
Interaction of Candida Species with the Skin

1
Department of Molecular Biotechnology, Fraunhofer Institute for Interfacial Engineering and Biotechnology, 70569 Stuttgart, Germany
2
Institute of Interfacial Process Engineering and Plasma Technology, University of Stuttgart, 70569 Stuttgart, Germany
*
Author to whom correspondence should be addressed.
Microorganisms 2017, 5(2), 32; https://doi.org/10.3390/microorganisms5020032
Submission received: 4 April 2017
/
Revised: 30 May 2017
/
Accepted: 2 June 2017
/
Published: 7 June 2017
(This article belongs to the Special Issue Fungal Pathogenesis and Immune Defense)
{kind=link}
Abstract
:The human skin is commonly colonized by diverse fungal species. Some Candida species, especially C. albicans, do not only reside on the skin surface as commensals, but also cause infections by growing into the colonized tissue. However, defense mechanisms at the skin barrier level are very efficient, involving residential non-immune and immune cells as well as immune cells specifically recruited to the site of infection. Therefore, the skin is an effective barrier against fungal infection. While most studies about commensal and pathogenic interaction of Candida species with host epithelia focus on the interaction with mucosal surfaces such as the vaginal and gastrointestinal epithelia, less is known about the mechanisms underlying Candida interaction with the skin. In this review, we focus on the ecology and molecular pathogenesis of Candida species on the skin and give an overview of defense mechanisms against C. albicans in this context. We also discuss new research avenues in dermal infection, including the involvement of neurons, fibroblasts, and commensal bacteria in both mouse and human model systems.
1. Dermal Candida Ecology and Epidemiology
Colonization of human skin with bacterial and fungal communities has been observed for decades, but much remains to be understood about host-fungus interactions at this surface. Nowadays, different fungi including Malassezia, Cryptococcus, Rhodotorula, and Candida species have been identified as human skin commensals. In 2013, Findlay and coworkers analyzed the fungal communities of various human skin sites of healthy individuals. The authors found that 11 out of 14 tested sites were predominantly colonized by Malassezia species, with diversity only at the species level, while fungal communities of three sites on the feet—namely planar heel, toenail, and toe web— were much more diverse [1]. It has also been shown that under conditions of primary immunodeficiencies, fungal diversity—and in particular the abundance of Candida species—on the skin increases [2].
Although fungi are part of the commensal skin microbiota, various species are also pathogenic. It has been estimated that 20–25% of the world population is affected by fungal skin infections [3]. Fungal skin pathogens can be divided into two classes: dermatophytes and yeast, with Candida species belonging to the latter. Among the 200 known Candida species, only a few, including C. tropicalis, C. parapsilosis, and C. orthopsilosis, commonly found on healthy skin, can become pathogenic [1,4]. C. albicans is the Candida species most often responsible for symptomatic skin infections [3]. Common symptoms of Candida skin infections include thickening of the skin, hyperkeratosis, and erythema [5].
Several physical and immunological factors influence Candida skin infections, which display a preference for occluded regions of skin where humidity and CO2 accumulate and the skin is constantly experiencing friction [6]. For example, such conditions can be found while infants are in diapers, where the combination of elevated pH and the presence of lipases and proteases from feces commonly leads to secondary Candida infections, mainly caused by C. albicans from the gastrointestinal tract, resulting in diaper dermatitis, as reviewed in [7]. Recurrent Candida infections of the skin and mucosal surfaces are referred to as chronic mucocutaneous candidiasis (CMC), which occurs mainly in individuals with primary or acquired immunodeficiencies [5], upon antibiotic treatment or injury with varying individual risk factors dependent on the epithelial surfaces [8]. Increased Candida colonization has also been associated with other skin disorders, including atopic dermatitis and psoriasis [9,10]. In addition to colonization, the risk for superficial Candida infections is increased for individuals suffering from psoriasis. In part, this may also be favored by the treatment of psoriasis with corticosteroids or novel immunosuppressive agents such as tumor necrosis factor alpha (TNFα) or interleukin-17 (IL-17) inhibitors, as these pharmacological interventions specifically affect antifungal immune responses [11]. Interestingly, psoriasin, which is produced in psoriatic plaques, has been shown to have broad antifungal activity by inducing apoptosis in several filamentous fungi, but it is not active against C. albicans [12].
2. Candida Pathogenesis at Epithelial Surfaces
Most of what is known about C. albicans pathogenesis on epithelial surfaces has been shown for the oral and vaginal mucosa, while less is known about skin invasion. The distinction between commensal colonization and invasive growth of C. albicans into epithelial surfaces is not clear cut, as adhesion to the host epithelium or even initial invasion may be relevant for establishment or maintenance of both states, while host cell damage is a distinct feature of pathogenic invasion [13,14]. Adhesion to epithelial cells has been studied for many Candida spp. and on different epithelial cell types, including fibroblasts, Caco-2 cells, buccal epithelial cells, and many others. Interestingly, cell surface adhesins in Candida spp. that colonize human epithelia like Candida glabrata or Candida albicans have developed in large gene families [15,16], indicating a specialization for colonization of the host. Extensive studies on binding motifs of lectin-like epithelial adhesins revealed that the different binding domains have well defined specificity for different di- or oligomeric carbohydrate combinations, enabling the colonization of a wide variety of host tissue [17,18]. For C. albicans, attachment to epidermal keratinocytes was also shown by direct interaction between moonlighting proteins [19] and host proteins, like the fungal protein Gpm1 (Phosphoglycerate mutase) and host cell vitronectin [20]. Two general phenomena can contribute to invasion of host epithelia: induced endocytosis and active penetration. C. albicans has been shown to induce its own passive uptake into normally non-phagocytic cells by a clathrin-dependent mechanism [21] that is dependent on Als3 or Ssa1 interaction with E-cadherin or N-cadherin expressed on epithelial host cells [22,23] or alternative pathways [24]. The contribution of induced endocytosis to epithelial invasion is, however, tissue or cell-type dependent. While induced endocytosis may potentially contribute to initial invasion of stratified epithelia such as the oral mucosa, intestinal enterocytes and Caco-2 cells do not internalize C. albicans by using this process. Here the fungus needs to actively penetrate the tissue [25,26]. In either case, active penetration is the predominant process of epithelial invasion [24]. In this case, the force is generated through a combination of hyphal extension and directional growth of the fungus [25]. Since the epidermis of skin is a cornified stratified epithelium shielded by a dense layer of dead keratinocytes, this barrier can likely only be breached by active penetration. Consistent with this notion, a non-hyphal C. albicans strain deleted for Efg1 and Cph1, the two major transcription factors involved in hyphae formation, is not able to invade intestinal or epidermal reconstituted epithelia [27]. In addition, this mutant cannot adhere to tissue due to a massive change in cell wall protein composition, and lacks expression of some key adhesins like ALS3 or HWP1 [26]. Besides being required for both active penetration and efficient induced endocytosis [25], hyphae formation is also a prerequisite for host tissue damage, a hallmark of C. albicans pathogenesis in epithelial tissues [8]. Physical force generated through hyphal extension as well as molecular factors such as secreted aspartyl proteases (SAPs) have been proposed to cause damage in an oral epithelial model, in particular Sap1-3 [28], while Sap1-2 have been associated with damage during invasion of a reconstituted vaginal epithelium [29]. Several Saps have also been shown to be overexpressed during C. albicans invasion of an epidermal model [30]. However, the precise contribution of Saps to C. albicans pathogenicity has been controversial. Additional results indicate that Saps are not required for invasion into reconstituted human oral or vaginal epithelia and that Sap1–6 are dispensable for virulence in a mouse model of disseminated candidiasis [31,32,33]. More recently, Candidalysin, a pore forming toxin, generated from the ECE1 gene product, which is expressed specifically during filamentous growth of C. albicans, was shown to contribute to damage of the oral epithelium during C. albicans invasion. Most interestingly, peptide 3, a short peptide derived from the Ece1p precursor molecule, has been shown to induce cell death by perforating the host cell membranes [34].
3. Initial Fungal Recognition
Candida invasion of the skin is rapidly recognized by innate immune receptors, such as pattern recognition receptors (PRRs), which initiate an efficient immune response (Figure 1). Different fungal structures such as the cell wall components β-glucans, mannans, and phospholipomannans can be sensed by receptors belonging to various classes of PRRs including Toll-like receptors (TLRs) and C-type lectins [35]. Their contribution to host defense against Candida species is, however, tissue specific and dependent on fungal morphology. Dectin-1 is a C-type lectin expressed on various cell types including monocytes, macrophages, dendritic cells, and neutrophils [36]. It recognizes the fungal cell wall component β-glucan and is involved in epithelial antifungal defense by Th17 induction in a Syk- and Card9-dependant manner [37]. β-glucan has been reported to primarily be surface accessible at the bud scars in the yeast form of C. albicans [38]. Hyphae are therefore thought to be invisible to dectin-1. C. albicans moreover reduces recognition by shielding the bud scars with a mannoprotein layer, thereby further limiting accessibility of β-glucan [38]. On the other hand, C. albicans-induced degranulation and TNF-α, IL-6, IL-10, CCL3, and CCL4 production by mast cells, which are also present in the skin, has been shown to be dectin-1-dependent, irrespective of the morphological state of C. albicans, while IL-1β production by mast cells was observed only in response to yeast cells [39]. Langerin, which is specifically expressed on epidermal Langerhans cells, was additionally proposed to play a central role in initial recognition of a broad spectrum of fungi including C. albicans on the skin. The authors could show that this C-type lectin binds not only to β-glucan, but also to mannose structures of fungal origin [40]. Activation of the inflammasome seems hyphae specific and likely involves recognition of mannan. C. albicans hyphae induce, for example, NLRP3-dependent inflammasome activation and IL-1β secretion by macrophages in the skin [8,41]. MDA5, an intracellular receptor for double-stranded RNA, has also recently been associated with C. albicans hyphal recognition by macrophages, and its induction upon fungal stimulation was reduced in peripheral blood mononuclear cells (PBMCs) from CMC patients [42]. It remains to be determined if there is a direct role of MDA5 in skin defense against C. albicans and what may be the fungal ligand for this receptor.
The precise role of individual TLRs in skin defense against C. albicans is not yet completely understood. The fungal cell wall components O-linked mannans and phospholipomannans (PLM) are detected by TLR4 and TLR2, respectively, and result in the activation of pro-inflammatory processes by signaling through MyD88 and NF-κB [43,44]. TLR2 and TLR4 are expressed on the surface of myeloid and lymphoid cells but also on epithelial cells. On keratinocytes, TLR2 expression has been shown repeatedly and TLR2-dependent activation of NF-κB and p38MapK in response to C. albicans PLM has been reported [45,46]. Moreover, in mouse epidermal Langerhans cells, MyD88 is necessary to respond to C. albicans invasion [47]. We have recently shown that among TLRs, TLR2 is most highly expressed in dermal fibroblasts in a CD4+ T cell supplemented human 3D skin model. We could further show that TLR2 is involved in dermal protection of this tissue model against C. albicans invasion in an IL-1β-dependent manner [48,49]. Moreover, TLR3, which recognizes nucleic acids, has also been associated with protection against cutaneous candidiasis [50,51]. TLR2 and TLR4 signaling is also involved in mucosal defense. TLR4 is induced in oral epithelial cells upon C. albicans infection of a reconstituted human epithelium in the presence of polymorphonuclear leukocytes and results in a direct epithelial antifungal defense [52]. In contrast, polymorphisms in TLR2, but not in TLR4 were associated with recurrent vulvovaginal candidiasis in humans [53].
4. Immune Networks Involved in Skin Defense against Candida Species
The subsequent defense reaction to a Candia infection also varies between different tissues [5]. In the skin, IL-17 is a central cytokine shaping immunity against C. albicans invasion of the epidermis. Mutations in five genes related to IL-17 and IL-17 signaling have been found to result in CMC in humans. These include deficiencies in IL-17 receptors: IL-17RA and IL-17RC, which are expressed on various cell types including epithelial cells [54] as well as IL-17F [55] which, like the prototypic IL-17A, binds to the heterodimeric IL-17RA/IL17-RC receptor [56]. In addition, deficiency of the adapter molecule ACT1 [57], and gain of function mutations in STAT1, lead to increased Th1 and Th2 responses and to a decrease in Th17 cells [58]. IL-17C is another IL-17 family member that can be detected in the skin. It is expressed by keratinocytes and together with IL-17A and IL-17F is involved in the pathology of psoriasis [59]. However, in contrast to IL-17RC, IL-17C and its receptor IL-17RE is not required for skin, oral, and systemic defense against C. albicans infection, as shown using Il17c−/−, Il17re−/−, and Il17rc−/− mouse models [59,60]. IL-17C or IL-17RE may therefore be interesting targets for the treatment of psoriasis without increasing susceptibility to CMC, as has been reported for IL-17A directed therapies, albeit at low incidence [61]. Th17 cells, which express IL-17A and IL-17F, play a crucial role in human defense against CMC. They are induced by C. albicans and are regulated by the proinflammatory cytokine IL-1β [62]. Antigen-presenting Langerhans cells (LCs) which reside in the epidermis have been shown to be sufficient to promote CD4+ T cell differentiation to Th17 cells in mice [63]. Upon skin infection with C. albicans, LCs are activated through TLR/MyD88 and dectin-1 signaling and secrete IL-6 which promotes Th17 differentiation [5,47]. While LCs are necessary for the development of antigen-specific Th17 cells, they are not crucial in the clearance of primary infections in an epicutaneous mouse model of candidiasis [64]. IL-17A and IL-17F are not solely produced by recruited Th17 cells, but also by skin resident γδ T cells which can sense invading pathogens through TLR2 and dectin-1 and allow for a fast defense reaction [54,65]. This has been shown to depend on IL-1β and IL-23, which are secreted by dendritic cells (DC) and lead to the expansion and secretion of IL-17A and IL-17F by γδ T cells [66,67]. IL-23-dependent IL-17A production has been shown to be critical for defense against C. albicans skin infection in an epicutaneous infection model using Il17af−/− and Il23a−/− mice [68], and humans with deficiencies in IL-23R signaling show increased susceptibility to CMC [69]. Interestingly, IL-23 secretion by CD301b+ dermal dendritic cells was found to depend on the neuropeptide CGRPα, which is secreted by nociceptive neurons directly sensing C. albicans in the skin [68]. This finding links the neuronal system to immunity against fungal skin infections.
While skin defense against C. albicans in the epidermis is predominantly governed by IL-17A/IL-17F-producing cells, protection of deeper tissue such as the dermis and prevention of systemic fungal dissemination has been reported to rely on a Th1 response [64]. Secretion of IL-6 by Langerhans cells and subsequent Th17 polarization depends on fungal recognition by dectin-1, which has been reported to be restricted to detect the yeast form of C. albicans only [64]. In contrast, neither LCs nor CD11b+ dermal DCs respond to C. albicans in its filamentous form, which is present in the dermis in a mouse skin infection model. Instead, the pseudohyphae are sensed by CD102+ dermal dendritic cells, leading to an induction of Th1 cells and protection against systemic C. albicans dissemination [64]. Using an immune cell supplemented human 3D skin model [48], we could show that activated CD4+ T cells contribute to dermal protection by inducing an antimicrobial response of dermal fibroblasts in the presence of C. albicans. Notably, this response in the dermal skin compartment was independent of IL-17A and IL-17RA signaling but relied on the induction of IL-1β secretion by dermal fibroblasts [49]. Besides Th17 and Th1 responses, skin resident and recruited Th9 cells have been reported to play a role in early defense against C. albicans skin infection, as reviewed in [70].
5. Antifungal Defense Execution by Antimicrobial Agents
Skin resident immune and non-immune cells as well as recruited immune cells are involved in the clearance of C. albicans in the skin. The first line of antifungal defense is carried out by keratinocytes, which produce antimicrobial peptides (AMPs) such as Cathelicidine/LL-37 and β-defensins and S100 proteins. LL-37 and human β-defensins 1 to 3 alter membrane integrity of C. albicans and provoke ATP release, but, in addition, alternative mechanisms of antifungal action have been described, as reviewed by [71]. Calprotectin, a dimer of S100A8 and S100A9, is able to inhibit C. albicans growth [72]. This effect is reversible by the addition of 30 µM zinc, indicating that the antifungal activity of calprotectin is likely through zinc chelation [73]. RNase7, which is released by keratinocytes, has been shown to exert candidacidal activity at low micromolar concentrations by membrane destabilization and targeting of fungal RNA upon internalization [74]. Antimicrobial peptides can be constitutively expressed in keratinocytes or regulated directly in response to PRR signaling or through inflammatory cytokines [75]. For example, IL-17F and mainly IL-17A induce the production of human β-defensin 2 and S100 proteins by primary human keratinocytes, and IL-22, which is also produced by Th17 cells, synergizes with these IL-17 cytokines in AMP induction by keratinocytes [76]. Moreover, together with TNF-α, IL-22 induces S100A7, human β-defensin 2, and the antimicrobial chemokines CXCL9, 10, and 11 in keratinocytes in vitro [77,78], yet the role of IL-22 in the defense of CMC is controversial [79]. CXCL9, 10, and 11 are also highly expressed in dermal fibroblasts during C. albicans infection of a CD4+ T cell supplemented human skin model [49], and CXCL10 has been shown to directly inhibit C. albicans growth in the radial diffusion assay [80].
IL-17 also contributes to Candida clearance at epithelial surfaces through recruitment of neutrophils, which kill fungal cells by releasing high amounts of antimicrobial peptides, by direct phagocytosis, or by formation of neutrophil extracellular traps (NETs) [5,81,82], but the role of neutrophils in epithelial antifungal defense has been reported to be tissue specific and therefore may not be generalized [83].
6. Fungal-Microbiome Interaction
It is becoming increasingly clear that local commensal microbial communities highly influence the mode of interaction of C. albicans—whether commensal or pathogenic—with epithelial surfaces. Treatment of mice with several classes of antibiotics has been shown to increase colonization of the intestine with Candida species and the intestinal bacterium Bacteroides thetaiotamicron has been observed to limit C. albicans colonization by increasing HIF-1α-dependent LL-37 expression [84]. Other bacteria, especially Lactobacilli, influence C. albicans colonization and invasion in the gastrointestinal tract and vaginal mucosa by various mechanisms including pH changes and modulation of the immune response [8]. Modulation of Candida infection of the skin is likely also highly influenced by skin commensals. For example, differentiated primary human keratinocytes were shown to sense secreted factors of the commensal bacterium Staphylococcus epidermidis through TLR2, leading to NF-κB activation. In combination with pathogenic Staphylococcus aureus, this results in the synergistic induction of the antimicrobial peptides β-defensin 3 and RNase7 [85]. Since both AMPs also have antifungal activity, it is likely that these staphylococci in combination also influence C. albicans skin infection. Using specific pathogen free mice, it was recently shown that colonization with S. epidermidis is also sensed by dermal dendritic cells. These resident immune cells then initiate the development of a CD8+ and IL-17+ T cell population. These cells are subsequently recruited to the epidermis, where they boost S100A8 and S100A9 production by keratinocytes, resulting in increased resistance against C. albicans invasion in the absence of inflammation [86].
7. Conclusions
The skin is similar to mucosal surfaces such as the oral and vaginal mucosa, as they are all stratified squamous epithelia. Yet there are also substantial differences, like different degrees of cornification in these tissues, and, importantly, their local environment. The mechanisms underlying skin colonization and invasion by Candida species as well as host defense mechanisms are consequently overlapping with those found for mucosal surfaces, but considerable differences such as the precise mode of pathogenic invasion, the role of different immune cell types in defense against C. albicans, and the influence of commensal microorganisms on Candida pathogenesis exist. Much has been learned from the analysis of genetic disorders in humans resulting in chronic mucocutaneous candidiasis and also from mouse models. These findings have revealed both mechanisms of fungal recognition as well as immunological networks required to control these infections. Simple, often unicellular in vitro models furthermore have revealed initial mechanisms of pathogenesis in Candida infections. Recently, complex 3D tissue models of human origin containing defined elements of the immune system have been shown to be particularly amenable for identifying and modeling aspects of antifungal defense of the human skin. These recent advances in setting up complex 3D-tissue models have shown great promise already and therefore will most likely offer additional approaches to dissect and study Candida-skin interactions in great detail in the near future.
Acknowledgments
We would like to thank Lilliana Radoshevich for her critical reading of the manuscript. This work was supported by the FP7-PEOPLE-2013-ITN—Marie-Curie Action: “Initial Training Networks”: Molecular Mechanisms of Human Fungal Pathogen Host Interaction, ImResFun, Project ID: 606786.
Author Contributions
A.K. wrote the initial draft and S.R. rewrote and edited major sections of the review, A.B.-K critically read and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; NIH Intramural Sequencing Center Comparative Sequencing Program; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar]
- Oh, J.; Freeman, A.F.; Program, N.C.S.; Park, M.; Sokolic, R.; Candotti, F.; Holland, S.M.; Segre, J.A.; Kong, H.H. The altered landscape of the human skin microbiome in patients with primary immunodeficiencies. Genome Res. 2013, 23, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Havlickova, B.; Czaika, V.A.; Friedrich, M. Epidemiological trends in skin mycoses worldwide. Mycoses 2008, 51 (Suppl. 4), 2–15. [Google Scholar] [CrossRef] [PubMed]
- Mohandas, V.; Ballal, M. Distribution of Candida species in different clinical samples and their virulence: Biofilm formation, proteinase and phospholipase production: A study on hospitalized patients in southern India. J. Glob. Infect. Dis. 2011, 3, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Kaplan, D.H. Skin immunity to Candida albicans. Trends Immunol. 2016, 37, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.M.; King, R.D. Occlusion, carbon dioxide, and fungal skin infections. Lancet 1978, 1, 360–362. [Google Scholar] [CrossRef]
- Bonifaz, A.; Rojas, R.; Tirado-Sanchez, A.; Chavez-Lopez, D.; Mena, C.; Calderon, L.; Maria, P.O. Superficial mycoses associated with diaper dermatitis. Mycopathologia 2016, 181, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Hofs, S.; Mogavero, S.; Hube, B. Interaction of Candida albicans with host cells: Virulence factors, host defense, escape strategies, and the microbiota. J. Microbiol. 2016, 54, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Tanaka, T.; Tajima, M.; Tsuboi, R.; Nishikawa, A.; Sugita, T. Characterization of the skin fungal microbiota in patients with atopic dermatitis and in healthy subjects. Microbiol. Immunol. 2011, 55, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Taheri Sarvtin, M.; Shokohi, T.; Hajheydari, Z.; Yazdani, J.; Hedayati, M.T. Evaluation of candidal colonization and specific humoral responses against Candida albicans in patients with psoriasis. Int. J. Dermatol. 2014, 53, e555–e560. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.W.; Bukhalo, M.; Blauvelt, A. A clinician’s guide to the diagnosis and treatment of candidiasis in patients with psoriasis. Am. J. Clin. Dermatol. 2016, 17, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Hein, K.Z.; Takahashi, H.; Tsumori, T.; Yasui, Y.; Nanjoh, Y.; Toga, T.; Wu, Z.; Grotzinger, J.; Jung, S.; Wehkamp, J.; et al. Disulphide-reduced psoriasin is a human apoptosis-inducing broad-spectrum fungicide. Proc. Natl. Acad. Sci. USA 2015, 112, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Pirofski, L.A.; Casadevall, A. The damage-response framework of microbial pathogenesis and infectious diseases. Adv. Exp. Med. Biol. 2008, 635, 135–146. [Google Scholar] [PubMed]
- Gow, N.A.; Hube, B. Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 2012, 15, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, L.L.; Cota, E. Candida albicans agglutinin-like sequence (als) family vignettes: A review of als protein structure and function. Front. Microbiol. 2016, 7, 280. [Google Scholar] [CrossRef] [PubMed]
- Gabaldon, T.; Martin, T.; Marcet-Houben, M.; Durrens, P.; Bolotin-Fukuhara, M.; Lespinet, O.; Arnaise, S.; Boisnard, S.; Aguileta, G.; Atanasova, R.; et al. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genom. 2013, 14, 623. [Google Scholar] [CrossRef] [PubMed]
- Maestre-Reyna, M.; Diderrich, R.; Veelders, M.S.; Eulenburg, G.; Kalugin, V.; Bruckner, S.; Keller, P.; Rupp, S.; Mosch, H.U.; Essen, L.O. Structural basis for promiscuity and specificity during Candida glabrata invasion of host epithelia. Proc. Natl. Acad. Sci. USA 2012, 109, 16864–16869. [Google Scholar] [CrossRef] [PubMed]
- Diderrich, R.; Kock, M.; Maestre-Reyna, M.; Keller, P.; Steuber, H.; Rupp, S.; Essen, L.O.; Mosch, H.U. Structural hot spots determine functional diversity of the Candida glabrata epithelial adhesin family. J. Biol. Chem. 2015, 290, 19597–19613. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.; Xiong, X.; Sohn, K.; Schroppel, K.; Brunner, H.; Rupp, S. The moonlighting protein tsa1p is implicated in oxidative stress response and in cell wall biogenesis in Candida albicans. Mol. Microbiol. 2005, 57, 1318–1341. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.M.; Wallich, R.; Riesbeck, K.; Skerka, C.; Zipfel, P.F. Candida albicans uses the surface protein gpm1 to attach to human endothelial cells and to keratinocytes via the adhesive protein vitronectin. PLoS ONE 2014, 9, e90796. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ruiz, E.; Galan-Diez, M.; Zhu, W.; Fernandez-Ruiz, E.; d'Enfert, C.; Filler, S.G.; Cossart, P.; Veiga, E. Candida albicans internalization by host cells is mediated by a clathrin-dependent mechanism. Cell Microbiol. 2009, 11, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.N.; Solis, N.V.; Phan, Q.T.; Bajwa, J.S.; Kashleva, H.; Thompson, A.; Liu, Y.; Dongari-Bagtzoglou, A.; Edgerton, M.; Filler, S.G. Host cell invasion and virulence mediated by Candida albicans ssa1. PLoS Pathog. 2010, 6, e1001181. [Google Scholar] [CrossRef] [PubMed]
- Phan, Q.T.; Myers, C.L.; Fu, Y.; Sheppard, D.C.; Yeaman, M.R.; Welch, W.H.; Ibrahim, A.S.; Edwards, J.E., Jr.; Filler, S.G. Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 2007, 5, e64. [Google Scholar] [CrossRef] [PubMed]
- Wachtler, B.; Citiulo, F.; Jablonowski, N.; Forster, S.; Dalle, F.; Schaller, M.; Wilson, D.; Hube, B. Candida albicans-epithelial interactions: Dissecting the roles of active penetration, induced endocytosis and host factors on the infection process. PLoS ONE 2012, 7, e36952. [Google Scholar] [CrossRef] [PubMed]
- Dalle, F.; Wachtler, B.; L’Ollivier, C.; Holland, G.; Bannert, N.; Wilson, D.; Labruere, C.; Bonnin, A.; Hube, B. Cellular interactions of Candida albicans with human oral epithelial cells and enterocytes. Cell Microbiol. 2010, 12, 248–271. [Google Scholar] [CrossRef] [PubMed]
- Sohn, K.; Senyurek, I.; Fertey, J.; Konigsdorfer, A.; Joffroy, C.; Hauser, N.; Zelt, G.; Brunner, H.; Rupp, S. An in vitro assay to study the transcriptional response during adherence of Candida albicans to different human epithelia. FEMS Yeast Res. 2006, 6, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, C.; Schandar, M.; Noll, M.; Johannes, F.J.; Brunner, H.; Graeve, T.; Rupp, S. In vitro reconstructed human epithelia reveal contributions of Candida albicans efg1 and cph1 to adhesion and invasion. Microbiology 2002, 148, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Korting, H.C.; Schafer, W.; Bastert, J.; Chen, W.; Hube, B. Secreted aspartic proteinase (sap) activity contributes to tissue damage in a model of human oral candidosis. Mol. Microbiol. 1999, 34, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Bein, M.; Korting, H.C.; Baur, S.; Hamm, G.; Monod, M.; Beinhauer, S.; Hube, B. The secreted aspartyl proteinases sap1 and sap2 cause tissue damage in an in vitro model of vaginal candidiasis based on reconstituted human vaginal epithelium. Infect. Immun. 2003, 71, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Schackert, C.; Korting, H.C.; Januschke, E.; Hube, B. Invasion of Candida albicans correlates with expression of secreted aspartic proteinases during experimental infection of human epidermis. J. Investig. Dermatol. 2000, 114, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Lermann, U.; Morschhauser, J. Secreted aspartic proteases are not required for invasion of reconstituted human epithelia by Candida albicans. Microbiology 2008, 154, 3281–3295. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.; Lermann, U.; Teixeira, L.; Cerca, F.; Botelho, S.; da Costa, R.M.; Sampaio, P.; Gartner, F.; Morschhauser, J.; Vilanova, M.; et al. Limited role of secreted aspartyl proteinases sap1 to sap6 in Candida albicans virulence and host immune response in murine hematogenously disseminated candidiasis. Infect. Immun. 2010, 78, 4839–4849. [Google Scholar] [CrossRef] [PubMed]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Moyes, D.L.; Wilson, D.; Richardson, J.P.; Mogavero, S.; Tang, S.X.; Wernecke, J.; Hofs, S.; Gratacap, R.L.; Robbins, J.; Runglall, M.; et al. Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016, 532, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Brown, G.D.; Kullberg, B.J.; Gow, N.A. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 2008, 6, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Willment, J.A.; Marshall, A.S.; Reid, D.M.; Williams, D.L.; Wong, S.Y.; Gordon, S.; Brown, G.D. The human beta-glucan receptor is widely expressed and functionally equivalent to murine dectin-1 on primary cells. Eur J. Immunol. 2005, 35, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- LeibundGut-Landmann, S.; Gross, O.; Robinson, M.J.; Osorio, F.; Slack, E.C.; Tsoni, S.V.; Schweighoffer, E.; Tybulewicz, V.; Brown, G.D.; Ruland, J.; et al. Syk- and card9-dependent coupling of innate immunity to the induction of t helper cells that produce interleukin 17. Nat. Immunol. 2007, 8, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Underhill, D.M. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005, 24, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Patlan, A.; Campillo-Navarro, M.; Rodriguez-Cortes, O.; Munoz-Cruz, S.; Wong-Baeza, I.; Estrada-Parra, S.; Estrada-Garcia, I.; Serafin-Lopez, J.; Chacon-Salinas, R. Recognition of Candida albicans by dectin-1 induces mast cell activation. Immunobiology 2015, 220, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.A.; Vriend, L.E.; Theelen, B.; Taylor, M.E.; Fluitsma, D.; Boekhout, T.; Geijtenbeek, T.B. C-type lectin langerin is a beta-glucan receptor on human langerhans cells that recognizes opportunistic and pathogenic fungi. Mol. Immunol. 2010, 47, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; Ma, N.; Sadler, J.J.; Soll, D.R.; Cassel, S.L.; Sutterwala, F.S. Cutting edge: Candida albicans hyphae formation triggers activation of the nlrp3 inflammasome. J. Immunol. 2009, 183, 3578–3581. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; van der Lee, R.; Cheng, S.C.; Johnson, M.D.; Kumar, V.; Ng, A.; Plantinga, T.S.; Smeekens, S.P.; Oosting, M.; Wang, X.; et al. The rig-i-like helicase receptor mda5 (ifih1) is involved in the host defense against candida infections. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Gow, N.A.; Munro, C.A.; Bates, S.; Collins, C.; Ferwerda, G.; Hobson, R.P.; Bertram, G.; Hughes, H.B.; Jansen, T.; et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and toll-like receptors. J. Clin. Investig. 2006, 116, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Jouault, T.; Ibata-Ombetta, S.; Takeuchi, O.; Trinel, P.A.; Sacchetti, P.; Lefebvre, P.; Akira, S.; Poulain, D. Candida albicans phospholipomannan is sensed through toll-like receptors. J. Infect. Dis. 2003, 188, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pivarcsi, A.; Bodai, L.; Rethi, B.; Kenderessy-Szabo, A.; Koreck, A.; Szell, M.; Beer, Z.; Bata-Csorgoo, Z.; Magocsi, M.; Rajnavolgyi, E.; et al. Expression and function of toll-like receptors 2 and 4 in human keratinocytes. Int. Immunol. 2003, 15, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, Q.; Shen, Y.; Liu, W. Candida albicans phospholipomannan triggers inflammatory responses of human keratinocytes through toll-like receptor 2. Exp. Dermatol. 2009, 18, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Haley, K.; Igyarto, B.Z.; Ortner, D.; Bobr, A.; Kashem, S.; Schenten, D.; Kaplan, D.H. Langerhans cells require myd88-dependent signals for Candida albicans response but not for contact hypersensitivity or migration. J. Immunol. 2012, 188, 4334–4339. [Google Scholar] [CrossRef] [PubMed]
- Kuhbacher, A.; Sohn, K.; Burger-Kentischer, A.; Rupp, S. Immune cell-supplemented human skin model for studying fungal infections. Methods Mol. Biol. 2017, 1508, 439–449. [Google Scholar] [PubMed]
- Kuhbacher, A.; Henkel, H.; Stevens, P.; Grumaz, C.; Finkelmeier, D.; Burger-Kentischer, A.; Sohn, K.; Rupp, S. Dermal fibroblasts play a central role in skin model protection against C. albicans invasion. J. Infect. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Nahum, A.; Dadi, H.; Bates, A.; Roifman, C.M. The biological significance of tlr3 variant, l412f, in conferring susceptibility to cutaneous candidiasis, CMV and autoimmunity. Autoimmun. Rev. 2012, 11, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Nahum, A.; Dadi, H.; Bates, A.; Roifman, C.M. The l412f variant of toll-like receptor 3 (tlr3) is associated with cutaneous candidiasis, increased susceptibility to cytomegalovirus, and autoimmunity. J. Allergy Clin. Immunol. 2011, 127, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Weindl, G.; Naglik, J.R.; Kaesler, S.; Biedermann, T.; Hube, B.; Korting, H.C.; Schaller, M. Human epithelial cells establish direct antifungal defense through tlr4-mediated signaling. J. Clin. Investig. 2007, 117, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Rosentul, D.C.; Delsing, C.E.; Jaeger, M.; Plantinga, T.S.; Oosting, M.; Costantini, I.; Venselaar, H.; Joosten, L.A.; van der Meer, J.W.; Dupont, B.; et al. Gene polymorphisms in pattern recognition receptors and susceptibility to idiopathic recurrent vulvovaginal candidiasis. Front. Microbiol. 2014, 5, 483. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Ramirez-Carrozzi, V.; Sambandam, A. The interleukin-17 cytokine family: Critical players in host defence and inflammatory diseases. Immunology 2011, 134, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Cypowyj, S.; Bustamante, J.; Wright, J.F.; Liu, L.Y.; Lim, H.K.; Migaud, M.; Israel, L.; Chrabieh, M.; Audry, M.; et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 2011, 332, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Cypowyj, S.; Aytekin, C.; Galicchio, M.; Camcioglu, Y.; Nepesov, S.; Ikinciogullari, A.; Dogu, F.; Belkadi, A.; Levy, R.; et al. Inherited il-17rc deficiency in patients with chronic mucocutaneous candidiasis. J. Exp. Med. 2015, 212, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Boisson, B.; Wang, C.; Pedergnana, V.; Wu, L.; Cypowyj, S.; Rybojad, M.; Belkadi, A.; Picard, C.; Abel, L.; Fieschi, C.; et al. An act1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 2013, 39, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Boisson-Dupuis, S.; Kong, X.F.; Okada, S.; Cypowyj, S.; Puel, A.; Abel, L.; Casanova, J.L. Inborn errors of human stat1: Allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Curr. Opin. Immunol. 2012, 24, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. 2009, 160, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Whibley, N.; Coleman, B.M.; Garg, A.V.; Jaycox, J.R.; Gaffen, S.L. Signaling through il-17c/il-17re is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS ONE 2015, 10, e0122807. [Google Scholar] [CrossRef] [PubMed]
- Saunte, D.M.; Mrowietz, U.; Puig, L.; Zachariae, C. Candida infections in psoriasis and psoriatic arthritis patients treated with il-17 inhibitors and their practical management. Br. J. Dermatol. 2016. [Google Scholar] [CrossRef]
- Zielinski, C.E.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, M.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-induced human th17 cells produce ifn-gamma or il-10 and are regulated by il-1beta. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Igyarto, B.Z.; Haley, K.; Ortner, D.; Bobr, A.; Gerami-Nejad, M.; Edelson, B.T.; Zurawski, S.M.; Malissen, B.; Zurawski, G.; Berman, J.; et al. Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific t helper cell responses. Immunity 2011, 35, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Igyarto, B.Z.; Gerami-Nejad, M.; Kumamoto, Y.; Mohammed, J.; Jarrett, E.; Drummond, R.A.; Zurawski, S.M.; Zurawski, G.; Berman, J.; et al. Candida albicans morphology and dendritic cell subsets determine t helper cell differentiation. Immunity 2015, 42, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-producing gammadelta t cells selectively expand in response to pathogen products and environmental signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.O.; Dunne, K.; Comerford, R.; O’Dea, S.; Loy, A.; Woo, J.; Rogers, T.R.; Mulcahy, F.; Dunne, P.J.; Doherty, D.G. Candida albicans stimulates il-23 release by human dendritic cells and downstream il-17 secretion by vdelta1 t cells. J. Immunol. 2015, 194, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and il-23 induce innate il-17 production from gammadelta t cells, amplifying th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Kashem, S.W.; Riedl, M.S.; Yao, C.; Honda, C.N.; Vulchanova, L.; Kaplan, D.H. Nociceptive sensory fibers drive interleukin-23 production from cd301b+ dermal dendritic cells and drive protective cutaneous immunity. Immunity 2015, 43, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; Plantinga, T.S.; van de Veerdonk, F.L.; Heinhuis, B.; Hoischen, A.; Joosten, L.A.; Arkwright, P.D.; Gennery, A.; Kullberg, B.J.; Veltman, J.A.; et al. Stat1 hyperphosphorylation and defective il12r/il23r signaling underlie defective immunity in autosomal dominant chronic mucocutaneous candidiasis. PLoS ONE 2011, 6, e29248. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Schlapbach, C. Th9 cells in skin disorders. Semin. Immunopathol. 2017, 39, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Swidergall, M.; Ernst, J.F. Interplay between Candida albicans and the antimicrobial peptide armory. Eukaryot. Cell 2014, 13, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Sohnle, P.G.; Hahn, B.L.; Santhanagopalan, V. Inhibition of Candida albicans growth by calprotectin in the absence of direct contact with the organisms. J. Infect. Dis. 1996, 174, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Sohnle, P.G.; Hunter, M.J.; Hahn, B.; Chazin, W.J. Zinc-reversible antimicrobial activity of recombinant calprotectin (migration inhibitory factor-related proteins 8 and 14). J. Infect. Dis. 2000, 182, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Salazar, V.A.; Arranz-Trullen, J.; Navarro, S.; Blanco, J.A.; Sanchez, D.; Moussaoui, M.; Boix, E. Exploring the mechanisms of action of human secretory rnase 3 and rnase 7 against Candida albicans. Microbiologyopen 2016, 5, 830–845. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.S.; Falconer, A.; Ikram, M.; Bissett, C.E.; Cerio, R.; Quinn, A.G. Expression of the peptide antibiotics human beta defensin-1 and human beta defensin-2 in normal human skin. J. Investig. Dermatol. 2001, 117, 106–111. [Google Scholar] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (il)-22 and il-17 are coexpressed by th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Hau, C.S.; Tada, Y.; Kanda, N.; Watanabe, S. Immunoresponses in dermatomycoses. J. Dermatol. 2015, 42, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Wagener, J.; Wenzel, V.; Scarponi, C.; Pennino, D.; Albanesi, C.; Schaller, M.; Behrendt, H.; Ring, J.; Schmidt-Weber, C.B.; et al. Il-22 and tnf-alpha represent a key cytokine combination for epidermal integrity during infection with Candida albicans. Eur J. Immunol. 2011, 41, 1894–1901. [Google Scholar] [CrossRef] [PubMed]
- Kagami, S.; Rizzo, H.L.; Kurtz, S.E.; Miller, L.S.; Blauvelt, A. Il-23 and il-17a, but not il-12 and il-22, are required for optimal skin host defense against Candida albicans. J. Immunol. 2010, 185, 5453–5462. [Google Scholar] [CrossRef] [PubMed]
- Holdren, G.O.; Rosenthal, D.J.; Yang, J.; Bates, A.M.; Fischer, C.L.; Zhang, Y.; Brogden, N.K.; Brogden, K.A. Antimicrobial activity of chemokine cxcl10 for dermal and oral microorganisms. Antibiotics (Basel) 2014, 3, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Trautwein-Weidner, K.; Gladiator, A.; Nur, S.; Diethelm, P.; LeibundGut-Landmann, S. Il-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol. 2015, 8, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Coughlin, L.A.; Neubauer, M.M.; Kim, J.; Kim, M.S.; Zhan, X.; Simms-Waldrip, T.R.; Xie, Y.; Hooper, L.V.; Koh, A.Y. Activation of hif-1alpha and ll-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 2015, 21, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Wanke, I.; Steffen, H.; Christ, C.; Krismer, B.; Gotz, F.; Peschel, A.; Schaller, M.; Schittek, B. Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J. Investig. Dermatol. 2011, 131, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Bouladoux, N.; Linehan, J.L.; Han, S.J.; Harrison, O.J.; Wilhelm, C.; Conlan, S.; Himmelfarb, S.; Byrd, A.L.; Deming, C.; et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015, 520, 104–108. [Google Scholar] [CrossRef] [PubMed]
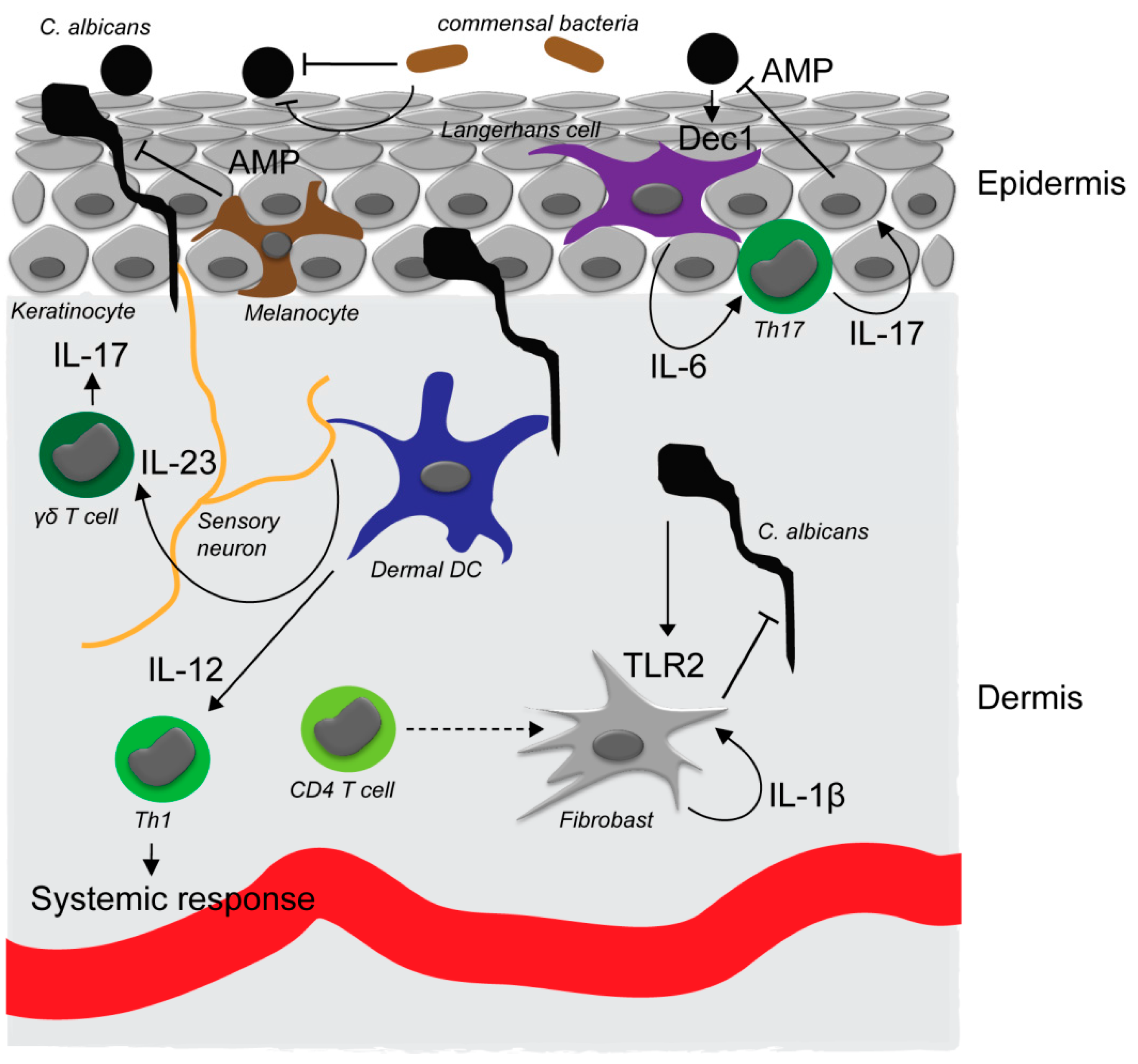
Figure 1.
C. albicans interaction with the skin. In the commensal, noninvasive state, C. albicans exists in the yeast form on the tissue surface. Yeast cells can be detected through the dectin-1 receptor (Dec1) that is expressed by Langerhans cells in the epidermis. Activation of Langerhans cells (LCs) results in an interleukin-6 (IL-6)-dependent Th17 response, antimicrobial peptide production by keratinocytes and superficial antifungal defense. Commensal skin bacteria are also involved in preventing invasion of C. albicans into the skin by direct and indirect mechanisms. C. albicans invading the epidermis can moreover be detected by sensory neurons that promote IL-23 secretion by dermal dendritic cells and subsequent proliferation and IL-17 secretion of skin resident γδ T cells. Upon breaching of the epidermis, C. albicans is detected in the dermis by dermal dendritic cells which induce an IL-12-dependent Th1 response required for systemic immunity against the fungus. Dermal fibroblasts support direct antimicrobial defense in the dermis upon activation through TLR2 by C. albicans and IL-1β secretion and auto-activation. Secretion of IL-1β by dermal fibroblasts requires an additional yet unidentified signal from CD4+ T cells.
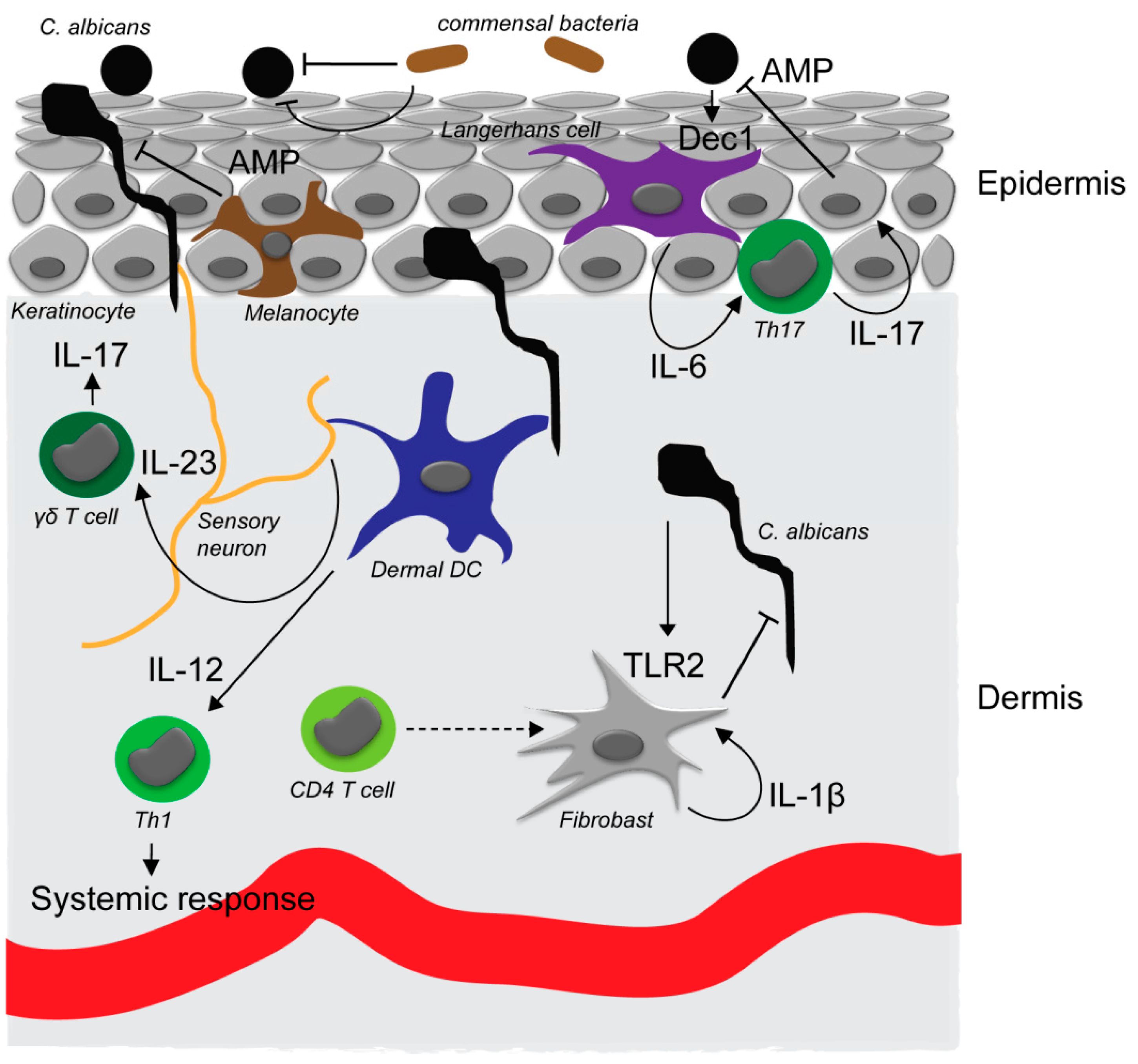
Figure 1.
C. albicans interaction with the skin. In the commensal, noninvasive state, C. albicans exists in the yeast form on the tissue surface. Yeast cells can be detected through the dectin-1 receptor (Dec1) that is expressed by Langerhans cells in the epidermis. Activation of Langerhans cells (LCs) results in an interleukin-6 (IL-6)-dependent Th17 response, antimicrobial peptide production by keratinocytes and superficial antifungal defense. Commensal skin bacteria are also involved in preventing invasion of C. albicans into the skin by direct and indirect mechanisms. C. albicans invading the epidermis can moreover be detected by sensory neurons that promote IL-23 secretion by dermal dendritic cells and subsequent proliferation and IL-17 secretion of skin resident γδ T cells. Upon breaching of the epidermis, C. albicans is detected in the dermis by dermal dendritic cells which induce an IL-12-dependent Th1 response required for systemic immunity against the fungus. Dermal fibroblasts support direct antimicrobial defense in the dermis upon activation through TLR2 by C. albicans and IL-1β secretion and auto-activation. Secretion of IL-1β by dermal fibroblasts requires an additional yet unidentified signal from CD4+ T cells.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kühbacher, A.; Burger-Kentischer, A.; Rupp, S. Interaction of Candida Species with the Skin. Microorganisms 2017, 5, 32. https://doi.org/10.3390/microorganisms5020032
AMA Style
Kühbacher A, Burger-Kentischer A, Rupp S. Interaction of Candida Species with the Skin. Microorganisms. 2017; 5(2):32. https://doi.org/10.3390/microorganisms5020032
Chicago/Turabian StyleKühbacher, Andreas, Anke Burger-Kentischer, and Steffen Rupp. 2017. "Interaction of Candida Species with the Skin" Microorganisms 5, no. 2: 32. https://doi.org/10.3390/microorganisms5020032
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.