The Role of the Mammalian Prion Protein in the Control of Sleep


1
The Roslin Institute and Royal (Dick) School of Veterinary Sciences, University of Edinburgh, Easter Bush Veterinary Centre, Edinburgh EH25 9RG, UK
2
School of Chemistry, Joseph Banks Laboratories, University of Lincoln, Green Lane, Lincoln, Lincolnshire LN6 7DL, UK
*
Author to whom correspondence should be addressed.
Pathogens 2017, 6(4), 58; https://doi.org/10.3390/pathogens6040058
Submission received: 28 September 2017
/
Revised: 8 November 2017
/
Accepted: 13 November 2017
/
Published: 17 November 2017
(This article belongs to the Special Issue PrPSc prions: state of the art)
{kind=link}
{kind=link}
Abstract
:Sleep disruption is a prevalent clinical feature in many neurodegenerative disorders, including human prion diseases where it can be the defining dysfunction, as in the case of the “eponymous” fatal familial insomnia, or an early-stage symptom as in certain types of Creutzfeldt-Jakob disease. It is important to establish the role of the cellular prion protein (PrPC), the key molecule involved in prion pathogenesis, within the sleep-wake system in order to understand fully the mechanisms underlying its contribution to both healthy circadian rhythmicity and sleep dysfunction during disease. Although severe disruption to the circadian rhythm and melatonin release is evident during the pathogenic phases of some prion diseases, untangling whether PrPC plays a role in circadian rhythmicity, as suggested in mice deficient for PrPC expression, is challenging given the lack of basic experimental research. We provide a short review of the small amount of direct literature focused on the role of PrPC in melatonin and circadian rhythm regulation, as well as suggesting mechanisms by which PrPC might exert influence upon noradrenergic and dopaminergic signaling and melatonin synthesis. Future research in this area should focus upon isolating the points of dysfunction within the retino-pineal pathway and further investigate PrPC mediation of pinealocyte GPCR activity.
1. Introduction
The highly conserved prion protein (PrP) is encoded by the PRNP gene (human cytogenic location 20p12) [1] and exists predominately in two conformationally different isoforms [2]. The cellular prion protein (PrPC) has an α-helical structure and is expressed highly within the central and peripheral nervous systems [3] and localized to neuronal and glial cell membranes [4]. PrPC expression has also been identified in tissues beyond the nervous systems, including the intestine, heart and lymph nodes [5]. This widespread distribution of apparently functional PrPC, coupled with its highly conserved nature, suggests that it has either one important role or multiple, context-dependent roles throughout the body. Total knockout of PrPC is not associated with major deleterious phenotypes [6], suggesting that the latter hypothesis is more likely to be true. Indeed, PrPC has been implicated in, among other things, immune response modulation [7], myelin maintenance [8], mitochondrial homeostasis [9] and signal transduction as reviewed recently by Castle and Gill [4]. A final possibility is that PrPC does indeed have a single role in a key biochemical pathway that impacts other physiological processes that PrPC is proposed to control; in this possibility, PrPC may be involved only in modulating activity of the top-level system rather than turning it on or off, thereby explaining its apparent redundancy and context-dependent functions. Of relevance to this review is the finding that, in models of PrPC dysfunction or knockout, marked alterations to circadian rhythms occur [10] suggesting a role for PrPC in one of the most essential processes for life: the sleep/wake (or active/rest) cycle.
The prion protein is one of a handful of naturally expressed proteins that can misfold into specific pathogenic isoforms and, hence, become integral to neurodegenerative phenotypes. The prion protein’s conformationally-altered isoform is known as PrPSc, which is enriched in β-sheet structure, is insoluble and which assembles into amyloid plaques and fibrils [11]. The conversion from PrPC to PrPSc and the subsequent aggregation and oligomerization of PrPSc results in severe, and ultimately fatal, neurodegenerative prion diseases, also known as transmissible spongiform encephalopathies or TSEs [11,12]. Prion diseases can occur sporadically, due to infection, or as the result of genetic influence [13]. The exact mechanism underlying sporadic conversion from PrPC to PrPSc is unknown, but it is believed that this mechanism is expedited when PrPC contains a disease-initiating mutation. However, following the initiation of an infection (e.g., by ingesting contaminated meat) exogenous PrPSc is believed to be used as a template, upon which the functional endogenous PrPC misfolds in a process of autocatalytic conversion [11,12]. The incubation period between inoculation and disease onset is prolonged in prion diseases [11], providing a potential window for neuroprotective interventions [14], but this relies on knowledge of the molecular mechanisms responsible for neuronal loss so that intervention can be targeted effectively and specifically. Identifying such mechanisms is complicated by the fact that different prion disease types (or strains) target pathology to rather different areas of the brain and the information that causes neuropathological targeting is believed to be encoded in the structure of PrPSc. Although multimeric forms of PrPSc may be toxic to neurons directly, by activating signaling pathways that lead to apoptosis or necrosis, it is also possible that the neuropathology that arises with PrPSc propagation occurs in combination with loss of function of PrPC [15]. Indeed, during pathogenesis of a prion disease, PrPC expression has been suggested to be downregulated [15,16]. Herein we review whether there may be role for PrPC loss of function in the sleep-wake cycle, a key biological system that is compromised to different extents during various prion diseases.
2. Sleep Dysfunction during Prion Disease Pathogenesis
Observations of human prion disease patients provided the first links between PrPC/PrPSc and sleep. In the genetic prion disease fatal familial insomnia (FFI), the “eponymous” clinical feature is severely disturbed sleep, characterized by anxiolytic-resistant insomnia, circadian rhythm dysfunction, sleep fragmentation and altered arousal. Polysomnographic studies show a decrease in total sleep time, decreased REM sleep and loss of REM atonia [17]. Central sleep apneas and decreased slow wave sleep are also common features of FFI clinicopathology [18]. These extensive symptoms are likely to be the result of dysfunction across the various systems regulating sleep.
To variable degrees, sleep disturbances are also evident in other prion diseases, including various human prion diseases, although they are not currently considered part of the clinical diagnostic criteria. Nevertheless, in a recent study almost 90% of sporadic Creutzfeldt-Jakob disease (spCJD) patients reported sleep dysfunction during clinical evaluation, making sleep disturbance more prevalent than any other diagnostic criteria for CJD [19]. Sleep disturbances were also prevalent clinical complaints in all familial CJD patients examined during a separate study [20]. Certain animal models of prion disease also result in disrupted sleep patterns; rats inoculated with various prion strains have pronounced slow wave sleep decreases [21], rhesus monkeys infected with the human prion disease kuru show complete loss of REM sleep and disrupted sleep stage cycling [22], whilst mice inoculated with the murine prion disease RML show alterations in rest period activity from extremely early in the incubation period [23]. Interestingly, it has been reported that patients with Gerstmann-Sträussler-Sheinker disease (a genetic prion disease characterized predominantly by ataxia and pyramidal dysfunction) do not exhibit sleep alterations [24,25], suggesting that sleep dysfunction is specific to particular prion strains. Given that neuropathology in different prion strains is targeted to different regions of the brain, it follows that molecular or cellular alterations in specific brain regions may underlie some of the prion-induced sleep abnormalities. It is pertinent, therefore, to consider the brain areas that control the different aspects of normal sleep and how these are affected during prion pathogenesis.
The slow wave oscillations of deep, non-REM (NREM sleep) are generated, synchronized and stabilized by the thalamocortical network [26,27], whilst fluctuations in thalamocortical excitability produces the hallmark deflections visualized on EEG traces during NREM sleep: the K-complex and sleep spindle [28]. Cortically-generated K-complexes occur both spontaneously and in response to sensory stimulation, acting to monitor environmental stimuli during reduced states of consciousness and to enhance sleep stability [28]. The K-complex waveform is reflected in thalamic activity, with thalamic neurons reinforcing the K-complex slow oscillation [26]. In response to the K-complex, thalamic reticular neurons generate their own oscillation—known as the sleep spindle—which is thought to be instrumental in thalamocortical plasticity, cognition and memory function [29]. One of the most striking polysomnographic observations in FFI and fCJD patients is a reduction or absence of the thalamocortical K-complex and sleep spindle oscillations [19,30,31,32]. Prion-induced dysregulation of slow wave sleep, sleep instability and the loss of K-complex and sleep spindle oscillations (as well as the memory and cognitive impairments seen in prion diseases) are therefore likely due to the gross neurodegeneration of the thalamocortical network as the disease process reaches the clinical phase [33]. Support for this rationale comes from mouse models of FFI: mice expressing PrPC carrying the D178N mutation (that causes FFI in humans in combination with methionine expressed at codon 129) develop thalamic pathology and exhibit disrupted circadian rhythmicity of sleep and motor activity. Both over-expressing [34] and knock-in [35] FFI transgenic mice are phenotypically similar to human FFI patients, with the overexpressing mice exhibiting abnormal REM-sleep transitioning, loss of sleep spindles, reduced slow wave activity and decreased sleep continuity. Anatomically, these mice also exhibited thalamic degeneration, which, as in human patients, is responsible for the observed breakdown of sleep architecture. However, changes in circadian-regulated motor activity of these mice, including decreased dark-phase activity compared to controls [34], are not readily rationalized by thalamic degeneration and knock-in FFI mice also show significant decreases in dark-phase motor activity compared to controls [35].
Symptoms of insomnia reported by prion disease patients can be explained by the reduction in both sleep stability and sleep maintenance associated with thalamocortical degeneration. It is also likely that the dysregulation of circadian rhythmicity seen in prion disease contributes to symptoms of insomnia. A key organ responsible for the modulation of sleep patterns through synthesis of the circadian hormone melatonin is the pineal gland, and it has been demonstrated that non-prion patients with insomnia exhibit reduced pineal gland volume [36] as well as significantly decreased nocturnal plasma melatonin levels [37]. The pineal gland is a site of high level expression of PrPC [4]. Whilst this may render the pineal gland highly susceptible to infection by prions, it also raises the possibility that reduced levels of functional PrPC during a prion infection [15] may be involved in sleep dysfunction. Thus, the mechanisms of circadian dysfunction evident in prion diseases require further basic science investigation beyond clinical reporting, as currently there is little known of how a prion infection can dysregulate such an essential system.
3. Circadian Rhythm, Homeostatic Sleep Pressure and Melatonin
The sleep-wake cycle is driven by two factors: circadian rhythm and homeostatic sleep pressure [38,39]. Homeostatic sleep pressure is best described as the feeling of sleepiness, such that the longer one goes without sleep, the more tired one becomes. The driving mechanism of homeostatic sleep pressure is thought to be the result of chemical build-up, such as increased levels of adenosine [40], within the brain. Slow wave oscillations during wakefulness and sleep are regulated homeostatically, with slow wave activity decreasing over the sleep period and increasing as time awake increases [41,42].
Similar to homeostatic sleep pressure, circadian rhythmicity is the result of endogenous processes, with every cell exhibiting its own intrinsic, oscillatory, circadian rhythm [43]. The oscillations in cellular gene expression, caused by a negative feedback transcription/translation loop, have a natural period of around 24 h, but are then synchronized to the exogenous light/dark cycle [43]. The circadian rhythm is also modulated, to a degree, by other external factors such as exercise and food consumption [44] as well as internal factors including core body temperature and the menstrual cycle [45].
The main regulatory hormone of the circadian rhythm is melatonin [39]. Melatonin is part of the tryptophan metabolic pathway [42] and it is synthesized and secreted by both central nervous system (CNS) tissues and organs throughout the body [46]. Within the CNS, melatonin production is entrained to the light/dark cycle, with the hormone acting as a mediator between hypothalamic nuclei and target tissues, relaying diurnal and seasonal timings to the body [47]. As with PrPC, the variety of organs capable of melatonin synthesis is suggestive of important, contextual roles for this hormone, including regulation of seasonal and circadian rhythms, reproductive function, modulation of neurotransmission and antioxidant effects [47].
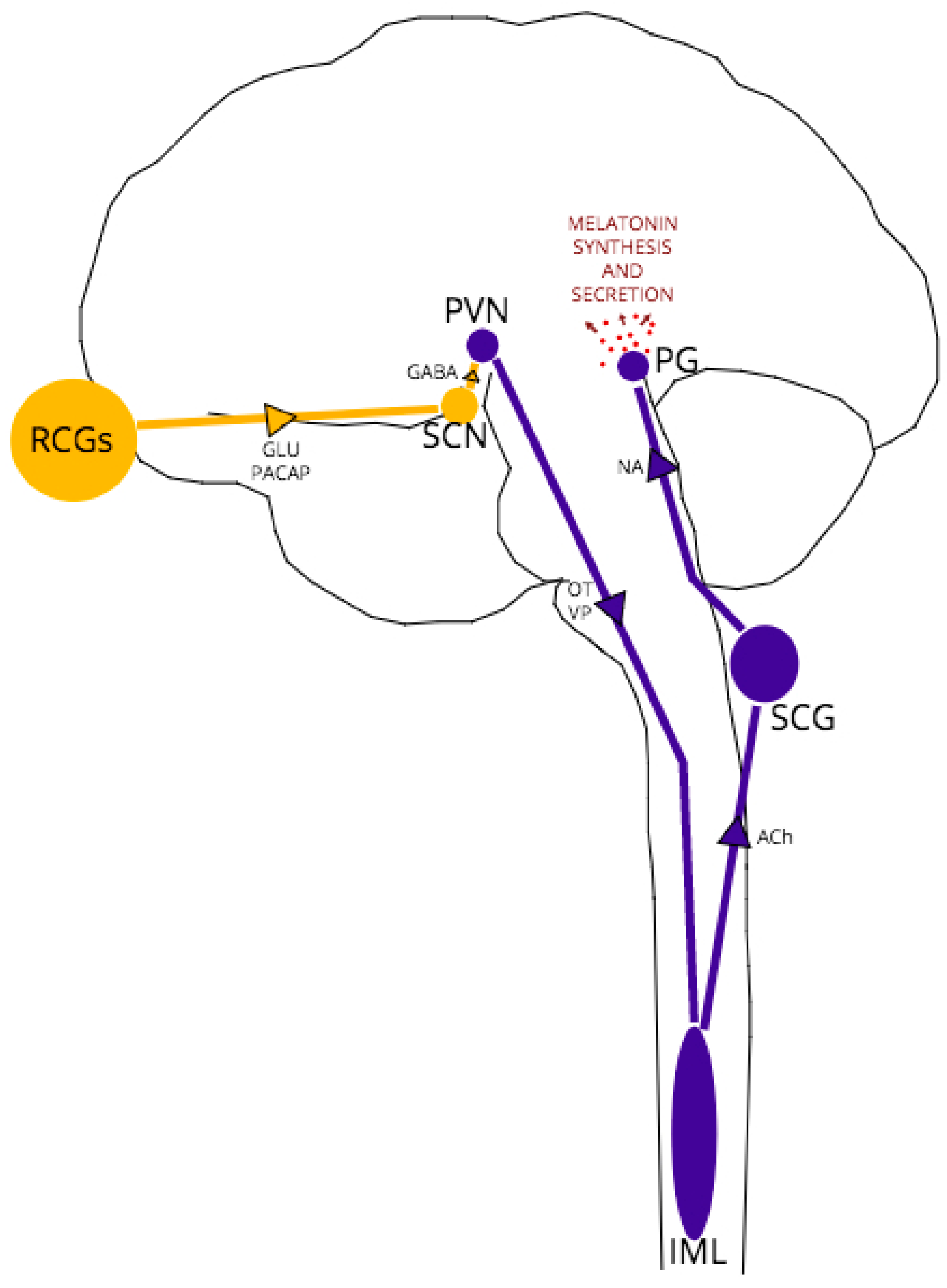
The complex, multi-nuclei pathway for melatonin synthesis, depicted schematically in Figure 1, begins with blue light excitation of photosensitive, melanopsin-containing retinal ganglion cells (RGCs). The RGC axons form the retino-hypothalamic tract which projects to the hypothalamic suprachiasmatic nucleus (SCN), providing glutamatergic excitation of SCN neurons. The SCN is the central pacemaker that fine-tunes the body’s circadian rhythm, conveying the light/dark fluctuations of the external environment to various tissues, including the brain, via oscillatory activity in order to coordinate metabolic function and homeostasis accordingly [48]. These SCN oscillations are generated in response to RCG neurotransmission by a transcriptional-translational feedback loop between clock genes [49].
In all melatonin-synthesizing organisms, light acts as a zeitgeber (environmental cue) for melatonin secretion, with peak melatonin levels arising in the middle of the night’s sleep at approximately 100 times the daytime melatonin level [39]. In daylight, GABAergic SCN projections synapse to paraventricular (PVN) neurons, inhibiting further excitation of the pathway. At night, without RCG/SCN excitation, PVN neurons are disinhibited, leading in turn to excitation of thoracic intermediolateral (IML) neurons and superior cervical ganglia (SCG) neurons. Noradrenergic SCG neurons project to the pineal gland, which expresses both α1 and β1-adrenoceptors [50]. Noradrenaline-adrenoceptor binding then activates adenylate cyclase and the melatonin synthesis cascade within pinealocytes.
Pinealocytes provide the majority of circulating melatonin, with immediate melatonin release into the blood (bypassing the blood brain barrier) and into CSF circulation at the point of the pineal recess and third ventricle [51]. Melatonin travels via CSF and blood to target tissues expressing G-protein coupled melatonin receptors MT1 and MT2 [52]. MT1 binding has been associated with metabolic regulation, whilst MT2 binding is associated with circadian rhythmicity [46]. Expression of both melatonin receptors is widespread throughout the brain, with localization to neurons of the SCN, thalamus and hippocampus, amongst other structures [41].
The pineal gland is not the only source of melatonin; extrapineal melatonin synthesis is thought to occur in a number of organs that express melatonin-synthesizing enzymes, including the heart, liver and placenta [46]. Indeed, the gut expresses melatonin levels 400 times greater than that of the pineal gland, and extrapineal melatonin synthesis has been shown to occur independently of the photoperiod, with an absence of diurnal fluctuations [53]. In contrast to the pineal-synthesized melatonin, which is immediately released into CSF and blood circulation, melatonin synthesized in tissues beyond the CNS is retained within cells [46]. Intracellular melatonin is correlated with its local anti-inflammatory/oxidative effects; in neonates with respiratory distress syndrome, melatonin treatment significantly reduces pro-inflammatory cytokine and nitrate levels [54], whilst melatonin levels in the elderly are inversely correlated with Charlson Comorbidity Index score, indicating a protective role against chronic disease [55].
Abolition of endogenous CNS melatonin has far-reaching consequences for the brain and body; reports of impaired wound healing [56], disrupted circadian organization of the SCN [57], abolition of clock gene expression in adipose tissue [58] as well as altered bone metabolism leading to bone loss [59] provide evidence for the protective function of melatonin in health. The effects of pinealectomy upon sleep are equally as wide-ranging. Long-term changes post-pinealectomy in humans include reduced sleep efficiency and increased switching between sleep stages, indicating a role for melatonin in sleep stability [60]. Pinealectomized rats show a significant decrease in REM-sleep theta power compared to controls [61]. Intriguingly, there are also reports of the loss of hippocampal CA1 and CA3 neurons post pinealectomy in rats [62], which can be reversed by administering exogenous melatonin; since MT1/MT2-expressing hippocampal CA1 and CA3 neurons [63] generate cortical theta oscillations, the theta reduction in pinealectomized rats may be, in part, the result of the hippocampal pyramidal neuron loss.
Extrapolating the influence of melatonin upon the CNS and body is challenging, given how intertwined the circadian rhythm is with whole-system function. It is important to identify whether prion disease-related sleep/wake dysfunctions are the result of circadian rhythm anomalies caused by PrPC loss of function from the pineal gland, whether anomalies can be explained by neurodegeneration caused by PrPSc-mediated neurotoxicity or whether both affects play a role. This requires a consideration of the link between PrPC, circadian rhythms and melatonin signaling.
4. A Role for PrPC in the Regulation of Melatonin Synthesis
Early research revealed that PrP mRNA expression peaks during the circadian dark phase at approximately 14 h (zeitgeber time) [64]. This significant increase in PrP mRNA expression precedes melatonin synthesis, with pineal melatonin levels increasing to a peak from 18 to 20 h (zeitgeber time) [65]. Whilst the apparent correlation between PrPC and melatonin levels may be just that—correlative rather than causative—there is some evidence that PrPC levels impact directly or indirectly on melatonin levels and that the high level of PrPC expression in the pineal glands may play a role in regulating melatonin synthesis.
Early studies of PrP knockout (PrP−/−) mice highlighted similar sleep disruptions to those reported in human prion disease cases, such as increased sleep fragmentation and altered slow wave activity [10]. It was noted that PrP−/− mice exhibited a significantly longer circadian phase than controls (23.9 h vs. 23.3 h, respectively), as well as demonstrating inverse dark phase activity compared to controls; control mice carried out the majority of their wheel-running during the first half of the dark phase, whilst PrP−/− mice had increased activity towards the end of the dark phase [10]. These findings suggest PrP−/− mice experience a phase shift in their circadian timings, akin to that seen in delayed phase sleep disorders, with circadian period elongation and delay in activity timings [66]. It was also noted that PrP−/− mice exhibited alterations to intrinsic circadian rhythms. Normally, in the complete absence of light, the circadian rhythm deregulates and becomes “free-running”, which manifests in incremental shifts in peak melatonin release from night to day [67], since intrinsic circadian rhythms in mice are slightly greater than 24 h. When placed in complete darkness, control mice exhibited this free-running circadian rhythm, using motor activity as an indicator, whilst, confusingly, the PrP−/− mice maintained 24 h rhythmicity despite no light entrainment [10]. Further to this, PrP−/− mice demonstrate non-cyclical, phase-shifted melatonin release in comparison to controls, with no increase in nighttime plasma melatonin level but increased levels of melatonin during the day relative to controls [68].
The above observations beg the question of whether PrPC functions as part of the pathway that turns light cues into melatonin and suggest that disruption of this pathway might be responsible, in part, for circadian abnormalities during prion disease pathogenesis. Progressive circadian disruption is observed in FFI patients, evidenced by decreasing plasma melatonin levels with disease advancement and complete loss of circadian rhythm by end-stage FFI [18,69]. A case study of one FFI patient reported normal function of the SCN, as evidenced by normal core body temperature rhythms and appropriate sleep stage timing, but dissociation of the circadian rhythms for melatonin and cortisol [69]. This suggests normal light/dark entrainment and SCN oscillatory activity, but a “functional interference” somewhere in the retino-pineal tract between the SCN and pineal gland function [69].
Reports of circadian phase-shifts and dysregulated melatonin expression in PrPC dysfunction, supported by the above experimental and clinical findings [10,68], suggest a delay in the synthesis or release of melatonin. Establishing the point at which PrPC interacts with the melatonin-synthesis pathway, whether at the pinealocyte or at structure upstream in the circuit, is therefore essential for revealing the mechanism underlying the abnormal circadian cycling.
As indicated earlier and depicted in Figure 1, functionally, between the SCN and pineal gland lie three structures: the hypothalamic paraventricular nucleus (PVN), the intermediolateral nucleus (IML) of the thoracic spinal column and the superior cervical ganglion (SCG) at the top of the sympathetic chain. The main neurotransmitters released by each structure are vasopressin/oxytocin, acetylcholine and noradrenaline respectively [47]. The fact that melatonin is still synthesized at healthy levels in models of PrP−/− and early-stage TSEs, albeit with an altered expression pattern, suggests that the structures are capable of neurotransmission of reasonable fidelity, but the phasing may be compromised in some way. It has been demonstrated that noradrenaline dysregulation is implicated in TSE pathology; for example, after oral and intraperitoneal prion inoculation, PrPSc neuroinvasion follows a selective route from the periphery via sympathetic nerves to the brain [70,71]. Noradrenergic cell death [72] and dramatically altered noradrenaline levels in specific brain regions (cerebral cortex, cerebellum, pons) and plasma following intraperitoneal or intracerebral inoculation [71,72,73,74] have also been demonstrated in experimental models of prion disease. However, these effects on noradrenaline expression and neurotransmission have not been demonstrated in PrP−/− models, which suggests the specific targeting of the noradrenergic system is the result of PrPSc pathology, not a loss of PrPC function in disease.
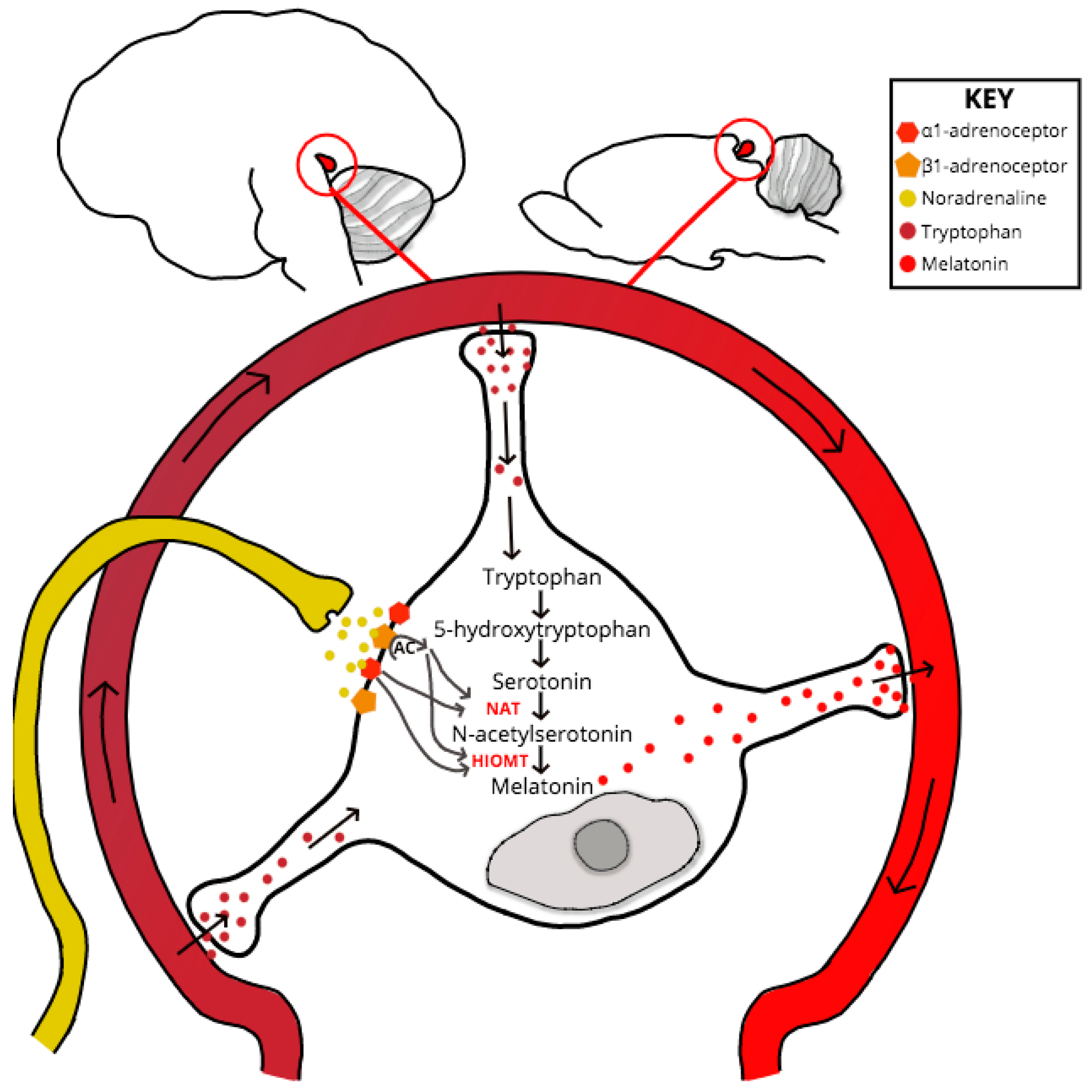
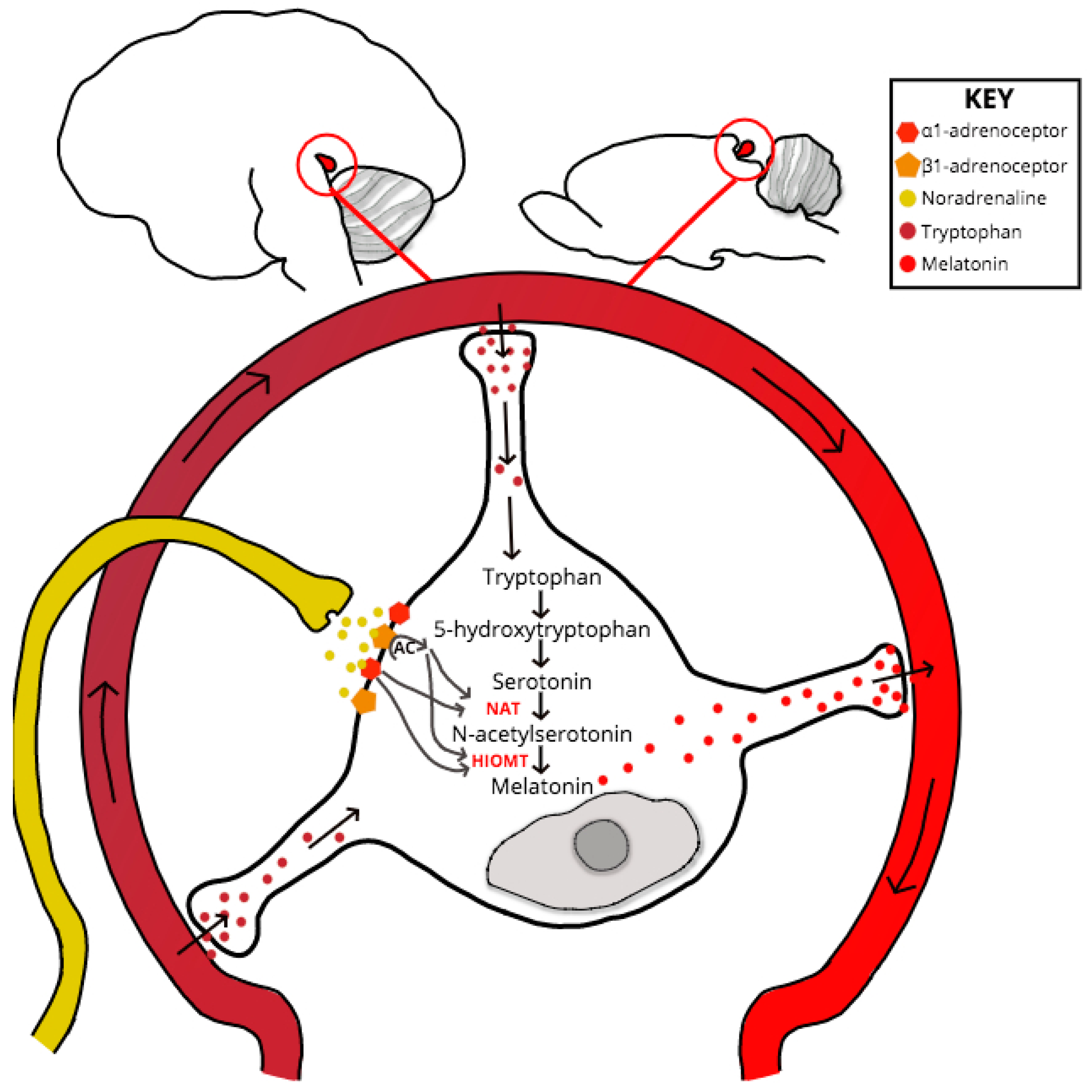
If neurotransmission throughout the retino-pineal tract is unchanged in PrP−/−, then the point of melatonin dysregulation or synthesis delay must be at the SCG-pinealocyte synapse, or due to a dysfunction at some point downstream and within the melatonin-synthesis signaling pathway (see Figure 2). Despite experimental evidence for PrP mRNA upregulation within the pinealocyte during the subjective night [75], no studies have specifically explored PrPC interactions or co-localization at the pinealocyte. One scenario that might explain the melatonin phase shift exhibited by clinical and experimental models of prion dysfunction is if compromised PrPC function causes dysregulation at the point of serotonin acetylation within the melatonin synthesis pathway. Serotonin is acetylated by the enzyme aralkylamine N-acetyltransferase (AA-NAT), whose activity acts as the rate-limiting step of melatonin synthesis [76]. The AA-NAT gene contains a cAMP-responsive element (cre) within its promoter region which, when stimulated by increased levels of cAMP, drives enzymatic expression and activity [77]. Levels of pinealocyte cAMP increase in response to noradrenergic, and to a lesser degree, dopaminergic, binding respectively with adrenergic and D1 receptors [77,78].
AA-NAT activity determines melatonin synthesis lag [79], and a significant association between AA-NAT polymorphisms and delayed phase sleep disorder has been shown [76]. Intriguingly, PrPC has continually been proposed to act as a cell-surface scaffolding protein and in this role it may influence AA-NAT activity indirectly through modulating the activity of pinealocyte G protein-coupled receptor (GPCR) complexes. The GPCRs can be divided according to whether they couple with the Gαi protein subunit, which inhibits cAMP production and adenylyl cyclase activity, or with the Gαs subunit, which inversely stimulates cAMP and adenylyl cyclase activity. Of the main GPCRs expressed by the pinealocyte, only the β-adrenergic and D1 GPCRs are Gαs-coupled [80,81]; it is, therefore, plausible that PrPC associates with one or other of these two GPCRs (or both) as a scaffold protein and influences their signaling cascade in pinealocytes. Supporting this, it has been demonstrated recently that PrPC co-localizes with D1 receptors, and there is selective impairment of cAMP signaling in response to D1 stimulation in PrP−/− mice [78]. A lack of PrPC may act to reduce adenylyl cyclase activity, resulting in an increased time for cAMP concentrations to reach a supra-threshold level. This would result in dysregulation of AA-NAT, since the length of time taken to achieve optimal acetylation of serotonin increases due to decreased AA-NAT enzymatic activity. Future investigations for a role of PrPC within the circadian rhythm and regulation of melatonin synthesis should therefore investigate protein-protein interactions of PrPC with noradrenergic and dopaminergic receptors, as well as analysis of analytes and catalysts within the melatonin synthesis pathway, with a particular focus on levels of serotonin, N-acetylserotonin and AA-NAT.
5. Conclusions
Clinical observation and experimental models of prion disease demonstrate clear circadian dysfunction, suggesting a role for PrPC within the synthesis or regulation of melatonin release in health. Drawing upon the limited literature, we propose that PrPC acts as a scaffold protein at the pinealocyte, associating with the Gαs-coupled β-adrenergic and D1 GPCRs. In this hypothesis, PrPC regulates the cAMP signaling cascade downstream of β-adrenergic/D1 activation, and in the absence of PrPC (as evidenced by PrP−/− models), a phase shift in melatonin synthesis results due to delayed AA-NAT enzymatic activity.
Acknowledgments
We acknowledge scientists at Roslin Institute for useful discussion and Sandra McCutcheon for proof reading the text. No specific funding was used for the review.
Author Contributions
A.R. performed the literature review, formulated hypotheses and wrote the first draft of the manuscript. A.C.G. supervised the work, edited the text and generated a final manuscript version. Both authors agreed the final manuscript content.
Conflicts of Interest
The authors declare no conflicts of interest, financial or otherwise.
References
- Sparkes, R.S.; Simon, M.; Cohn, V.H.; Fournier, R.E.; Lem, J.; Klisak, I.; Heinzmann, C.; Blatt, C.; Lucero, M.; Mohandas, T.; et al. Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc. Natl. Acad. Sci. USA 1986, 83, 7358–7362. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Wulf, M.A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Peralta, O.A.; Eyestone, W.H. Quantitative and qualitative analysis of cellular prion protein (prp(c)) expression in bovine somatic tissues. Prion 2009, 3, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface prp protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Bakkebo, M.K.; Mouillet-Richard, S.; Espenes, A.; Goldmann, W.; Tatzelt, J.; Tranulis, M.A. The cellular prion protein: A player in immunological quiescence. Front. Immunol. 2015, 6, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.A.; Weis, J.; et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, U310–U319. [Google Scholar] [CrossRef] [PubMed]
- Miele, G.; Jeffrey, M.; Turnbull, D.; Manson, J.; Clinton, M. Ablation of cellular prion protein expression affects mitochondrial numbers and morphology. Biochem. Biophys. Res. Commun. 2002, 291, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rulicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Annus, A.; Csati, A.; Vecsei, L. Prion diseases: New considerations. Clin. Neurol. Neurosurg. 2016, 150, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Jaunmuktane, Z. Prion disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 197–222. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.R. What is the basis of transmissible spongiform encephalopathy induced neurodegeneration and can it be repaired? Neuropathol. Appl. Neurobiol. 2002, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; van der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Investig. 2014, 124, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Ansoleaga, B.; Garcia-Esparcia, P.; Zafar, S.; Grau-Rivera, O.; Lopez-Gonzalez, I.; Blanco, R.; Carmona, M.; Yague, J.; Nos, C.; et al. Prp mrna and protein expression in brain and prp(c) in csf in creutzfeldt-jakob disease mm1 and vv2. Prion 2013, 7, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Zarranz, J.J.; Fischer, A.; Zerr, I.; Ferrer, I. Fatal familial insomnia: Clinical aspects and molecular alterations. Curr. Neurol. Neurosci. Rep. 2017, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Portaluppi, F.; Cortelli, P.; Avoni, P.; Vergnani, L.; Maltoni, P.; Pavani, A.; Sforza, E.; Degli Uberti, E.C.; Gambetti, P.; Lugaresi, E. Progressive disruption of the circadian rhythm of melatonin in fatal familial insomnia. J. Clin. Endocrinol. Metab. 1994, 78, 1075–1078. [Google Scholar] [PubMed]
- Kang, P.; de Bruin, G.S.; Wang, L.H.; Ward, B.A.; Ances, B.M.; Lim, M.M.; Bucelli, R.C. Sleep pathology in creutzfeldt-jakob disease. J. Clin. Sleep Med. 2016, 12, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Givaty, G.; Maggio, N.; Cohen, O.S.; Blatt, I.; Chapman, J. Early pathology in sleep studies of patients with familial creutzfeldt-jakob disease. J. Sleep Res. 2016, 25, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Bassant, M.H.; Cathala, F.; Court, L.; Gourmelon, P.; Hauw, J.J. Experimental scrapie in rats: First electrophysiological observations. Electroencephalogr. Clin. Neurophysiol. 1984, 57, 541–547. [Google Scholar] [CrossRef]
- Bert, J.; Vuillon-Cacciuttolo, G.; Balzamo, E.; De Micco, P.; Gambarelli, D.; Tamalet, J.; Gastaut, H. Experimental kuru in the rhesus monkey: A study of eeg modifications in the waking state and during sleep. Electroencephalogr. Clin. Neurophysiol. 1978, 45, 611–620. [Google Scholar] [CrossRef]
- Steele, A.D.; Jackson, W.S.; King, O.D.; Lindquist, S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of huntington’s and prion diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Pierangeli, G.; Bono, F.; Aguglia, U.; Maltoni, P.; Montagna, P.; Lugaresi, E.; Quattrone, A.; Cortelli, P. Normal sleep-wake and circadian rhythms in a case of gerstmann-straussler-sheinker (gss) disease. Clin. Auton. Res. 2004, 14, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Provini, F.; Vetrugno, R.; Pierangeli, G.; Cortelli, P.; Rizzo, G.; Filla, A.; Strisciuglio, C.; Gallassi, R.; Montagna, P. Sleep and temperature rhythms in two sisters with p102l gerstmann-straussler-scheinker (gss) disease. Sleep Med. 2009, 10, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Amzica, F.; Steriade, M. Cellular substrates and laminar profile of sleep k-complex. Neuroscience 1998, 82, 671–686. [Google Scholar] [CrossRef]
- McCormick, D.A.; Bal, T. Sleep and arousal: Thalamocortical mechanisms. Annu. Rev. Neurosci. 1997, 20, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, K.; von Wegner, F.; Morzelewski, A.; Borisov, S.; Maischein, M.; Steinmetz, H.; Laufs, H. To wake or not to wake? The two-sided nature of the human k-complex. Neuroimage 2012, 59, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Clawson, B.C.; Durkin, J.; Aton, S.J. Form and function of sleep spindles across the lifespan. Neural Plast. 2016, 2016, 6936381. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.S.; Chapman, J.; Korczyn, A.D.; Warman-Alaluf, N.; Orlev, Y.; Givaty, G.; Nitsan, Z.; Appel, S.; Rosenmann, H.; Kahana, E.; et al. Characterization of sleep disorders in patients with e200k familial creutzfeldt-jakob disease. J. Neurol. 2015, 262, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Ferrillo, F.; Plazzi, G.; Nobili, L.; Beelke, M.; De Carli, F.; Cortelli, P.; Tinuper, P.; Avoni, P.; Vandi, S.; Gambetti, P.; et al. Absence of sleep eeg markers in fatal familial insomnia healthy carriers: A spectral analysis study. Clin. Neurophysiol. 2001, 112, 1888–1892. [Google Scholar] [CrossRef]
- Gemignani, A.; Laurino, M.; Provini, F.; Piarulli, A.; Barletta, G.; d’Ascanio, P.; Bedini, R.; Lodi, R.; Manners, D.N.; Allegrini, P.; et al. Thalamic contribution to sleep slow oscillation features in humans: A single case cross sectional EEG study in fatal familial insomnia. Sleep Med. 2012, 13, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.S.; Kakita, A.; Watanabe, H.; Kitamoto, T.; Takahashi, H. Sporadic fatal insomnia with spongiform degeneration in the thalamus and widespread prpsc deposits in the brain. Neuropathology 2005, 25, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Bouybayoune, I.; Mantovani, S.; Del Gallo, F.; Bertani, I.; Restelli, E.; Comerio, L.; Tapella, L.; Baracchi, F.; Fernandez-Borges, N.; Mangieri, M.; et al. Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS Pathog. 2015, 11, e1004796. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.S.; Borkowski, A.W.; Faas, H.; Steele, A.D.; King, O.D.; Watson, N.; Jasanoff, A.; Lindquist, S. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 2009, 63, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Bumb, J.M.; Schilling, C.; Enning, F.; Haddad, L.; Paul, F.; Lederbogen, F.; Deuschle, M.; Schredl, M.; Nolte, I. Pineal gland volume in primary insomnia and healthy controls: A magnetic resonance imaging study. J. Sleep Res. 2014, 23, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Hajak, G.; Rodenbeck, A.; Staedt, J.; Bandelow, B.; Huether, G.; Ruther, E. Nocturnal plasma melatonin levels in patients suffering from chronic primary insomnia. J. Pineal Res. 1995, 19, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Borbely, A.A. A two process model of sleep regulation. Hum. Neurobiol. 1982, 1, 195–204. [Google Scholar] [PubMed]
- Carter, M.D.; Juurlink, D.N. Melatonin. Can. Med. Assoc. J. 2012, 184, 1923. [Google Scholar] [CrossRef] [PubMed]
- Bjorness, T.E.; Dale, N.; Mettlach, G.; Sonneborn, A.; Sahin, B.; Fienberg, A.A.; Yanagisawa, M.; Bibb, J.A.; Greene, R.W. An adenosine-mediated glial-neuronal circuit for homeostatic sleep. J. Neurosci. 2016, 36, 3709–3721. [Google Scholar] [CrossRef] [PubMed]
- Achermann, P.; Borbely, A.A. Sleep homeostasis and models of sleep regulation. In Principles and Practice of Sleep Medicine, 5th ed.; Kryger, M.H., Roth, T., Dement, W.C., Eds.; Elsevier: Saint Louis, MO, USA, 2011; pp. 431–444. [Google Scholar]
- Bersagliere, A.; Achermann, P. Slow oscillations in human non-rapid eye movement sleep electroencephalogram: Effects of increased sleep pressure. J. Sleep Res. 2010, 19, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.R.; Ceglia, N.; Zeller, M.; Eckel-Mahan, K.; Sassone-Corsi, P.; Baldi, P. The pervasiveness and plasticity of circadian oscillations: The coupled circadian-oscillators framework. Bioinformatics 2015, 31, 3181–3188. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Sasaki, H.; Ikeda, Y. Chrono-nutrition and chrono-exercise. Nihon Rinsho 2013, 71, 2194–2199. [Google Scholar] [PubMed]
- Baker, F.C.; Driver, H.S. Circadian rhythms, sleep, and the menstrual cycle. Sleep Med. 2007, 8, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Venegas, C.; Diaz-Casado, M.E.; Lima-Cabello, E.; Lopez, L.C.; Rosales-Corral, S.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef] [PubMed]
- Simonneaux, V.; Ribelayga, C. Generation of the melatonin endocrine message in mammals: A review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol. Rev. 2003, 55, 325–395. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.H.; Brancaccio, M.; Maywood, E.S. Circadian pacemaking in cells and circuits of the suprachiasmatic nucleus. J. Neuroendocrinol. 2014, 26, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Bonmati-Carrion, M.A.; Arguelles-Prieto, R.; Martinez-Madrid, M.J.; Reiter, R.; Hardeland, R.; Rol, M.A.; Madrid, J.A. Protecting the melatonin rhythm through circadian healthy light exposure. Int. J. Mol. Sci. 2014, 15, 23448–23500. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.B.; Spessert, R.; Vollrath, L. Molecular components and mechanism of adrenergic signal transduction in mammalian pineal gland: Regulation of melatonin synthesis. Indian J. Exp. Biol. 2005, 43, 115–149. [Google Scholar] [PubMed]
- Leston, J.; Harthe, C.; Mottolese, C.; Mertens, P.; Sindou, M.; Claustrat, B. Is pineal melatonin released in the third ventricle in humans? A study in movement disorders. Neurochirurgie 2015, 61, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. Mt1 and mt2 melatonin receptors: A therapeutic perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Q.; Fichna, J.; Bashashati, M.; Li, Y.Y.; Storr, M. Distribution, function and physiological role of melatonin in the lower gut. World J. Gastroenterol. 2011, 17, 3888–3898. [Google Scholar] [CrossRef] [PubMed]
- Gitto, E.; Reiter, R.J.; Cordaro, S.P.; La Rosa, M.; Chiurazzi, P.; Trimarchi, G.; Gitto, P.; Calabro, M.P.; Barberi, I. Oxidative and inflammatory parameters in respiratory distress syndrome of preterm newborns: Beneficial effects of melatonin. Am. J. Perinatol. 2004, 21, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, M.; Flores-Obando, R.; Mazzuco, S.; Ongaro, F.; Di Giorgi, E.; Boldrini, P.; Durante, E.; Frigato, A.; Albani, D.; Forloni, G.; et al. Melatonin and the charlson comorbidity index (cci): The treviso longeva (trelong) study. Int. J. Biol. Markers 2014, 29, e253–e260. [Google Scholar] [CrossRef] [PubMed]
- Ozler, M.; Simsek, K.; Ozkan, C.; Akgul, E.O.; Topal, T.; Oter, S.; Korkmaz, A. Comparison of the effect of topical and systemic melatonin administration on delayed wound healing in rats that underwent pinealectomy. Scand. J. Clin. Lab. Investig. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.D.; Rusak, B.; Piggins, H.D. Regulation of melatonin-sensitivity and firing-rate rhythms of hamster suprachiasmatic nucleus neurons: Constant light effects. Brain Res. 1993, 602, 191–199. [Google Scholar] [CrossRef]
- De Farias Tda, S.; de Oliveira, A.C.; Andreotti, S.; do Amaral, F.G.; Chimin, P.; de Proenca, A.R.; Leal, F.L.; Sertie, R.A.; Campana, A.B.; Lopes, A.B.; et al. Pinealectomy interferes with the circadian clock genes expression in white adipose tissue. J. Pineal Res. 2015, 58, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Egermann, M.; Gerhardt, C.; Barth, A.; Maestroni, G.J.; Schneider, E.; Alini, M. Pinealectomy affects bone mineral density and structure—An experimental study in sheep. BMC Musculoskelet. Disord. 2011, 12, 271. [Google Scholar] [CrossRef] [PubMed]
- Slawik, H.; Stoffel, M.; Riedl, L.; Vesely, Z.; Behr, M.; Lehmberg, J.; Pohl, C.; Meyer, B.; Wiegand, M.; Krieg, S.M. Prospective study on salivary evening melatonin and sleep before and after pinealectomy in humans. J. Biol. Rhythm. 2016, 31, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.P.; Sugden, D. Endogenous melatonin is not obligatory for the regulation of the rat sleep-wake cycle. Sleep 2010, 33, 833–840. [Google Scholar] [CrossRef] [PubMed]
- De Butte, M.; Pappas, B.A. Pinealectomy causes hippocampal ca1 and ca3 cell loss: Reversal by melatonin supplementation. Neurobiol. Aging 2007, 28, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, B.; Angeloni, D.; Dominguez-Lopez, S.; Calderoni, S.; Mauro, A.; Fraschini, F.; Descarries, L.; Gobbi, G. Anatomical and cellular localization of melatonin mt1 and mt2 receptors in the adult rat brain. J. Pineal Res. 2015, 58, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Cagampang, F.R.; Whatley, S.A.; Mitchell, A.L.; Powell, J.F.; Campbell, I.C.; Coen, C.W. Circadian regulation of prion protein messenger rna in the rat forebrain: A widespread and synchronous rhythm. Neuroscience 1999, 91, 1201–1204. [Google Scholar] [CrossRef]
- Nakahara, D.; Nakamura, M.; Iigo, M.; Okamura, H. Bimodal circadian secretion of melatonin from the pineal gland in a living cba mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 9584–9589. [Google Scholar] [CrossRef] [PubMed]
- Micic, G.; de Bruyn, A.; Lovato, N.; Wright, H.; Gradisar, M.; Ferguson, S.; Burgess, H.J.; Lack, L. The endogenous circadian temperature period length (tau) in delayed sleep phase disorder compared to good sleepers. J. Sleep Res. 2013, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Verwey, M.; Robinson, B.; Amir, S. Recording and analysis of circadian rhythms in running-wheel activity in rodents. J. Vis. Exp. 2013, 113, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Nicholas, R.S.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Dauvilliers, Y.; Cervena, K.; Carlander, B.; Espa, F.; Bassetti, C.; Claustrat, B.; Laplanche, J.L.; Billiard, M.; Touchon, J. Dissociation in circadian rhythms in a pseudohypersomnia form of fatal familial insomnia. Neurology 2004, 63, 2416–2418. [Google Scholar] [CrossRef] [PubMed]
- Beekes, M.; Baldauf, E.; Diringer, H. Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie. J. Gen. Virol. 1996, 77 Pt 8, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Pollera, C.; Bondiolotti, G.; Formentin, E.; Puricelli, M.; Mantegazza, P.; Bareggi, S.; Poli, G.; Ponti, W. Plasma noradrenalin as marker of neuroinvasion in prion diseases. Vet. Res. Commun. 2007, 31 (Suppl. 1), 249–252. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.W.; Choi, E.K.; Ju, W.K.; Ahn, M.S.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Extensive degeneration of catecholaminergic neurons to scrapie agent 87v in the brains of im mice. Mol. Chem. Neuropathol. 1998, 34, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Bassant, M.H.; Fage, D.; Dedek, J.; Cathala, F.; Court, L.; Scatton, B. Monoamine abnormalities in the brain of scrapie-infected rats. Brain Res. 1984, 308, 182–185. [Google Scholar] [CrossRef]
- Bondiolotti, G.; Rossoni, G.; Puricelli, M.; Formentin, E.; Lucchini, B.; Poli, G.; Ponti, W.; Bareggi, S.R. Changes in sympathetic activity in prion neuroinvasion. Neurobiol. Dis. 2010, 37, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Hohjoh, H.; Takasu, M.; Shishikura, K.; Takahashi, Y.; Honda, Y.; Tokunaga, K. Significant association of the arylalkylamine n-acetyltransferase (aa-nat) gene with delayed sleep phase syndrome. Neurogenetics 2003, 4, 151–153. [Google Scholar] [PubMed]
- Baler, R.; Covington, S.; Klein, D.C. The rat arylalkylamine n-acetyltransferase gene promoter. Camp activation via a camp-responsive element-ccaat complex. J. Biol. Chem. 1997, 272, 6979–6985. [Google Scholar] [CrossRef] [PubMed]
- Beckman, D.; Santos, L.E.; Americo, T.A.; Ledo, J.H.; de Mello, F.G.; Linden, R. Prion protein modulates monoaminergic systems and depressive-like behavior in mice. J. Biol. Chem. 2015, 290, 20488–20498. [Google Scholar] [CrossRef] [PubMed]
- Gauer, F.; Poirel, V.J.; Garidou, M.L.; Simonneaux, V.; Pevet, P. Molecular cloning of the arylalkylamine-n-acetyltransferase and daily variations of its mrna expression in the syrian hamster pineal gland. Brain Res. Mol. Brain Res. 1999, 71, 87–95. [Google Scholar] [CrossRef]
- Green, S.A.; Holt, B.D.; Liggett, S.B. Beta 1- and beta 2-adrenergic receptors display subtype-selective coupling to Gs. Mol. Pharmacol. 1992, 41, 889–893. [Google Scholar] [PubMed]
- Konig, B.; Gratzel, M. Site of dopamine d1 receptor binding to Gs protein mapped with synthetic peptides. Biochim. Biophys. Acta 1994, 1223, 261–266. [Google Scholar] [CrossRef]
Figure 1.
The Retino-Pineal Pathway. Structures and pathways in yellow represent light-active regions, those in dark blue represent dark-active regions. Arrows represent direction of signaling; associated text represents main neurotransmitters in the pathway. Region abbreviations: RCGs retinal ganglion cells; SCN suprachiasmatic nucleus; PVN paraventricular nucleus; IML intermediolateral nucleus; SCG superior cervical ganglion; PG pineal gland. Neurotransmitter abbreviations: GLU glutamate; PACAP pituitary adenylate-cyclase activating peptide; GABA gamma-aminobutyric acid; OT oxytocin; VP vasopressin; ACh acetylcholine; NA noradrenaline.
Figure 1.
The Retino-Pineal Pathway. Structures and pathways in yellow represent light-active regions, those in dark blue represent dark-active regions. Arrows represent direction of signaling; associated text represents main neurotransmitters in the pathway. Region abbreviations: RCGs retinal ganglion cells; SCN suprachiasmatic nucleus; PVN paraventricular nucleus; IML intermediolateral nucleus; SCG superior cervical ganglion; PG pineal gland. Neurotransmitter abbreviations: GLU glutamate; PACAP pituitary adenylate-cyclase activating peptide; GABA gamma-aminobutyric acid; OT oxytocin; VP vasopressin; ACh acetylcholine; NA noradrenaline.
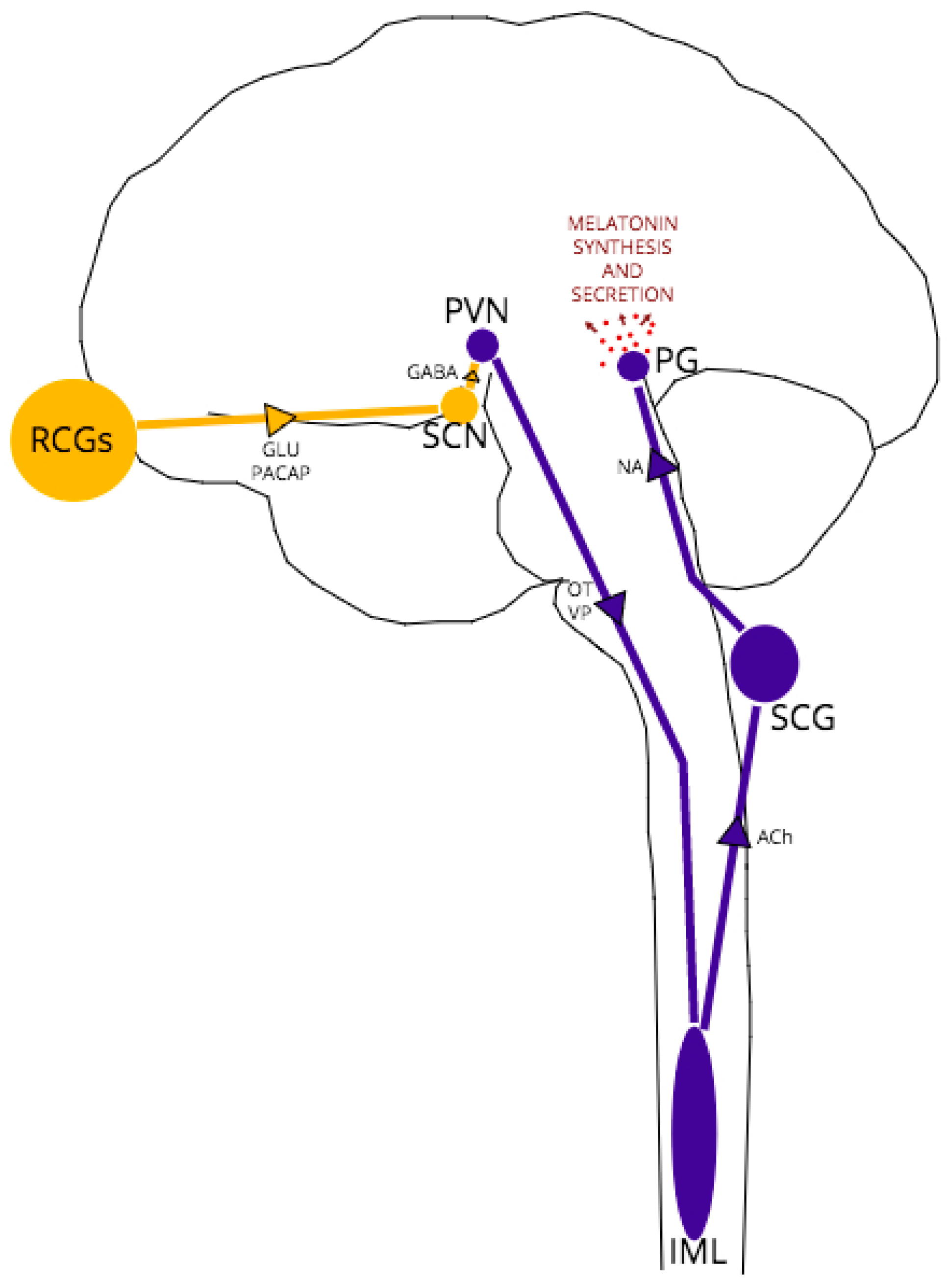
Figure 2.
Location of the Pineal Gland in the Human (left) and Mouse (right) Brain and Pinealocyte Melatonin Synthesis. The essential amino acid tryptophan is uptaken by pinealocytes from the surrounding vasculature and is converted to 5-hydroxytryptophan via a process of hydroxylation. Decarboxylation of 5-hydroxytryptophan gives rise to serotonin. SCG afferents, which project to the pineal adjacent to capillaries, release noradrenaline into the pineal perivascular space, where the neurotransmitter binds to α1B and β1-adrenoceptors on the pinealocyte membrane. Coincident activation of adrenoceptors increases cAMP levels, in turn inducing enzyme expression increases of N-acetyltransferase (NAT)—the rate-limiting step of the synthesis pathway- and acetylserotonin O-methyltransferase (HIOMT). Melatonin synthesis is dependent upon serotonin acetylation by NAT and HIOMT methylation activity of N-acetylserotonin. Once synthesized the indolamine is secreted into the circulation, where it has a biological half-life of ~45 min in humans and ~20 min in rats, before hepatic metabolism and urinal metabolite excretion.
Figure 2.
Location of the Pineal Gland in the Human (left) and Mouse (right) Brain and Pinealocyte Melatonin Synthesis. The essential amino acid tryptophan is uptaken by pinealocytes from the surrounding vasculature and is converted to 5-hydroxytryptophan via a process of hydroxylation. Decarboxylation of 5-hydroxytryptophan gives rise to serotonin. SCG afferents, which project to the pineal adjacent to capillaries, release noradrenaline into the pineal perivascular space, where the neurotransmitter binds to α1B and β1-adrenoceptors on the pinealocyte membrane. Coincident activation of adrenoceptors increases cAMP levels, in turn inducing enzyme expression increases of N-acetyltransferase (NAT)—the rate-limiting step of the synthesis pathway- and acetylserotonin O-methyltransferase (HIOMT). Melatonin synthesis is dependent upon serotonin acetylation by NAT and HIOMT methylation activity of N-acetylserotonin. Once synthesized the indolamine is secreted into the circulation, where it has a biological half-life of ~45 min in humans and ~20 min in rats, before hepatic metabolism and urinal metabolite excretion.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Roguski, A.; Gill, A.C. The Role of the Mammalian Prion Protein in the Control of Sleep. Pathogens 2017, 6, 58. https://doi.org/10.3390/pathogens6040058
AMA Style
Roguski A, Gill AC. The Role of the Mammalian Prion Protein in the Control of Sleep. Pathogens. 2017; 6(4):58. https://doi.org/10.3390/pathogens6040058
Chicago/Turabian StyleRoguski, Amber, and Andrew C. Gill. 2017. "The Role of the Mammalian Prion Protein in the Control of Sleep" Pathogens 6, no. 4: 58. https://doi.org/10.3390/pathogens6040058
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.