What Is Our Current Understanding of PrPSc-Associated Neurotoxicity and Its Molecular Underpinnings?


1
MRC Toxicology Unit, Hodgkin Building, University of Leicester, Lancaster Road, Leicester LE1 9HN, UK
2
Department of Clinical Neurosciences, University of Cambridge, Cambridge Biomedical Campus, Cambridge CB2 0AH, UK
*
Author to whom correspondence should be addressed.
Pathogens 2017, 6(4), 63; https://doi.org/10.3390/pathogens6040063
Submission received: 29 September 2017
/
Revised: 21 November 2017
/
Accepted: 27 November 2017
/
Published: 1 December 2017
(This article belongs to the Special Issue PrPSc prions: state of the art)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The prion diseases are a collection of fatal, transmissible neurodegenerative diseases that cause rapid onset dementia and ultimately death. Uniquely, the infectious agent is a misfolded form of the endogenous cellular prion protein, termed PrPSc. Despite the identity of the molecular agent remaining the same, PrPSc can cause a range of diseases with hereditary, spontaneous or iatrogenic aetiologies. However, the link between PrPSc and toxicity is complex, with subclinical cases of prion disease discovered, and prion neurodegeneration without obvious PrPSc deposition. The toxic mechanisms by which PrPSc causes the extensive neuropathology are still poorly understood, although recent advances are beginning to unravel the molecular underpinnings, including oxidative stress, disruption of proteostasis and induction of the unfolded protein response. This review will discuss the diseases caused by PrPSc toxicity, the nature of the toxicity of PrPSc, and our current understanding of the downstream toxic signaling events triggered by the presence of PrPSc.
1. Introduction
The Prion diseases, or transmissible spongiform encephalopathies, are a group of fatal neurodegenerative diseases characterized by extensive neuronal death, spongiform change and gliosis. Uniquely among neurodegenerative disease, the prion diseases are transmissible, with infection between members of the same species, and in some cases between different species, possible. Since the discovery that the infectious component of prions is comprised solely of protein, more specifically a misfolded form of the cellular prion protein (PrPC), a great deal of research has focused on how this misfolded protein can cause such extensive and catastrophic damage to neurons. The major histopathological feature is the accumulation of extracellular amyloid plaques, comprised of misfolded PrPC (termed PrPSc for PrP scrapie). However, it is not known how PrPSc causes neurodegeneration, or indeed what the exact toxic species is, as the link between PrPSc, infectivity and toxicity is not sharply defined. The role of cellular PrPC also remains elusive, further clouding the investigation into disease processes. Due to its insolubility, the structure of PrPSc has not been definitively proven, hampering rational drug discovery efforts, although recent advances have allowed various models to be proposed [1]. Unfortunately, and largely due to our poor understanding of these molecular mechanisms, therapeutic treatments for prion disease remain elusive.
Recent research has begun to unravel the role PrPSc plays in the neurodegeneration associated with prion disease. This review will discuss the diseases caused by PrPSc toxicity, the nature of the toxicity of PrPSc, and our current understanding of the downstream toxic signaling events triggered by the presence of PrPSc.
1.1. PrPC
The human prion protein is highly conserved in mammals, suggesting an essential role for the protein [2]. It is a small glycoprotein found mainly on the outer leaflet of the plasma membrane, held in place by a C-terminal glycosylphosphatidylinositol (GPI) anchor [3]. Although highly expressed in the tissues of the central nervous system (CNS), PrPC is also expressed to varying degrees in most tissues in the body. Expression begins early in embryogenesis, and in adults the highest levels are in neurons, with moderate expression observed in glial cells and the peripheral nervous system [4]. Human PrPC is synthesized as a 231 amino acid polypeptide (after removal of a 22 residue signal peptide [5]), which is processed through the endoplasmic reticulum (ER) and golgi apparatus. Post-translational modifications, including the removal of a signal sequence from the C-terminal end of PrPC, result in a mature protein of 208 amino acids in length. The main structural features are a globular C-terminal domain made up of three alpha helices with a small antiparallel beta sheet composed of two separate strands, and a largely unstructured flexible N-terminal tail [6].
The exact cellular function of PrPC remains unclear, with several distinct and overlapping roles suggested. One of the most important proposed roles of PrPC is the maintenance of myelination in the peripheral nervous system [7]. Interestingly, neuron-specific PrPC expression is enough to maintain myelination, as although PrPC is expressed in Schwann cells, it appears to not be essential there [7]. It is unknown what effect PrPC has on CNS myelin. The first proposed role for PrPC was in Cu2+ homeostasis due it its high affinity binding [8], although a functional physiological role for this affinity remains elusive. PrPC has a putative role in protecting against stress, especially oxidative and some apoptotic stresses [9,10,11]. There is also evidence it helps to regulate neuronal excitability and memory [12,13]. Interestingly, it is also involved in the regulation of the circadian rhythm [14] and cellular differentiation [15,16]. The wide variety of biological roles has led to the suggestion that PrPC is a scaffold protein that regulates the formation of a number of multi-protein complexes, but it is unlikely that the entirety of its physiological roles have been discovered [2].
1.2. PrPSc
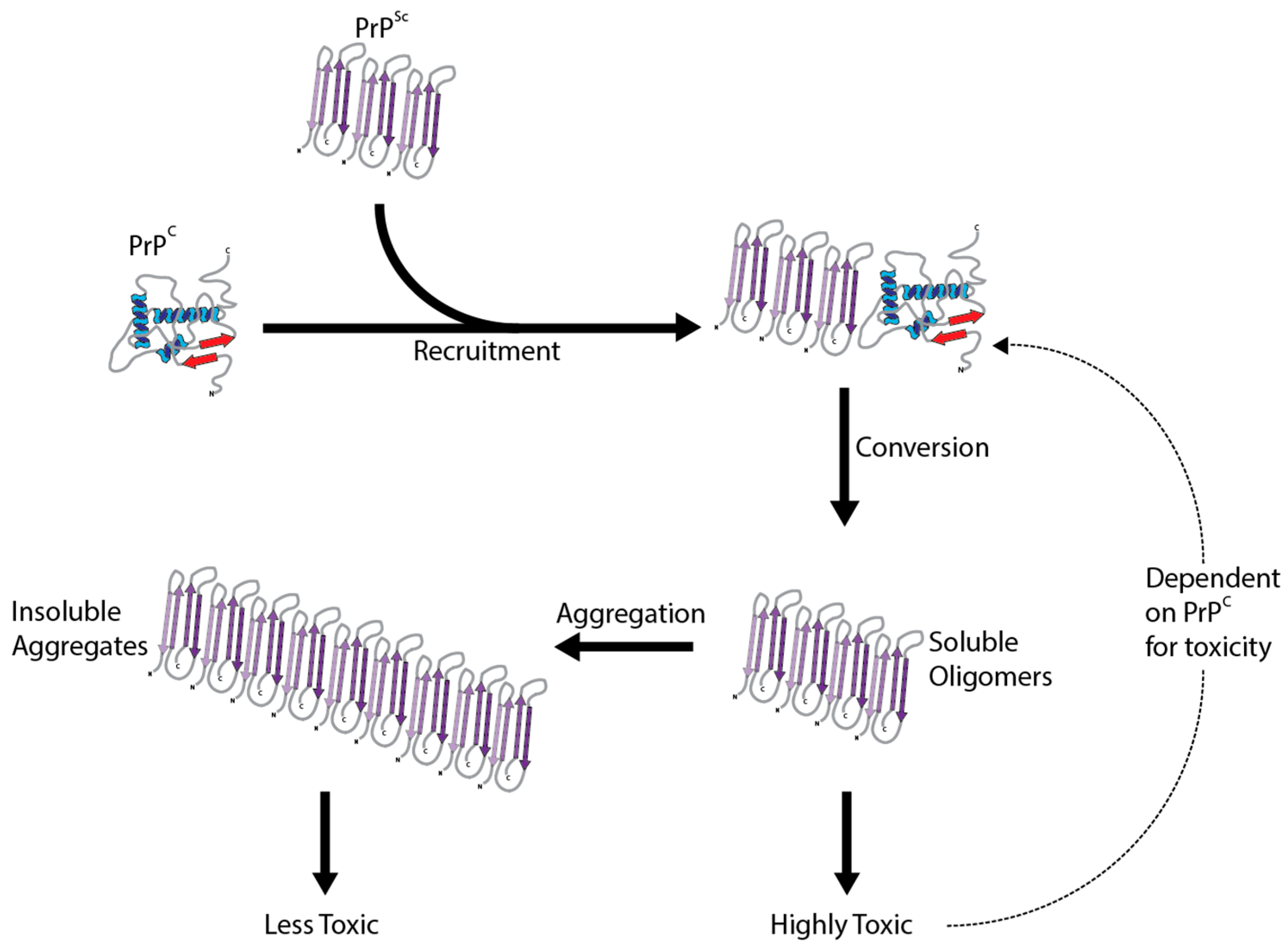
The crucial event in the development of prion disease is the structural and conformational change of PrPC to the disease associated misfolded form, PrPSc. This conversion changes PrPC from a protein characterized by alpha-helices to a partially protease-resistant misfolded protein categorized by beta sheets [17]. Proteinase K (PK) partially digests PrPSc and is often used to determine the presence of misfolded PrPSc [18]. Despite this conversion being essential for the pathogenesis of prion disease, the molecular underpinnings are still not understood. The transformation is thought to be a post-translational change in conformation which initiates the catalytic conversion of PrPC into more PrPSc, by the interaction of existing PrPSc molecules (Figure 1). Continuous synthesis of PrPC in the brain only provides more substrate for the pathological conversion to PrPSc. While this mainly occurs after exposure to already misfolded PrPSc, conversion can occur spontaneously in rare cases without exposure or a genetic basis. There are no primary sequence differences between PrPC and PrPSc, so the change is mediated by different secondary structures and a propensity to aggregate. This pathological change involving only the prion protein is summarized in the protein only hypothesis [19]. Strong evidence supporting PrPSc as being the main cause of prion disease comes from the production of infectious PrPSc in vitro [20]. Whatever the mechanism, this conversion is the basis for all the prion diseases.
1.3. The PRNP Gene
PrPC is encoded by the PRNP gene found on chromosome 20 in humans, and chromosome 2 in mice. It is significantly conserved across mammalian species and even vertebrates as a whole. It contains three exons, but the entire open reading frame lies within exon 3 [21,22], with all of the disease-associated mutations discovered so far located within exon 3 [23]. The PRNP gene encodes a nonapeptide region followed by four octarepeats; this motif is thought to be important for its copper binding ability. More than 30 disease-causing mutations in PRNP have been discovered, leading to a single amino acid substitution, the addition of superfluous residues or an early truncation of the protein [24]. A number of insertion mutations have also been discovered in the octarepeat region. Many of these mutations are believed to facilitate the conversion of PrPC to PrPSc, linking these mutations to disease. There are also polymorphisms in the PRNP gene that can influence the risk of developing prion disease. The most important is at codon 129, as it predisposes to sporadic, iatrogenic and variant Creutzfeldt–Jakob Disease (CJD-see below) [25]. Codon 129 codes for either methionine (M) or valine (V), and M/M homozygosity predisposes to an earlier and more rapid onset of disease, while heterozygosity is protective. A glutamate to lysine substitution at codon 219 also appears to confer a protective effect against prion disease [26]. The shortest incubation times for prion disease occur when PrPSc and the host PrPC share the same sequence, and when inoculation occurs intracerebrally instead of peripherally [27]. If the inoculating prion differs to the host PrPC, incubation times can be greatly increased, or clinical signs of disease never develop. This can prevent transmission between species, and is known as the species barrier.
1.4. Human Prion Disease
Human prion diseases are characterized by the presence of spongiform change, gliosis, amyloidosis and neuronal loss. Spongiosis appears as a series of vacuoles in fixed brain tissue. Astrocyte proliferation and neuronal cell death are other common features, and insoluble amyloid plaques containing aggregates of protease resistant prion protein (PrPSc) are often correlated with prion diseases. Uniquely in the field of neurodegeneration, prion diseases are transmissible between members of the same species, and often between (mammalian) species, although not freely as species barriers do exist. They can be sporadic, familial or acquired in origin. The most common is CJD; others include Kuru, Fatal Familial Insomnia (FFI) and Gerstmann–Straussler–Scheinker (GSS) disease. Although all are caused by the misfolding of PrPC, these diseases often display startlingly different pathological and biochemical characteristics. These diseases can also affect different regions of the brain, causing further differences in disease course and symptoms.
Mutations in PRNP cause inherited prion disease that accounts for approximately 15% of prion disease cases, producing a wide spectrum of clinical phenotypes [28]. Inherited prion diseases generally have an earlier onset, but slower disease progression than sporadic cases. These mutations are autosomal dominant, and can result in either an expanded octapeptide repeat in the normal sequence of the prion protein, a non-conservative point mutation or a stop codon insertion in the PRNP open reading frame (ORF). This can lead to familial CJD (fCJD), GSS and FFI. fCJD causes a rapidly progressive dementia with myoclonus and abnormal electroencephalogram (EEG) recordings, GSS is characterized by a slow progression of ataxia and late onset dementia, and FFI is unique with its refractory insomnia, dysautonomia and motor dysfunction. These disease syndromes are not absolute; however, the same mutation can lead to highly divergent phenotypic and pathological variation between individuals [29].
Sporadic CJD (sCJD) accounts for 85% of cases of human prion disease, occurring in around one in a million people over the age of 65. Early onset cases are extremely rare. The disease presents with a rapidly progressive dementia with myoclonus and development of movement disorders such as tremor and rigidity. Associated neurological symptoms include cerebellar ataxia, pyramidal and extra pyramidal signs, and cortical blindness. Most cases have a characteristic EEG that includes periodic sharp-wave complexes. Death occurs after an average of 4 months from diagnosis, making it one the most aggressive forms of neurodegeneration [30].
Acquired prion diseases include Kuru, iatrogenic CJD (iCJD) and vCJD. Kuru is caused by the eating of infected brain tissue, and is characterized by progressive cerebellar ataxia, mood and personality changes, and a late onset dementia [31]. Death occurs approximately one year after the emergence of clinical symptoms. iCJD is rare, and has occurred after the exposure of patients to contaminated medical treatments or equipment. Contaminated dura matter and corneal grafts, inoculation with human pituitary-derived growth hormone and gonadotrophins have all been reported [32]. Improperly sterilized surgical equipment has also led to iCJD after brain surgery. iCJD caused by intracerebral infection is relatively rapid in onset and duration, with prominent early dementia. In contrast, peripheral inoculation is associated with a prolonged incubation time and late onset dementia.
In the mid-1990s, in the wake of the Bovine Spongiform Encephalopathy (BSE) epidemic, a new neurodegenerative illness emerged in the UK. Clinically and pathologically, it resembled sCJD, but the disease had a longer duration with a protracted neuropsychiatric syndrome, and critically, mainly affected young people [33,34]. After the realization, it was a new disease, it was termed new variant, or variant CJD (vCJD) [35]. The age of onset was much earlier than sporadic CJD, with a mean age of 29, and patients as young as 16. The initial symptoms are mainly behavioural, followed by ataxia and movement disorders. Dementia occurs at a much later point in the disease than CJD, with EEG abnormalities frequently absent. It also progresses slower than sporadic CJD, with a mean duration of 14 months. As none of the patients had PRNP mutations and were at a very low risk of iatrogenic exposure, BSE was considered to be the most likely cause. Molecular studies on vCJD tissue showed that the biochemical properties of the protease-resistant prion protein found in these patients were distinct from other human prion diseases [36], but similar to that of BSE [37], leading to the acceptance that BSE exposure causes vCJD.
1.5. Animal Prion Diseases
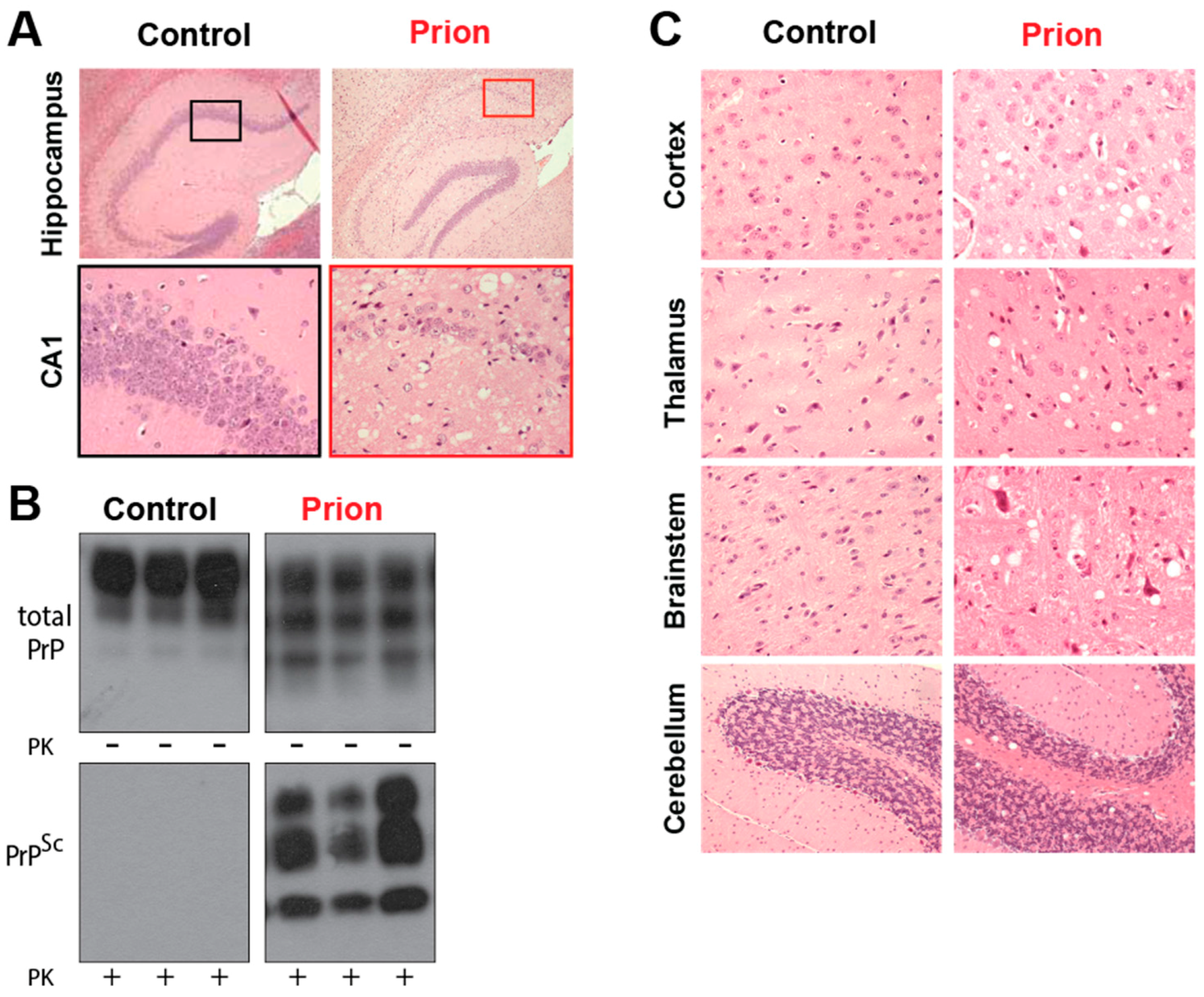
Several mammalian species are also afflicted by prion disease, including scrapie in sheep, Chronic Wasting Disease (CWD), which mainly affects deer and elk in North America and transmissible mink encephalopathy, which affects mink feeding on infected livestock. Unlike in humans, most cases are infectious in origin, although the increased surveillance for prion disease after the BSE outbreak is identifying increasing numbers of spontaneous cases. The histopathological features are grossly similar between human and animal prion disease. BSE affects the brainstem of cattle causing ataxia, with a presymptomatic incubation time of 5 years [38]. CWD is thought to be highly contagious due to the high number of animals infected despite the free-ranging habits of deer and elk. It has spread through much of North America and been detected in South Korea [39,40]. CWD has recently been observed in free ranging reindeer in Norway, with the origin of this outbreak currently unknown [41]. Despite not being natural carriers of prion disease, mice have been used extensively to model prion disease. They have been infected with sheep scrapie, and genetic modification of the carried Prnp gene allows prion disease from other hosts to be replicated (Figure 2).
1.6. Selective Neuronal Vulnerability in Prion Diseases
The same disease agent, PrPSc, is associated with all the prion diseases, however, the signs and symptoms of each disease can differ dramatically. This may be due to the regional differences of PrPSc accumulation in the brain and the neurons affected. This is thought to be a result of many factors including specific interactions of different protein conformers as well as region-specific micro-environments which contain a different combination of metals, chaperone proteins and translational machinery [42].
For instance, in FFI, neurodegeneration occurs in the thalamus, accounting for the insomnia associated with this prion disease due to the involvement of the thalamus in sleep regulation [30,43]. In Kuru, the damage often occurs in the cerebellum, leading to defects in coordination, while GSS has a wider range of clinical manifestations ranging from cerebellar ataxia to spastic parapesis, often in combination with dementia [43]. In CJD, the cerebral cortex is the main affected brain region [44], which results in mental impairments, mood change and various visual disturbances.
2. Is the Prion Protein Directly Responsible for Prion Disease?
Despite initial resistance, the protein only hypothesis of prion disease is now widely accepted. However, many unanswered questions remain. The most pressing, and to this day still the most elusive, is how exactly does the conversion of PrPC to PrPSc cause prion disease? There are several possibilities that have been suggested to be a cause: the conversion of PrPC to PrPSc causes a toxic loss of function in the PrPC protein; PrPSc, or aggregates of PrPSc, are directly toxic to neurons; the conversion process itself is somehow toxic, or there are transient intermediaries formed that mediate the toxicity. In addition to the main cause, multiple toxic downstream processes are likely to be engaged, with neuroprotective responses failing or behaving ineffectively.
2.1. PrPC is Essential for Prion Disease
It is now known that PrPC loss of function is not the main cause of prion disease. Knockout mice models are grossly normal and display no obvious phenotypes [12,45,46]. In species where prion disease is naturally occurring, such as cows and goats, PrP knockout is again non-toxic [47,48]. Therefore, loss of PrPC function does not cause prion-induced neurodegeneration. However, since the first reports of PrPC knockout, many subtle phenotypes have been described, including some related to neuroprotective pathways such as neurogenesis and stress protection [49]. Although not a direct cause, impairment of some of these processes might contribute to disease under specific stress insults.
PrPC does appear to have some directly neuroprotective abilities. PrPC is upregulated in neurons after ischaemic stroke in humans, and knocking out PrPC was shown to greatly increase infarct size in an animal model [50]. PrPC is involved in the maintenance of myelin in the mouse peripheral nervous system [7]. PrPC is also involved in protecting against the neurotoxicity induced by the artificial expression of its closest homologue, doppel (Dpl). This was first discovered due to the accidental expression in the brain of Dpl by a group trying to delete the Prnp gene in mice [51], caused by the fusion of the PrP promoter to the Dpl open reading frame and its subsequent ectopic expression in the brain [52]. Dpl expression caused Purkinje cell death [51] and a late onset ataxia [53] that could be rescued by PrPC expression in a dose-dependent manner [54]. However, Dpl is not normally expressed in the CNS in any significant quantities, and levels are not increased during prion disease [55], so the physiological extent of neuroprotection by PrPC cannot be inferred.
The most important observation from PrPC knock out experiments is the absolute and total requirement of the presence of PrPC for any PrPSc induced pathology. This was first demonstrated in mice lacking PrPC, which were resistant to prion infection [56]. A set of elegant experiments further proved this effect. Brandner and colleagues grafted neural tissue overexpressing PrPC into the brain of PrP null mice [57]. After inoculation with prions, the grafts accumulated high levels of PrPSc and developed the severe histopathological changes characteristic of prion disease. Substantial amounts of graft-derived PrPSc migrated into the surrounding areas of the host brain, but even 16 months after inoculation no pathological changes were seen in PrP null tissue [57]. Therefore, in addition to being resistant to scrapie infection, brain tissue devoid of PrPC is not damaged by exogenous PrPSc, providing further evidence that PrPSc is not directly toxic in vivo. This was further demonstrated in experiments in which PrPC was depleted during the course of prion infection [58]. Double transgenic mice were generated that had floxed PrP transgenes, from which the PrP coding sequence is deleted by neuronal Cre expression at 9 weeks of age. When these mice are inoculated with prions before PrP knockout, they develop the initial stages of prion disease, including spongiosis and hippocampal shrinkage. When the Cre-mediated excision of the Prnp gene occurred, prion disease was prevented from developing and the early spongiform changes were reversed, despite continued prion replication in non-neuronal cells and further astrocytic extra-neuronal PrPSc deposition. The mice lived for the normal lifespan, and remarkably, they never developed further clinical disease [58]. These results also argue against direct neurotoxicity of PrPSc, because the continued non-neuronal replication and accumulation of PrPSc throughout the brains of scrapie-infected mice was not pathogenic.
2.2. The Weak Links between PrPSc and Neurotoxicity
A number of other experiments have also demonstrated the complicated relationship between PrPSc and toxicity. Interestingly, the GPI-anchor has been demonstrated to be required for PrPSc induced toxicity. Prion inoculated mice expressing anchor-less PrP which is released into the extracellular space instead of being tethered to the plasma membrane, freely replicate PrPSc but do not develop disease [59]. This again suggests that PrPSc is not directly toxic to neurons, and also that either conversion at the membrane or subsequent internalization mediated by the GPI anchor is required for toxicity. In concert with this, the levels of PrPSc deposition are poorly correlated with disease progression, with subclinical cases of prion infection and prion disease observed with low prion titers observed in both animal and human cases [60]. Several studies have noted neurodegeneration without amyloidgenic PrPSc when passaging BSE prions into mice or rats [61,62]. In humans, several inherited mutations cause neurodegeneration without plaques [63,64,65]. A transmembrane form of the prion protein has been demonstrated to cause GSS without any detectable PrPSc [66]. Studies using hamster prions injection into mice have demonstrated cases of substantial PrPSc replication without the emergence of clinical signs [67,68]. These experiments have profound implications for the development, diagnosis and treatment of prion disease. It may be that PrPSc is a better marker for prion infection rather than prion-induced neurodegeneration, and again demonstrates the relative lack of neurotoxicity of PrPSc. Subclinical infection also poses a public health risk, as PrPSc from non-symptomatic individuals can still be infectious to other, perhaps more susceptible individuals [69].
Unfortunately, the identity of the actual infective agent even in purified scrapie infectious fractions remains a source for debate. Only 1 in 105 particles appears to be infectious [70], so the structure of the infectious agent cannot be definitively inferred. The most infectious particles have been shown to be non-fibrillar in nature, and comprised of 14–28 PrP molecules, with infectivity significantly reduced in oligomers larger or smaller than this [71]. PrPSc is partially protease resistant by definition, but infectious PK-sensitive forms of PrPSc have also been detected [72,73,74]. These PK-sensitive forms share structural features with PK-resistant PrPSc, and are similarly infective [74]. In contrast, high levels of PK-resistant PrPSc are not always correlated with disease [75], or infectivity [76]. Partial protease resistance is not consistently correlated with infectivity, and non-infective protease resistant PrP can be produced [77]. Furthermore, infectious PrPSc can be comprised of both protease sensitive and resistant fractions. All of these studies highlight the complexity and heterogeneity of the toxic agent in prion disease, obfuscating a better understanding of the mechanisms of disease.
2.3. PrPSc Structure and Toxicity
A puzzling characteristic of prion disease is the existence of different strains/isolates of PrPSc that when inoculated into model organisms can produce drastically different incubation times, clinical signs and pathology. Biochemically, the strains display different immunohistopathological characteristics and protease sensitivities [78]. As prions are comprised only of protein and are generated by the conversion of host PrPC, the prion strain phenomenon cannot be attributed to genetic variability. Instead, prion strains are likely to result from distinct conformational changes, that are maintained during the conversion process [79]. This suggests that the structure of PrPSc may help to explain the associated neurotoxicity. Unfortunately, difficulties in purifying and determining the structure of PrPSc have hindered investigations.
The toxic conversion results in PrPSc characterized by extensive β-sheet secondary structure [17], a protease resistant core [80] and new epitopes not shared with PrPC [81]. From here, monomers, oligomers, protofibrils and insoluble fibrils of PrPSc then accumulate, creating a heterogeneous assortment of structures. It is believed the β-sheet is essential for the aggregation of PrPSc into amyloid fibrils [82]. Several models of PrPSc amyloid have been suggested, including short compact fibrils or parallel β-sheets (see [1] for review). It is believed that fibrils dictate infectivity and the species barrier effect [1], as fibril load correlates with infectivity, but not toxicity [83]. Even if they are not the toxic species, their presence may catalyze the formation of a more toxic species [84], with growing evidence pointing towards oligomers of PrPSc as being the culprit. They display increased toxicity compared to fibrils both in vitro and in vivo [85,86,87]. These results are in agreement with the current evidence from other protein misfolding neurodegenerative diseases, where smaller oligomer molecules are thought to be the most toxic [88]. Determining the exact structure of the various PrPSc species remains elusive, and thus so does the exact relationship between PrPSc structure and toxicity. There is evidence that the disordered N-terminal domain of PrPC mediates the toxicity of PrPSc [89]; it is likely that a better understanding of similar relationships between structure and function will improve our understanding of PrPSc neurotoxicity.
3. The Molecular Underpinnings of PrPSc Toxicity
Despite the weak evidence for the direct neurotoxicity of PrPSc, there are numerous reported detrimental effects of PrPSc formation/aggregation that explain at least some of the toxicity of prion disease. This includes the induction or inhibition of a range of cellular processes, discussed below.
3.1. Autophagy
Autophagy is the cell’s main clearance mechanism for aggregated or dysfunctional proteins, which delivers cytoplasmic macromolecules or organelles to be degraded to the lysosomes. The structure to be degraded is enclosed in a double membrane structure termed the autophagic vacuole (or autophagosome), which then fuses with a lysosome containing hydrolases and digestive enzymes. Autophagic processes can ultimately initiate a form of cell death similar to apoptosis if levels of aggregated protein are deemed to be too high. Due to the large buildup of aggregated proteins in the protein misfolding neurodegenerative diseases, it is no surprise that dysfunction of this pathway is often observed, and increasingly autophagy is being explored as a possible therapeutic target. The links between autophagy and prion disease were first described in a hamster model of prion disease [90], where large autophagy vacuoles were observed, which increased in size as the disease progressed. Since then, autophagic vacuoles have been observed in many experimental models and human patients [91,92,93,94].
Pharmacological treatments that induce autophagy have conferred neuroprotection in various models of prion disease. Lithium, astemizole and the experimental compound FK506 have all been reported to induce autophagy and prolong survival in prion infected mice, with the authors attributing the neuroprotective effects to an increased removal and degradation of PrPSc [95,96,97]. The mTOR inhibitor rapamycin, which also induces autophagy, was shown to be neuroprotective in a mouse model of GSS [98]. Despite these results, it is still unclear if reduced or dysfunctional autophagy is a direct toxic effect of PrPSc, or if inducing autophagy confers neuroprotection by increasing clearance of PrPSc preventing other toxic downstream effects from occurring.
3.2. The Induction of Apoptosis
Apoptosis is the programmed death of cells, and is characterized by cell shrinkage, condensation of the chromatin and fragmentation of the nucleus. It is an active process requiring gene transcription and protein translation, and markedly different from the uncontrolled necrosis [99]. Apoptosis occurs in both experimental prion disease and human patients [100,101,102,103,104]. However, it is likely that the apoptosis observed in prion disease is a downstream effect of the prion infection, rather than the direct cause of PrPSc toxicity. Genetic deletion of the major pro-apoptotic protein Bax has no effect on prion disease progression [105], and over expression of the anti-apoptotic Bcl-2 again has no effect [106]. Deletion of caspase-12, a pro-apoptotic protein induced by ER stress, also failed to protect neurons in prion diseased mice [107].
3.3. Proteasome/Ubiquitin Inhibition
The Ubiquitin Proteasome (UPS) system is the main route for targeted protein degradation in mammalian cells. Degradation of proteins by the UPS occurs over two stages; firstly, the protein to be degraded is conjugated with multiple ubiquitin (Ub) molecules, targeting it for destruction. The Ub tagged protein is then degraded by the 26S proteasome [108] and broken down into its constituent peptides, which can be recycled for future protein synthesis.
Numerous studies suggest that disruption of the UPS by PrPSc in prion disease is a contributing factor to pathogenesis. An increase in ubiquitin immuno-reactivity, which is indicative of UPS dysregulation, has been reported in prion-infected mice in the early stages of disease and precedes behavioural deficits as well as correlating with PrPSc deposition [109].
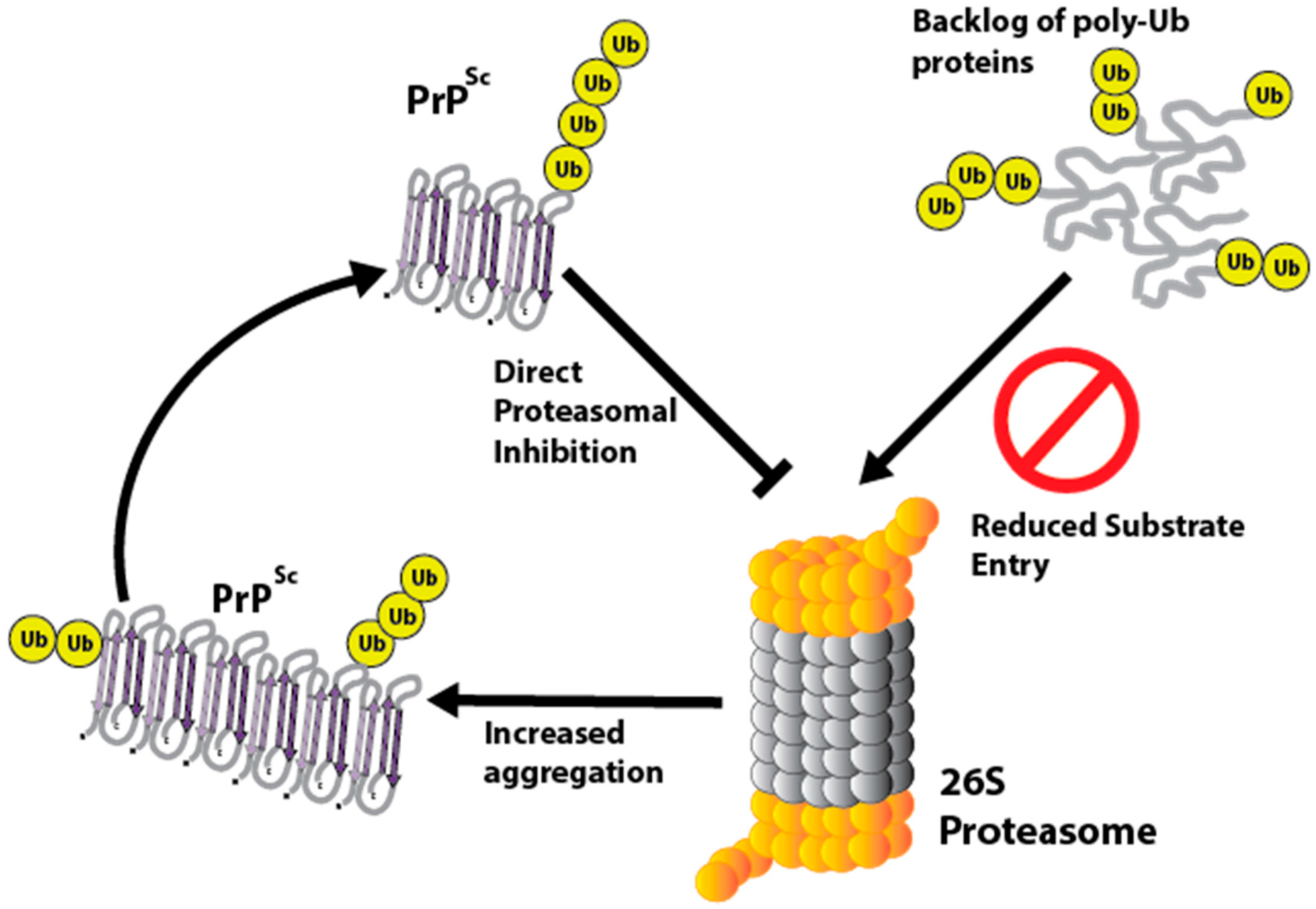
PrP isoforms have been shown to directly inhibit the 26S proteasome [109,110,111,112,113]. In vitro data using purified proteasomes and three different cell lines show that PrP oligomers directly inhibit the 26S proteasome [112]. It is thought that PrP molecules rich in β-sheet conformations such as PrPSc inhibit the 26S proteasome by reducing gate opening of the 20S subunit and hence limiting substrate entry [111]. This has been inferred because β-sheet rich PrP does not inhibit mutant proteasomes with a constitutively open gate [110]. Inhibition of the proteasome can also increase aggregation of PrPSc, in addition to affecting the processing of PrPC [114,115]. This could result in a positive feedback mechanism where PrPSc induces proteasomal malfunction, which in turn leads to increased aggregation of PrPSc and a backlog of poly-ubiquitinated proteins (Figure 3).
Proteasomal dysfunction is a common feature of many neurodegenerative diseases and given that PrPSc can directly inhibit the UPS, this could make activation of the UPS a possible therapeutic target in prion disease. McKinnon et al., found that activation of the UPS by a small molecule inhibitor resulted in enhanced clearance of poly-ubiquitinated substrates and reduced PrPSc load in prion infected cells [109]. Reducing PrPSc and its effects on proteostasis in this manner could be doubly beneficial.
3.4. The Unfolded Protein Response
Recent studies have implicated aberrant signaling by the unfolded protein response (UPR) as a major pathological player in prion disease progression and neuronal cell death [116,117,118,119,120,121,122]. The UPR is made up of three signaling cascades all beginning in the endoplasmic reticulum membrane. They are controlled by PKR-like endoplasmic reticulum kinase (PERK), Inositol-requiring enzyme 1 (IRE1) and Activating transcription factor 6 (ATF6) [123,124,125], and all are activated in response to an unfolded protein load within the ER. PERK phosphorylation attenuates protein translation via the phosphorylation of eIF2α at serine 51, in an attempt to prevent additional unfolded protein production, and a number of stress response genes are upregulated by ATF6, XBP1 (a transcription factor downstream of IRE1) and ATF4 (a transcription factor induced by eIF2α phosphorylation) [126]. After the resolution of unfolded protein stress, GADD34, the eIF2α-targeting component of protein phosphatase 1, reduces eIF2α phosphorylation and restores translation [127].
A series of studies using Tg37 hemizygous mice infected with Rocky Mountain Laboratory (RML) prions identified aberrant UPR signaling as a pathogenic mechanism in prion disease. These mice overexpress wild type murine PrP around 3-fold normal levels and follow a well-documented disease progression after prion inoculation. Loss of synapse regeneration is evident at 7 weeks post inoculation (wpi), and PERK activation and phosphorylation of eIF2α at 9 wpi precedes neuronal loss, which is apparent at 10 wpi [122]. Lentivirally expressing GADD34 reduces eIF2α phosphorylation, restores translation and prevents neurodegeneration. In contrast, administration of salubrinal (an inhibitor of eIF2α dephosphorylation) accelerates disease in these mice. Interestingly, targeting the PERK pathway in this model confers neuroprotection without affecting levels of PrPC or PrPSc. In a subsequent study, prion-infected Tg37 hemizygous mice were treated with GSK2606414 [121] a potent and bioavailable PERK inhibitor [128]. This compound effectively prevented neurodegeneration, even when dosed after the emergence of early neurological disease indicators. Again, this protection was independent of PrPSc levels.
Another compound, ISRIB (integrated stress response inhibitor), which acts downstream of eIF2α phosphorylation, has been used to successfully delay neurodegeneration in prion-infected mice [119]. ISRIB binds to eIF2B, a guanine exchange factor essential for supplying the energy needed for the initiation of translation, stabilizing it in its dimeric form [129]. This allows eIF2B to act as a guanine exchange factor even in the presence of eIF2α-P, which normally inhibits this action. The result is a partial restoration of protein synthesis in ER-stressed conditions. Administration of ISRIB to prion mice resulted in an increase in survival and marked neuroprotection of hippocampal neurons, with the treated mice also performing better in behavioural tests [119]. These studies validate PERK and the PERK-eIF2α pathway as therapeutic targets, with the restoration of translation proposed to be the neuroprotective process.
A further study aimed at repurposing drugs for use in neurodegeneration identified two molecules, trazodone and dibenzoylmethane (DBM), that partially restore protein synthesis rates during ER stress [120]. Both were neuroprotective in prion disease and a model of fronto-temporal dementia [120]. Trazodone is a serotonin agonist and reuptake inhibitor that can be rapidly repurposed, and DBM is a curcumin analogue under investigation as an anti-cancer agent.
The results of these studies suggest that the decline in protein synthesis rates mediated by PERK pathway signaling is a main contributing factor to prion disease associated neurodegeneration (Figure 4). Neuroprotection was observed despite the continued replication of PrPSc, further demonstrating the lack of direct toxicity of PrPSc and instead implicating downstream processes as mediating the toxicity in prion disease.
3.5. Oxidative Stress
Increased oxidative stress and prion disease have been linked in a plethora of previous studies. However, it is unclear if prion disease progression causes increased oxidative stress or if prion-associated pathology results from oxidative stress.
Conversion of PrPC to PrPSc as an effect of oxidative stress has been reported in various in vitro studies [130,131,132]. It has been postulated that prion misfolding in disease, particularly those of a sporadic nature, could be triggered by oxidative stress. It has also been suggested that PrPC plays a role in cellular resilience to oxidative stress via copper metabolism associated with PrPC and superoxide dismutase (SOD) activity [133,134]. Loss of the antioxidant functions of PrPC after conversion to PrPSc could hence contribute to pathogenesis of prion diseases. Studies performed in scrapie-infected mice and hamsters show an increase in markers of oxidative stress such as heme oxidase 1 [135] during disease progression. Alterations in free radical metabolism and increased oxidative stress can cause mitochondrial dysfunction in the brains of scrapie-infected animals, suggesting that this mitochondrial dysfunction is a contributing factor to prion disease progression [136,137].
3.6. Synaptic Dysfunction
One of the starkest phenotypes of prion disease is the gradual but continuous loss of synapses as the disease progresses. In mice, the number of synapses progressively decreases across a number of brain regions, including the hippocampus, with both pre- and post-synaptic processes affected [138,139,140,141,142]. This loss of synapses is correlated with a number of behavioural phenotypes [122,143,144], and precedes neuronal loss, suggesting that it is the loss of synapses that is causing the behavioral phenotypes. In human disease, there is also evidence for large quantities of synaptic loss and neurological deficits without extensive neuronal loss [145], but examples are limited due to the necessity of waiting until a post-mortem for investigation, where neuronal loss leaves a much more catastrophic signature than synaptic dysfunction.
PrP can modulate the activity of N-methyl-d-aspartate (NMDA) receptors, an important ion channel involved in long-term potentiation, but also to the detriment of neurons during excitotoxicity. In prion disease, an increase in activity through NMDA receptors is observed facilitating neuronal death, which can be attenuated by expressing PrPC [146,147,148]. There is also evidence for PrPSc directly forming pores in the plasma membrane, disrupting the electrochemical balance of the neuron [149]. Here, soluble oligomers of misfolded PrP insert into the plasma membrane forming ion channel like structures that allow the passage of ions, causing homeostatic disruption [150].
3.7. Microglia
Microglial accumulation is observed in a range of neurodegenerative diseases, including prion disease. Microglia are the macrophages of the CNS and release cytokines in response to a wide variety of stressors. There is some evidence that microglia can actually be involved in the dissemination of PrPSc throughout the brain. Baker and colleagues demonstrated that purified microglia from CJD-infected mice show similar infectivity to crude homogenate, despite having 50x less PrPSc [151]. Microglia have been shown to be required for the toxicity of a human PrP fragment in vitro, while release of reactive oxygen species (ROS) from activated microglia is suggested as the cause of neuronal apoptosis [152,153].
However, one study [154] suggests a protective role of Cx3cl1/Cx3cr1 signaling in prion disease. The chemokine Cx3cl1 is expressed by neurons and its receptor Cxcr1 is solely expressed by microglia. Prion incubation times in Cx3cr1 null mice were significantly reduced, with no observed changes in microglial activation or chemokine/cytokine expression.
4. Discussion
The prion diseases display a unique pathological and biochemical profile characterized by spongiosis, astrocytosis and neuronal death. These disorders are accompanied by a unique set of biological features, and remain among some of the most puzzling and inscrutable diseases known. Uniquely for a transmissible disease, the infectious agent is a protein encoded by the host’s own genome. The protein only hypothesis of prion disease is now widely accepted, but many unanswered questions still remain. At the center of the prion phenomenon is the conversion of PrPC to PrPSc, usually followed by the widespread aggregation of PrPSc throughout the brain. One of the first major milestones of prion disease research was the discovery that disease was not caused by the loss of function of PrPC [56]. Subsequent research has uncovered a number of proposed cellular roles for PrPC, including some neuroprotective processes [2], so the loss of function of PrPC cannot be completely ruled out as a contributor to pathology, especially as levels drop during the disease course [157]. Another interesting finding, and one that has yet to be fully explored, is that PrPC is required for the toxicity of prion disease to manifest [57,58]. Uncovering why will surely be a major step in the search for a cure for prion disease.
If the loss of function of PrPC is not the main pathological cause, then the aggregation of PrPSc becomes the most likely suspect for the mediator of toxicity. Unfortunately, prion disease is rarely so simple. Aggregates of PrPSc correlate poorly with disease progression, subclinical cases of prion disease with large amounts of PrPSc have been discovered, and prion disease without aggregates of PrPSc has also been observed [68,69]. In addition, the extra-neuronal replication of PrPSc by glial cells is not toxic to neurons if they no longer express PrPC [58]. Even the infectivity of PrPSc is not fully understood, as only a tiny proportion of PrPSc appears to be infectious, and protease resistance, usually the main marker of the presence of PrPSc, is poorly correlated with infectivity [71,75,76].
At the heart of prion disease is the structural change from α-helical PrPC to β-sheet rich PrPSc [17]. This, along with the puzzling prion strain phenomenon, suggests that determining the structure of PrPSc and its various misfolded states will greatly illuminate the molecular mechanisms of PrPSc associated toxicity. Unfortunately, this has proven extremely difficult due to the insolubility of PrPSc, hampering our understanding. However, growing evidence is suggesting that it is oligomers of PrPSc that are most toxic, compared to the larger fibrils of PrPSc or single monomers [158]. Why PrPC is more prone to misfolding than other proteins, and exactly how this happens, especially in sporadic cases where no template of PrPSc is present, is an extremely important topic of research. It has been suggested that there are cofactors that may contribute to the misfolding [159], but so far no definitive explanation has been found. Another suggestion is that an intermediate between PrPC and PrPSc may be the most toxic species, but as even stable PrPSc is proving difficult to determine the structure for, more temporary species are likely to be even harder to uncover.
Despite the evidence that argues against a direct toxic role of PrPSc, a number of detrimental processes are associated with the misfolded protein. Among the best characterized are inhibition of the proteasome and over-activation of the UPR, but a number of other processes including synaptic disruptions, the initiation of apoptosis and induction of oxidative stress are also observed. A consensus on the molecular underpinnings of PrPSc associated toxicity is far from clear, and the heterogeneity of PrPSc may mean some of these processes are strain/structure dependent. It is also possible that the mere presence of an aggregating misfolded protein, rather than the intrinsic properties of PrPSc, explain some of the associated toxicity. Similar damaging pathways are activated in other neurodegenerative diseases such as Alzheimer’s and Parkinson’s, where the identity of the misfolding proteins are different, but many similarities remain [88,160].
Unfortunately, treatment options for the prion diseases are extremely limited, but several strategies have been suggested. Reducing levels of PrPC will remove the substrate of the conversion to PrPSc [161,162], but the increasing number of beneficial cellular roles attributed to PrPC reduces the attractiveness of this approach. Another is to prevent the conversion of PrPC to PrPSc by either stabilizing PrPC or inhibiting the conversion process [163]. This has been the most popular approach, as it is amenable to small molecule or therapeutic antibody intervention, while preserving the function of PrPC [164,165]. A third method is to accelerate clearance of PrPSc from the brain [166]. A new approach is to target the downstream effects of prion replication, by inhibiting processes such as the UPR. This is surprisingly effective, as the underlying prion conversion and aggregation is unaffected, but extensive neuroprotection is still observed [119,120,121]. The effectiveness of UPR inhibitors again illustrates the complicated relationship between PrPSc and neurotoxicity; the elucidation of these underlying molecular mechanisms will undoubtedly improve our ability to treat these devastating diseases.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rodriguez, J.A.; Jiang, L.; Eisenberg, D.S. Toward the atomic structure of PrPSc. Cold Spring Harb. Perspect. Biol. 2017, 9, a031336. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Peralta, O.A.; Huckle, W.R.; Eyestone, W.H. Developmental expression of the cellular prion protein (prp(c)) in bovine embryos. Mol. Reprod. Dev. 2012, 79, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Bamborough, P.; Wille, H.; Telling, G.C.; Yehiely, F.; Prusiner, S.B.; Cohen, F.E. Prion protein structure and scrapie replication: Theoretical, spectroscopic, and genetic investigations. Cold Spring Harb. Symp. Quant. Biol. 1996, 61, 495–509. [Google Scholar] [PubMed]
- Wulf, M.A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.A.; Weis, J.; et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, C.; Takeuchi, A.M.; Nishimura, T.; Haraguchi, K.; Kubosaki, A.; Matsumoto, Y.; Saeki, K.; Matsumoto, Y.; Yokoyama, T.; Itohara, S.; et al. Prions prevent neuronal cell-line death. Nature 1999, 400, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Bounhar, Y.; Zhang, Y.; Goodyer, C.G.; LeBlanc, A. Prion protein protects human neurons against bax-mediated apoptosis. J. Biol. Chem. 2001, 276, 39145–39149. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Watt, N.T.; Walmsley, A.R.; Hooper, N.M. Tethering the n-terminus of the prion protein compromises the cellular response to oxidative stress. J. Neurochem. 2003, 84, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.R.; Ratte, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.; Collinge, J. Post-natal knockout of prion protein alters hippocampal ca1 properties, but does not result in neurodegeneration. EMBO J. 2002, 21, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Katamine, S.; Shigematsu, K.; Nakatani, A.; Sakamoto, N.; Hasegawa, S.; Nakaoke, R.; Atarashi, R.; Kataoka, Y.; Miyamoto, T. Prion protein is necessary for latent learning and long-term memory retention. Cell. Mol. Neurobiol. 1997, 17, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alavez, M.; Conti, B.; Moroncini, G.; Criado, J.R. Contributions of neuronal prion protein on sleep recovery and stress response following sleep deprivation. Brain Res. 2007, 1158, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Hajj, G.N.; Muras, A.G.; Mancini, G.L.; Castro, R.M.; Ribeiro, K.C.; Brentani, R.R.; Linden, R.; Martins, V.R. Interaction of cellular prion and stress-inducible protein 1 promotes neuritogenesis and neuroprotection by distinct signaling pathways. J. Neurosci. 2005, 25, 11330–11339. [Google Scholar] [CrossRef] [PubMed]
- Loubet, D.; Dakowski, C.; Pietri, M.; Pradines, E.; Bernard, S.; Callebert, J.; Ardila-Osorio, H.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O.; et al. Neuritogenesis: The prion protein controls beta1 integrin signaling activity. FASEB J. 2012, 26, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.J.; Vazquez-Fernandez, E.; Onisko, B.; Requena, J.R. Proteinase k and the structure of PrPSc: The good, the bad and the ugly. Virus Res. 2015, 207, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Laurent, M. Prion diseases and the ‘protein only’ hypothesis: A theoretical dynamic study. Biochem. J. 1996, 318, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Saa, P.; Hetz, C.; Soto, C. In vitro generation of infectious scrapie prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.C.; Lebo, R.V.; Clawson, G.A.; Smuckler, E.A. Human prion protein cdna: Molecular cloning, chromosomal mapping, and biological implications. Science 1986, 233, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, H.A.; Stowring, L.E.; Westaway, D.; Stubblebine, W.H.; Prusiner, S.B.; Dearmond, S.J. Molecular cloning of a human prion protein cdna. DNA 1986, 5, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Prion diseases of humans and animals: Their causes and molecular basis. Annu. Rev. Neurosci. 2001, 24, 519–550. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef] [PubMed]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic creutzfeldt-jakob disease. Nature 1991, 352, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Hizume, M.; Kobayashi, A.; Teruya, K.; Ohashi, H.; Ironside, J.W.; Mohri, S.; Kitamoto, T. Human prion protein (prp) 219k is converted to PrPSc but shows heterozygous inhibition in variant creutzfeldt-jakob disease infection. J. Biol. Chem. 2009, 284, 3603–3609. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Trabattoni, G.; Hainfellner, J.A.; Ironside, J.W.; Knight, R.S.; Budka, H. Mutations of the prion protein gene phenotypic spectrum. J. Neurol. 2002, 249, 1567–1582. [Google Scholar] [PubMed]
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559. [Google Scholar] [CrossRef] [PubMed]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial cjd: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef] [PubMed]
- Gajdusek, D.C.; Zigas, V. Degenerative disease of the central nervous system in new guinea: The endemic occurrence of kuru in the native population. N. Engl. J. Med. 1957, 257, 974–978. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.J., Jr.; Joy, A.; Heffner, R.; Franko, M.; Miyazaki, M.; Asher, D.M.; Parisi, J.E.; Brown, P.W.; Gajdusek, D.C. Clinical and pathological features and laboratory confirmation of creutzfeldt-jakob disease in a recipient of pituitary-derived human growth hormone. N. Engl. J. Med. 1985, 313, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Britton, T.C.; Al-Sarraj, S.; Shaw, C.; Campbell, T.; Collinge, J. Sporadic creutzfeldt-jakob disease in a 16-year-old in the UK. Lancet 1995, 346, 1155. [Google Scholar] [CrossRef]
- Bateman, D.; Hilton, D.; Love, S.; Zeidler, M.; Beck, J.; Collinge, J. Sporadic creutzfeldt-jakob disease in a 18-year-old in the UK. Lancet 1995, 346, 1155–1156. [Google Scholar] [CrossRef]
- Will, R.G.; Ironside, J.W.; Zeidler, M.; Cousens, S.N.; Estibeiro, K.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hofman, A.; Smith, P.G. A new variant of creutzfeldt-jakob disease in the uk. Lancet 1996, 347, 921–925. [Google Scholar] [CrossRef]
- Collinge, J.; Sidle, K.C.; Meads, J.; Ironside, J.; Hill, A.F. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ cjd. Nature 1996, 383, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vcjd and bse. Nature 1997, 389, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, N.; Wilesmith, J.; Griot, C. Bovine spongiform encephalopathy (bse): Causes and consequences of a common source epidemic. Am. J. Epidemiol. 1997, 145, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Williams, E.S.; McCarty, C.W.; Spraker, T.R.; Kreeger, T.J.; Larsen, C.T.; Thorne, E.T. Epizootiology of chronic wasting disease in free-ranging cervids in colorado and wyoming. J. Wildl. Dis. 2000, 36, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Hoover, E.A. Chronic wasting disease of cervids: Current knowledge and future perspectives. Annu. Rev. Anim. Biosci. 2015, 3, 305–325. [Google Scholar] [CrossRef] [PubMed]
- Benestad, S.L.; Mitchell, G.; Simmons, M.; Ytrehus, B.; Vikoren, T. First case of chronic wasting disease in europe in a norwegian free-ranging reindeer. Vet. Res. 2016, 47, 88. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.S. Selective vulnerability to neurodegenerative disease: The curious case of prion protein. Dis. Model. Mech. 2014, 7, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; McLean, C.A.; Masters, C.L. Gerstmann-straussler-scheinker syndrome, fatal familial insomnia, and kuru: A review of these less common human transmissible spongiform encephalopathies. J. Clin. Neurosci. 2001, 8, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Montagna, P.; Gambetti, P.; Cortelli, P.; Lugaresi, E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003, 2, 167–176. [Google Scholar] [CrossRef]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface prp protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/ola mice carrying a null mutation in prp that abolishes mrna production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Chen, J.; Yu, H.; Liu, S.; Chen, J.; Xu, X.; Sha, H.; Zhang, X.; Wu, G.; Xu, S.; et al. Functional disruption of the prion protein gene in cloned goats. J. Gen. Virol. 2006, 87, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Richt, J.A.; Kasinathan, P.; Hamir, A.N.; Castilla, J.; Sathiyaseelan, T.; Vargas, F.; Sathiyaseelan, J.; Wu, H.; Matsushita, H.; Koster, J.; et al. Production of cattle lacking prion protein. Nat. Biotechnol. 2007, 25, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Lindquist, S.; Aguzzi, A. The prion protein knockout mouse: A phenotype under challenge. Prion 2007, 1, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Katamine, S.; Nishida, N.; Moriuchi, R.; Shigematsu, K.; Sugimoto, T.; Nakatani, A.; Kataoka, Y.; Houtani, T.; Shirabe, S.; et al. Loss of cerebellar purkinje cells in aged mice homozygous for a disrupted prp gene. Nature 1996, 380, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Lee, I.Y.; Silverman, G.L.; Harrison, P.M.; Strome, R.; Heinrich, C.; Karunaratne, A.; Pasternak, S.H.; Chishti, M.A.; Liang, Y.; et al. Ataxia in prion protein (prp)-deficient mice is associated with upregulation of the novel prp-like protein doppel. J. Mol. Biol. 1999, 292, 797–817. [Google Scholar] [CrossRef] [PubMed]
- Katamine, S.; Nishida, N.; Sugimoto, T.; Noda, T.; Sakaguchi, S.; Shigematsu, K.; Kataoka, Y.; Nakatani, A.; Hasegawa, S.; Moriuchi, R.; et al. Impaired motor coordination in mice lacking prion protein. Cell. Mol. Neurobiol. 1998, 18, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Sakaguchi, S.; Shigematsu, K.; Okimura, N.; Katamine, S. Doppel-induced purkinje cell death is stoichiometrically abrogated by prion protein. Biochem. Biophys. Res. Commun. 2004, 319, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Peoc’h, K.; Volland, H.; De Gassart, A.; Beaudry, P.; Sazdovitch, V.; Sorgato, M.C.; Creminon, C.; Laplanche, J.L.; Lehmann, S. Prion-like protein doppel expression is not modified in scrapie-infected cells and in the brains of patients with creutzfeldt-jakob disease. FEBS Lett. 2003, 536, 61–65. [Google Scholar] [CrossRef]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of prp are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal prp in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Chesebro, B.; Trifilo, M.; Race, R.; Meade-White, K.; Teng, C.; LaCasse, R.; Raymond, L.; Favara, C.; Baron, G.; Priola, S.; et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005, 308, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Collinge, J. Subclinical prion infection in humans and animals. Br. Med. Bull. 2003, 66, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Lasmezas, C.I.; Deslys, J.P.; Robain, O.; Jaegly, A.; Beringue, V.; Peyrin, J.M.; Fournier, J.G.; Hauw, J.J.; Rossier, J.; Dormont, D. Transmission of the bse agent to mice in the absence of detectable abnormal prion protein. Science 1997, 275, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Manuelidis, L.; Fritch, W.; Xi, Y.G. Evolution of a strain of cjd that induces bse-like plaques. Science 1997, 277, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Nitrini, R.; Rosemberg, S.; Passos-Bueno, M.R.; da Silva, L.S.; Iughetti, P.; Papadopoulos, M.; Carrilho, P.M.; Caramelli, P.; Albrecht, S.; Zatz, M.; et al. Familial spongiform encephalopathy associated with a novel prion protein gene mutation. Ann. Neurol. 1997, 42, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.; Boyd, A.; Fletcher, A.; Byron, K.; Harper, C.; McLean, C.A.; Masters, C.L. Novel prion protein gene mutation in an octogenarian with creutzfeldt-jakob disease. Arch. Neurol. 2000, 57, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Grasbon-Frodl, E.; Lorenz, H.; Mann, U.; Nitsch, R.M.; Windl, O.; Kretzschmar, H.A. Loss of glycosylation associated with the t183a mutation in human prion disease. Acta Neuropathol. 2004, 108, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Mastrianni, J.A.; Scott, M.R.; DeFea, K.A.; Tremblay, P.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998, 279, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Joiner, S.; Linehan, J.; Desbruslais, M.; Lantos, P.L.; Collinge, J. Species-barrier-independent prion replication in apparently resistant species. Proc. Natl. Acad. Sci. USA 2000, 97, 10248–10253. [Google Scholar] [CrossRef] [PubMed]
- Race, R.; Raines, A.; Raymond, G.J.; Caughey, B.; Chesebro, B. Long-term subclinical carrier state precedes scrapie replication and adaptation in a resistant species: Analogies to bovine spongiform encephalopathy and variant creutzfeldt-jakob disease in humans. J. Virol. 2001, 75, 10106–10112. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Collinge, J. Subclinical prion infection. Trends Microbiol. 2003, 11, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Bolton, D.C.; Bendheim, P.E. Purification of scrapie agents: How far have we come? Curr. Top. Microbiol. Immunol. 1991, 172, 39–55. [Google Scholar] [PubMed]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The most infectious prion protein particles. Nature 2005, 437, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Tzaban, S.; Friedlander, G.; Schonberger, O.; Horonchik, L.; Yedidia, Y.; Shaked, G.; Gabizon, R.; Taraboulos, A. Protease-sensitive scrapie prion protein in aggregates of heterogeneous sizes. Biochemistry 2002, 41, 12868–12875. [Google Scholar] [PubMed]
- Pastrana, M.A.; Sajnani, G.; Onisko, B.; Castilla, J.; Morales, R.; Soto, C.; Requena, J.R. Isolation and characterization of a proteinase k-sensitive PrPSc fraction. Biochemistry 2006, 45, 15710–15717. [Google Scholar] [CrossRef] [PubMed]
- Sajnani, G.; Silva, C.J.; Ramos, A.; Pastrana, M.A.; Onisko, B.C.; Erickson, M.L.; Antaki, E.M.; Dynin, I.; Vazquez-Fernandez, E.; Sigurdson, C.J.; et al. Pk-sensitive prp is infectious and shares basic structural features with pk-resistant prp. PLoS Pathog. 2012, 8, e1002547. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Raeber, A.; Sailer, A.; Fischer, M.; Aguzzi, A.; Weissmann, C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted prp gene. Mol. Med. 1994, 1, 19–30. [Google Scholar] [PubMed]
- Biasini, E.; Seegulam, M.E.; Patti, B.N.; Solforosi, L.; Medrano, A.Z.; Christensen, H.M.; Senatore, A.; Chiesa, R.; Williamson, R.A.; Harris, D.A. Non-infectious aggregates of the prion protein react with several PrPSc-directed antibodies. J. Neurochem. 2008, 105, 2190–2204. [Google Scholar] [CrossRef] [PubMed]
- Riesner, D.; Kellings, K.; Post, K.; Wille, H.; Serban, H.; Groth, D.; Baldwin, M.A.; Prusiner, S.B. Disruption of prion rods generates 10-nm spherical particles having high alpha-helical content and lacking scrapie infectivity. J. Virol. 1996, 70, 1714–1722. [Google Scholar] [PubMed]
- Morales, R.; Abid, K.; Soto, C. The prion strain phenomenon: Molecular basis and unprecedented features. Biochim. Biophys. Acta 2007, 1772, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.; Wille, H.; Itri, V.; Groth, D.; Serban, H.; Torchia, M.; Cohen, F.E.; Prusiner, S.B. Eight prion strains have prp(sc) molecules with different conformations. Nat. Med. 1998, 4, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.K.; McKinley, M.P.; Bowman, K.A.; Braunfeld, M.B.; Barry, R.A.; Prusiner, S.B. Separation and properties of cellular and scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1986, 83, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Korth, C.; Stierli, B.; Streit, P.; Moser, M.; Schaller, O.; Fischer, R.; Schulz-Schaeffer, W.; Kretzschmar, H.; Raeber, A.; Braun, U.; et al. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 1997, 390, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; Clarke, A.R.; Collinge, J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011, 470, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Udgaonkar, J.B. Molecular mechanism of the misfolding and oligomerization of the prion protein: Current understanding and its implications. Biochemistry 2015, 54, 4431–4442. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskaite, J.; Young, A.; Gardner, C.E.; Macpherson, J.V.; Venien-Bryan, C.; Pinheiro, T.J. An unusual soluble beta-turn-rich conformation of prion is involved in fibril formation and toxic to neuronal cells. Biochem. Biophys. Res. Commun. 2005, 328, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Novitskaya, V.; Bocharova, O.V.; Bronstein, I.; Baskakov, I.V. Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons. J. Biol. Chem. 2006, 281, 13828–13836. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, S.; Rezaei, H.; Sales, N.; Kaiser-Schulz, G.; Lefebvre-Roque, M.; Vidal, C.; Fournier, J.G.; Comte, J.; Wopfner, F.; Grosclaude, J.; et al. In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 2007, 3, e125. [Google Scholar] [CrossRef] [PubMed]
- Ugalde, C.L.; Finkelstein, D.I.; Lawson, V.A.; Hill, A.F. Pathogenic mechanisms of prion protein, amyloid-beta and alpha-synuclein misfolding: The prion concept and neurotoxicity of protein oligomers. J. Neurochem. 2016, 139, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Resenberger, U.K.; Harmeier, A.; Woerner, A.C.; Goodman, J.L.; Muller, V.; Krishnan, R.; Vabulas, R.M.; Kretzschmar, H.A.; Lindquist, S.; Hartl, F.U.; et al. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011, 30, 2057–2070. [Google Scholar] [CrossRef] [PubMed]
- Boellaard, J.W.; Schlote, W.; Tateishi, J. Neuronal autophagy in experimental creutzfeldt-jakob’s disease. Acta Neuropathol. 1989, 78, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Boellaard, J.W.; Kao, M.; Schlote, W.; Diringer, H. Neuronal autophagy in experimental scrapie. Acta Neuropathol. 1991, 82, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Liberski, P.P.; Yanagihara, R.; Gibbs, C.J., Jr.; Gajdusek, D.C. Neuronal autophagic vacuoles in experimental scrapie and creutzfeldt-jakob disease. Acta Neuropathol. 1992, 83, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, B.; Liberski, P.P.; Giraud, P.; Kopp, N.; Brown, P. Autophagy is a part of ultrastructural synaptic pathology in creutzfeldt-jakob disease: A brain biopsy study. Int. J. Biochem. Cell Biol. 2004, 36, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tian, C.; Wang, S.B.; Xie, W.L.; Guo, Y.; Zhang, J.; Shi, Q.; Chen, C.; Dong, X.P. Activation of the macroautophagic system in scrapie-infected experimental animals and human genetic prion diseases. Autophagy 2012, 8, 1604–1620. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.; Aguib, Y.; Riemer, C.; Baier, M.; Schatzl, H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J. Neurochem. 2009, 109, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Karapetyan, Y.E.; Sferrazza, G.F.; Zhou, M.; Ottenberg, G.; Spicer, T.; Chase, P.; Fallahi, M.; Hodder, P.; Weissmann, C.; Lasmezas, C.I. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc. Natl. Acad. Sci. USA 2013, 110, 7044–7049. [Google Scholar] [CrossRef] [PubMed]
- Nakagaki, T.; Satoh, K.; Ishibashi, D.; Fuse, T.; Sano, K.; Kamatari, Y.O.; Kuwata, K.; Shigematsu, K.; Iwamaru, Y.; Takenouchi, T.; et al. Fk506 reduces abnormal prion protein through the activation of autolysosomal degradation and prolongs survival in prion-infected mice. Autophagy 2013, 9, 1386–1394. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; Qin, K.; Cook, J.; Solanki, A.; Mastrianni, J.A. Rapamycin delays disease onset and prevents prp plaque deposition in a mouse model of gerstmann-straussler-scheinker disease. J. Neurosci. 2012, 32, 12396–12405. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Groschup, M.H.; Hess, B.; Kretzschmar, H.A. Neuronal cell death in scrapie-infected mice is due to apoptosis. Brain Pathol. 1995, 5, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Jesionek-Kupnicka, D.; Buczynski, J.; Kordek, R.; Liberski, P.P. Neuronal loss and apoptosis in experimental creutzfeldt-jakob disease in mice. Folia Neuropathol. 1999, 37, 283–286. [Google Scholar] [PubMed]
- Gray, F.; Chretien, F.; Adle-Biassette, H.; Dorandeu, A.; Ereau, T.; Delisle, M.B.; Kopp, N.; Ironside, J.W.; Vital, C. Neuronal apoptosis in creutzfeldt-jakob disease. J. Neuropathol. Exp. Neurol. 1999, 58, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Budka, H. Distribution of apoptosis-related proteins in sporadic creutzfeldt-jakob disease. Brain Res. 2010, 1323, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Drew, S.C.; Haigh, C.L.; Klemm, H.M.; Masters, C.L.; Collins, S.J.; Barnham, K.J.; Lawson, V.A. Optical imaging detects apoptosis in the brain and peripheral organs of prion-infected mice. J. Neuropathol. Exp. Neurol. 2011, 70, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Coulpier, M.; Messiaen, S.; Hamel, R.; de Marco, M.F.; Lilin, T.; Eloit, M. Bax deletion does not protect neurons from bse-induced death. Neurobiol. Dis. 2006, 23, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; King, O.D.; Jackson, W.S.; Hetz, C.A.; Borkowski, A.W.; Thielen, P.; Wollmann, R.; Lindquist, S. Diminishing apoptosis by deletion of bax or overexpression of bcl-2 does not protect against infectious prion toxicity in vivo. J. Neurosci. 2007, 27, 13022–13027. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Hetz, C.; Yi, C.H.; Jackson, W.S.; Borkowski, A.W.; Yuan, J.; Wollmann, R.H.; Lindquist, S. Prion pathogenesis is independent of caspase-12. Prion 2007, 1, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Goold, R.; Andre, R.; Devoy, A.; Ortega, Z.; Moonga, J.; Linehan, J.M.; Brandner, S.; Lucas, J.J.; Collinge, J.; et al. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin-proteasome system. Acta Neuropathol. 2016, 131, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Andre, R.; Tabrizi, S.J. Misfolded prp and a novel mechanism of proteasome inhibition. Prion 2012, 6, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Deriziotis, P.; Andre, R.; Smith, D.M.; Goold, R.; Kinghorn, K.J.; Kristiansen, M.; Nathan, J.A.; Rosenzweig, R.; Krutauz, D.; Glickman, M.H.; et al. Misfolded prp impairs the ups by interaction with the 20s proteasome and inhibition of substrate entry. EMBO J. 2011, 30, 3065–3077. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.; Deriziotis, P.; Dimcheff, D.E.; Jackson, G.S.; Ovaa, H.; Naumann, H.; Clarke, A.R.; van Leeuwen, F.W.; Menendez-Benito, V.; Dantuma, N.P.; et al. Disease-associated prion protein oligomers inhibit the 26s proteasome. Mol. Cell 2007, 26, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Zhao, D.; Yang, L. Interaction between misfolded prp and the ubiquitin-proteasome system in prion-mediated neurodegeneration. Acta Biochim. Biophys. Sin. 2013, 45, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Dron, M.; Dandoy-Dron, F.; Farooq Salamat, M.K.; Laude, H. Proteasome inhibitors promote the sequestration of PrPSc into aggresomes within the cytosol of prion-infected cad neuronal cells. J. Gen. Virol. 2009, 90, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Ishibashi, D.; Nakagaki, T.; Satoh, K.; Sano, K.; Atarashi, R.; Nishida, N. Increased expression of p62/sqstm1 in prion diseases and its association with pathogenic prion protein. Sci. Rep. 2014, 4, 4504. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Russelakis-Carneiro, M.; Maundrell, K.; Castilla, J.; Soto, C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003, 22, 5435–5445. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Matamala, J.M.; Duran-Aniotz, C.; Cornejo, V.H.; Foley, A.; Hetz, C. Er stress signaling and neurodegeneration: At the intersection between alzheimer's disease and prion-related disorders. Virus Res. 2015, 207, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Radford, H.; Mallucci, G.R. Prions: Generation and spread versus neurotoxicity. J. Biol. Chem. 2014, 289, 19862–19868. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule isrib prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Radford, H.; Zents, K.A.M.; Molloy, C.; Moreno, J.A.; Verity, N.C.; Smith, E.; Ortori, C.A.; Barrett, D.A.; Bushell, M.; et al. Repurposed drugs targeting eif2alpha-p-mediated translational repression prevent neurodegeneration in mice. Brain 2017, 140, 1768–1783. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eif2alpha-p mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under er stress and beyond. Nat. Rev. Mol. Cell. Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Schroder, M. The unfolded protein response. Mol. Biotechnol. 2006, 34, 279–290. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by gadd34-mediated dephosphorylation of eif2alpha. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1h-indol-5-yl)-7h-p yrrolo[2,3-d]pyrimidin-4-amine (gsk2606414), a potent and selective first-in-class inhibitor of protein kinase r (pkr)-like endoplasmic reticulum kinase (perk). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Zyryanova, A.; Crespillo-Casado, A.; Fischer, P.M.; Harding, H.P.; Ron, D. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science 2015, 348, 1027–1030. [Google Scholar] [CrossRef] [PubMed]
- Doronina, V.A.; Staniforth, G.L.; Speldewinde, S.H.; Tuite, M.F.; Grant, C.M. Oxidative stress conditions increase the frequency of de novo formation of the yeast [psi+] prion. Mol. Microbiol. 2015, 96, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Redecke, L.; Binder, S.; Elmallah, M.I.; Broadbent, R.; Tilkorn, C.; Schulz, B.; May, P.; Goos, A.; Eich, A.; Rubhausen, M.; et al. Uv-light-induced conversion and aggregation of prion proteins. Free Radic. Biol. Med. 2009, 46, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Yang, L.; Zhang, Z.; Wu, W.; Zhou, X.; Yin, X.; Zhao, D. Cellular prion protein (PrPC) of the neuron cell transformed to a pk-resistant protein under oxidative stress, comprising main mitochondrial damage in prion diseases. J. Mol. Neurosci. 2013, 51, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Besinger, A. Prion protein expression and superoxide dismutase activity. Biochem. J. 1998, 334 Pt 2, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Sakudo, A.; Lee, D.C.; Li, S.; Nakamura, T.; Matsumoto, Y.; Saeki, K.; Itohara, S.; Ikuta, K.; Onodera, T. Prp cooperates with sti1 to regulate sod activity in prp-deficient neuronal cell line. Biochem. Biophys. Res. Commun. 2005, 328, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.G.; Kim, J.I.; Lee, H.P.; Jin, J.K.; Choi, E.K.; Carp, R.I.; Kim, Y.S. Induction of heme oxygenase-1 in the brains of scrapie-infected mice. Neurosci. Lett. 2000, 289, 173–176. [Google Scholar] [CrossRef]
- Choi, S.I.; Ju, W.K.; Choi, E.K.; Kim, J.; Lea, H.Z.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 k scrapie agent. Acta Neuropathol. 1998, 96, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Sohn, H.O.; Lim, H.B.; Lee, Y.G.; Kim, Y.S.; Carp, R.I.; Wisniewski, H.M. Alteration of free radical metabolism in the brain of mice infected with scrapie agent. Free Radic. Res. 1999, 30, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Belichenko, P.V.; Brown, D.; Jeffrey, M.; Fraser, J.R. Dendritic and synaptic alterations of hippocampal pyramidal neurones in scrapie-infected mice. Neuropathol. Appl. Neurobiol. 2000, 26, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; Halliday, W.G.; Bell, J.; Johnston, A.R.; MacLeod, N.K.; Ingham, C.; Sayers, A.R.; Brown, D.A.; Fraser, J.R. Synapse loss associated with abnormal prp precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol. Appl. Neurobiol. 2000, 26, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Belichenko, P.; Sales, J.; Jeffrey, M.; Fraser, J.R. Early loss of dendritic spines in murine scrapie revealed by confocal analysis. Neuroreport 2001, 12, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.C.; Siskova, Z.; Perry, V.H.; O'Connor, V. Selective presynaptic degeneration in the synaptopathy associated with me7-induced hippocampal pathology. Neurobiol. Dis. 2009, 35, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.R.; White, M.D.; Farmer, M.; Dickinson, A.; Khatun, H.; Powell, A.D.; Brandner, S.; Jefferys, J.G.; Collinge, J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007, 53, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Deacon, R.; Wells, H.; Boche, D.; Waters, S.; Diniz, C.P.; Scott, H.; Rawlins, J.N.; Perry, V.H. Synaptic changes characterize early behavioural signs in the me7 model of murine prion disease. Eur. J. Neurosci. 2003, 17, 2147–2155. [Google Scholar] [CrossRef] [PubMed]
- Chiti, Z.; Knutsen, O.M.; Betmouni, S.; Greene, J.R. An integrated, temporal study of the behavioural, electrophysiological and neuropathological consequences of murine prion disease. Neurobiol. Dis. 2006, 22, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Clinton, J.; Forsyth, C.; Royston, M.C.; Roberts, G.W. Synaptic degeneration is the primary neuropathological feature in prion disease: A preliminary study. Neuroreport 1993, 4, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Sassoon, J.; Daniels, M.; Brown, D.R. Astrocytic regulation of nmda receptor subunit composition modulates the toxicity of prion peptide prp106-126. Mol. Cell. Neurosci. 2004, 25, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Ratte, S.; Prescott, S.A.; Collinge, J.; Jefferys, J.G. Hippocampal bursts caused by changes in nmda receptor-dependent excitation in a mouse model of variant cjd. Neurobiol. Dis. 2008, 32, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting nmda receptors. J. Cell Biol. 2008, 181, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.A.; Martin, D.; Manuelidis, L. Microglia from creutzfeldt-jakob disease-infected brains are infectious and show specific mrna activation profiles. J. Virol. 2002, 76, 10905–10913. [Google Scholar] [CrossRef] [PubMed]
- Giese, A.; Brown, D.R.; Groschup, M.H.; Feldmann, C.; Haist, I.; Kretzschmar, H.A. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998, 8, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Schmidt, B.; Kretzschmar, H.A. Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 1996, 380, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Grizenkova, J.; Akhtar, S.; Brandner, S.; Collinge, J.; Lloyd, S.E. Microglial cx3cr1 knockout reduces prion disease incubation time in mice. BMC Neurosci. 2014, 15, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia in prion diseases. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.M.; Field, R.H.; Perry, V.H.; Murray, C.L.; Cunningham, C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon lps stimulation. Glia 2010, 58, 2017–2030. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; van der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Investig. 2014, 124, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Lian, F.; Wen, Y.; Guo, C.; Lin, D. Prion protein oligomer and its neurotoxicity. Acta Biochim. Biophys. Sin. 2013, 45, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, L.; Hinrichs, W.; Groschup, M.H. Prion protein misfolding. Curr. Mol. Med. 2009, 9, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Vats, A.; Taneja, V. Toxic species in amyloid disorders: Oligomers or mature fibrils. Ann. Indian Acad. Neurol. 2015, 18, 138–145. [Google Scholar] [PubMed]
- Safar, J.G.; DeArmond, S.J.; Kociuba, K.; Deering, C.; Didorenko, S.; Bouzamondo-Bernstein, E.; Prusiner, S.B.; Tremblay, P. Prion clearance in bigenic mice. J. Gen. Virol. 2005, 86, 2913–2923. [Google Scholar] [CrossRef] [PubMed]
- White, M.D.; Farmer, M.; Mirabile, I.; Brandner, S.; Collinge, J.; Mallucci, G.R. Single treatment with rnai against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc. Natl. Acad. Sci. USA 2008, 105, 10238–10243. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Kawagoe, K.; Chen, C.J.; Teruya, K.; Sakasegawa, Y.; Doh-ura, K. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J. Virol. 2007, 81, 12889–12898. [Google Scholar] [CrossRef] [PubMed]
- Trevitt, C.R.; Collinge, J. A systematic review of prion therapeutics in experimental models. Brain 2006, 129, 2241–2265. [Google Scholar] [CrossRef] [PubMed]
- Sim, V.L.; Caughey, B. Recent advances in prion chemotherapeutics. Infect. Disord. Drug Targets 2009, 9, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Supattapone, S.; Nguyen, H.O.; Cohen, F.E.; Prusiner, S.B.; Scott, M.R. Elimination of prions by branched polyamines and implications for therapeutics. Proc. Natl. Acad. Sci. USA 1999, 96, 14529–14534. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The conversion of PrPC to PrPSc. The protein only hypothesis of prion conversion posits that misfolded PrPSc acts a catalyst, directly binding to PrPC and causing its conversion to PrPSc. This self-perpetuating recruitment leads to large aggregates of PrPSc, and underlies its infectious potential. Surprisingly, aggregated PrPSc appears to be minimally toxic, with smaller, soluble oligomers of PrPSc likely mediating the majority of the observed neurodegeneration. Importantly, PrPC is required for both the conversion process and for the toxicity of PrPSc to manifest.
Figure 1.
The conversion of PrPC to PrPSc. The protein only hypothesis of prion conversion posits that misfolded PrPSc acts a catalyst, directly binding to PrPC and causing its conversion to PrPSc. This self-perpetuating recruitment leads to large aggregates of PrPSc, and underlies its infectious potential. Surprisingly, aggregated PrPSc appears to be minimally toxic, with smaller, soluble oligomers of PrPSc likely mediating the majority of the observed neurodegeneration. Importantly, PrPC is required for both the conversion process and for the toxicity of PrPSc to manifest.
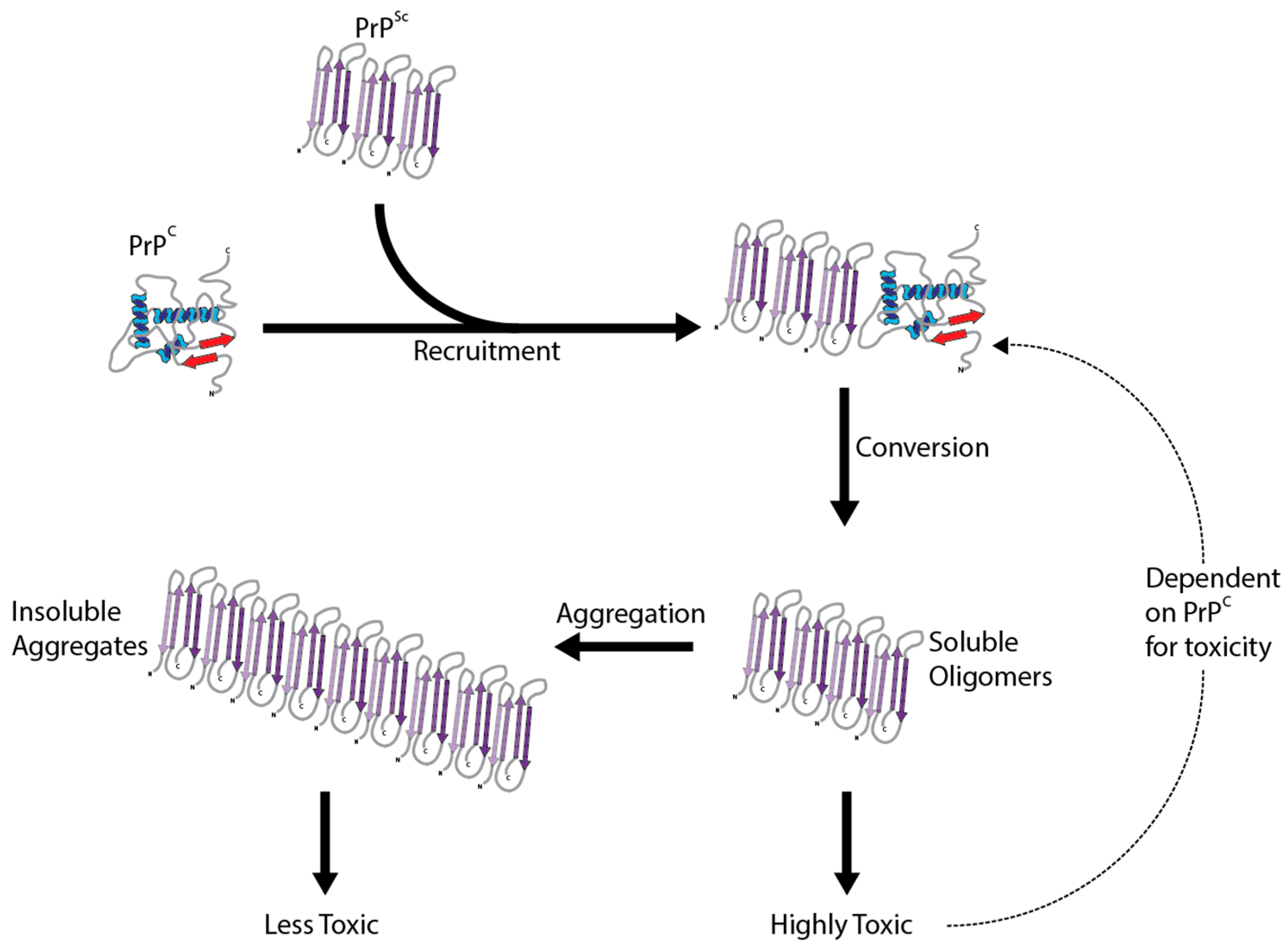
Figure 2.
The neuropathology of prion disease. (A). The Rocky Mountain Laboratory (RML) strain of prion disease induces extensive neurodegeneration in mice, especially in the hippocampus where considerable neuronal death is observed in the CA1 region (hematoxylin and eosin stained hippocampal sections from uninfected control mice, or prion-infected terminal tg37+/− mice) (B). Prion infection is associated with the accumulation of PrPSc, which is often detected by its partial proteinase resistance to digestion by proteinase K (total PrP and PrPSc levels, detected with and without proteinase K digestion) (C). Spongiosis, an intracellular oedema that appears as holes in histological slices after fixation, is observed throughout the brain in both human and animal cases of prion disease (hematoxylin and eosin stained hippocampal sections from uninfected control mice, or prion-infected terminal tg37+/− mice).
Figure 2.
The neuropathology of prion disease. (A). The Rocky Mountain Laboratory (RML) strain of prion disease induces extensive neurodegeneration in mice, especially in the hippocampus where considerable neuronal death is observed in the CA1 region (hematoxylin and eosin stained hippocampal sections from uninfected control mice, or prion-infected terminal tg37+/− mice) (B). Prion infection is associated with the accumulation of PrPSc, which is often detected by its partial proteinase resistance to digestion by proteinase K (total PrP and PrPSc levels, detected with and without proteinase K digestion) (C). Spongiosis, an intracellular oedema that appears as holes in histological slices after fixation, is observed throughout the brain in both human and animal cases of prion disease (hematoxylin and eosin stained hippocampal sections from uninfected control mice, or prion-infected terminal tg37+/− mice).
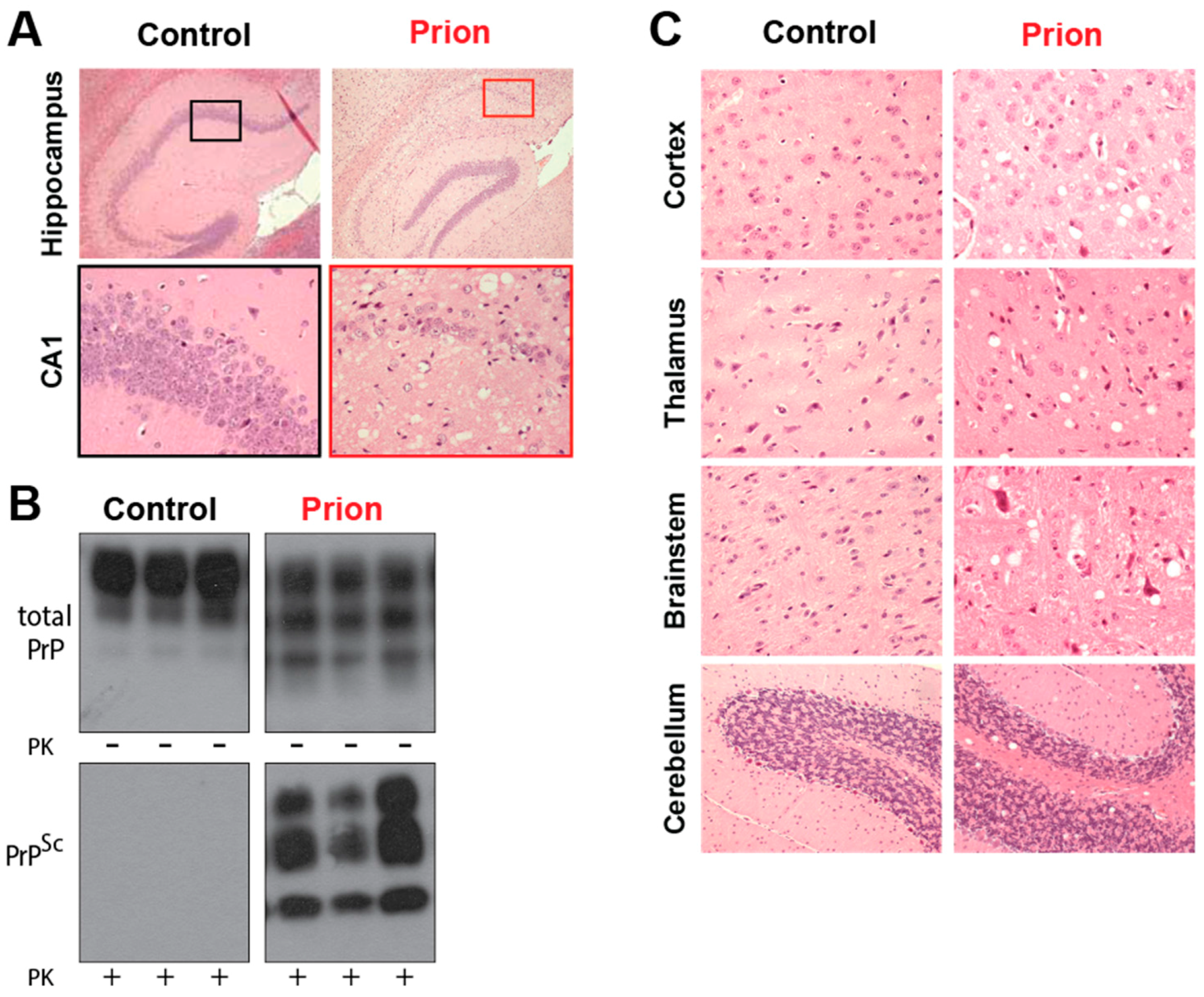
Figure 3.
Disruption of the ubiquitin proteasome system by PrPSc. PrPSc can directly inhibit the 26S proteasome by binding to the 20S subunit, preventing substrate entry. The causes increased PrPSc aggregation and accumulation of poly-ubiquitinated proteins in the cytoplasm.
Figure 3.
Disruption of the ubiquitin proteasome system by PrPSc. PrPSc can directly inhibit the 26S proteasome by binding to the 20S subunit, preventing substrate entry. The causes increased PrPSc aggregation and accumulation of poly-ubiquitinated proteins in the cytoplasm.

Figure 4.
The role of the unfolded protein response (UPR) in prion neurodegeneration. Aggregates of PrPSc activate PERK signaling, leading to a reduction in protein synthesis rates mediated by the phosphorylation of eIF2α. This starves neurons of essential proteins, leading to neurodegeneration. Restoring translation rates via lentiviral expression of GADD34 or treatment with a variety of small molecule inhibitors increases translation and is substantially neuroprotective.
Figure 4.
The role of the unfolded protein response (UPR) in prion neurodegeneration. Aggregates of PrPSc activate PERK signaling, leading to a reduction in protein synthesis rates mediated by the phosphorylation of eIF2α. This starves neurons of essential proteins, leading to neurodegeneration. Restoring translation rates via lentiviral expression of GADD34 or treatment with a variety of small molecule inhibitors increases translation and is substantially neuroprotective.
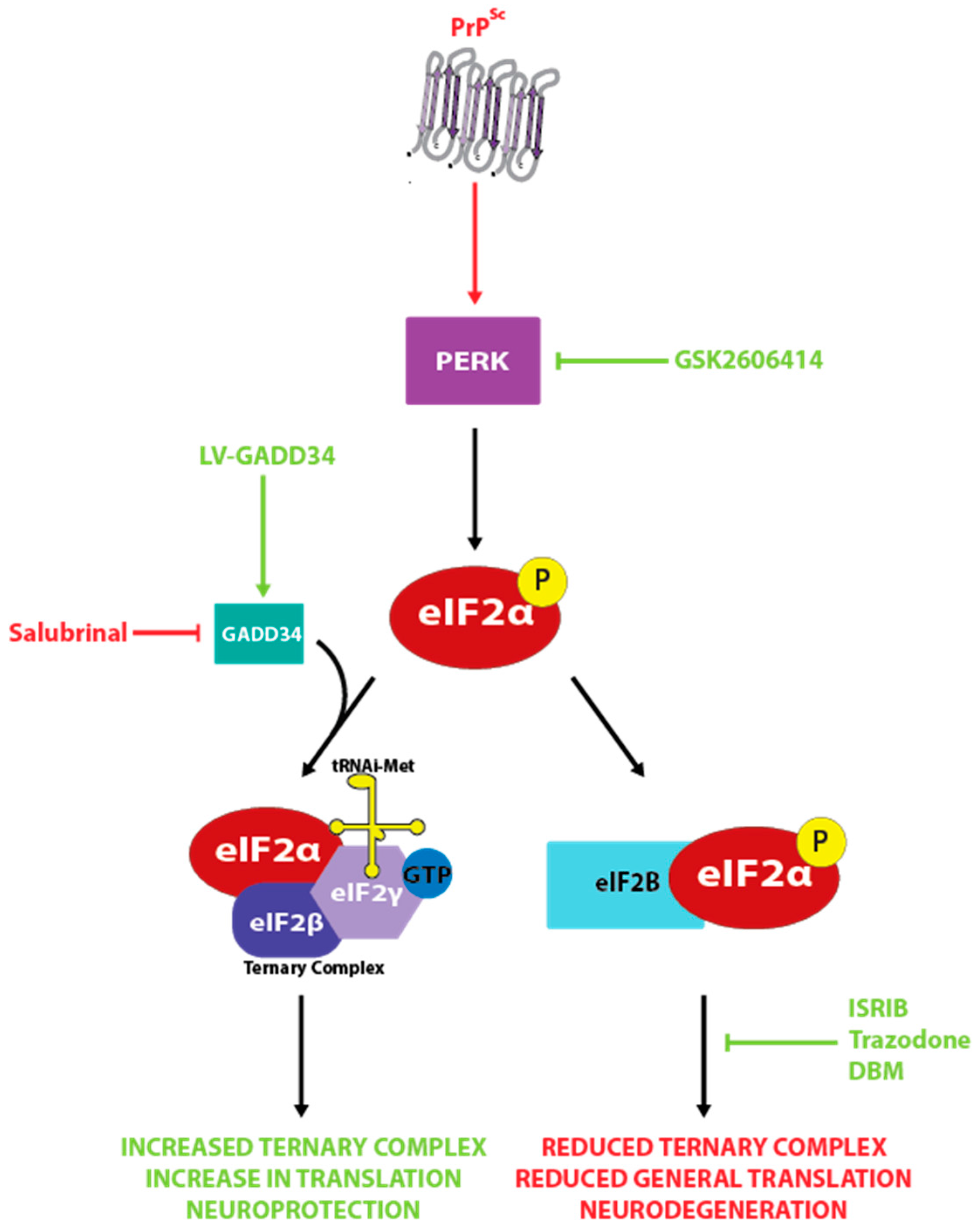
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hughes, D.; Halliday, M. What Is Our Current Understanding of PrPSc-Associated Neurotoxicity and Its Molecular Underpinnings? Pathogens 2017, 6, 63. https://doi.org/10.3390/pathogens6040063
AMA Style
Hughes D, Halliday M. What Is Our Current Understanding of PrPSc-Associated Neurotoxicity and Its Molecular Underpinnings? Pathogens. 2017; 6(4):63. https://doi.org/10.3390/pathogens6040063
Chicago/Turabian StyleHughes, Daniel, and Mark Halliday. 2017. "What Is Our Current Understanding of PrPSc-Associated Neurotoxicity and Its Molecular Underpinnings?" Pathogens 6, no. 4: 63. https://doi.org/10.3390/pathogens6040063
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.