On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts

1
School of Life Sciences, Arizona State University, Tempe, AZ 85281, USA
2
Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA 01655, USA
*
Author to whom correspondence should be addressed.
Pathogens 2018, 7(1), 16; https://doi.org/10.3390/pathogens7010016
Submission received: 10 January 2018
/
Revised: 23 January 2018
/
Accepted: 25 January 2018
/
Published: 28 January 2018
(This article belongs to the Special Issue Cytomegalovirus Infection)
Abstract
:Human cytomegalovirus (HCMV) is a member of the -herpesvirus subfamily within Herpesviridae that is nearly ubiquitous in human populations, and infection generally results only in mild symptoms. However, symptoms can be severe in immunonaive individuals, and transplacental congenital infection of HCMV can result in serious neurological sequelae. Recent work has revealed much about the demographic and selective forces shaping the evolution of congenitally transmitted HCMV both on the level of hosts and within host compartments, providing insight into the dynamics of congenital infection, reinfection, and evolution of HCMV with important implications for the development of effective treatments and vaccines.
1. Introduction
Human cytomegalovirus (HCMV) is a -herpesvirus in the Herpesviridae family of dsDNA viruses. It possesses the largest genome of any known human virus (~235,000 bp), and is nearly ubiquitous in human populations, with a seroprevalence of 30–90% in the United States and >90% in adults outside of the developed world [1,2]. Though HCMV infection is generally asymptomatic or mild in most individuals, it can lead to severe symptoms in immunocompromised patients and neonates [3,4]. HCMV is the most common cause of birth defects resulting from an infectious agent, affecting ~0.7% of life births in the U.S. (30,000 per year), with 20% of congenitally infected infants exhibiting permanent neurological sequelae, including deafness, blindness, and/or mental disability [4].
HCMV is exquisitely adapted to its human host, and cytomegaloviruses in general demonstrate a high level of host-specificity, with non-human CMV being absent even in populations with high levels of opportunity for zoonotic transmission [5]. The long-term evolution of HCMV has been well documented [6,7], but the short-term evolution of HCMV within hosts has only recently been elucidated, and much remains unknown. We review recent work that has been devoted to characterizing the heretofore unanticipated abundance of within-host variation and the complex demographic, genetic, and selective forces that shape the populations of HCMV within congenitally infected newborns. We argue that further research into the population-level processes of transplacental congenital infection and subsequent dispersal and compartmentalization is necessary for progress to be made toward successful treatments or preventative vaccines.
2. HCMV Diversity within Hosts
A large body of work has previously shown that HCMV exhibits high levels of genetic diversity within individuals [8,9,10,11,12], a potentially unexpected finding, given that HCMV encodes a DNA polymerase with proofreading activity [13]. Most research has historically focused on the sequencing of just a few loci, such as glycoproteins gB, gN, gH, and g0 [14,15,16,17,18,19,20], and has generally suffered from technological limitations related to genome coverage and sequencing depth. However, several recent studies have applied modern high-throughput sequencing methods to analyze whole genomes of longitudinally time-sampled populations [21,22,23,24,25,26,27,28], yielding novel insights into the full extent of HCMV diversity by measuring the frequencies of even rare variants within populations of HCMV and elucidating patterns of population-level evolution following initial infection.
Renzette et al. performed population genomic sequencing from urine samples collected within two weeks of birth from three congenitally HCMV-infect neonates and found that intrahost pairwise diversity across all known open reading frames (ORFs) averaged 0.22%, similar to levels observed in genome-wide analyses of human immunodeficiency virus (HIV) and ORF-specific analyses of other RNA viruses, such as dengue and West Nile [21,29]. At the time, however, it remained unclear how this high level of diversity arose in light of the low mutation rates of herpesviruses relative to those of RNA viruses.
Further work by Renzette et al. used similar population genomic analysis of urine and plasma samples from five congenitally infected infants to investigate divergence between individuals as well as within individuals across sequentially sampled time points and between different tissue compartments [22]. HCMV is known to disseminate across many tissues and organs of the body, and there is some evidence of variable levels of diversity within compartments [22,30] and genetic differentiation between specific tissues, possibly owing to cellular tropism [9,31,32]. Remarkably, they found that FST, a measure of differentiation between populations, was roughly the same between populations from the urine and plasma compartments of the same patient as between populations from the urine compartments of two unrelated individuals, suggesting evolutionary forces of sufficient strength to generate rapid divergence between compartments within a single host [22]. Comparatively, within-host divergence of a single compartment appeared largely stable over time. Hage et al. likewise found, from analysis of longitudinal population genomic samples, that a majority of 20 transplant recipients and congenitally-infected neonates included in the study displayed stability within the plasma compartment, but that nearly every individual exhibited evidence of mixed infection [24]. Additionally, one patient from this study suffering from full blown HCMV retinitis exhibited major differentiation between samples from the plasma compartment and the vitreous humour, providing additional evidence of the potential for extreme compartmentalization with HCMV.
Having established that there are high levels of within-host genomic diversity in HCMV across compartments, we must next seek to understand what combination of neutral and selective processes are generating these patterns, given that the observed levels of genetic diversity are on par with those of high-mutation rate RNA viruses.
3. Demographic Process Underlying Congenital Infection
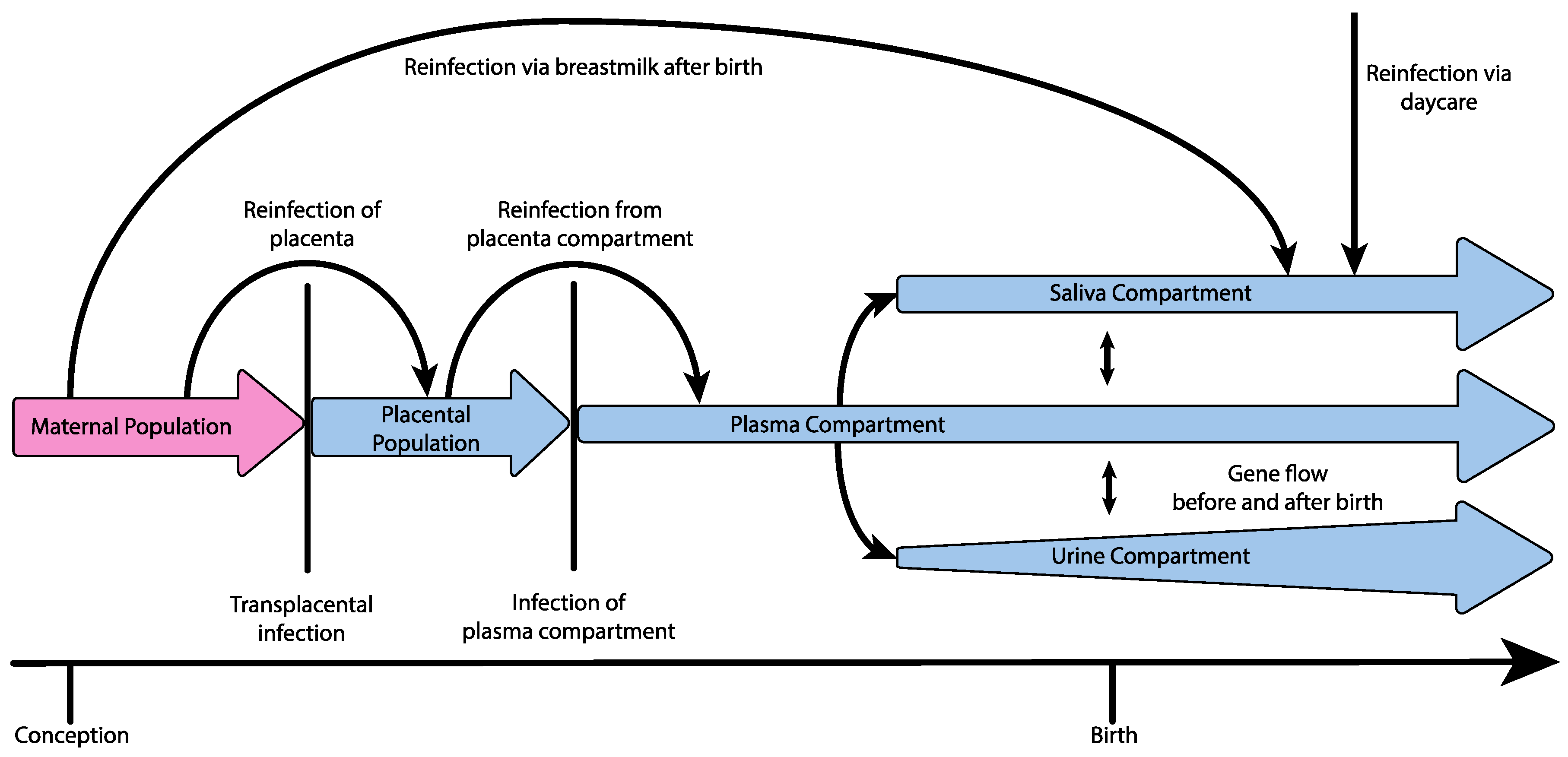
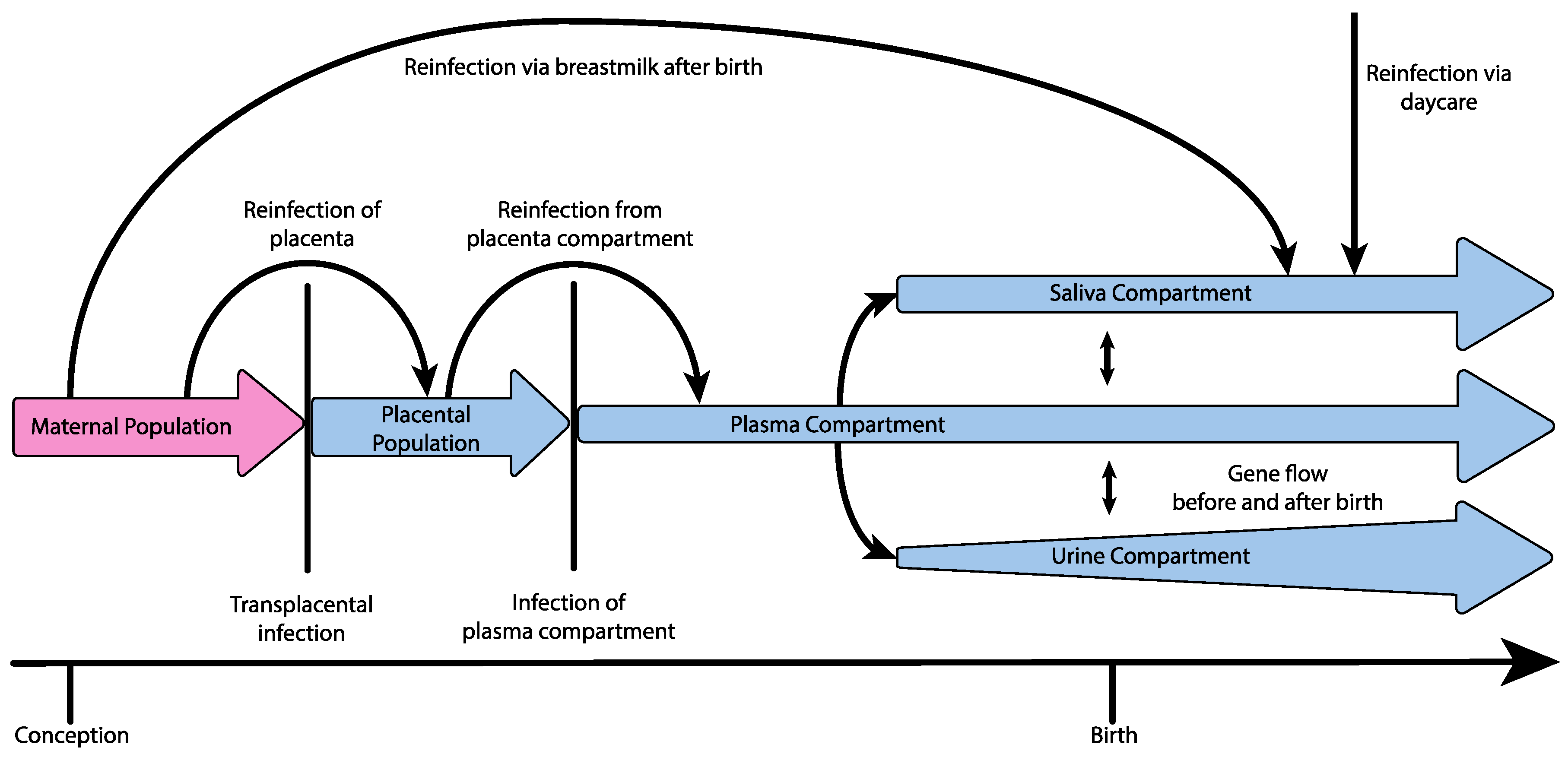
One principle explanation for the high levels of observed diversity may be the demographic processes themselves underlying infection and dissemination within the host (i.e., the viral population history of size change, structure, and migration). Applying demographic modeling methods to time-sampled genomic HCMV data from five infants, Renzette et al. found support for a demographic model of transplacental infection and dissemination, wherein HCMV populations in congenitally infected neonates experience at least two bottlenecks en route to the kidney compartment, followed by subsequent population expansions within each compartment (illustrated in Figure 1, and see Table 1 for definitions of population genetic terminology). Specifically, at least one population bottleneck was inferred to occur during movement of the virus from the maternal compartment to the plasma compartment. Additionally, in one pair of twins, two bottlenecks were inferred during this step, including a bottleneck between 13–18 weeks of gestational age, possibly during transmission from the mother to the placenta, and a second bottleneck two weeks later during infection of the fetal plasma compartment, although it could not be determined whether the bottleneck during placental infection was an anomaly of that particular pregnancy [33,34,35]. After successful colonization of the plasma compartment, an additional bottleneck was inferred to occur during transmission from the plasma compartment to the urine compartment [22]. Perhaps most importantly, demographic models indicated that the bottleneck during maternal transmission across the placenta may involve as many as hundreds of unique HCMV genomes, generating a much greater initial pool of diversity than observed during RNA virus infection, which is generally thought to involve single or few virions [22,25,36,37]. Thus, the population history of HCMV infection may partly explain why measures of diversity in HCMV populations are comparable to those of RNA viruses, despite the lower mutation rate, although some evidence suggests that oral HCMV infection of infants may primarily occur via a single virus [38].
Multiple studies have also found support for the presence of gene flow between compartments after initial dissemination and colonization throughout the host as well as recurrent maternal admixture or reinfection, possibly seeding new diversity into individual compartments [22,28]. Pokalyuk et al. found, with longitudinal sampling of the plasma, urine, and saliva compartments, that admixture from maternal HCMV populations can take place after initial infection and compartmentalization either through additional transplacental infection or reinfection of the fetus over a period of several weeks or months from a population of HCMV multiplying within the placenta, and reinfection is also known to occur after birth by way of the mother’s milk [28,39,40]. Furthermore, Renzette et al. found support for a demographic model including gene flow between compartments from longitudinal sampling of one patient’s urine and plasma, suggesting that, following collapse of the plasma compartment population due to the effect of the host’s adaptive immune response or antiviral therapy, the plasma compartment could be reinfected by genetically differentiated HCMV from other compartments [22]. If this mechanism of gene flow or periodic reinfection from various compartments is common, effective treatment design will need to account not only for epitope diversity within the targeted compartments, but for diversity across the other host compartments as well.
4. The Role of Variable Mutation, Recombination, and Selection in HCMV Evolution
In addition, an important factor contributing to variation in genome-wide diversity observed in HCMV appears to be attributable to genome-wide variation in mutation and recombination rates. Relatedly, there is accumulating evidence for variable levels of linked selection as well—namely, background selection effects modifying levels of variation owing to the recurrent removal of linked deleterious variation, and genetic hitchhiking effects modifying variation owing to linkage with positively selected alleles [41,42,43]. Renzette et al. calculated genome-wide rates of genetic diversity, as well as rates of mutation and recombination in 500 bp windows across the genome for 48 longitudinal HCMV-positive samples from 18 patients, estimating a genome-wide average mutation rate of 2.0 × 10−7 new mutations per base pair per generation, ten-fold higher than estimates for herpes simplex virus type 1, but comparable to estimates for murine cytomegalovirus [23,44,45]. Variable single nucleotide polymorphism (SNP) diversity was found to correlate across the genome with both mutation rate and recombination rate. SNP diversity was also strongly correlated with protein function, with genes encoding DNA-processing enzymes, capsid, and tegument proteins being highly conserved and genes encoding envelope and glycoproteins being on average more diverse. They also found that samples from the plasma compartment were more highly constrained than those in the saliva or urine compartments and were enriched for SNPs within glycoproteins and regulatory proteins, a possible response to cellular tropism or host immune response [23]. Related studies also found evidence for positive selection acting on glycoprotein mutations, as well as a high degree of longitudinal variability in glycoprotein SNPs [22,24]. This result is consistent with the biological understanding of glycoproteins in contributing to pathogenesis through differential properties conferred by polymorphisms, which are known to facilitate immune evasion via antigenic variation and which may also play important roles in tissue tropism and protection from neutralizing antibodies [46,47].
Given that the genomic impacts of selection are largely determined by local recombination rates [41,42], recent work has found support for background selection being the dominant mode of linked selection governing SNP diversity in HCMV, particularly in regions of low recombination rate. Conversely, genetic hitchhiking owing to positive selection appears dominant at only a handful of loci, including those associated with virulence and the transition from acute to persistent infection [26,48,49]. In total, 23–28% of the HCMV genome appears to be strongly conserved as a result of purifying selection [23]. Renzette et al. suggested more generally that the relatively small genome sizes characterizing most viruses may shift the distribution of mutational fitness effects such that a greater proportion of new mutations are deleterious and therefore subject to purifying selection, as compared to humans for example. This, combined with the generally high virus mutation rates, results in widespread background selection effects [26].
5. Clinical Implications
High levels of diversity and multiple-strain infection have been found to be associated with higher viral loads [30,50,51], delayed clearance [52], coinfection with other herpesviruses [15], and faster disease progression in HCMV disease [15] and AIDS patients [53], as well as lethal outcomes during gestation [54]. Thus, characterizing the forces shaping the diversity of HCMV populations discussed above is a critical step in the development of therapies and effective vaccines. Continued research into the magnitude and mechanisms of population-level variation should elucidate the connections between strain diversity and increased pathogenicity, and the identification of sites or epitopes that are conserved across all compartments will aid in novel vaccine development.
Hage et al. recently demonstrated the ability to detect the presence of mutations conferring resistance to antiviral drugs in clinically sampled HCMV populations, suggesting the possibility of tracking the presence and frequency of a suite of known resistance mutations during the course of treatment during HCMV infection [24]. They also found compartmentalization between blood and the vitreal humour on the level of resistance mutations in a patient suffering from HCMV retinitis, indicating that compartments unaffected by antiviral drugs may provide reservoirs of viral diversity during periods of treatment. Combined with persistent gene flow or periodic admixture between compartments [22], this would permit re-settlement of drug-resistant or naïve HCMV variants within targeted compartments during and after treatment.
Given that no current methods exist to prevent maternal-fetal transmission or reduce the severity of fetal infection [55], acting to modulate the population level processes underlying HCMV transmission may be the key to improved treatment. For example, clinically imposing stronger and more severe population bottlenecks during pregnancy (i.e., during congenital infection) may reduce genomic HCMV variability in the fetus, thus limiting the pool of standing variation on which selection may act and thereby potentially improving treatment outcomes. Advances in immunotherapy have produced treatments capable of reducing the rate of maternal transmission of other viruses, such as heptatitis B and HIV, and may prove a promising route for either preventing transmission or imposing more severe bottlenecks in HCMV as well [56,57]
6. Conclusions and Future Directions
HCMV generally persists in highly diverse populations, with diversity ranging by up to two orders of magnitude across the genome [26]. Complex demographic histories featuring multiple population bottlenecks, rapid population growth, gene flow between compartments, and admixture events during reinfection—as well as variable effects of positive and negative selection and variable rates of mutation and recombination across the genome—all contribute to variable diversity within and between hosts, compartments, and time points. Via high-throughput genomic sequencing of HCMV populations, much has been recently elucidated regarding these processes of HCMV infection, dissemination and evolution, but much yet remains to be studied. In particular, new data pairing HCMV samples from congenitally infected neonates with samples from the viral populations of their mothers across multiple compartments would shed light on the ways in which diversity is passed from mother to child during infection, and the degree to which observed variation in patients is a result of new mutations versus the transmission of pre-existing diversity owing to multiple virion infections or subsequent admixture.
Given what we now know regarding the frequency of mixed infection, reinfection, and gene flow between compartments, the development of successful vaccines and treatments for HCMV may be very difficult and require a knowledge and incorporation of variants not just from the targeted compartment, but from all compartments contributing to within-patient diversity. Furthermore, Pokalyuk et al. demonstrated that genetic sampling at individual loci can result in the misclassification of mixed infections as single infections, and Garrigue et al. have also recently shown that even when looking at individual loci, traditional Sanger sequencing can miss even relatively high-frequency resistance mutations, highlighting the utility of high-throughput, genome-level characterization of HCMV infection. This is of particular importance given the emerging links between genetic diversity and patient outcome [28,58]. Yet, given the rapidly declining costs of high-throughput sequencing methods, genome-wide multi-compartment patient sequencing paired with newly developed virus-specific population genetic analyses may soon be accessible and affordable as part of routine medical care, and there is ample reason to be optimistic that improved treatment strategies may be on the near horizon.
Acknowledgments
A.S. is funded by grants from the Swiss National Science Foundation and the U.S. Department of Defense (PR140464) to J.J. T.K. is funded by the NIH (AI109001) and the U.S. Department of Defense (PR140464).
Author Contributions
A.S., S.P., T.K., and J.J. conceived of, outlined, and designed the review, and A.S. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest. The funding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Boeckh, M.; Geballe, A.P. Science in medicine Cytomegalovirus: Pathogen, paradigm, and puzzle. J. Clin. Investig. 2011, 121, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “Silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Hassan, J.; Connell, J. Translational Mini-Review Series on Infectious Disease: Congenital cytomegalovirus infection: 50 Years on. Clin. Exp. Immunol. 2007, 149, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Anoh, A.E.; Akoua-Koffi, C.; Couacy-Hymann, E.; Pauly, M.; Schubert, G.; Mossoun, A.; Weiss, S.; Leendertz, S.A.J.; Jarvis, M.A.; Leendertz, F.H.; et al. Genetic identification of cytomegaloviruses in a rural population of Côte d’Ivoire. Virol. J. 2015, 12, 1–5. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann. N. Y. Acad. Sci. 2011, 1230, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Hirata, K.K.; Neuman, T.R. Identification of Multiple Cytomegalovirus Strains in Homosexual Men with Acquired Immunodeficiency Syndrome. J. Infect. Dis. 1984, 150, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Meyer-König, U.; Ebert, K.; Schrage, B.; Pollak, S.; Hufert, F. Simultaneous infection of healthy people with multiple human cytomegalovirus strain. Lancet 1998, 352, 1280. [Google Scholar] [CrossRef]
- Drew, W.L.; Sweet, E.S.; Miner, R.C.; Mocarski, E.S. Multiple infections by cytomegalovirus in patients with acquired immunodeficiency syndrome: Documentation by Southern blot hybridization. J. Infect. Dis. 1984, 150, 952–953. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Meyer-Konig, U.; Hufert, F.T. Variation within the glycoprotein B gene of human cytomegalovirus is due to homologous recombination. J. Gen. Virol. 1999, 80 Pt 6, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Faure-Della Corte, M.; Samot, J.; Garrigue, I.; Magnin, N.; Reigadas, S.; Couzi, L.; Dromer, C.; Velly, J.F.; Déchanet-Merville, J.; Fleury, H.J.A.; et al. Variability and recombination of clinical human cytomegalovirus strains from transplantation recipients. J. Clin. Virol. 2010, 47, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Maeno, K.; Yoshida, S. Characterization of human cytomegalovirus-induced DNA polymerase and the associated 3′-to-5′, exonuclease. Virology 1983, 124, 221–231. [Google Scholar] [CrossRef]
- Trincado, D.E.; Scott, G.M.; White, P.A.; Hunt, C.; Rasmussen, L.; Rawlinson, W.D. Human cytomegalovirus strains associated with congenital and perinatal infections. J. Med. Virol. 2000, 61, 481–487. [Google Scholar] [CrossRef]
- Coaquette, A.; Bourgeois, A.; Dirand, C.; Varin, A.; Chen, W.; Herbein, G. Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin. Infect. Dis. 2004, 39, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, S.; Dal Monte, P.; Rossini, G.; Chou, S.; Gojobori, T.; Hanada, K.; Guo, J.J.; Rawlinson, W.; Britt, W.; Mach, M.; et al. Human cytomegalovirus glycoprotein N (gpUL73-gN) genomic variants: Identification of a novel subgroup, geographical distribution and evidence of positive selective pressure. J. Gen. Virol. 2003, 84, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Novak, Z.; Pati, S.; Patro, R.K.; Blumenthal, J.; Danthuluri, V.R.; Ahmed, A.; Michaels, M.G.; Sánchez, P.J.; Bernstein, D.I.; et al. Mixed infection and strain diversity in congenital cytomegalovirus infection. J. Infect. Dis. 2011, 204, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Zweygberg, W.B.; Brytting, M.; Linde, A.; Wahren, B.; Grillner, L. Sequence variation within three important cytomegalovirus gene regions in isolates from four different patient populations. J. Clin. Microbiol. 1998, 36, 3662–3669. [Google Scholar]
- Rasmussen, L.; Geissler, A.; Winters, M. Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J. Infect. Dis. 2003, 187, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Lasry, S.; Dény, P.; Asselot, C.; Rauzy, M.; Boucher, J.; Guyot, C.; Leroux, M.C.; Livartowski, A.; Reinert, P.; Nicolas, J.C. Interstrain variations in the cytomegalovirus (CMV) glycoprotein B gene sequence among CMV-infected children attending six day care centers. J. Infect. Dis. 1996, 174, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive Genome-Wide Variability of Human Cytomegalovirus in Congenitally Infected Infants. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid Intrahost Evolution of Human Cytomegalovirus Is Shaped by Demography and Positive Selection. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Pokalyuk, C.; Gibson, L.; Bhattacharjee, B.; Schleiss, M.R.; Hamprecht, K.; Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Britt, W.J.; Jensen, J.D.; et al. Limits and patterns of cytomegalovirus genomic diversity in humans. Proc. Natl. Acad. Sci. USA 2015, 112, E4120–E4128. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Wilkie, G.S.; Linnenweber-Held, S.; Dhingra, A.; Suárez, N.M.; Schmidt, J.J.; Kay-Fedorov, P.C.; Mischak-Weissinger, E.; Heim, A.; Schwarz, A.; et al. Characterization of Human Cytomegalovirus Genome Diversity in Immunocompromised Hosts by Whole-Genome Sequencing Directly From Clinical Specimens. J. Infect. Dis. 2017, 215, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Gibson, L.; Jensen, J.D.; Kowalik, T.F. Human cytomegalovirus intrahost evolution—A new avenue for understanding and controlling herpesvirus infections. Curr. Opin. Virol. 2014, 8, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Kowalik, T.F.; Jensen, J.D. On the relative roles of background selection and genetic hitchhiking in shaping human cytomegalovirus genetic diversity. Mol. Ecol. 2016, 25, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Pfeifer, S.P.; Matuszewski, S.; Kowalik, T.F.; Jensen, J.D. On the Analysis of Intrahost and Interhost Viral Populations: Human Cytomegalovirus as a Case Study of Pitfalls and Expectations. J. Virol. 2017, 91, e01976-16. [Google Scholar] [CrossRef] [PubMed]
- Pokalyuk, C.; Renzette, N.; Irwin, K.K.; Pfeifer, S.P.; Gibson, L.; Britt, W.J.; Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Kowalik, T.F.; Jensen, J.D. Characterizing human cytomegalovirus reinfection in congenitally infected infants: An evolutionary perspective. Mol. Ecol. 2017, 26, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Sowmya, P.; Madhavan, H.N. Analysis of mixed infections by multiple genotypes of human cytomegalovirus in immunocompromised patients. J. Med. Virol. 2009, 81, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Görzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stöckl, E. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol. 2010, 84, 7195–7203. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Grefte, A.; Plachter, B.; Gouw, A.S.; The, T.H.; Jahn, G. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 1995, 76 Pt 4, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Maidji, E.; McDonagh, S.; Genbacev, O.; Fisher, S. Human cytomegalovirus transmission from the uterus to the placenta correlates with the presence of pathogenic bacteria and maternal immunity. J. Virol. 2003, 77, 13301–13314. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, T.; Gabrielli, L.; Foschini, M.P.; Lanari, M.; Guerra, B.; Eusebi, V.; Landini, M.P. Congenital cytomegalovirus infection in twin pregnancies: Viral load in the amniotic fluid and pregnancy outcome. Pediatrics 2003, 112, E133–E137. [Google Scholar] [CrossRef]
- Stagno, S. Primary Cytomegalovirus Infection in Pregnancy. JAMA 1986, 256, 1904. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Ganusov, V.V.; Giorgi, E.E.; Hraber, P.T.; Keele, B.F.; Leitner, T.; Han, C.S.; Gleasner, C.D.; Green, L.; Lo, C.C.; et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.T.; Krantz, E.M.; Swan, D.; Ferrenberg, J.; Simmons, K.; Selke, S.; Huang, M.; Casper, C.; Corey, L.; Wald, A.; et al. Transient Oral Human Cytomegalovirus Infections Indicate Inefficient Viral Spread from Very Few Initially Infected Cells. J. Virol. 2017, 91, e00380-17. [Google Scholar] [CrossRef] [PubMed]
- Enders, G.; Daiminger, A.; Bäder, U.; Exler, S.; Enders, M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 2011, 52, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Numazaki, K. Human cytomegalovirus infection of breast milk. FEMS Immunol. Med. Microbiol. 1997, 18, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Begun, D.J.; Aquadro, C.F. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 1992, 356, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Morgan, M.T.; Charlesworth, D. The effect of deleterious mutations on neutral molecular variation. Genetics 1993, 134, 1289–1303. [Google Scholar] [PubMed]
- Maynard Smith, J.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar] [CrossRef]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Hwang, C.B.C. On the mutation rate of herpes simplex virus type 1. Genetics 2005, 170, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Kropff, B.; Burkhardt, C.; Schott, J.; Nentwich, J.; Fisch, T.; Britt, W.; Mach, M. Glycoprotein N of Human Cytomegalovirus Protects the Virus from Neutralizing Antibodies. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.J.; Tortorella, D. Virion Glycoprotein-Mediated Immune Evasion by Human Cytomegalovirus: A Sticky Virus Makes a Slick Getaway. Microbiol. Mol. Biol. Rev. 2016, 80, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Kulesza, C.A.; Shenk, T. Murine cytomegalovirus encodes a stable intron that facilitates persistent replication in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 18302–18307. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Humar, A.; Preiksaitis, J.K. Concurrent genotyping and quantitation of cytomegalovirus gB genotypes in solid-organ-transplant recipients by use of a real-time PCR assay. J. Clin. Microbiol. 2008, 46, 4004–4010. [Google Scholar] [CrossRef] [PubMed]
- Puchhammer-Stockl, E.; Gorzer, I. Human cytomegalovirus: An enormous variety of strains and their possible clinical significance in the human host. Future Virol. 2011, 6, 259–271. [Google Scholar] [CrossRef]
- Manuel, O.; Åsberg, A.; Pang, X.; Rollag, H.; Emery, V.C.; Preiksaitis, J.K.; Kumar, D.; Pescovitz, M.D.; Bignamini, A.A.; Hartmann, A.; et al. Impact of Genetic Polymorphisms in Cytomegalovirus Glycoprotein B on Outcomes in Solid-Organ Transplant Recipients with Cytomegalovirus Disease. Clin. Infect. Dis. 2009, 49, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Leach, C.T.; Detels, R.; Hennessey, K.; Liu, Z.; Visscher, B.R.; Dudley, J.P.; Cherry, J.D. A longitudinal study of cytomegalovirus infection in human immunodeficiency virus type 1-seropositive homosexual men: Molecular epidemiology and association with disease progression. J. Infect. Dis. 1994, 170, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Arav-Boger, R.; Willoughby, R.E.; Pass, R.F.; Zong, J.-C.; Jang, W.-J.; Alcendor, D.; Hayward, G.S. Polymorphisms of the cytomegalovirus (CMV)-encoded tumor necrosis factor-alpha and beta-chemokine receptors in congenital CMV disease. J. Infect. Dis. 2002, 186, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J. Congenital Human Cytomegalovirus Infection and the Enigma of Maternal Immunity. J. Virol. 2017, 91, e02392-16. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.C.; Kao, J.H. Elimination of Hepatitis B: Is It a Mission Possible? BMC Med. 2017, 15, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Voronin, Y.; Jani, I.; Graham, B.S.; Cunningham, C.K.; Mofenson, L.M.; Musoke, P.M.; Permar, S.R.; Scarlatti, G. Recent progress in immune-based interventions to prevent HIV-1 transmission to children. J. Int. AIDS Soc. 2017, 20, e25038. [Google Scholar] [CrossRef] [PubMed]
- Garrigue, I.; Moulinas, R.; Recordon-Pinson, P.; Delacour, M.L.; Essig, M.; Kaminski, H.; Rerolle, J.P.; Merville, P.; Fleury, H.; Alain, S. Contribution of next generation sequencing to early detection of cytomegalovirus UL97 emerging mutants and viral subpopulations analysis in kidney transplant recipients. J. Clin. Virol. 2016, 80, 74–81. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hypothetical demography of congenital HCMV infection, including bottlenecks during infection of the placenta, infection of the plasma compartment from the placenta, and during the subsequent infection of additional compartments from the plasma (bottlenecks indicated by vertical lines). Evidence additionally suggests reinfection or continuous gene flow to the placenta from the mother over long periods of time, as well as reinfection of the plasma compartment from a stable placental population, gene flow between compartments within the new host before and after birth, rapid population growth within the urine compartment, and reinfection of the neonate via daycare or breastmilk.
Figure 1.
Hypothetical demography of congenital HCMV infection, including bottlenecks during infection of the placenta, infection of the plasma compartment from the placenta, and during the subsequent infection of additional compartments from the plasma (bottlenecks indicated by vertical lines). Evidence additionally suggests reinfection or continuous gene flow to the placenta from the mother over long periods of time, as well as reinfection of the plasma compartment from a stable placental population, gene flow between compartments within the new host before and after birth, rapid population growth within the urine compartment, and reinfection of the neonate via daycare or breastmilk.

{kind=link}
Table 1.
Summary of terms and definitions.
| Term | Definition |
|---|---|
| Population bottleneck | A sharp reduction in the size of a population, followed by recovery |
| Gene flow | The transfer of genetic variation between structured populations |
| Genetic hitchhiking | Changes in allele frequency at sites linked to positively selected loci |
| Background selection | Reduction of genetic diversity at sites linked to negatively selected loci |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sackman, A.M.; Pfeifer, S.P.; Kowalik, T.F.; Jensen, J.D. On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts. Pathogens 2018, 7, 16. https://doi.org/10.3390/pathogens7010016
AMA Style
Sackman AM, Pfeifer SP, Kowalik TF, Jensen JD. On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts. Pathogens. 2018; 7(1):16. https://doi.org/10.3390/pathogens7010016
Chicago/Turabian StyleSackman, Andrew M., Susanne P. Pfeifer, Timothy F. Kowalik, and Jeffrey D. Jensen. 2018. "On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts" Pathogens 7, no. 1: 16. https://doi.org/10.3390/pathogens7010016
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.