection by its influence on neutrophil function in vivo. PLoS ONE 2014, 9, e99029. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.C.; Reins, R.Y.; Gallo, R.L.; McDermott, A.M. Cathelicidin-deficient (Cnlp−/−) mice show increased susceptibility to Pseudomonas aeruginosa keratitis. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4498–4508. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Hou, Y.; Sun, F.; Yang, Z.; Li, C. Dysregulated chemokine signaling in cystic fibrosis lung disease: A potential therapeutic target. Curr. Drug Targets 2016, 17, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Colombo, C.; Costantini, D.; Rocchi, A.; Cariani, L.; Garlaschi, M.L.; Tirelli, S.; Calori, G.; Copreni, E.; Conese, M. Cytokine levels in sputum of cystic fibrosis patients before and after antibiotic therapy. Pediatr. Pulmonol. 2005, 40, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Hamblett, N.; Aitken, M.L.; Accurso, F.J.; Kronmal, R.A.; Konstan, M.W.; Burns, J.L.; Sagel, S.D.; Ramsey, B.W. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Okamoto, K.; Rubin, B.K. Pulmonary function is negatively correlated with sputum inflammatory markers and cough clearability in subjects with cystic fibrosis but not those with chronic bronchitis. Chest 2006, 129, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Osika, E.; Cavaillon, J.M.; Chadelat, K.; Boule, M.; Fitting, C.; Tournier, G.; Clement, A. Distinct sputum cytokine profiles in cystic fibrosis and other chronic inflammatory airway disease. Eur. Respir J. 1999, 14, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zoumot, Z.; Wilson, R. Respiratory infection in noncystic fibrosis bronchiectasis. Curr. Opin. Infect. Dis. 2010, 23, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Bodini, A.; D’Orazio, C.; Peroni, D.; Corradi, M.; Folesani, G.; Baraldi, E.; Assael, B.M.; Boner, A.; Piacentini, G.L. Biomarkers of neutrophilic inflammation in exhaled air of cystic fibrosis children with bacterial airway infections. Pediatr. Pulmonol. 2005, 40, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Copreni, E.; Di Gioia, S.; De Rinaldis, P.; Fumarulo, R. Neutrophil recruitment and airway epithelial cell involvement in chronic cystic fibrosis lung disease. J. Cyst. Fibros 2003, 2, 129–135. [Google Scholar] [CrossRef]
- Rada, B.; Gardina, P.; Myers, T.G.; Leto, T.L. Reactive oxygen species mediate inflammatory cytokine release and EGFR-dependent mucin secretion in airway epithelial cells exposed to pseudomonas pyocyanin. Mucosal Immunol. 2011, 4, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Pyocyanin effects on respiratory epithelium: Relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol. 2013, 21, 73–81. [Google Scholar] [CrossRef] [PubMed]
- DiMango, E.; Zar, H.J.; Bryan, R.; Prince, A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J. Clin. Investig. 1995, 96, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.P.; Inoue, H.; Richman-Eisenstat, J.; Grunberger, D.; Jorens, P.G.; Housset, B.; Pittet, J.F.; Wiener-Kronish, J.P.; Nadel, J.A. Novel pseudomonas product stimulates interleukin-8 production in airway epithelial cells in vitro. J. Clin. Investig. 1994, 93, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Yoshimura, K.; McElvaney, N.G.; Crystal, R.G. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J. Clin Investig. 1992, 89, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Konstan, M.W.; Berger, M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J. Allergy Clin. Immunol. 1999, 104, 72–78. [Google Scholar] [CrossRef]
- Tabary, O.; Zahm, J.M.; Hinnrasky, J.; Couetil, J.P.; Cornillet, P.; Guenounou, M.; Gaillard, D.; Puchelle, E.; Jacquot, J. Selective up-regulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am. J. Pathol. 1998, 153, 921–930. [Google Scholar] [CrossRef]
- Tabary, O.; Escotte, S.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Jacquot, J. Genistein inhibits constitutive and inducible nfkappab activation and decreases IL-8 production by human cystic fibrosis bronchial gland cells. Am. J. Pathol. 1999, 155, 473–481. [Google Scholar] [CrossRef]
- Tang, A.C.; Saferali, A.; He, G.; Sandford, A.J.; Strug, L.J.; Turvey, S.E. Endoplasmic reticulum stress regulates chemokine production in cystic fibrosis airway cells through STAT3 modulation. J. Infect. Dis. 2017, 215, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Stecenko, A.A.; King, G.; Torii, K.; Breyer, R.M.; Dworski, R.; Blackwell, T.S.; Christman, J.W.; Brigham, K.L. Dysregulated cytokine production in human cystic fibrosis bronchial epithelial cells. Inflammation 2001, 25, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.; Stecenko, A.A.; King, G.; Blackwell, T.R.; Brigham, K.L.; Christman, J.W.; Blackwell, T.S. Exaggerated activation of nuclear factor-kappab and altered ikappab-beta processing in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2000, 23, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Mackerness, K.J.; Jenkins, G.R.; Bush, A.; Jose, P.J. Characterisation of the range of neutrophil stimulating mediators in cystic fibrosis sputum. Thorax 2008, 63, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Hopken, U.E.; Lu, B.; Gerard, N.P.; Gerard, C. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature 1996, 383, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Fick, R.B., Jr.; Robbins, R.A.; Squier, S.U.; Schoderbek, W.E.; Russ, W.D. Complement activation in cystic fibrosis respiratory fluids: In vivo and in vitro generation of C5a and chemotactic activity. Pediatr. Res. 1986, 20, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, C.W.; Tambourgi, D.V.; Clark, H.W.; Hoong, S.J.; Spiller, O.B.; McGreal, E.P. Mechanism of neutrophil dysfunction: Neutrophil serine proteases cleave and inactivate the C5a receptor. J. Immunol. 2014, 192, 1787–1795. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Sharma, A.; Jen, R.; Hirschfeld, A.F.; Chilvers, M.A.; Lavoie, P.M.; Turvey, S.E. Inflammasome-mediated IL-1beta production in humans with cystic fibrosis. PLoS ONE 2012, 7, e37689. [Google Scholar]
- Bakele, M.; Joos, M.; Burdi, S.; Allgaier, N.; Poschel, S.; Fehrenbacher, B.; Schaller, M.; Marcos, V.; Kummerle-Deschner, J.; Rieber, N.; et al. Localization and functionality of the inflammasome in neutrophils. J. Biol. Chem. 2014, 289, 5320–5329. [Google Scholar] [CrossRef] [PubMed]
- Cromwell, O.; Walport, M.J.; Morris, H.R.; Taylor, G.W.; Hodson, M.E.; Batten, J.; Kay, A.B. Identification of leukotrienes d and b in sputum from cystic fibrosis patients. Lancet 1981, 2, 164–165. [Google Scholar] [CrossRef]
- Cromwell, O.; Walport, M.J.; Taylor, G.W.; Morris, H.R.; O’Driscoll, B.R.; Kay, A.B. Identification of leukotrienes in the sputum of patients with cystic fibrosis. Adv. Prostaglandin Thromboxane Leukot. Res. 1982, 9, 251–257. [Google Scholar] [CrossRef]
- Konstan, M.W.; Walenga, R.W.; Hilliard, K.A.; Hilliard, J.B. Leukotriene B4 markedly elevated in the epithelial lining fluid of patients with cystic fibrosis. Am. Rev. Respir. Dis. 1993, 148, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Dayer Pastore, F.; Schlegel-Haueter, S.E.; Belli, D.C.; Rochat, T.; Dudez, T.S.; Suter, S. Chemotactic factors in bronchial secretions of cystic fibrosis patients. J. Infect. Dis. 1998, 177, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.H.; Sorrelli, T.C. Decreased polymorphonuclear leucocyte chemotactic response to leukotriene B4 in cystic fibrosis. Clin. Exp. Immunol. 1992, 89, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Bayes, H.K.; Ritchie, N.D.; Evans, T.J. IL-17 is required for control of chronic lung infection caused by Pseudomonas aeruginosa. Infect. Immun. 2016, 84, 3507–3516. [Google Scholar] [CrossRef] [PubMed]
- Decraene, A.; Willems-Widyastuti, A.; Kasran, A.; De Boeck, K.; Bullens, D.M.; Dupont, L.J. Elevated expression of both mRNA and protein levels of IL-17a in sputum of stable cystic fibrosis patients. Respir. Res. 2010, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; Kolls, J.K. IL-23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am. J. Physiol Lung Cell. Mol. Physiol. 2007, 292, L519–L528. [Google Scholar] [CrossRef] [PubMed]
- Brodlie, M.; McKean, M.C.; Johnson, G.E.; Anderson, A.E.; Hilkens, C.M.; Fisher, A.J.; Corris, P.A.; Lordan, J.L.; Ward, C. Raised interleukin-17 is immunolocalised to neutrophils in cystic fibrosis lung disease. Eur. Respir. J. 2011, 37, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Bonfield, T.L.; Chmiel, J.F.; Pearlman, E. Neutrophils from F508del cystic fibrosis patients produce IL-17a and express IL-23—Dependent IL-17RC. Clin. Immunol. 2016, 170, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; Kolls, J.K. Il-17 in cystic fibrosis: More than just Th17 cells. Am. J. Respir. Crit. Care Med. 2011, 184, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Dubin, P.J.; McAllister, F.; Kolls, J.K. Is cystic fibrosis a Th17 disease? Inflamm. Res. 2007, 56, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Lee, M.; Bae, Y.S. Formyl peptide receptors in cellular differentiation and inflammatory diseases. J. Cell Biochem. 2017, 9999, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Balloy, V.; Thevenot, G.; Bienvenu, T.; Morand, P.; Corvol, H.; Clement, A.; Ramphal, R.; Hubert, D.; Chignard, M. Flagellin concentrations in expectorations from cystic fibrosis patients. BMC Pulm. Med. 2014, 14, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutterwala, F.S.; Mijares, L.A.; Li, L.; Ogura, Y.; Kazmierczak, B.I.; Flavell, R.A. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 2007, 204, 3235–3245. [Google Scholar] [CrossRef] [PubMed]
- Hansch, G.M.; Prior, B.; Brenner-Weiss, G.; Obst, U.; Overhage, J. The pseudomonas quinolone signal (PQS) stimulates chemotaxis of polymorphonuclear neutrophils. J. Appl. Biomater. Funct. Mater. 2014, 12, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.K.; Geiszt, M.; Kaldi, K.; Timar, C.; Ligeti, E. Dual role of phagocytic nadph oxidase in bacterial killing. Blood 2004, 104, 2947–2953. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Utomo, A.; Cullere, X.; Choi, M.M.; Milner, D.A., Jr.; Venkatesh, D.; Yun, S.H.; Mayadas, T.N. The beta-glucan receptor dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011, 10, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.W.; McVicar, D.W. TREM and Trem-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.; Girardin, S.E.; Boneca, I.G.; Lemos, R.; Bozza, M.T.; Domingues, R.C.; Coyle, A.J.; Bertin, J.; Philpott, D.J.; et al. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J. Biol. Chem. 2005, 280, 36714–36718. [Google Scholar] [CrossRef]
- Pollard, A.J.; Heale, J.P.; Tsang, A.; Massing, B.; Speert, D.P. Nonopsonic phagocytosis of pseudomonas aeruginoas: Insights from an infant with leukocyte adhesion deficiency. Pediatr. Infect. Dis. J. 2001, 20, 452–454. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Patankar, Y.R.; Berwin, B. Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am. J. Physiol Lung Cell. Mol. Physiol. 2014, 306, L591–L603. [Google Scholar] [CrossRef] [PubMed]
- Heale, J.P.; Pollard, A.J.; Stokes, R.W.; Simpson, D.; Tsang, A.; Massing, B.; Speert, D.P. Two distinct receptors mediate nonopsonic phagocytosis of different strains of Pseudomonas aeruginosa. J. Infect. Dis. 2001, 183, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Amiel, E.; Lovewell, R.R.; O’Toole, G.A.; Hogan, D.A.; Berwin, B. Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect. Immun. 2010, 78, 2937–2945. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family nadph oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [PubMed]
- Rada, B.; Hably, C.; Meczner, A.; Timar, C.; Lakatos, G.; Enyedi, P.; Ligeti, E. Role of nox2 in elimination of microorganisms. Semin. Immunopathol. 2008, 30, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.K.; Geiszt, M.; Hably, C.; Ligeti, E. Consequences of the electrogenic function of the phagocytic nadph oxidase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002, 416, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Schultz, H.; Weiss, J.P. The bactericidal/permeability-increasing protein (BPI) in infection and inflammatory disease. Clin. Chim. Acta 2007, 384, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, I.; Schonefeld, M.; Aichele, D.; Groer, G.; Gessner, A.; Schnare, M. Murine bactericidal/permeability-increasing protein inhibits the endotoxic activity of lipopolysaccharide and gram-negative bacteria. J. Immunol. 2008, 180, 7546–7552. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol 2006, 6, 541–550. [Google Scholar] [CrossRef]
- Hirche, T.O.; Benabid, R.; Deslee, G.; Gangloff, S.; Achilefu, S.; Guenounou, M.; Lebargy, F.; Hancock, R.E.; Belaaouaj, A. Neutrophil elastase mediates innate host protection against Pseudomonas aeruginosa. J. Immunol. 2008, 181, 4945–4954. [Google Scholar] [CrossRef] [PubMed]
- Sedor, J.; Hogue, L.; Akers, K.; Boslaugh, S.; Schreiber, J.; Ferkol, T. Cathepsin-G interferes with clearance of Pseudomonas aeruginosa from mouse lungs. Pediatr. Res. 2007, 61, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Tai, K.P.; Kamdar, K.; Yamaki, J.; Le, V.V.; Tran, D.; Tran, P.; Selsted, M.E.; Ouellette, A.J.; Wong-Beringer, A. Microbicidal effects of alpha- and theta-defensins against antibiotic-resistant Staphylococcus aureus and Pseudomonas aeruginosa. Innate Immun. 2015, 21, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, T.; Liu, M.; Hardej, D.; Liu, X.; Cantor, J. Aerosolized recombinant human lysozyme ameliorates Pseudomonas aeruginosa-induced pneumonia in hamsters. Exp. Lung Res. 2010, 36, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Cole, A.M.; Thapa, D.R.; Gabayan, V.; Liao, H.I.; Liu, L.; Ganz, T. Decreased clearance of Pseudomonas aeruginosa from airways of mice deficient in lysozyme M. J. Leuk. Biol. 2005, 78, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Winn, M.; Pang, L.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L.; Rada, B. Release of cystic fibrosis airway inflammatory markers from Pseudomonas aeruginosa-stimulated human neutrophils involves nadph oxidase-dependent extracellular DNA trap formation. J. Immunol. 2014, 192, 4728–4738. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Kuijpers, T.W.; Wirawan, E.; Lippens, S.; Vandenabeele, P.; Vanden Berghe, T. Dying for a cause: Netosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011, 18, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.G.; Floyd, M.; Winn, M.; Moskowitz, S.M.; Rada, B. Net formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol. Lett. 2014, 160, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, M.; Nur, E.; Biemond, B.J.; van Mierlo, G.J.; Solati, S.; Brandjes, D.P.; Otten, H.M.; Schnog, J.J.; Zeerleder, S.; Curama Study, G. Nucleosomes and neutrophil activation in sickle cell disease painful crisis. Haematologica 2013, 98, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Nakazawa, D.; Shida, H.; Miyoshi, A.; Kusunoki, Y.; Tomaru, U.; Ishizu, A. Netosis markers: Quest for specific, objective, and quantitative markers. Clin. Chim. Acta 2016, 459, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Stoiber, W.; Obermayer, A.; Steinbacher, P.; Krautgartner, W.D. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules 2015, 5, 702–723. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Jendrysik, M.A.; Pang, L.; Hayes, C.P.; Yoo, D.G.; Park, J.J.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L. Pyocyanin-enhanced neutrophil extracellular trap formation requires the nadph oxidase. PLoS ONE 2013, 8, e54205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Hayes, C.P.; Buac, K.; Yoo, D.G.; Rada, B. Pseudogout-associated inflammatory calcium pyrophosphate dihydrate microcrystals induce formation of neutrophil extracellular traps. J. Immunol. 2013, 190, 6488–6500. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Leshner, M.; Wang, S.; Lewis, C.; Zheng, H.; Chen, X.A.; Santy, L.; Wang, Y. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front. Immunol. 2012, 3, 307. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M.; Kubes, P. Pondering neutrophil extracellular traps with healthy skepticism. Cell. Microbiol. 2016, 18, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Halverson, T.W.; Wilton, M.; Poon, K.K.; Petri, B.; Lewenza, S. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 2015, 11, e1004593. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. Netosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Albrett, A.M.; Kettle, A.J.; Winterbourn, C.C. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J. Leuk. Biol. 2012, 91, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and netosis in autoimmune and renal diseases. Nat. Rev. Nephrol 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [PubMed]
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819. [Google Scholar] [CrossRef] [PubMed]
- Schonrich, G.; Raftery, M.J. Neutrophil extracellular traps go viral. Front. Immunol. 2016, 7, 366. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.K.; Cedervall, J. Netosis in cancer—Platelet-neutrophil crosstalk promotes tumor-associated pathology. Front. Immunol. 2016, 7, 373. [Google Scholar] [CrossRef] [PubMed]
- Timar, C.I.; Lorincz, A.M.; Csepanyi-Komi, R.; Valyi-Nagy, A.; Nagy, G.; Buzas, E.I.; Ivanyi, Z.; Kittel, A.; Powell, D.W.; McLeish, K.R.; et al. Antibacterial effect of microvesicles released from human neutrophilic granulocytes. Blood 2013, 121, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.L., III; Kuethe, J.W.; Caldwell, C.C. Neutrophil derived microvesicles: Emerging role of a key mediator to the immune response. Endocr. Metab. Immune Disord. Drug Targets 2014, 14, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, J.; Wolach, O.; Gavrieli, R.; Wolach, B. Infections associated with chronic granulomatous disease: Linking genetics to phenotypic expression. Expert Rev. Anti-Infect. Ther. 2012, 10, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C. Disorders of neutrophil function: An overview. Methods Mol. Biol. 2014, 1124, 501–515. [Google Scholar] [PubMed]
- Nauseef, W.M. Myeloperoxidase deficiency. Hematol. Pathol. 1990, 4, 165–178. [Google Scholar] [PubMed]
- Speert, D.P.; Bond, M.; Woodman, R.C.; Curnutte, J.T. Infection with pseudomonas cepacia in chronic granulomatous disease: Role of nonoxidative killing by neutrophils in host defense. J. Infect. Dis. 1994, 170, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Mathee, K.; Ciofu, O.; Sternberg, C.; Lindum, P.W.; Campbell, J.I.; Jensen, P.; Johnsen, A.H.; Givskov, M.; Ohman, D.E.; Molin, S.; et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: A mechanism for virulence activation in the cystic fibrosis lung. Microbiology 1999, 145 Pt 6, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.I.; Mukherjee, S. Treatment for chronic methicillin-sensitive staphylococcus aureus pulmonary infection in people with cystic fibrosis. Cochrane Database Syst. Rev. 2016, 3, CD011581. [Google Scholar] [PubMed]
- Buvelot, H.; Posfay-Barbe, K.M.; Linder, P.; Schrenzel, J.; Krause, K.H. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Regelmann, W.E.; Siefferman, C.M.; Herron, J.M.; Elliott, G.R.; Clawson, C.C.; Gray, B.H. Sputum peroxidase activity correlates with the severity of lung disease in cystic fibrosis. Pediatr. Pulmonol. 1995, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Garner, H.P.; Phillips, J.R.; Herron, J.G.; Severson, S.J.; Milla, C.E.; Regelmann, W.E. Peroxidase activity within circulating neutrophils correlates with pulmonary phenotype in cystic fibrosis. J. Lab. Clin. Med. 2004, 144, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef]
- Davis, S.D.; Ferkol, T. Identifying the origins of cystic fibrosis lung disease. N. Engl. J. Med. 2013, 368, 2026–2028. [Google Scholar] [CrossRef] [PubMed]
- Watt, A.P.; Courtney, J.; Moore, J.; Ennis, M.; Elborn, J.S. Neutrophil cell death, activation and bacterial infection in cystic fibrosis. Thorax 2005, 60, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Waters, V.J.; Stanojevic, S.; Sonneveld, N.; Klingel, M.; Grasemann, H.; Yau, Y.C.; Tullis, E.; Wilcox, P.; Freitag, A.; Chilvers, M.; et al. Factors associated with response to treatment of pulmonary exacerbations in cystic fibrosis patients. J. Cyst. Fibros 2015, 14, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, K.; Nakamura, H.; Trapnell, B.C.; Chu, C.S.; Dalemans, W.; Pavirani, A.; Lecocq, J.P.; Crystal, R.G. Expression of the cystic fibrosis transmembrane conductance regulator gene in cells of non-epithelial origin. Nucleic Acids Res. 1991, 19, 5417–5423. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.G.; Bonvillain, R.W.; Valentine, V.G.; Lombard, G.A.; LaPlace, S.G.; Nauseef, W.M.; Wang, G. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J. Leuk. Biol. 2008, 83, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.P.; Zhou, Y.; Song, K.; Hodges, C.A.; Drumm, M.L.; Wang, G. Neutrophil-mediated phagocytic host defense defect in myeloid CFTR-inactivated mice. PLoS ONE 2014, 9, e106813. [Google Scholar] [CrossRef] [PubMed]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by cftr potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Graff, I.; Schram-Doumont, A.; Szpirer, C. Defective protein kinase C-mediated actions in cystic fibrosis neutrophils. Cell. Signal. 1991, 3, 259–266. [Google Scholar] [CrossRef]
- Kemp, T.; Schram-Doumont, A.; van Geffel, R.; Kram, R.; Szpirer, C. Alteration of the N-formyl-methionyl-leucyl-phenylalanine-induced response in cystic fibrosis neutrophils. Pediatr. Res. 1986, 20, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Cabrini, G.; De Togni, P. Increased cytosolic calcium in cystic fibrosis neutrophils effect on stimulus-secretion coupling. Life Sci. 1985, 36, 1561–1567. [Google Scholar] [CrossRef]
- Galant, S.P.; Norton, L.; Herbst, J.; Wood, C. Impaired beta adrenergic receptor binding and function in cystic fibrosis neutrophils. J. Clin. Investig. 1981, 68, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Young, R.L.; Malcolm, K.C.; Kret, J.E.; Caceres, S.M.; Poch, K.R.; Nichols, D.P.; Taylor-Cousar, J.L.; Saavedra, M.T.; Randell, S.H.; Vasil, M.L.; et al. Neutrophil extracellular trap (net)-mediated killing of Pseudomonas aeruginosa: Evidence of acquired resistance within the CF airway, independent of cftr. PLoS ONE 2011, 6, e23637. [Google Scholar] [CrossRef] [PubMed]
- McKeon, D.J.; Cadwallader, K.A.; Idris, S.; Cowburn, A.S.; Pasteur, M.C.; Barker, H.; Haworth, C.S.; Bilton, D.; Chilvers, E.R.; Condliffe, A.M. Cystic fibrosis neutrophils have normal intrinsic reactive oxygen species generation. Eur. Respir. J. 2010, 35, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Dinauer, M.C. Primary immune deficiencies with defects in neutrophil function. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Ralhan, A.; Hartl, D. Neutrophils in cystic fibrosis. Biol. Chem. 2016, 397, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Gifford, A.M.; Chalmers, J.D. The role of neutrophils in cystic fibrosis. Curr. Opin. Hematol. 2014, 21, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Hayes, E.; Pohl, K.; McElvaney, N.G.; Reeves, E.P. The cystic fibrosis neutrophil: A specialized yet potentially defective cell. Arch. Immunol. Ther. Exp. 2011, 59, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Laval, J.; Touhami, J.; Herzenberg, L.A.; Conrad, C.; Taylor, N.; Battini, J.L.; Sitbon, M.; Tirouvanziam, R. Metabolic adaptation of neutrophils in cystic fibrosis airways involves distinct shifts in nutrient transporter expression. J. Immunol. 2013, 190, 6043–6050. [Google Scholar] [CrossRef] [PubMed]
- Makam, M.; Diaz, D.; Laval, J.; Gernez, Y.; Conrad, C.K.; Dunn, C.E.; Davies, Z.A.; Moss, R.B.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Activation of critical, host-induced, metabolic and stress pathways marks neutrophil entry into cystic fibrosis lungs. Proc. Natl. Acad. Sci. USA 2009, 106, 5779–5783. [Google Scholar] [CrossRef] [PubMed]
- Tirouvanziam, R.; Gernez, Y.; Conrad, C.K.; Moss, R.B.; Schrijver, I.; Dunn, C.E.; Davies, Z.A.; Herzenberg, L.A.; Herzenberg, L.A. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc. Natl. Acad. Sci. USA 2008, 105, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- Petit-Bertron, A.F.; Tabary, O.; Corvol, H.; Jacquot, J.; Clement, A.; Cavaillon, J.M.; Adib-Conquy, M. Circulating and airway neutrophils in cystic fibrosis display different TLR expression and responsiveness to interleukin-10. Cytokine 2008, 41, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Tabary, O.; Corvol, H.; Boncoeur, E.; Chadelat, K.; Fitting, C.; Cavaillon, J.M.; Clement, A.; Jacquot, J. Adherence of airway neutrophils and inflammatory response are increased in CF airway epithelial cell-neutrophil interactions. Am. J. Physiol Lung Cell. Mol. Physiol. 2006, 290, 588–596. [Google Scholar] [CrossRef]
- Conese, M.; Castellani, S.; Lepore, S.; Palumbo, O.; Manca, A.; Santostasi, T.; Polizzi, A.M.; Copetti, M.; Di Gioia, S.; Casavola, V.; et al. Evaluation of genome-wide expression profiles of blood and sputum neutrophils in cystic fibrosis patients before and after antibiotic therapy. PLoS ONE 2014, 9, e104080. [Google Scholar] [CrossRef] [PubMed]
- Houston, N.; Stewart, N.; Smith, D.S.; Bell, S.C.; Champion, A.C.; Reid, D.W. Sputum neutrophils in cystic fibrosis patients display a reduced respiratory burst. J. Cyst. Fibros 2013, 12, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Koller, B.; Kappler, M.; Latzin, P.; Gaggar, A.; Schreiner, M.; Takyar, S.; Kormann, M.; Kabesch, M.; Roos, D.; Griese, M.; et al. TLR expression on neutrophils at the pulmonary site of infection: TLR1/TLR2-mediated up-regulation of TLR5 expression in cystic fibrosis lung disease. J. Immunol. 2008, 181, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.L.; Scott, S.F.; Knight, R.A.; Marriott, C.; Ranasinha, C.; Hodson, M.E. In vivo effects of recombinant human DNase I on sputum in patients with cystic fibrosis. Thorax 1996, 51, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Shak, S.; Capon, D.J.; Hellmiss, R.; Marsters, S.A.; Baker, C.L. Recombinant human DNase I reduces the viscosity of cystic fibrosis sputum. Proc. Natl. Acad. Sci. USA 1990, 87, 9188–9192. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.L.; Gibson, P.G.; Carty, K.; Cai, Y.; Francis, J.L. Airway inflammation after treatment with aerosolized deoxyribonuclease in cystic fibrosis. Pediatr. Pulmonol. 1998, 26, 97–100. [Google Scholar] [CrossRef]
- Bucki, R.; Cruz, K.; Pogoda, K.; Eggert, A.; Chin, L.; Ferrin, M.; Imbesi, G.; Hadjiliadis, D.; Janmey, P.A. Enhancement of pulmozyme activity in purulent sputum by combination with poly-aspartic acid or gelsolin. J. Cyst. Fibros 2015, 14, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.I.; Bush, A.; Canny, G.J.; Colin, A.A.; Fuchs, H.J.; Geddes, D.M.; Johnson, C.A.; Light, M.C.; Scott, S.F.; Tullis, D.E.; et al. Recombinant human DNase I in cystic fibrosis patients with severe pulmonary disease: A short-term, double-blind study followed by six months open-label treatment. Eur. Respir. J. 1995, 8, 954–958. [Google Scholar] [PubMed]
- Riethmueller, J.; Vonthein, R.; Borth-Bruhns, T.; Grassme, H.; Eyrich, M.; Schilbach, K.; Stern, M.; Gulbins, E. DNA quantification and fragmentation in sputum after inhalation of recombinant human deoxyribonuclease. Cell. Physiol. Biochem. 2008, 22, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Lethem, M.I.; James, S.L.; Marriott, C.; Burke, J.F. The origin of DNA associated with mucus glycoproteins in cystic fibrosis sputum. Eur. Respir. J. 1990, 3, 19–23. [Google Scholar] [PubMed]
- Manzenreiter, R.; Kienberger, F.; Marcos, V.; Schilcher, K.; Krautgartner, W.D.; Obermayer, A.; Huml, M.; Stoiber, W.; Hector, A.; Griese, M.; et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros 2012, 11, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving dnase therapy. PLoS ONE 2011, 6, e28526. [Google Scholar] [CrossRef] [PubMed]
- Carswell, F.; Robinson, D.W.; Ward, C.C.; Waterfield, M.R. Deoxyribonucleic acid output in the sputum from cystic fibrosis patients. Eur. J. Respir. Dis. 1984, 65, 53–57. [Google Scholar] [PubMed]
- Dubois, A.V.; Gauthier, A.; Brea, D.; Varaigne, F.; Diot, P.; Gauthier, F.; Attucci, S. Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am. J. Respir. Cell Mol. Biol. 2012, 47, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Torphy, T.J.; Allen, J.; Cantin, A.M.; Konstan, M.W.; Accurso, F.J.; Joseloff, E.; Ratjen, F.A.; Chmiel, J.F.; Antiinflammatory Therapy Working Group. Considerations for the conduct of clinical trials with antiinflammatory agents in cystic fibrosis. A cystic fibrosis foundation workshop report. Ann. Am. Thorac. Soc. 2015, 12, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Dinwiddie, R. Anti-inflammatory therapy in cystic fibrosis. J. Cyst. Fibros 2005, 4 (Suppl. 2), 45–48. [Google Scholar] [CrossRef]
- Lands, L.C.; Stanojevic, S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2016, 4, CD001505. [Google Scholar] [PubMed]
- Frerichs, C.; Smyth, A. Treatment strategies for cystic fibrosis: What’s in the pipeline? Expert Opin. Pharmacother. 2009, 10, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Balfour-Lynn, I.M. The protease-antiprotease battle in the cystic fibrosis lung. J. R. Soc. Med. 1999, 92 (Suppl. 37), 23–30. [Google Scholar] [PubMed]
- Tsai, Y.F.; Hwang, T.L. Neutrophil elastase inhibitors: A patent review and potential applications for inflammatory lung diseases (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Lee, D.M.; Kolbinger, F.; Antoni, C. Effect of IL-17a blockade with secukinumab in autoimmune diseases. Ann. Rheum. Dis. 2013, 72 (Suppl. 2), ii116–ii123. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Hayes, S.M.; O’Toole, G.A.; Berwin, B. Pseudomonas aeruginosa flagellar motility activates the phagocyte PI3K/AKT pathway to induce phagocytic engulfment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Floyd, M.; Winn, M.; Cullen, C.; Sil, P.; Chassaing, B.; Yoo, D.G.; Gewirtz, A.T.; Goldberg, J.B.; McCarter, L.L.; Rada, B. Swimming motility mediates the formation of neutrophil extracellular traps induced by flagellated Pseudomonas aeruginosa. PLoS Pathog. 2016, 12, e1005987. [Google Scholar] [CrossRef] [PubMed]
- Mahenthiralingam, E.; Campbell, M.E.; Speert, D.P. Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 1994, 62, 596–605. [Google Scholar] [PubMed]
- Luzar, M.A.; Montie, T.C. Avirulence and altered physiological properties of cystic fibrosis strains of Pseudomonas aeruginosa. Infect. Immun. 1985, 50, 572–576. [Google Scholar] [PubMed]
- Luzar, M.A.; Thomassen, M.J.; Montie, T.C. Flagella and motility alterations in Pseudomonas aeruginosa strains from patients with cystic fibrosis: Relationship to patient clinical condition. Infect. Immun. 1985, 50, 577–582. [Google Scholar] [PubMed]
- Lopez-Boado, Y.S.; Espinola, M.; Bahr, S.; Belaaouaj, A. Neutrophil serine proteinases cleave bacterial flagellin, abrogating its host response-inducing activity. J. Immunol. 2004, 172, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, A.; Jyot, J.; During, R.; Ramphal, R. Neutrophil elastase, an innate immunity effector molecule, represses flagellin transcription in Pseudomonas aeruginosa. Infect. Immun. 2006, 74, 6682–6689. [Google Scholar] [CrossRef] [PubMed]
- Leid, J.G.; Kerr, M.; Selgado, C.; Johnson, C.; Moreno, G.; Smith, A.; Shirtliff, M.E.; O’Toole, G.A.; Cope, E.K. Flagellum-mediated biofilm defense mechanisms of Pseudomonas aeruginosa against host-derived lactoferrin. Infect. Immun. 2009, 77, 4559–4566. [Google Scholar] [CrossRef] [PubMed]
- Pritt, B.; O’Brien, L.; Winn, W. Mucoid pseudomonas in cystic fibrosis. Am. J. Clin. Pathol. 2007, 128, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Kragh, K.N.; Alhede, M.; Jensen, P.O.; Moser, C.; Scheike, T.; Jacobsen, C.S.; Seier Poulsen, S.; Eickhardt-Sorensen, S.R.; Trostrup, H.; Christoffersen, L.; et al. Polymorphonuclear leukocytes restrict growth of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Infect. Immun. 2014, 82, 4477–4486. [Google Scholar] [CrossRef] [PubMed]
- Caceres, S.M.; Malcolm, K.C.; Taylor-Cousar, J.L.; Nichols, D.P.; Saavedra, M.T.; Bratton, D.L.; Moskowitz, S.M.; Burns, J.L.; Nick, J.A. Enhanced in vitro formation and antibiotic resistance of nonattached Pseudomonas aeruginosa aggregates through incorporation of neutrophil products. Antimicrob. Agents Chemother. 2014, 58, 6851–6860. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Schaefer, A.L.; Parsek, M.R.; Moninger, T.O.; Welsh, M.J.; Greenberg, E.P. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature 2000, 407, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T.; Jensen, P.O.; Fiandaca, M.J.; Pedersen, J.; Hansen, C.R.; Andersen, C.B.; Pressler, T.; Givskov, M.; Hoiby, N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Klebensberger, J.; Rui, O.; Fritz, E.; Schink, B.; Philipp, B. Cell aggregation of Pseudomonas aeruginosa strain pao1 as an energy-dependent stress response during growth with sodium dodecyl sulfate. Arch. Microbiol. 2006, 185, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Schleheck, D.; Barraud, N.; Klebensberger, J.; Webb, J.S.; McDougald, D.; Rice, S.A.; Kjelleberg, S. Pseudomonas aeruginosa PAO1 preferentially grows as aggregates in liquid batch cultures and disperses upon starvation. PLoS ONE 2009, 4, e5513. [Google Scholar] [CrossRef] [PubMed]
- Fuxman Bass, J.I.; Russo, D.M.; Gabelloni, M.L.; Geffner, J.R.; Giordano, M.; Catalano, M.; Zorreguieta, A.; Trevani, A.S. Extracellular DNA: A major proinflammatory component of Pseudomonas aeruginosa biofilms. J. Immunol. 2010, 184, 6386–6395. [Google Scholar] [CrossRef] [PubMed]
- Alhede, M.; Kragh, K.N.; Qvortrup, K.; Allesen-Holm, M.; van Gennip, M.; Christensen, L.D.; Jensen, P.O.; Nielsen, A.K.; Parsek, M.; Wozniak, D.; et al. Phenotypes of non-attached Pseudomonas aeruginosa aggregates resemble surface attached biofilm. PLoS ONE 2011, 6, e27943. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Kutty, S.K.; Tavallaie, R.; Ibugo, A.I.; Panchompoo, J.; Sehar, S.; Aldous, L.; Yeung, A.W.; Thomas, S.R.; Kumar, N.; et al. Phenazine virulence factor binding to extracellular DNA is important for Pseudomonas aeruginosa biofilm formation. Sci. Rep. 2015, 5, 8398. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Manefield, M. Phenazine production enhances extracellular DNA release via hydrogen peroxide generation in Pseudomonas aeruginosa. Commun. Integr. Biol. 2013, 6, e23570. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Sehar, S.; Koop, L.; Wong, Y.K.; Ahmed, S.; Siddiqui, K.S.; Manefield, M. Influence of calcium in extracellular DNA mediated bacterial aggregation and biofilm formation. PLoS ONE 2014, 9, e91935. [Google Scholar] [CrossRef] [PubMed]
- Klebensberger, J.; Lautenschlager, K.; Bressler, D.; Wingender, J.; Philipp, B. Detergent-induced cell aggregation in subpopulations of Pseudomonas aeruginosa as a preadaptive survival strategy. Environ. Microbiol. 2007, 9, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Kahle, N.A.; Brenner-Weiss, G.; Overhage, J.; Obst, U.; Hansch, G.M. Bacterial quorum sensing molecule induces chemotaxis of human neutrophils via induction of p38 and leukocyte specific protein 1 (LSP1). Immunobiology 2013, 218, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Wagner, C.; Muller, W.; Brenner-Weiss, G.; Hug, F.; Prior, B.; Obst, U.; Hansch, G.M. Induction of neutrophil chemotaxis by the quorum-sensing molecule N-(3-oxododecanoyl)-l-homoserine lactone. Infect. Immun. 2006, 74, 5687–5692. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B.; Coleman, F.; Grout, M.; Franklin, M.; Ohman, D.E. Role of alginate o acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infect. Immun. 2001, 69, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Krieg, D.P.; Helmke, R.J.; German, V.F.; Mangos, J.A. Resistance of mucoid Pseudomonas aeruginosa to nonopsonic phagocytosis by alveolar macrophages in vitro. Infect. Immun. 1988, 56, 3173–3179. [Google Scholar] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Kharazmi, A.; Garred, P.; Kronborg, G.; Fomsgaard, A.; Mollnes, T.E.; Hoiby, N. Complement activation by Pseudomonas aeruginosa biofilms. Microb. Pathog. 1993, 15, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.T.; Kharazmi, A.; Hoiby, N.; Costerton, J.W. Some bacterial parameters influencing the neutrophil oxidative burst response to Pseudomonas aeruginosa biofilms. APMIS 1992, 100, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Alhede, M.; Bjarnsholt, T.; Jensen, P.O.; Phipps, R.K.; Moser, C.; Christophersen, L.; Christensen, L.D.; van Gennip, M.; Parsek, M.; Hoiby, N.; et al. Pseudomonas aeruginosa recognizes and responds aggressively to the presence of polymorphonuclear leukocytes. Microbiology 2009, 155, 3500–3508. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.O.; Bjarnsholt, T.; Phipps, R.; Rasmussen, T.B.; Calum, H.; Christoffersen, L.; Moser, C.; Williams, P.; Pressler, T.; Givskov, M.; et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 2007, 153, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Kharami, A.; Bibi, Z.; Nielsen, H.; Hoiby, N.; Doring, G. Effect of Pseudomonas aeruginosa rhamnolipid on human neutrophil and monocyte function. APMIS 1989, 97, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Van Gennip, M.; Christensen, L.D.; Alhede, M.; Phipps, R.; Jensen, P.O.; Christophersen, L.; Pamp, S.J.; Moser, C.; Mikkelsen, P.J.; Koh, A.Y.; et al. Inactivation of the rhla gene in Pseudomonas aeruginosa prevents rhamnolipid production, disabling the protection against polymorphonuclear leukocytes. APMIS 2009, 117, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Jesaitis, A.J.; Franklin, M.J.; Berglund, D.; Sasaki, M.; Lord, C.I.; Bleazard, J.B.; Duffy, J.E.; Beyenal, H.; Lewandowski, Z. Compromised host defense on Pseudomonas aeruginosa biofilms: Characterization of neutrophil and biofilm interactions. J. Immunol. 2003, 171, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Van Gennip, M.; Christensen, L.D.; Alhede, M.; Qvortrup, K.; Jensen, P.O.; Hoiby, N.; Givskov, M.; Bjarnsholt, T. Interactions between polymorphonuclear leukocytes and Pseudomonas aeruginosa biofilms on silicone implants in vivo. Infect. Immun. 2012, 80, 2601–2607. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Sil, P.; Hayes, C.P.; Reaves, B.J.; Breen, P.; Quinn, S.; Sokolove, J.; Rada, B. P2Y6 receptor antagonist MRS2578 inhibits neutrophil activation and aggregated neutrophil extracellular trap formation induced by gout-associated monosodium urate crystals. J. Immunol. 2017, 198, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Parks, Q.M.; Young, R.L.; Poch, K.R.; Malcolm, K.C.; Vasil, M.L.; Nick, J.A. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: Human f-actin and DNA as targets for therapy. J. Med. Microbiol. 2009, 58, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Walker, T.S.; Tomlin, K.L.; Worthen, G.S.; Poch, K.R.; Lieber, J.G.; Saavedra, M.T.; Fessler, M.B.; Malcolm, K.C.; Vasil, M.L.; Nick, J.A. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect. Immun. 2005, 73, 3693–3701. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.M.; Parks, Q.M.; Young, R.L.; Kret, J.; Poch, K.R.; Malcolm, K.C.; Nichols, D.P.; Nichols, M.; Zhu, M.; Cavanagh, H.D.; et al. Disruption of contact lens-associated Pseudomonas aeruginosa biofilms formed in the presence of neutrophils. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2844–2850. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme of complex interactions between polymorphonuclear neutrophil granulocytes (PMNs) and Pseudomonas aeruginosa in cystic fibrosis (CF) airways with a special emphasis on neutrophil extracellular traps (NETs). ROS: reactive oxygen species.
Figure 1.
Scheme of complex interactions between polymorphonuclear neutrophil granulocytes (PMNs) and Pseudomonas aeruginosa in cystic fibrosis (CF) airways with a special emphasis on neutrophil extracellular traps (NETs). ROS: reactive oxygen species.
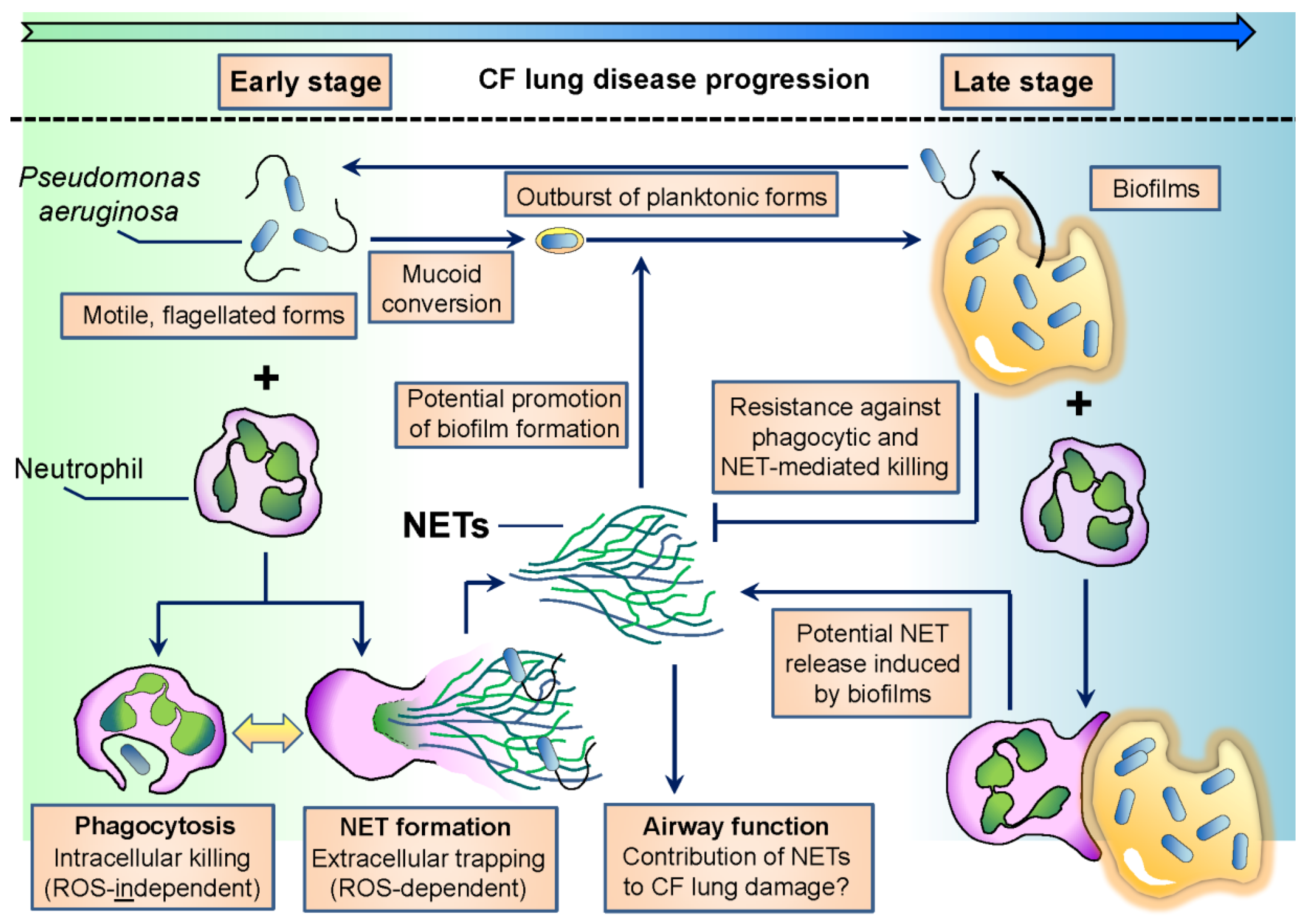
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rada, B. Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis. Pathogens 2017, 6, 10. https://doi.org/10.3390/pathogens6010010
AMA Style
Rada B. Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis. Pathogens. 2017; 6(1):10. https://doi.org/10.3390/pathogens6010010
Chicago/Turabian StyleRada, Balázs. 2017. "Interactions between Neutrophils and Pseudomonas aeruginosa in Cystic Fibrosis" Pathogens 6, no. 1: 10. https://doi.org/10.3390/pathogens6010010
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.