Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation

Sorbonne Université, CNRS-INSU, Institut des Sciences de la Terre Paris, ISTeP UMR 7193, F-75005 Paris, France
Geosciences 2018, 8(6), 198; https://doi.org/10.3390/geosciences8060198
Submission received: 4 May 2018
/
Revised: 22 May 2018
/
Accepted: 26 May 2018
/
Published: 31 May 2018
(This article belongs to the Special Issue Volcanic Plumes: Impacts on the Atmosphere and Insights into Volcanic Processes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The impact of volcanic eruptions on the climate has been studied over the last decades and the role played by sulfate aerosols appears to be major. S-bearing volcanic gases are oxidized in the atmosphere into sulfate aerosols that disturb the radiative balance on earth at regional to global scales. This paper discusses the use of the oxygen and sulfur multi-isotope systematics on volcanic sulfates to understand their formation and fate in more or less diluted volcanic plumes. The study of volcanic aerosols collected from air sampling and ash deposits at different distances from the volcanic systems (from volcanic vents to the Earth poles) is discussed. It appears possible to distinguish between the different S-bearing oxidation pathways to generate volcanic sulfate aerosols whether the oxidation occurs in magmatic, tropospheric, or stratospheric conditions. This multi-isotopic approach represents an additional constraint on atmospheric and climatic models and it shows how sulfates from volcanic deposits could represent a large and under-exploited archive that, over time, have recorded atmospheric conditions on human to geological timescales.
1. Introduction
Globally on Earth, each year, volcanoes release an average of 10–20 Mt of sulfur-bearing gases, and occasionally much more during super eruptions (e.g., [1,2]). Atmospheric sulfur plays a paramount role in the terrestrial radiative balance. Consequently, determining its source and understanding its physico-chemical transformations and fate in the atmosphere appear crucial in predicting its impact on the atmosphere and climate. Indeed, when released into the atmosphere S-bearing gases (mainly SO2: ≤25%; H2S:≤ 10%; and COS and CS2: ≤0.01% of the volume of the emitted gases; [3,4,5] and references therein) are oxidized, being thus transformed into sulfate aerosols that directly and indirectly lead to a negative radiative forcing. First, sulfate aerosols directly reflect part of the solar radiations, thus decreasing the amount of sun energy reaching the Earth’s surface. On the other hand, sulfate aerosols absorb part of the incoming solar radiation in the IR wavelengths and this results in a warming of the aerosol-bearing atmospheric layer associated to a temperature decrease between it and the Earth’s surface. Additionally, volcanic aerosols play the role of cloud nucleation leading to more cloud formation, which reinforces the albedo and therefore causes the Earth’s surface to cool (e.g., [6,7]). Such “volcanic winters”, that can be more or less severe and global depending on the strength and location of the eruption, have been observed after several major eruptions such as those at Mt Agung in 1963, El Chichón in 1983, and Pinatubo in 1991 (e.g., [8,9,10,11]).
Atmospheric modeling aims at predicting climate evolution on Earth. However, including the sulfur volcanic aerosol formation processes would increase the models’ accuracy regarding the volcanic forcing climate. This paper discusses the use of oxygen and sulfur multi-isotopes in improving our understanding of sulfate aerosol formation, fate, and sources. The 18O/16O and 17O/16O as well as 34S/32S, 33S/32S, and 36S/32S are measured and expressed as δ18O and δ17O as well as δ34S, δ33S and, δ36S respectively (Equation (1)). During any process, the isotopes fractionate, changing the isotopic ratios, depending on the mass differences between heavy and light isotopes. For instance, the mass difference between 18O and 16O is twice as much as between 17O and 16O, leading to the relation 17O/16O ~ 0.5 * 18O/16O, commonly expressed as δ17O = (δ18O + 1)0.524 − 1. This very widespread rule suffers some exceptions, where isotopic fractionation does not depend on the mass differences between isotopes, it is referred to as Mass Independent Fractionation (MIF) (e.g., [12,13,14,15]). The difference to the isotopic mass dependent fractionation relation is quantified by Δ17O, Δ33S and Δ36S (Equations (2)–(4)). Therefore, isotopic mass dependent fractionation results in a Δ = 0‰ whilst MIF has Δ ≠ 0‰.
considering that: δA: isotopic composition of a sample (e.g., δ18O); RA: isotope ratio of the measured sample (e.g., 18O/16O); Rst: isotope ratio of a standard. The mass dependent coefficients 0.524, 0.515, and 1.89 are from references [16,17].
δA = (RA/Rst) − 1; for instance δ18O = (18O/16O(sample)/18O/16O(standard)) − 1
Δ17O = δ17O − [(δ18O + 1)0.524 − 1]
Δ33S = δ33S − [(δ34S + 1)0.515 − 1]
Δ36S = δ36S − [(δ34S + 1)1.89 − 1]
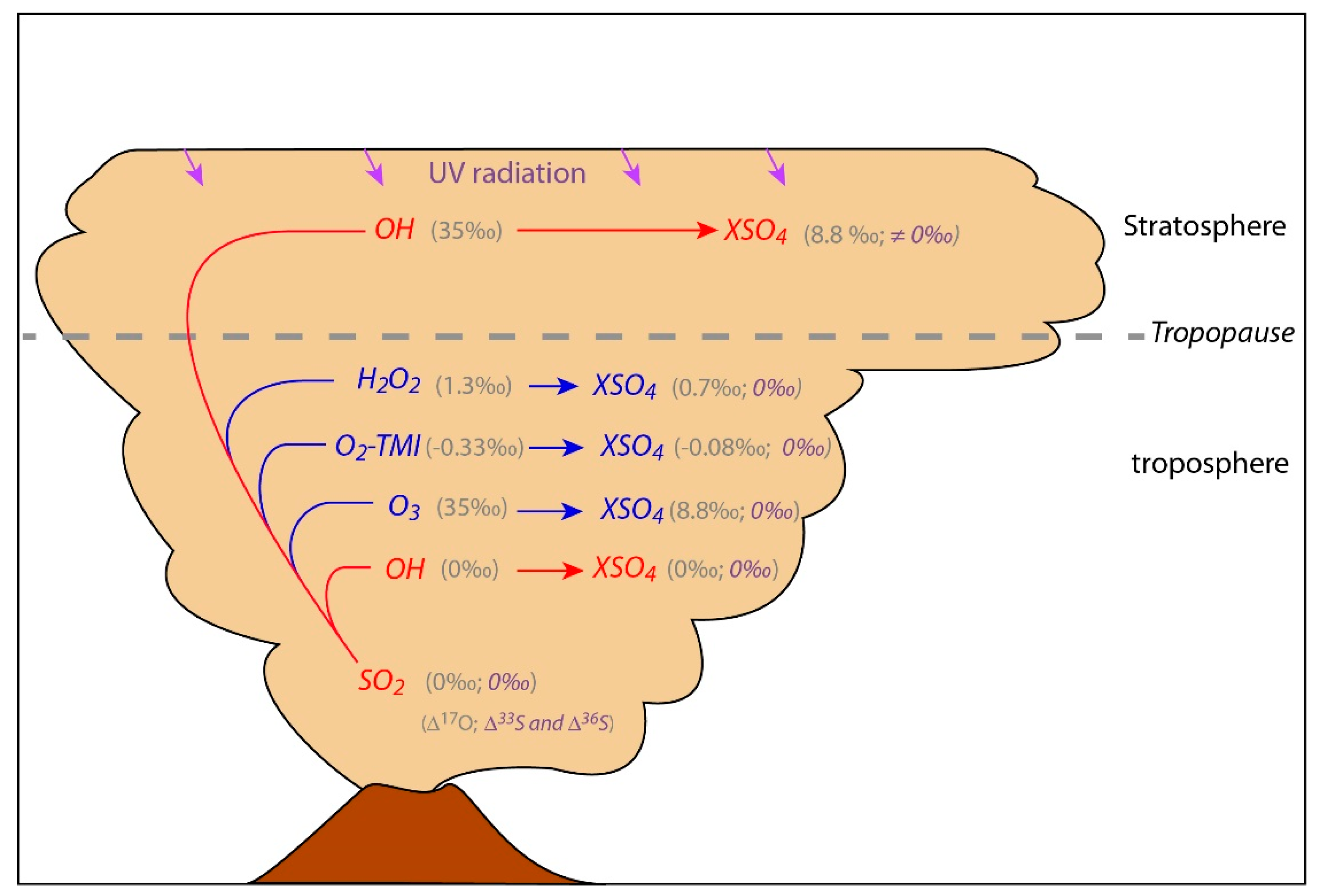
In the atmosphere, sulfate aerosols are generated by oxidation of S-bearing gases through different possible reactions or channels (Ch1, Ch2, Ch3, and Ch4; e.g., [18]). The multi O-isotopic composition of the different atmospheric oxidants is relatively well-known such that the resulting sulfate composition can be estimated for each oxidation channel (Figure 1). Therefore, coupling O- and S-isotopic composition with atmospheric chemistry allows the oxidation channel(s) from which they formed to be retraced. This not only helps to elucidate the sulfate aerosol formation in the atmosphere but also permits us to probe into the composition of the atmosphere and more specifically its oxidant capacity during specific volcanic eruptions.
where M: any inert molecule that removes excess energy without participating in the reaction; TMI: transition metal ion; for (Ch4); SO2(aq) is in aqueous phase and can be present as SO2 · H2O, , or (for pH from 2 to 7, which is most likely the case in volcanic plumes, dominates). Ch1, Ch2, Ch3 and Ch4 are the same as in Figure 1.
| Ch1: | |
| Ch2: | |
| Ch3: | |
| Ch4: |
This isotopic approach opens new perspectives when considering volcanic deposits formed by different eruptions all around the world during the whole history of Earth. As volcanic activity has always been present, sulfates from volcanic deposits represent an underexploited archive that, over time, has recorded atmospheric conditions on human to geological timescales.
This paper reviews the different methods used for sulfate aerosol sampling, as well as for O- and S-isotopic measurements, the final purpose being a discussion of the mechanisms of formation of the volcanic sulfate during the different kind of volcanic eruptions, from passive degassing to large caldera-forming eruptions (super-eruptions).
2. Methods
Several laboratories use a variety of methods to extract sulfate from natural samples and to analyze their isotopic composition. Some methods are still in development, nonetheless the present paper describes the methods classically used nowadays.
2.1. Volcanic SO2 and H2S Sampling
Gas sampling at volcanic vents have been used for the last 40 years [23] and is based on the fact that acidic volcanic gases, including SO2 and H2S can be trapped in alkaline solutions. The most quantitative method appears to be the Giggenbach bottle system [24], but it requires approaching the volcanic vent, making the sampling and isotopic characterization of volcanic S-bearing gases difficult. The Giggenbach bottle system consists of pumping the volcanic gases, which are collected into a bottle containing an alkaline solution. The acid gases are dissolved in the solution and the low solubility gases trapped in the headspace of the bottle. The development of such a method allows the simultaneous and separate collection of SO2 and H2S [25,26]. Indeed, both gases are trapped in the alkaline solution, but the presence of Zn-acetate for instance leads to the immediate precipitation of H2S into ZnS, allowing the physical separation of sulfur from SO2 and H2S. This method is rather quantitative, which is necessary especially when isotopic analyses and mass balances are considered. Another method is based on passive alkaline traps, and consists simply of plastic beakers containing alkaline solution close the volcanic vents [27]. This method permits the volcanic gases to be collected over a longer period of time, allowing a better estimation of the average volcanic gas fluxes, but it does not allow the separation of different gases (SO2 vs. H2S for instance). Furthermore, during the absorption of S-bearing gases, some isotopic fractionation may occur, which may lead to a significant bias for isotopic studies [28]. Finally, filters soaked in alkaline solution are also used, but no separation of SO2 and H2S is possible either and some isotopic fraction occurs when the SO2 flux is too high [29,30].
2.2. Sulfate Aerosol Sampling
Air sampling and ash leaching are the two classic methods for collecting volcanic sulfates. The first method consists of pumping the air near volcanic vents; the particles (aerosols) are then gathered on filters, usually polytetrafluoroethylene (PTFE) filter packs (e.g., [29]). Pumps with a flow rate of about 30 L min−1 are generally used, as they are able to collect—in a few hours—enough sulfate aerosols for isotopic measurements [30,31]. Filters are then leached with deionized water in order to extract and concentrate sulfate aerosols into solution. Special care must be taken as, even if the reason is not yet fully understood, there is some variability in the δ18O measured in sulfates collected on filters [29,30]; this could be linked to the collecting method as it cannot be explained by any magmatic processes. However, this variability is not observed for Δ17O and S-isotopes. The other method for sulfate sampling consists of directly leaching volcanic ash. Indeed, a few tens to thousands of ppm of sulfate can be present in volcanic ash deposits (e.g., [32,33,34]). With the exception of barite (BaSO4), sulfates are highly soluble in water, making their preservation in volcanic ash problematic or impossible in humid regions as well as in old volcanic deposits. Nevertheless, in arid to semi-arid environments, sulfates are at least partially preserved in volcanic ash layers up to tens of Ma [33,35]. Although the scavenging of sulfate is possible, isotopic exchange between water and sulfate is rather negligible in such environments [36]. Therefore, even if sulfate is partially removed by leaching or weathering from a volcanic deposit, its overall isotopic composition is preserved. In some cases, as in a sedimentary basin (an alkaline lake for instance), volcanic deposits can undertake the formation of authigenic sulfates that have a different isotopic composition compared to volcanic sulfates. Furthermore, long-term atmospheric deposition can also contribute to the sulfate from volcanic deposits, but it is rather negligible compared to the volcanic and authigenic sulfates [33]. The overall isotopic composition of sulfate from these volcanic deposits can possibly be progressively modified by dilution of the initial volcanic sulfate. However, using isotopic mixing models, it is possible to estimate the proportion of volcanic sulfate left in such deposits [33,35]. Deionized water is used for ash leaching and diluted HCl is added when carbonates were formed and could have trapped some sulfate. For efficient leaching, the preferred water/ash mass ratio is of about 1/20 [37].
2.3. Sulfate Chemical Composition and Preparation
Even if the volcanic sulfate aerosol mineralogy is very complex and not totally understood, it appears that at volcanic vents, dominating sulfates are commonly K-Na-sulfates [38,39]. Considering the high volatility of Na and K, the formation of these sulfate aerosol particles can be the result of the reaction between sulfuric acid and volatilized alkali-chlorides, the condensation of alkali-sulfate directly volatilized out of the magma or emitted by the hydrothermal system [40,41,42,43].
On the other hand, further away from the vent (≥1–2 km), where the plume is more diluted, it seems that Ca-sulfates dominate [38,39]. They are found as particles but also as well-formed crystals and coating on other particles such as volcanic ash. This testifies that Ca-sulfates are less likely to have been produced by mechanical aerosol formation like in volcanic conduits or vents. However, considering the low volatility of calcium, Ca-sulfates are most likely formed by the alteration of volcanic glass in the plume itself. This is also consistent with the fact that in volcanic deposits sampled far from the volcanic centers (between 5 and 25 km [30]; ≥500 km [33,35]), Ca-sulfates seem to be the dominant sulfate species. Ayris et al. [44] show that at high temperatures the high Ca2+ diffusivity in ash particles leads to CaSO4 formation in the volcanic conduit, which during the cooling down of the plume and its mixing with air, can be hydrated and generate gypsum as observed in volcanic ash deposits. However, in their experiments, K-Na-sulfates are not generated at high temperature by diffusion-driven mechanism. Therefore, this mechanism does not seem to explain the dominance of alkali-sulfate at volcanic vents.
In order to be able to measure the multi-isotopic composition of the collected sulfates, the latter is reacted with salts, which results in precipitates such as BaSO4, Ag2SO4, or Ag2S.
- -
- BaSO4: For about 30 years (e.g., [36,45]) sulfates were transformed into highly insoluble BaSO4 by adding BaCl2 in the leachate obtained from different kind of samples such as ash or filters. However, when precipitated from a multi-anion solution, barite (BaSO4) can occlude impurities such as nitrate, which can introduce an analytical bias in the measurement of O-isotopes [46]. For this reason, it is of paramount importance to purify the collected sulfate and make sure that no nitrate remains in the leachate. In order to purify barite, Bao et al. [47] developed the DDARP (diethylenetriaminepentaacetic acid dissolution and re-precipitation) method, while more recently, Le Gendre et al. [48] worked out a Resin Method for Sulfate Extraction and Purification (RMSEP), that passes the leachate solution through an anionic exchange resin, allowing the purification and concentration of sulfate. Then, by adding BaCl2, pure BaSO4 is precipitated.
- -
- Ag2SO4: Via anionic exchange resins, sulfate from the leachate is converted into Na2SO4. Then Ag2SO4 is produced by passing the Na2SO4 through another resin conditioned into Ag+ [49].
- -
2.4. Oxygen Isotope Measurements
Oxygen isotope ratios were conventionally determined via graphite-reduction techniques that generate CO2 from the oxygen extracted from the sulfates [52,53]. However, due to an isobaric interference from 13C (mass 45: 13C16O16O or 12C16O17O), only δ18O can be determined via these methods. More recently, Bao and Thiemens [54] developed a laser fluorination method, in order to precisely and simultaneously measure 18O/16O and 17O/16O from O2 extracted from sulfate samples. It is noteworthy that this method typically requires >20–30 µmol of sulfate (>4–7 mg of BaSO4), so that enough material remains available for duplicate or triplicate analyses. Using a CO2 laser, 2–3 mg of BaSO4 reacted with BrF5 at a high temperature, leading to O2 being released, which is then extracted and purified by successive cryogenic traps in an extraction line. Finally, it is trapped onto a molecular sieve and then sent to the mass spectrometer. However, O2 extraction from BaSO4 by fluorination is never total (only 30–45% yield) [33,54], which induces an isotopic fractionation. Typical fractionation of δ18O is of about +8‰, yet this fractionation is mass dependent, such that it has no effect on the Δ17O value. Overall, the method uncertainties (sulfate transformation into BaSO4 + O2 extraction line + mass spectrometer) are: δ18O ± 0.5‰ and Δ17O ± 0.1‰ (2σ).
Whilst the laser fluorination method requires a minimum of 8–10 µmol of sulfate for an O-isotopes analysis, samples as small as 0.2 µmol of sulfate can be analyzed by pyrohydrolysis of Ag2SO4 using an elemental analyzer [55,56]. This method is ideally adapted for analyzing small quantities of sulfate aerosols. However, the uncertainties (δ18O ± 2‰ and Δ17O ± 0.2–0.3‰ (2σ)) are greater than with the laser fluorination method.
2.5. Sulfur Isotope Measurements
While 34S/32S in sulfates can be easily and directly analyzed using an elemental analyzer, multi S-isotope measurements (34S/32S, 33S/32S and 36S/32S) require that Ag2S precipitate is converted into SF6 [51]. SF6 is obtained by fluorination (via F2) of Ag2S; it is then purified by gas phase chromatography, and subsequently concentrated in a cryogenic trap before being injected into the mass spectrometer. Recently, Au Yang et al. [57] developed a method allowing the analysis of less than 0.1 µmol of SF6. Overall, the method uncertainties (sulfate transformation into Ag2S + SF6 extraction line + mass spectrometer) are: δ34S ± 0.2‰; Δ33S ± 0.02‰ Δ36S ± 0.09‰ (2σ).
3. Volcanic Sulfate Formation
Sulfate aerosols can possibly be generated at high temperature in the magma itself in very oxidizing conditions or more likely in the volcanic conduit during the gas ascent. Indeed, as discussed above, they mostly consist of K-Na-sulfates and can result from the reaction between sulfuric acid and volatilized alkali-chlorides, from the condensation of alkali-sulfate directly volatilized out of the magma or emitted by the hydrothermal systems [40,41,42,43]. These sulfates are referred to as primary sulfates. Their proportion in volcanic plumes during passive magma degassing is usually low with a sulfate/SO2 ratio, usually <1% near the volcanic vents [25,58,59]. The S-bearing gases that are carried by the colder (dense or diluted) volcanic plumes and clouds produce secondary sulfate aerosols via different possible oxidation pathways in the atmosphere [20] and also possibly by chemical and photochemical reactions on mineral and dust surfaces [60]. As discussed above, these secondary sulfates consist most likely of Ca-sulfates due to interaction between sulfuric acid and volcanic glass. Below is detailed the possible mechanisms responsible for the formation of primary volcanic sulfates at high temperatures and the secondary volcanic sulfates in the troposphere, stratosphere, or during super-volcanic events.
3.1. Isotopic Composition of S-Bearing Gases
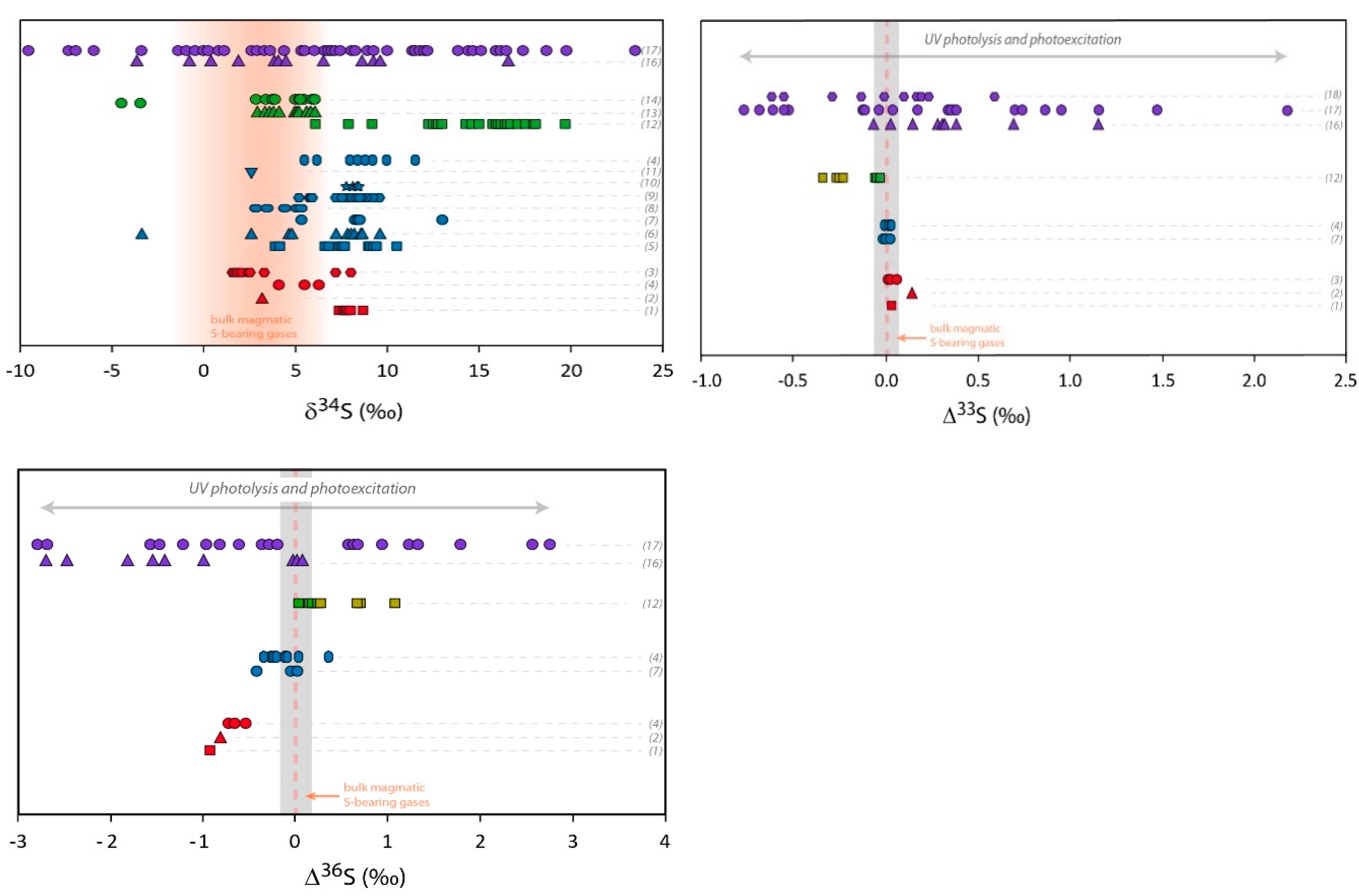
Due to obvious difficulties regarding a quantitative sampling close to volcanic vents, the previous determination of isotopic composition of volcanic S-bearing gases collected close to open degassing vents were only achieved at volcanic systems, where only minor eruptions occur [25,26,27,29,61,62,63,64,65,66,67,68]. In such open degassing systems, it appears that the δ34S of the bulk volcanic S-bearing gases (mainly SO2 + H2S) are rather similar to their magmatic sources, as expected since sulfur is almost thoroughly degassed (>90%) from degassing magmas [25]. However, Menyailov et al. [26] were able to measure lower δ34S for H2S than for SO2 by ~4‰ at 700–800 °C, ~12‰ at 500 °C and ≥16‰ at temperature ≤100 °C, which is consistent with the tendency previously observed [66] and the isotopic fractionation expected between the two S-bearing gases [69,70]. While the bulk S-bearing gas has a similar composition to its magmatic source, the latter strongly depends on the oxygen fugacity of the magma, with light isotopic ratios for low oxygen fugacity (MORB-like magmas: δ34S ~ −1‰ to 0‰; e.g., [71]) and heavier ratios when the magmatic conditions are more oxidizing (arc lavas: δ34S ~ 5‰; e.g., [72]). In Figure 2, the range of −1 to 6‰ is reported as representative of the bulk volcanic S-bearing gases. No measures of multi-S and -O isotopes ratios have been done on the S-bearing gases. However, as it has been observed for δ34S, the Δ33S, Δ36S, δ18O, and Δ17O of S-bearing gases is expected to be similar to the magmatic sources and the mantle values that are close to 0‰ for Δ33S, Δ36S, and Δ17O and 5.5 to 7‰ for δ18O depending on the geodynamic context ([17,71,72,73,74]; Figure 2).
3.2. High Temperature Primary Sulfates
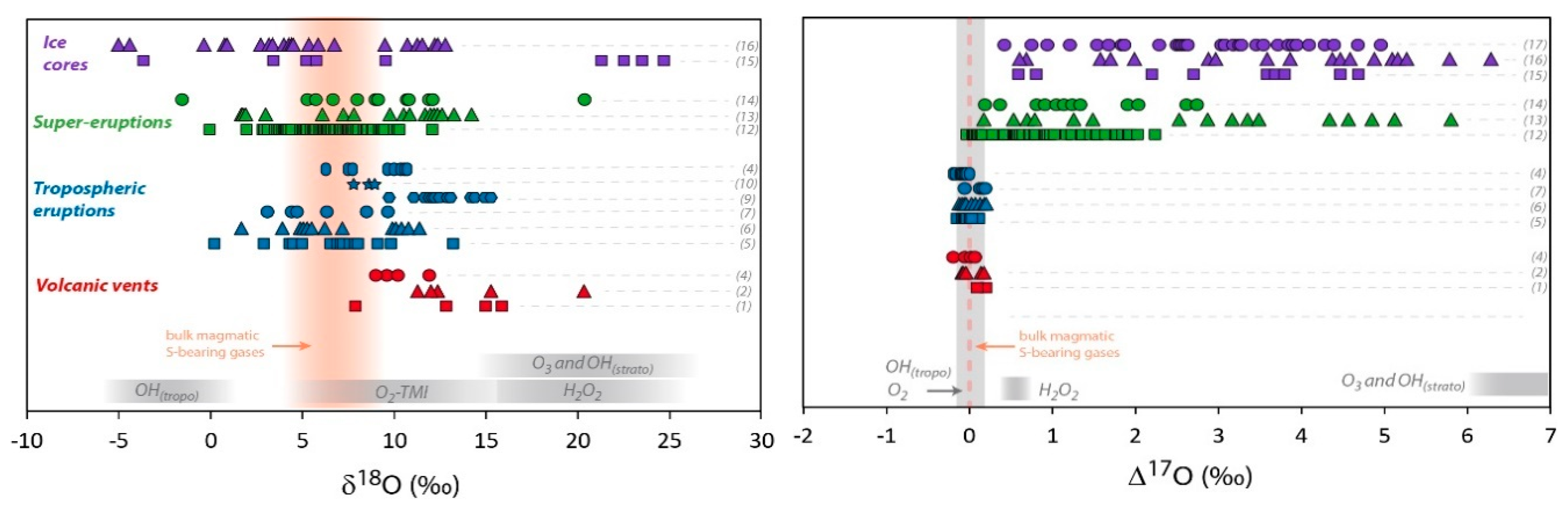
In order to study the primary volcanic sulfates, the sampling of aerosols must be performed as close as possible to the volcanic vents (up to a few hundred meters). Figure 2 shows a compilation of the isotopic composition of such primary sulfates. The variation range for O-isotopes is from 7‰ to 20‰ in δ18O and it becomes very narrow for Δ17O whose values are close to 0‰ (from −0.18 to +0.2‰). S-isotopes are more homogeneous with only 8‰ variation in δ34S (from 1 to 9‰), Δ33S very close to 0‰ or slightly positive (from 0.01 to 0.14‰) and Δ36S significantly negative values from −0.5 to −0.9‰.
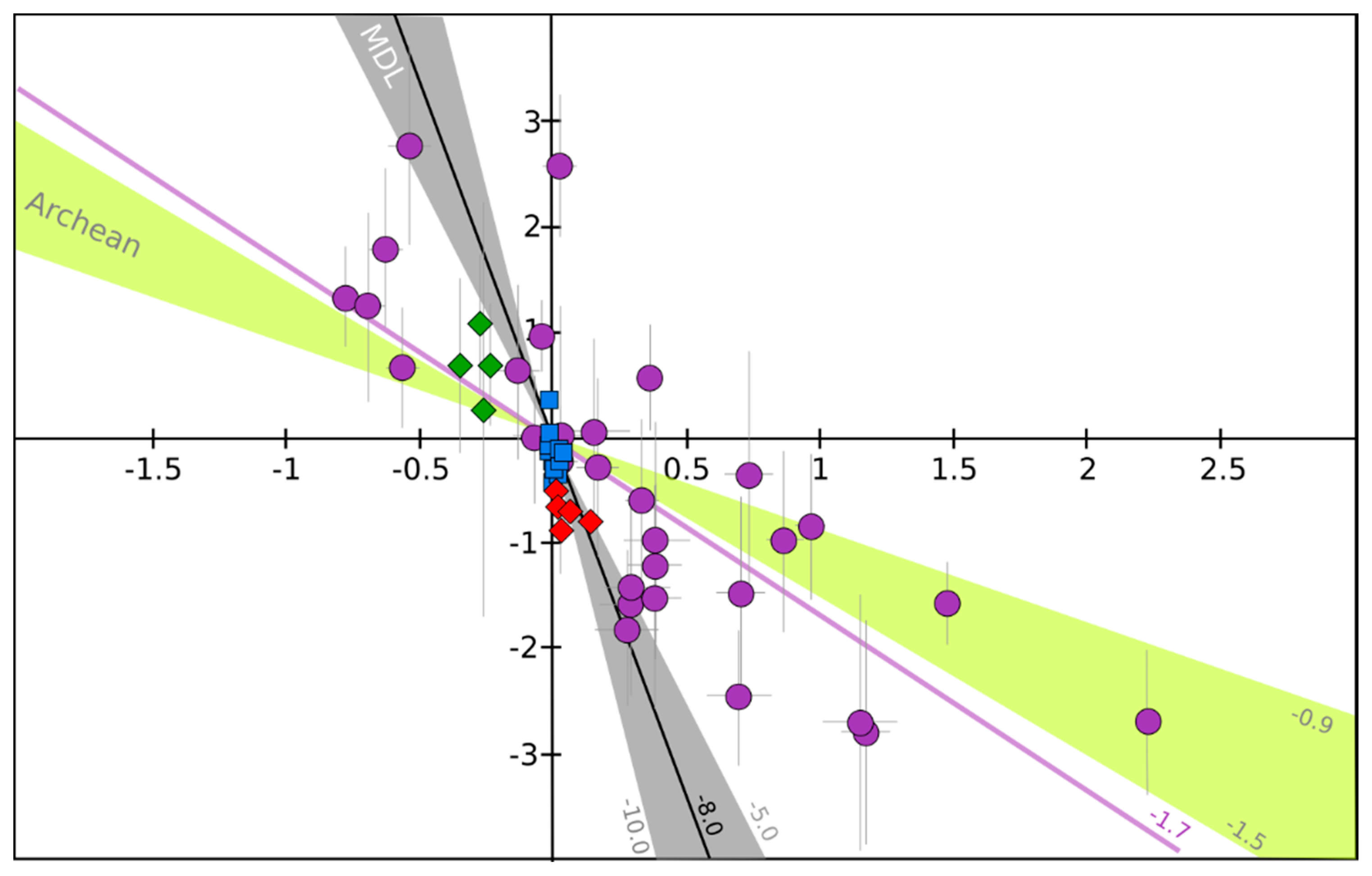
Overall, the aerosols δ34S tend to be higher than the bulk S-bearing gases, and when measured simultaneously the δ34S of bulk S-bearing gases is systematically a few permil lower than in sulfate [26], in agreement with the expected isotopic fractionation [84]. The Δ33S of ~0‰ is consistent with a direct oxidation of S-bearing gases at high temperature where, like in magmatic conditions, a mass dependent isotopic fractionation is expected (e.g., [85]). In contrast, the negative Δ36S values are consistent with non-MIF processes as small non-zero Δ33S (between 0 and 0.14‰; Figure 2) and Δ36S (between −0.5 and −0.9; Figure 2) can be generated by mass dependent processes. The observed Δ33S/Δ36S ratio of about −8 is indeed in the same range as the mass dependent fractionation line that has a Δ33S/Δ36S ratio between −5 and −10 [51,86,87] (Figure 3).
The measured δ18O values are systematically above the magmatic composition, which can be interpreted in terms of isotopic fractionation during distillation/condensation processes in the volcanic conduit and/or to sulfur oxidants that have systematically higher δ18O than the magma. Such oxidants could be atmospheric oxidants like O2 or H2O2, which could be responsible for these high δ18O as they can have δ18O higher than 20‰ (e.g., [20] and references therein; Figure 2). However, it is noteworthy that when sulfate aerosols are collected on filters, their δ18O seems to increase with the sulfate concentration [30,31]. This seems to show that some isotopic fractionation may occur on the filter during the sampling. In locations where such measurements have been done (Stromboli), LeGendre [30] also analyzed sulfate collected on volcanic ash and the δ18O is much more reproducible and corresponds to the lowest range of what was measured on filters (around 10‰; Figure 2). These values are closer than the magmatic values, but still slightly higher (Figure 2); therefore, we cannot rule out that atmospheric oxidants play a significant role in the high temperature chemistry at volcanic vents and therefore on the primary volcanic sulfate formation.
The Δ17O values are all very close to zero, indicating that the S-bearing gases oxidant is mass dependent, which is consistent with magmatic conditions, where all the compounds (volcanic gases, silicates minerals and glass) are mass dependent. If, as suggested above, some atmospheric oxidants play a significant role in the formation of the primary sulfate, these oxidants should have a Δ17O close to 0‰, which is only consistent with O2 (Δ17O = −0.33‰) and not H2O2. Indeed, even if Δ17O of atmospheric O2 is significantly negative, when combined with magmatic oxygen atoms in the resulting sulfate, the overall Δ17O should be very close to 0 ± 0.1‰ (Figure 1). Overall, we could expect sulfate δ18O to be magmatic-like or slightly higher if atmospheric O2 played a significant role and did not isotopically exchange with magmatic compounds.
It is very challenging to assess processes that occur at high temperatures, but some studies try to do that via equilibrium thermodynamic models [88,89,90,91]. They show that, considering volcanic gas mixtures with atmospheric air during the plume cooling, SO3 can be generated from S-bearing gases and subsequently form H2SO4 by co-condensation along with water. Combustion experiment studies also demonstrate that ash particles and their iron oxides are excellent catalyzers to the SO2 to SO3 conversion and that the resulting sulfate is isotopically mass dependent [92,93]. The models also predict the formation of H, OH, and OH2 radicals that are initially coming from thermal dissociation of H2O, which produces H and OH radicals reacting with atmospheric O2 to generate OH and OH2 [88]. The incorporation of atmospheric OH is also possible, but probably quickly depleted in the high temperature plume as the highly concentrated SO2 oxidation (via OH) would rapidly consume it.
3.3. Tropospheric Secondary Sulfates
Secondary sulfates are generated by oxidation of sulfur precursors in the atmosphere, either in the gas or condensed phases. When the volcanic plume does not reach the stratosphere, and remains in the troposphere, it is possible to address the formation of secondary sulfates in a more or less diluted volcanic tropospheric plume or cloud, by collecting sulfates from volcanic ashes deposited at distances even up to a few hundred kilometers from the volcanic vent [20]. Such sulfates measured all over the world show compositions ranging from 0 to 15‰ for both δ18O and δ34S; their Δ17O, Δ33S and Δ36S values are very homogeneous and very close to 0‰ as well (Figure 2). In detail, Δ36S/Δ33S of about −9.4 is similar to what is observed close to volcanic vents and close to the mass dependent fractionation line (Figure 3).
Models of tropospheric chemistry show that SO2 must be overwhelmingly oxidized by H2O2 when pH < 6, which is expected to be the case in volcanic plume. However, as all non-volcanic tropospheric sulfates have a Δ17O of about 0.7‰ (e.g., references [30,92,95,96,97]), this should also be the case for volcanic tropospheric sulfates (Figure 1). The fact that Δ17O of tropospheric volcanic sulfate is close to 0‰, clearly shows that H2O2 is not the main oxidant. Based on the fact that H2O2 concentration in the troposphere (H2O2 concentration = [H2O2] < 0.5 DU; Dobson unit; 1 DU = 2.68 × 1016 molecule cm−2) is much lower than the concentration of volcanic SO2 in the volcanic plumes ([SO2] = 10–100 DU; e.g., [98,99]), Martin et al. [20] proposed that H2O2 reacts and is rapidly consumed in the tropospheric column. The second main oxidants are OH (Ch1) or O2 (Ch4) that are non-MIF (or slightly negative for O2). Consequently, they logically produce sulfate aerosols with Δ17O very close to 0‰ (Figure 1). In tropospheric volcanic plumes, when tropospheric humidity is high (water condensation in the plume), heterogeneous aqueous oxidation reaction via O2-TMI is expected to be faster than homogeneous gas phase reaction via OH. However, in relatively low humidity conditions (no condensing plume), the oxidation via OH should dominate [100]. This is in agreement with observations made at Kilauea (Hawaii) where diurnal variations in the sulfate fraction ([]/([] + [SO2])) have been observed in the volcanic plume, indicating that photochemically produced oxidants play a major role in the SO2 oxidation [101]. OH and H2O2 are photochemically produced oxidants but, as discussed above, oxidation via H2O2 cannot explain the non-MIF isotopic composition of volcanic sulfate produced in the troposphere. Therefore, oxidation via OH seems to play the main role in relatively dry condition plumes.
Finally, collecting tropospheric secondary sulfates from volcanic ashes could be biased by the fact that primary sulfates adsorbed on ash particles are still present, which dilutes the secondary sulfate isotopic signature. Unfortunately, based only on petrographic observations and on O-isotopic signatures of these sulfates, distinguishing a primary vs. secondary origin (Figure 2) is a strenuous task. However, as measured at volcanic vents, the sulfate/SO2 ratio is usually <1% [25,58,59], indicating that further away from the vent, after oxidation of some SO2 in the volcanic plume, secondary sulfate should rapidly (in a few hours) dominate the primary ones. Therefore, in ash deposit collected at more than ~30–50 km from the volcanic vent, secondary sulfate should dominate. However, secondary sulfate could also be generated in volcanic plumes by other processes such as S-bearing gases oxidation by OH radicals produced on ash particles (e.g., [60,102,103,104,105]). Additionally, another way of producing secondary sulfate in the volcanic plume can be the SO2 reaction with halogen species (HOBr or HOCl) dissolved in the aqueous phase [106,107]. Unfortunately, yet very little is known on the conditions prevailing inside the dense and hot volcanic plumes. For instance, in such an environment, the halogen chemical behaviors are poorly constrained so it is difficult to estimate the role played in volcanic plumes by these oxidation channels.
In a tropospheric volcanic plume, the estimation of the conversion of SO2 into sulfate aerosols is a key parameter in order to have accurate volcanic S-spices fluxes, which is crucial in volcano monitoring (see [5] for a review). The SO2 oxidation rate depends on the oxidation pathways, which as discussed above, depend on the SO2 concentration, the relative humidity, the time of the day and the season of the year. In tropospheric volcanic plumes, the estimations of SO2 oxidation rates usually range from ~10−7 s−1 to ~10−4 s−1 ([101] and references therein). It is noteworthy that these estimations can be biased by the content of primary sulfate, loss of SO2 by non-oxidative processes, and the presence of aerosols other than sulfates. These parameters are not easy to quantify and are often not taken into account, leading to large uncertainties on the SO2 oxidation rate. Kroll et al. [101] measured in real-time the SO2 and sulfate concentrations in the Kilauea volcanic plume and inferred SO2 oxidation rates up to 2.5 × 10−6 s−1 at noon and an average over 24 h of 5.3 × 10−7 s−1, which is in the lower range of all the previous estimations. Values of about 10−7 s−1 are also inferred from atmospheric chemistry models that do not include halogen chemistry, which is comparable to the Kilauea case, where halogen emissions are rather low (Galeazzo, personal communication).
3.4. Stratospheric Secondary Sulfates
During major explosive volcanic eruptions, the volcanic plume can reach the stratosphere. There, S-bearing gases can be oxidized and form sulfate aerosols that, due to strong stratospheric winds, are able to travel long distances, to be potentially dispersed globally and finally to slowly sediment on the Earth’s surface. It is striking that, due to atmospheric circulation, a volcanic plume emitted in the tropics is easily spread out all over the globe, while at a higher latitude it remains in the same hemisphere. Furthermore, and for the same reasons, the concentration of volcanic sulfate aerosols is unlikely to be homogeneous on the deposition area.
This has been very well demonstrated for the 1991 Pinatubo eruption (e.g., [108]). These stratospheric volcanic sulfate aerosols can be ideally sampled in polar ice cores. Indeed, for the last hundred thousand years, ice accumulation at the poles has progressively recorded and archived atmosphere compositions. Thanks to its chemical stability (very low volatility and reactivity with other compounds), and taking into account some snow redistribution by the wind, the sulfate concentration in the ice has been used for decades to identify volcanic events (references [109,110,111,112,113] among others). In Antarctica, O- and S-isotope compositions of these sulfates have been studied [78,79,80,81]. It is noteworthy that background correction is necessary as concentration up to 100 ppb of mostly biogenic sulfate can be present. There, δ18O ranges from −5‰ to +25‰, and δ34S: from −5‰ to +20‰ (Figure 2). Furthermore, S- and O-MIF signatures (−0.8‰ < Δ33S < 2.2‰; −2.7‰ < Δ36S < 2.8‰ and Δ17O up 6.5‰; Figure 2) were recorded in the sulfates from more than ten stratospheric eruptions (e.g., Pinatubo, Agung, Krakatoa, Kuwae and different unknown eruptions). Note that the average Δ33S/Δ36S ratio from this dataset is about −1.7, which is clearly different from the mass dependent fractionation line ratio (−5 to −10; see [94] for a review; Figure 3).
The most accepted process for generating such S-MIF signatures consists of the photolysis and photoexcitation of SO2 by UV radiations, in an oxygen-poor atmosphere ([12,21,22,114,115,116] and [94] for a review). On the early Earth, before the great oxidation event (2.4–2.2 Ga), there was no atmospheric oxygen and consequently, no ozone layer. So, due to the lack of this shield, UV radiation reached the surface of the Earth, such that sulfur photolysis and photoexcitation could take place through the whole atmospheric column, resulting in possibly large S-MIF. Since then, the ozone layer has partially protected the Earth’s surface from UV radiation. Consequently, the only place where such processes can take place (and generate large S-MIF signatures) is in the stratosphere above or in the upper part of the ozone layer (≥25 km). Volcanic sulfate aerosols generated in the stratosphere are thus expected to have S-MIF signatures [114,117]. Therefore, volcanic sulfate aerosols having large S-MIF signatures clearly testify to their stratospheric origin.
In ice-cores, an evolution of Δ33S from negative to positive and Δ36S from positive to negative is recorded during a single volcanic event [80,82]. This can be explained by formation in the stratosphere of 33S enriched and 36S depleted sulfate aerosols at first and then, by mass balance, the resulting SO2 pool generates sulfates that are 33S depleted and 36S enriched [82]. However, in the stratosphere, the S-bearing gases oxidation after an eruption takes a few months, while the ice volcanic sulfate records are on a timescale of years. This requires a physical separation in space and time of the SO2 and generated sulfates right from the beginning of the SO2 oxidation in the stratosphere and maintained separated for a few years while traveling in the stratosphere and while depositing in formation-time order in the ice.
The large O-MIF signature (Δ17O up to 6.5‰) observed in the same stratospheric sulfates [78,79] can only be explained by the oxidation of S-bearing gases by OH radicals that have high Δ17O. Indeed, as OH results from O3 photo-dissociation, they carry the same isotopic anomaly as O3 (Figure 1). Due to isotopic exchange with non-MIF water, tropospheric OH is O-mass dependent (Δ17O = 0‰; Figure 1). The “cold trap” effect played by the tropopause prevents significant water transfers between troposphere and stratosphere. This lack of exchanges results in the low water content in the stratosphere (2–6 ppmv which is 3–4 orders of magnitude lower than in the troposphere) [118]. These relatively anhydrous stratospheric conditions favor homogeneous (gas phase) oxidation of SO2; in that case, the main oxidant of volcanic sulfur is OH (Figure 1). It is noteworthy that during major eruptions, the water-rich volcanic plumes reach the stratosphere where they could provide significant amounts of water (at least locally), such that heterogeneous gas phase oxidation by O3 could potentially play a significant role as well. Unfortunately, taken alone, O-isotopes do not allow precise quantification of the relative role played by O3 or OH in the stratospheric oxidation of sulfur. If the ozone plays a role in such diluted stratospheric volcanic plumes, it means that the conditions are rather basic and not acidic as in denser plumes. In the case where acidic conditions are still preserved, then the SO2 oxidation via O2-TMI should prevail, which should lead to a decrease of the sulfate Δ17O. This could explain the fact that some sulfates generated during large eruptions and collected in ice-core do not have high Δ17O signatures [78]. However, these low values seem to correlate with very large eruptions that inject more than 100 Mt of SO2 into the stratosphere. Savarino et al. [78] proposed that in such conditions, OH radicals could be rapidly exhausted and that oxidation via O(3P), which quickly reacts with O2 that has a Δ17O very close to 0‰, could be the main mechanism taking place in the stratosphere during these exceptional volcanic eruptions.
3.5. Ash Layer from Large Caldera-Forming Eruptions (Super-Eruptions)
Super-eruptions correspond to volcanic eruptions that emit more than 1000 km3 of fragmental material (mainly ash) and form large calderas. Sulfates extracted from these deposits display a wide range of isotopic compositions: δ18O and δ34S generally spread from 0‰ to 15‰, Δ17O from 0‰ up to 6‰, while Δ33S and Δ36S remain very close to 0‰ (Figure 2). The study of super-eruption deposits raises some questions that are still under debate [33,35]. During super-eruptions, such as those from Yellowstone (Lava Creek Tuff, 0.64 Ma; Huckleberry Ridge Tuff, 2.1 Ma) and the Long Valley eruption (Bishop tuff, 0.76 Ma), ashes have been deposited at distances up to 5000 km from their source calderas. As discussed above, at such distances, the fraction of primary sulfates should be negligible compared to secondary sulfates. Even if these old ash layers (hundreds of ka to tens of Ma), have been diluted by non-MIF sedimentary sulfates, O- and S-MIF signatures could still have been preserved. Indeed, Martin and Bindeman [33] have shown that up to 25% of O- and S-MIF volcanic sulfates are still preserved in some Yellowstone and Bishop Tuff volcanic deposits. Taking into account the effects of dilution, the authors were able to recalculate the Δ33S and Δ36S of the initial volcanic sulfates, which have an unambiguous S-MIF signature (Δ33S down to -0.4‰ and Δ36S up to 1.2‰; Figure 2). Therefore, even in deposits located within a radius of a few thousands of kilometers from the caldera, S-MIF sulfates are observed, which indicates that a significant amount of stratospheric sulfates is still present in super-eruption deposits. It is noteworthy that Bao et al. [119] proposed that the sulfate O-MIF signature from super-eruption deposits could be accounted by tropospheric oxidation via O3. This is possible in basic environments (pH > 6), however it is inconsistent with the acidic (low pH) character of dense volcanic plumes. Nonetheless, such conclusions raise new questions.
(A) The tropospheric origin of sulfates: If the O-MIF (up to 6‰) sulfates are generated in the troposphere, O3 must have played an important role (Figure 1), which seems unlikely considering acidic conditions of volcanic plumes. Furthermore, if S-MIF is also generated in the troposphere during super-eruptions, it has to be considered that the ozone layer was depleted, at least locally, such that a significant fraction of UV radiation was able to reach the troposphere, thus making SO2 photoexcitation and photolysis possible. It is noteworthy that such a process is compatible with the UV-B flux variations possibly linked to the large igneous provinces emplacements ([120,121] and reference therein).
(B) The stratospheric origin of sulfates: If sulfates are generated by O3 (aqueous phase oxidation) in the stratosphere, does it get at least locally hydrated? Such a scenario was observed after the 1982 El Chichon eruption [122,123], but the injection of SO2 in the stratosphere could also lead to the drying out of the stratosphere [124] which makes it difficult to give a general answer to this question. Indeed, the formation of sulfates in the stratosphere by OH oxidation would account for both O- and S-MIF signatures. In turn, this scenario implies that the watering of the stratosphere must be negligible, otherwise OH should react with H2O and dilute its positive Δ17O signature. It must be noted that in very high SO2 concentration conditions, OH can react significantly with SO2 before isotopic exchange with water (Galeazzo, personal communication). The fact that all the Δ17O measured is lower than 8.8‰, as expected by OH oxidation (Figure 1), can obviously be explained by non-MIF sediment sulfate dilution in the volcanic deposit, but it could also be explained by another oxidation pathway in the stratosphere. For large stratospheric eruptions, it has been suggested that, due to OH depletion, oxidation via O(3P) could generate non-MIF sulfate [78]. In any case, the stratospheric origin is unable to explain why such an amount of sulfate (up to a few hundreds of ppm) can be found in an area of a few hundreds to thousands of kilometers around the caldera while it should have been spread out all around the world (or at least hemispherically for high latitude eruptions) as it is observed for stratospheric eruptions.
(C) The mixed origin of sulfates: This raises questions about the physico-chemical properties of the tropopause during such volcanic events. After the 1982 El Chichon and the 1991 Pinatubo eruptions for instance the temperature of the low stratosphere increased up to 1.5 °C [9]. If a significant temperature increase had occurred at the tropopause, its “cold trap” effect would have been partially reduced, thus facilitating chemical fluxes (O3 and H2O for instance) between the troposphere and stratosphere. Even if the impact was global, the apogee of such an effect could have been restricted to a few hundreds to thousands of kilometers in the atmospheric column located above the volcanic systems.
4. Summary and Conclusions
Oxygen and sulfur isotopes provide good constraints on the formation and fate of volcanic sulfate aerosols and ultimately the real nature of the physico-chemical interactions between the atmosphere and volcanic eruptions. Multi O- and S-isotope analyses of sulfates collected by air sampling or volcanic ash leaching allow us to decipher the oxidation pathways of S-bearing gases in the atmosphere. They also discriminate between primary vs. secondary aerosols and give information about the place where they were generated (troposphere vs. stratosphere).
Some aspects need to be improved:
- -
- High temperature chemistry (including sulfate aerosol formation) is assessed by equilibrium thermodynamic models, but at volcanic vents the plume cools down and dilutes very quickly, therefore modeling using a kinetic approach should be more appropriate.
- -
- The isotopic approach alone can hardly differentiate between different possible mass-dependent processes responsible for sulfate formation. In volcanic plumes, sulfates can be generated by oxidation channels such as OH or O2-TMI oxidation, but the exploration of other possible oxidation processes is required. The role played by halogens and OH radicals generated from ash particles in the SO2 oxidation needs to be quantified as in volcanic plumes they may play a more preponderant role than expected. This would improve our understanding of sulfate aerosol formation in a relatively particle-dense plume or cloud.
- -
- In the stratosphere, the low Δ17O sulfates are not totally understood yet. Oxidation channels other than oxidation via OH need to be explored. Furthermore, the fact that in ice-cores the evolution of Δ33S (from negative to positive) and Δ36S (from positive to negative) is recorded during a single volcanic event on a year timescale, while the SO2 oxidation in the stratosphere takes a few months after an eruption, is still unexplained.
- -
- The impact of super-eruptions on the atmosphere and more specifically on the tropopause needs to be explored in more detail in order to better constrain the potential chemical fluxes between the troposphere and the stratosphere during such an event. This would have an impact on the sulfate aerosol formation and on the atmospheric and climatic impact of super-eruptions in general.
In dry environments, sulfates can be preserved in volcanic deposits for millions of years, thus they can be considered as a reliable archive having recorded the impact on the atmosphere of ancient volcanic eruptions. Furthermore, volcanoes are widespread all over the world, and they have erupted more or less regularly over the Earth’s history. Consequently, volcanic sulfates can also be used as proper proxies for the oxidant capacity of the atmosphere on a geological timescale. This would represent an additional constraint on the climatic models for the past but also the future periods of times. From this point of view, the study of volcanic sulfates could provide an open window on past and future climatic changes. Additionally, a better understanding of sulfate aerosol formation and fate in the atmosphere would provide a new parameter in our understanding of atmospheric chemistry evolution. A similar approach performed on anthropogenic S-bearing emissions would significantly improve our knowledge on present-day atmospheric chemistry. This appears to be a fundamental parameter for climatic modeling and for a better prediction of forthcoming climatic changes.
Acknowledgments
The author expresses his sincere thanks the “Emergence program” from the city of Paris, the Agence National de la recherche (ANR) via the contracts 14-CE33-0009-02-FOFAMIFS and 16-CE31-0010-PaleOX, the Sorbonne Universités and the KIC-Climate PhD programs for their support. He is also grateful to E. LeGendre, H. Martin, S. Bekki, I. Bindeman, T. Galeazzo, A. Aroskay, E. Gautier and L. Whiteley for their informal reviews, discussions and proof-reading of the manuscript. The author thanks the anonymous reviewers for their comments that certainly improved the manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Andres, R.J.; Kasgnoc, A.D. A time-averaged inventory of subaerial volcanic sulfur emissions. J. Geophys. Res. Atmos. 1998, 103, 25251–25261. [Google Scholar] [CrossRef]
- Graf, H.-F.; Feichter, J.; Langmann, B. Volcanic sulfur emissions: Estimates of source strength and its contribution to the global sulfate distribution. J. Geophys. Res. Atmos. 1997, 102, 10727–10738. [Google Scholar] [CrossRef]
- Textor, C.; Graf, H.-F.; Timmreck, C.; Robock, A. Emissions from volcanoes. In Emissions of Atmospheric Trace Compounds; Advances in Global Change Research; Springer: Dordrecht, The Netherlands, 2004; pp. 269–303. ISBN 978-90-481-6605-3. [Google Scholar]
- Delmelle, P.; Stix, J. Volcanic gases. In Encyclopedia of Volcanoes; Sigurdsson, H., Houghton, B., McNutt, S.R., Rymer, H., Stix, J., Eds.; Elsevier: New York, NY, USA, 2000; pp. 803–815. [Google Scholar]
- Oppenheimer, C.; Scaillet, B.; Martin, R.S. Sulfur Degassing From Volcanoes: Source Conditions, Surveillance, Plume Chemistry and Earth System Impacts. Rev. Mineral. Geochem. 2011, 73, 363–421. [Google Scholar] [CrossRef] [Green Version]
- Haywood, J.; Boucher, O. Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review. Rev. Geophys. 2000, 38, 513–543. [Google Scholar] [CrossRef]
- Robock, A. Volcanic eruption and climate. Rev. Geophys. 2000, 38, 191–219. [Google Scholar] [CrossRef]
- Hansen, J.E.; Wang, W.-C.; Lacis, A.A. Mount Agung Eruption Provides Test of a Global Climatic Perturbation. Science 1978, 199, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.E.; Wilson, H.; Jones, P.D.; Christy, J.R.; Folland, C.K. The impact of mount pinatubo on world-wide temperatures. Int. J. Climatol. 1996, 16, 487–497. [Google Scholar] [CrossRef]
- Parker, D.E.; Brownscombe, J.L. Stratospheric warming following the El Chichón volcanic eruption. Nature 1983, 301, 406–408. [Google Scholar] [CrossRef]
- Rampino, M.R.; Self, S. Historic Eruptions of Tambora (1815), Krakatau (1883), and Agung (1963), their Stratospheric Aerosols, and Climatic Impact. Quat. Res. 1982, 18, 127–143. [Google Scholar] [CrossRef]
- Farquhar, J.; Savarino, J.; Airieau, S.; Thiemens, M.H. Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere. Geophys. Res. 2001, 106, 32829–32839. [Google Scholar] [CrossRef]
- Farquhar, J.; Bao, H.; Thiemens, M. Atmospheric Influence of Earth’s Earliest Sulfur Cycle. Science 2000, 289, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Thiemens, M.H. History and applications of mass-independent isotope effects. Annu. Rev. Earth Planet. Sci. 2006, 34, 217–262. [Google Scholar] [CrossRef]
- Thiemens, M.H.; Heidenreich, J.E. The Mass-Independent Fractionation of Oxygen: A Novel Isotope Effect and Its Possible Cosmochemical Implications. Science 1983, 219, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, J.; Wing, B.A. Multiple sulfur isotopes and the evolution of the atmosphere. Earth Planet. Sci. Lett. 2003, 213, 1–13. [Google Scholar] [CrossRef]
- Rumble, D.; Miller, M.F.; Franchi, I.A.; Greenwood, R.C. Oxygen three-isotope fractionation lines in terrestrial silicate minerals: An inter-laboratory comparison of hydrothermal quartz and eclogitic garnet. Geochim. Cosmochim. Acta 2007, 71, 3592–3600. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Savarino, J.; Lee, C.C.W.; Thiemens, M.H. Laboratory oxygen isotopic study of sulfur (IV) oxidation: Origin of the mass-independent oxygen isotopic anomaly in atmospheric sulfates and sulfate mineral deposits on Earth. J. Geophys. Res. Atmos. 2000, 105, 29079–29088. [Google Scholar] [CrossRef]
- Martin, E.; Bekki, S.; Ninin, C.; Bindeman, I. Volcanic sulfate aerosol formation in the troposphere. J. Geophys. Res. Atmos. 2014, 119, 12:660–12:673. [Google Scholar] [CrossRef]
- Ono, S.; Whitehill, A.R.; Lyons, J.R. Contribution of isotopologue self-shielding to sulfur massindependent fractionation during sulfur dioxide photolysis. Ournal Geophys. Res. Atmos. 2013, 118, 2444–2454. [Google Scholar] [CrossRef]
- Whitehill, A.R.; Xie, C.; Hu, X.; Xie, D.; Guo, H.; Ono, S. Vibronic origin of sulfur mass-independent isotope effect in photoexcitation of SO2 and the implications to the early earth’s atmosphere. Proc. Natl. Acad. Sci. USA 2013, 110, 17697–17702. [Google Scholar] [CrossRef] [PubMed]
- Giggenbach, W.F. A simple method for the collection and analysis of volcanic gas samples. Bull. Volcanol. 1975, 39, 132–145. [Google Scholar] [CrossRef]
- Giggenbach, W.F.; Goguel, R.L. Methods for the Collection and Analysis of Geothermal and Volcanic Water and Gas Samples; Chemistry Division, Department of Scientific and Industrial Research: Petone, New Zealand, 1989; p. 53.
- De Moor, J.M.; Fischer, T.P.; Sharp, Z.D.; King, P.L.; Wilke, M.; Botcharnikov, R.E.; Cottrell, E.; Zelenski, M.; Marty, B.; Klimm, K.; et al. Sulfur degassing at Erta Ale (Ethiopia) and Masaya (Nicaragua) volcanoes: Implications for degassing processes and oxygen fugacities of basaltic systems: Sulfur Degassing at Basaltic Volcanoes. Geochem. Geophys. Geosyst. 2013, 14, 4076–4108. [Google Scholar] [CrossRef]
- Menyailov, I.A.; Nikitina, L.P.; Shapar, V.N.; Pilipenko, V.P. Temperature increase and chemical change of fumarolic gases at Momotombo Volcano, Nicaragua, in 1982–1985: Are these indicators of a possible eruption? J. Geophys. Res. Solid Earth 1986, 91, 12199–12214. [Google Scholar] [CrossRef]
- Goff, F.; Janik, C.J.; Delgado, H.; Werner, C.; Counce, D.; Stimac, J.A.; Siebe, C.; Love, S.P.; Williams, S.N.; Fischer, T.; et al. Geochemical surveillance of magmatic volatiles at Popocatépetl volcano, Mexico. GSA Bull. 1998, 110, 695–710. [Google Scholar] [CrossRef]
- Ohba, T.; Nogami, K.; Hirabayashi, J.-I.; Mori, T. Isotopic fractionation of SO2 and H2S gases during the absorption by KOH solution, with the application to volcanic gas monitoring at Miyakejima Island, Japan. Geochem. J. 2008, 42, 119–131. [Google Scholar] [CrossRef]
- Mather, T.A.; Pyle, D.M.; Heaton, T.H.E. Investigation of the use of filter packs to measure the sulphur isotopic composition of volcanic sulphur dioxide and the sulphur and oxygen isotopic composition of volcanic sulphate aerosol. Atmos. Environ. 2008, 42, 4611–4618. [Google Scholar] [CrossRef]
- Le Gendre, E. Étude des Anomalies Isotopiques de L’oxygène Etdusoufre Dans les Sulfates D’origine Volcanique et Anthropique; UPMC: Pittsburgh, PA, USA, 2016. [Google Scholar]
- Mather, T.A.; McCabe, J.R.; Rai, V.K.; Thiemens, M.H.; Pyle, D.M.; Heaton, T.H.E.; Sloane, H.J.; Fern, G.R. Oxygen and sulfur isotopic composition of volcanic sulfate aerosol at the point of emission. J. Geophys. Res. Atmos. 2006, 111, D18205. [Google Scholar] [CrossRef]
- Armienta, M.A.; De la Cruz-Reyna, S.; Soler, A.; Cruz, O.; Ceniceros, N.; Aguayo, A. Chemistry of ash-leachates to monitor volcanic activity: An application to Popocatépetl volcano, central Mexico. Appl. Geochem. 2010, 25, 1198–1205. [Google Scholar] [CrossRef]
- Martin, E.; Bindeman, I. Mass-independent isotopic signatures of volcanic sulfate from three supereruption ash deposits in Lake Tecopa, California. Earth Planet. Sci. Lett. 2009, 282, 102–114. [Google Scholar] [CrossRef]
- Risacher, F.; Alonso, H. Geochemistry of ash leachates from the 1993 Lascar eruption, northern Chile. Implication for recycling of ancient evaporites. J. Volcanol. Geotherm. Res. 2001, 109, 319–337. [Google Scholar] [CrossRef]
- Bao, H.; Thiemens, M.H.; Loope, D.B.; Yuan, X.-L. Sulfate oxygen-17 anomaly in an Oligocene ash bed in mid-North America: Was it the dry fogs? Geophys. Res. Lett. 2003, 30, 11–14. [Google Scholar] [CrossRef]
- Van Stempvoort, D.R.; Krouse, H.R. Controls of d18O in sulfate: Review of experimental data and application to specific environments. In Environmental Geochemistry of Sulfide Oxidation; Alpers, C.N., Blowes, D.W., Eds.; American Chemical Society: Washington, DC, USA, 1994; Volume 550. [Google Scholar]
- Witham, C.S.; Oppenheimer, C.; Horwell, C.J. Volcanic ash-leachates: A review and recommendations for sampling methods. J. Volcanol. Geotherm. Res. 2005, 141, 299–326. [Google Scholar] [CrossRef]
- Botter, C. Utomated Single-Particle SEM/EDS Analysis of Volcanic Aerosols from Stromboli, Aeolian Arc, Italy; Mineralogical Characterization of the PM10 Fraction; University of Fribourg: Fribourg, Switzerland, 2011. [Google Scholar]
- Botter, C.; Meier, M.; Wiedenmann, D.; Grobety, B.; Ricci, T. Single Particle Analysis of Volcanic Aerosols from Stromboli, Italy. In Proceedings of the Cities on Volcanoes 6 (COV6), Tenerife, Spain, 31 May–4 June 2010. [Google Scholar]
- Toutain, J.; Quisefit, J.; Briole, P.; Aloupogiannis, P.; Blanc, P.; Robaye, G. Mineralogy and chemistry of solid aerosols emitted from Mount Etna. Geochem. J. 1995, 29, 163–173. [Google Scholar] [CrossRef]
- Stoiber, R.E.; Rose, W.I. The Geochemistry of Central American Volcanic Gas Condensates. GSA Bull. 1970, 81, 2891–2912. [Google Scholar] [CrossRef]
- Symonds, R.B.; Reed, M.H.; Rose, W.I. Origin, speciation, and fluxes of trace-element gases at Augustine volcano, Alaska: Insights into magma degassing and fumarolic processes. Geochim. Cosmochim. Acta 1992, 56, 633–657. [Google Scholar] [CrossRef]
- Quisefit, J.P.; Bergametti, G.; Tedesco, D.; Pinart, J.; Colin, J.L. Origin of particulate potassium in Mt Etna emissions before and during the 1983 eruption. J. Volcanol. Geotherm. Res. 1988, 35, 111–119. [Google Scholar] [CrossRef]
- Ayris, P.M.; Lee, A.F.; Wilson, K.; Kueppers, U.; Dingwell, D.B.; Delmelle, P. SO2 sequestration in large volcanic eruptions: High-temperature scavenging by tephra. Geochim. Cosmochim. Acta 2013, 110, 58–69. [Google Scholar] [CrossRef]
- Van Stempvoort, D.R.; Reardon, E.J.; Fritz, P. Fractionation of sulfur and oxygen isotopes in sulfate by soil sorption. Geochim. Cosmochim. Acta 1990, 54, 2817–2826. [Google Scholar] [CrossRef]
- Michalski, G.; Kasem, M.; Rech, J.A.; Adieu, S.; Showers, W.S.; Genna, B.; Thiemens, M. Uncertainties in the oxygen isotopic composition of barium sulfate induced by coprecipitation of nitrate. Rapid Commun. Mass Spectrom. 2008, 22, 2971–2976. [Google Scholar] [CrossRef] [PubMed]
- Bao, H. Purifying barite for oxygen isotope measurement by dissolution and reprecipitation in a chelating solution. Anal. Chem. 2006, 78, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Le Gendre, E.; Martin, E.; Villemant, B.; Cartigny, P.; Assayag, N. A simple and reliable anion-exchange resin method for sulfate extraction and purification suitable for multiple O- and S-isotope measurements: Anion-exchange method for multiple O- and S-isotope analysis. Rapid Commun. Mass Spectrom. 2017, 31, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Schauer, A.J.; Kunasek, S.A.; Sofen, E.D.; Erbland, J.; Savarino, J.; Johnson, B.W.; Amos, H.M.; Shaheen, R.; Abaunza, M.; Jackson, T.L.; et al. Oxygen isotope exchange with quartz during pyrolysis of silver sulfate and silver nitrate: Oxygen isotope exchange during pyrolysis of Ag2SO4 and AgNO3. Rapid Commun. Mass Spectrom. 2012, 26, 2151–2157. [Google Scholar] [CrossRef] [PubMed]
- Pepkowitz, L.; Shirley, E. microdetection of sulfur. Anal. Chem. 1951, 23, 1709–1710. [Google Scholar] [CrossRef]
- Farquhar, J.; Peters, M.; Johnston, D.T.; Strauss, H.; Masterson, A.; Wiechert, U.; Kaufman, A.J. Isotopic evidence for Mesoarchaean anoxia and changing atmospheric sulphur chemistry. Nature 2007, 449, 706–709. [Google Scholar] [CrossRef] [PubMed]
- Rafter, T.A. Oxygen isotopic composition of sulfates. Part I. A method for the extraction of oxygen and its quantitative conversion to carbon dioxide for isotope radiation measurements. N. Z. J. Sci. 1967, 10, 493–510. [Google Scholar]
- Mizutani, Y. An improvement in the carbon-reduction method for the oxygen isotopic analysis of sulphates. Geochem. J. 1971, 5, 69–77. [Google Scholar] [CrossRef]
- Bao, H.; Thiemens, M.H. Generation of O2 from BaSO4 using a CO2-laser fluorination system for simultaneous analysis of d18O and d17O. Anal. Chem. 2000, 72, 4029–4032. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Schauer, A.J.; Kunasek, S.A.; Sofen, E.D.; Erbland, J.; Savarino, J.; Allman, D.J.; Sletten, R.S.; Alexander, B. Analysis of oxygen-17 excess of nitrate and sulfate at sub-micromole levels using the pyrolysis method: Analysis of oxygen-17 excess of nitrate and sulfate. Rapid Commun. Mass Spectrom. 2013, 27, 2411–2419. [Google Scholar] [CrossRef] [PubMed]
- Savarino, J.; Alexander, B.; Darmohusodo, V.; Thiemens, M.H. Sulfur and Oxygen Isotope Analysis of Sulfate at Micromole Levels Using a Pyrolysis Technique in a Continuous Flow System. Anal. Chem. 2001, 73, 4457–4462. [Google Scholar] [CrossRef] [PubMed]
- Au Yang, D.; Landais, G.; Assayag, N.; Widory, D.; Cartigny, P. Improved analysis of micro- and nanomole-scale sulfur multi-isotope compositions by gas source isotope ratio mass spectrometry: Improved analysis of S isotopes at micro/nanomole levels by IRMS. Rapid Commun. Mass Spectrom. 2016, 30, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.S.; Sawyer, G.M.; Spampinato, L.; Salerno, G.G.; Ramirez, C.; Ilyinskaya, E.; Witt, M.L.I.; Mather, T.A.; Watson, I.M.; Phillips, J.C.; et al. A total volatile inventory for Masaya Volcano, Nicaragua. J. Geophys. Res. 2010, 115. [Google Scholar] [CrossRef]
- Allen, A.G.; Oppenheimer, C.; Ferm, M.; Baxter, P.J.; Horrocks, L.A.; Galle, B.; McGonigle, A.J.S.; Duffell, H.J. Primary sulfate aerosol and associated emissions from Masaya Volcano, Nicaragua. J. Geophys. Res. Atmos. 2002, 107, 4682. [Google Scholar] [CrossRef]
- Cwiertny, D.M.; Young, M.A.; Grassian, V.H. Chemistry and Photochemistry of Mineral Dust Aerosol. Annu. Rev. Phys. Chem. 2008, 59, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Casadevall, T.J.; Moore, J.G. Chemistry and isotope ratios of sulfur in basalts and volcanic gases at Kilauea volcano, Hawaii. Geochim. Cosmochim. Acta 1982, 46, 729–738. [Google Scholar] [CrossRef]
- Allard, P. 13C/12C and 34S/32S ratios in magmatic gases from ridge volcanism in Afar. Nature 1979, 282, 56–58. [Google Scholar] [CrossRef]
- Sakai, H.; Gunnlaugsson, E.; Tòmasson, J.; Rouse, J.E. Sulfur isotope systematics in icelandic geothermal systems and influence of seawater circulation at Reykjanes. Geochim. Cosmochim. Acta 1980, 44, 1223–1231. [Google Scholar] [CrossRef]
- Goff, F.; McMurtry, G.M. Tritium and stable isotopes of magmatic waters. J. Volcanol. Geotherm. Res. 2000, 97, 347–396. [Google Scholar] [CrossRef]
- Taran, Y.; Gavilanes, J.C.; Cortés, A. Chemical and isotopic composition of fumarolic gases and the SO2 flux from Volcán de Colima, México, between the 1994 and 1998 eruptions. J. Volcanol. Geotherm. Res. 2002, 117, 105–119. [Google Scholar] [CrossRef]
- Sakai, H.; Matsubaya, O. Stable isotopic studies of japanese geothermal systems. Geothermics 1977, 5, 97–124. [Google Scholar] [CrossRef]
- Giggenbach, W. The chemical and isotopic composition of gas discharges from New Zealand andesitic volcanoes. Bull. Volcanol. 1982, 45, 253–255. [Google Scholar] [CrossRef]
- Williams, S.N.; Sturchio, N.C.; Calvache, V.M.L.; Mendez, F.R.; Londoño, C.A.; García, P.N. Sulfur dioxide from Nevado del Ruiz volcano, Colombia: Total flux and isotopic constraints on its origin. J. Volcanol. Geotherm. Res. 1990, 42, 53–68. [Google Scholar] [CrossRef]
- Richet, P.; Bottinga, Y.; Javoy, M. A Review of Hydrogen, Carbon, Nitrogen, Oxygen, Sulphur, and Chlorine Stable Isotope Fractionation Among Gaseous Molecules. Annu. Rev. Earth Planet. Sci. 1977, 5, 65–110. [Google Scholar] [CrossRef]
- Ohmoto, H.; Rye, R.O. Isotope of sulfur and carbon. In Geochemestry of Hydrothermal Deposits; Barnes, H.L., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 1979; pp. 509–567. [Google Scholar]
- Labidi, J.; Cartigny, P.; Birck, J.L.; Assayag, N.; Bourrand, J.J. Determination of multiple sulfur isotopes in glasses: A reappraisal of the MORB δ34S. Chem. Geol. 2012, 334, 189–198. [Google Scholar] [CrossRef]
- De Hoog, J.C.M.; Taylor, B.E.; van Bergen, M.J. Sulfur isotope systematics of basaltic lavas from Indonesia: Implications for the sulfur cycle in subduction zones. Earth Planet. Sci. Lett. 2001, 189, 237–252. [Google Scholar] [CrossRef]
- Martin, E.; Bindeman, I.; Grove, T. The origin of high-Mg magmas in Mt Shasta and Medicine Lake volcanoes, Cascade Arc (California): Higher and lower than mantle oxygen isotope signatures attributed to current and past subduction. Contrib. Miner. Petrol. 2011, 162, 945–960. [Google Scholar] [CrossRef]
- Eiler, J. Oxygen isotope variations of basaltic lavas and upper mantle rocks. In Reviews in Mineralogy and Geochemistry; Walley, J.W., Cole, D.R., Eds.; The Mineralogical Society of America: Washington, DC, USA, 2001; Volume 43, pp. 319–364. [Google Scholar]
- Bindeman, I.N.; Eiler, J.M.; Wing, B.A.; Farquhar, J. Rare sulfur and triple oxygen isotope geochemistry of volcanogenic sulfate aerosols. Geochim. Cosmochim. Acta 2007, 71, 2326–2343. [Google Scholar] [CrossRef]
- De Moor, J.M.; Fischer, T.P.; Hilton, D.R.; Hauri, E.; Jaffe, L.A.; Camacho, J.T. Degassing at Anatahan volcano during the May 2003 eruption: Implications from petrology, ash leachates, and SO2 emissions. J. Volcanol. Geotherm. Res. 2005, 146, 117–138. [Google Scholar] [CrossRef]
- Rye, R.O.; Luhr, J.F.; Wasserman, M.D. Sulfur and oxygen isotopic systematics of the 1982 eruptions of El Chichón Volcano, Chiapas, Mexico. J. Volcanol. Geotherm. Res. 1984, 23, 109–123. [Google Scholar] [CrossRef]
- Savarino, J.; Bekki, S.; Cole-Dai, J.H.; Thiemens, M.H. Evidence from sulfate mass independent oxygen isotopic compositions of dramatic changes in atmospheric oxidation following massive volcanic eruptions. J. Geophys. Res. Atmos. 2003, 108. [Google Scholar] [CrossRef]
- Baroni, M. Etude des Anomalies Isotopiques du Soufre et de L’oxygène Dans le Sulfate D’origine Volcanique Enregistré dans les Archives Glaciaires; Univeristé Joseph Fourier: Grenoble, France, 2006. [Google Scholar]
- Baroni, M.; Thiemens, M.H.; Delmas, R.J.; Savarino, J. Mass-Independent Sulfur Isotopic Compositions in Stratospheric Volcanic Eruptions. Science 2007, 315, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Savarino, J.; Cole-Dai, J.; Rai, V.K.; Thiemens, M.H. Anomalous sulfur isotope compositions of volcanic sulfate over the last millennium in Antarctic ice cores. J. Geophys. Res. Atmos. 2008, 113, D20112. [Google Scholar] [CrossRef]
- Gautier, E. Empreinte Isotopique et Histoire du Volcanisme Stratosphérique des 2600 Dernières Années, Enregistrées à Dome C, Antarctique; Université Grenoble Alpes: Grenoble, France, 2015. [Google Scholar]
- Cole-Dai, J.; Ferris, D.; Lanciki, A.; Savarino, J.; Baroni, M.; Thiemens, M.H. Cold decade (AD 1810–1819) caused by Tambora (1815) and another (1809) stratospheric volcanic eruption. Geophys. Res. Lett. 2009, 36. [Google Scholar] [CrossRef]
- Miyoshi, T.; Sakai, H.; Chiba, H. Experimental study of sulfur isotope fractionation factors between sulfate and sulfide in high temperature melts. Geochem. J. 1984, 18, 75–84. [Google Scholar] [CrossRef]
- Hoefs, J. Stable Isotope Geochemistry; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Farquhar, J.; Johnston, D.T.; Wing, B.A. Implications of conservation of mass effects on mass-dependent isotope fractionations: Influence of network structure on sulfur isotope phase space of dissimilatory sulfate reduction. Geochim. Cosmochim. Acta 2007, 71, 5862–5875. [Google Scholar] [CrossRef]
- Ono, S.; Wing, B.; Johnston, D.; Farquhar, J.; Rumble, D. Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles. Geochim. Cosmochim. Acta 2006, 70, 2238–2252. [Google Scholar] [CrossRef]
- Martin, R.S.; Mather, T.A.; Pyle, D.M. High-temperature mixtures of magmatic and atmospheric gases. Geochem. Geophys. Geosyst. 2006, 7, Q04006. [Google Scholar] [CrossRef]
- Roberts, T.J.; Braban, C.F.; Martin, R.S.; Oppenheimer, C.; Adams, J.W.; Cox, R.A.; Jones, R.L.; Griffiths, P.T. Modelling reactive halogen formation and ozone depletion in volcanic plumes. Chem. Geol. 2009, 263, 151–163. [Google Scholar] [CrossRef]
- Roberts, T.J.; Martin, R.S.; Jourdain, L. Reactive bromine chemistry in Mount Etna’s volcanic plume: The influence of total Br, high-temperature processing, aerosol loading and plume–air mixing. Atmos. Chem. Phys. 2014, 14, 11201–11219. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, T.M. Volcanic sources of tropospheric ozone-depleting trace gases. Geochem. Geophys. Geosyst. 2004, 5. [Google Scholar] [CrossRef]
- Lee, C.C.-W.; Savarino, J.H.; Cachier, H.; Thiemens, M.H. Sulfur (32S, 33S, 34S, 36S) and oxygen (16O,17O,18O) isotopic ratios of primary sulfate produced from combustion processes. Tellus B 2002, 54, 193–200. [Google Scholar] [CrossRef]
- Belo, L.P.; Elliott, L.K.; Stanger, R.J.; Spörl, R.; Shah, K.V.; Maier, J.; Wall, T.F. High-Temperature Conversion of SO2 to SO3: Homogeneous Experiments and Catalytic Effect of Fly Ash from Air and Oxy-fuel Firing. Energy Fuels 2014, 28, 7243–7251. [Google Scholar] [CrossRef]
- Ono, S. Photochemistry of Sulfur Dioxide and the Origin of Mass-Independent Isotope Fractionation in Earth’s Atmosphere. Annu. Rev. Earth Planet. Sci. 2017, 45, 301–329. [Google Scholar] [CrossRef]
- Jenkins, K.A.; Bao, H. Multiple oxygen and sulfur isotope compositions of atmospheric sulfate in Baton Rouge, LA, USA. Atmos. Environ. 2006, 40, 4528–4537. [Google Scholar] [CrossRef]
- Lee, C.C.-W.; Thiemens, M.H. The δ17O and δ18O measurements of atmospheric sulfate from a coastal and high alpine region: A mass-independent isotopic anomaly. J. Geophys. Res. Atmos. 2001, 106, 17359–17373. [Google Scholar] [CrossRef]
- Li, X.; Bao, H.; Gan, Y.; Zhou, A.; Liu, Y. Multiple oxygen and sulfur isotope compositions of secondary atmospheric sulfate in a mega-city in central China. Atmos. Environ. 2013, 81, 591–599. [Google Scholar] [CrossRef]
- Carn, S.A.; Clarisse, L.; Prata, A.J. Multi-decadal satellite measurements of global volcanic degassing. J. Volcanol. Geotherm. Res. 2016, 311, 99–134. [Google Scholar] [CrossRef]
- Yang, K.; Krotkov, N.A.; Krueger, A.J.; Carn, S.A.; Bhartia, P.K.; Levelt, P.F. Improving retrieval of volcanic sulfur dioxide from backscattered UV satellite observations. Geophys. Res. Lett. 2009, 36, L03102. [Google Scholar] [CrossRef]
- Galeazzo, T.; Bekki, S.; Martin, E. Modeling the Pathways of Generation of O-MIF in Tropospheric Sulfates; GOLDSCHMIDT: Paris, France, 2017. [Google Scholar]
- Kroll, J.H.; Cross, E.S.; Hunter, J.F.; Pai, S.; Wallace, L.M.M.; Croteau, P.L.; Jayne, J.T.; Worsnop, D.R.; Heald, C.L.; Murphy, J.G.; et al. Atmospheric Evolution of Sulfur Emissions from Kı̅lauea: Real-Time Measurements of Oxidation, Dilution, and Neutralization within a Volcanic Plume. Environ. Sci. Technol. 2015, 49, 4129–4137. [Google Scholar] [CrossRef] [PubMed]
- Dupart, Y.; King, S.M.; Nekat, B.; Nowak, A.; Wiedensohler, A.; Herrmann, H.; David, G.; Thomas, B.; Miffre, A.; Rairoux, P.; et al. Mineral dust photochemistry induces nucleation events in the presence of SO2. Proc. Natl. Acad. Sci. USA 2012, 109, 20842–20847. [Google Scholar] [CrossRef] [PubMed]
- Vallyathan, V.; Shi, X.; Dalal, N.S.; Irr, W.; Castranova, V. Generation of Free Radicals from Freshly Fractured Silica Dust: Potential Role in Acute Silica-induced Lung Injury. Am. Rev. Respir. Dis. 1988, 138, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Konecny, R. Reactivity of Hydroxyl Radicals on Hydroxylated Quartz Surface. 1. Cluster Model Calculations. J. Phys. Chem. B 2001, 105, 6221–6226. [Google Scholar] [CrossRef]
- Narayanasamy, J.; Kubicki, J.D. Mechanism of Hydroxyl Radical Generation from a Silica Surface: Molecular Orbital Calculations. J. Phys. Chem. B 2005, 109, 21796–21807. [Google Scholar] [CrossRef] [PubMed]
- Von Glasow, R.; Sander, R.; Bott, A.; Crutzen, P.J. Modeling halogen chemistry in the marine boundary layer 2. Interactions with sulfur and the cloud-covered MBL. J. Geophys. Res. Atmos. 2002, 107, 4323. [Google Scholar] [CrossRef]
- Roberts, T.J.; Jourdain, L.; Griffiths, P.T.; Pirre, M. Re-evaluating the reactive uptake of HOBr in the troposphere with implications for the marine boundary layer and volcanic plumes. Atmos. Chem. Phys. 2014, 14, 11185–11199. [Google Scholar] [CrossRef] [Green Version]
- McCormick, M.P.; Thomason, L.W.; Trepte, C.R. Atmospheric effects of the Mt Pinatubo eruption. Nature 1995, 373, 399–404. [Google Scholar] [CrossRef]
- Castellano, E.; Becagli, S.; Jouzel, J.; Migliori, A.; Severi, M.; Steffensen, J.P.; Traversi, R.; Udisti, R. Volcanic eruption frequency over the last 45 ky as recorded in Epica-Dome C ice core (East Antarctica) and its relationship with climatic changes. Glob. Planet. Chang. 2004, 42, 195–205. [Google Scholar] [CrossRef]
- Cole-Dai, J.; Mosley-Thompson, E.; Wight, S.P.; Thompson, L.G. A 4100-year record of explosive volcanism from an East Antarctica ice core. J. Geophys. Res. Atmos. 2000, 105, 24431–24441. [Google Scholar] [CrossRef]
- Delmas, R.J.; Kirchner, S.; Palais, J.M.; Petit, J.-R. 1000 years of explosive volcanism recorded at the South Pole. Tellus B 1992, 44, 335–350. [Google Scholar] [CrossRef]
- Langway, C.C.; Osada, K.; Clausen, H.B.; Hammer, C.U.; Shoji, H. A 10-century comparison of prominent bipolar volcanic events in ice cores. J. Geophys. Res. Atmos. 1995, 100, 16241–16247. [Google Scholar] [CrossRef]
- Legrand, M.; Delmas, R.J. A 220-year continuous record of volcanic H2SO4 in the Antarctic ice sheet. Nature 1987, 327, 671–676. [Google Scholar] [CrossRef]
- Hattori, S.; Schmidt, J.A.; Johnson, M.S.; Danielache, S.O.; Yamada, A.; Ueno, Y.; Yoshida, N. SO2 photoexcitation mechanism links mass-independent sulfur isotopic fractionation in cryospheric sulfate to climate impacting volcanism. Proc. Natl. Acad. Sci. USA 2013, 110, 17656–17661. [Google Scholar] [CrossRef] [PubMed]
- Danielache, S.O.; Hattori, S.; Johnson, M.S.; Ueno, Y.; Nanbu, S.; Yoshida, N. Photoabsorption cross-section measurements of32S, 33S, 34S, and 36S sulfur dioxide for the B1B1-X1A1 absorption band. J. Geophys. Res. Atmos. 2012, 117, D24301. [Google Scholar] [CrossRef]
- Danielache, S.O.; Eskebjerg, C.; Johnson, M.S.; Ueno, Y.; Yoshida, N. High-precision spectroscopy of 32S, 33S, and 34S sulfur dioxide: Ultraviolet absorption cross sections and isotope effects. J. Geophys. Res. Atmos. 2008, 113, D17314. [Google Scholar] [CrossRef]
- Whitehill, A.R.; Jiang, B.; Guo, H.; Ono, S. SO2 photolysis as a source for sulfur mass-independent isotope signatures in stratospehric aerosols. Atmos. Chem. Phys. 2015, 15, 1843–1864. [Google Scholar] [CrossRef]
- Brasseur, G.P.; Solomon, S. Aeronomy of the Middle Atmosphere, Chemistry and Physics of the Stratosphere and Mesosphere; Springer: Dordrecht, The Netherlands, 2005. [Google Scholar]
- Bao, H.; Yu, S.; Tong, D.Q. Massive volcanic SO2 oxidation and sulphate aerosol deposition in Cenozoic North America. Nature 2010, 465, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Fraser, W.; Lomax, B.; Beerling, D.; James, D.; Pyle, J.; Self, S.; Sephton, M.; Wellman, C. Episodic perturbations of end-Permian atmosphere recorded in plant spore chemistry. In Proceedings of the EGU General Assembly 2016, Vienna, Austria, 17–22 April 2016; Volume 18, p. EPSC2016-17251. [Google Scholar]
- Lomax, B.H.; Fraser, W.T. Palaeoproxies: Botanical monitors and recorders of atmospheric change. Palaeontology 2015, 58, 759–768. [Google Scholar] [CrossRef]
- Shepherd, T.G. Issues in stratosphere-troposphere coupling. J. Meteorol. Soc. Jpn. 2002, 80, 769–792. [Google Scholar] [CrossRef]
- Robock, A. Climatic impact of volcanic emissions. In State of the Planet; Sparks, R.S.J., Hawkesworth, C.J., Eds.; American Geophysical Union: Washington, DC, USA, 2004; Volume 19. [Google Scholar]
- Bekki, S. Oxidation of Volcanic SO2: A Sink for Stratospheric OH and H2O. Geophys. Res. Lett. 1995, 22, 913–916. [Google Scholar] [CrossRef]
Figure 1.
Volcanic SO2 main oxidation channels in the troposphere and stratosphere (gas phase reactions in red and aqueous reactions in blue). X can be two monovalents or one bivalent cation (e.g., H+, K+, Na+, or Ca2+). The average isotopic values of volcanic SO2, atmospheric oxidants and expected sulfates are indicated in grey values for Δ17O and purple for Δ33S and Δ36S. The detailed oxidation reactions are discussed in the text (Ch1, Ch2, Ch3 and Ch4). Note that sulfate oxygen atoms come partially from the atmospheric oxidants, at 25% from OH in Ch1, 50% from H2O2 in Ch2, 25% from O3 in for Ch3, and 25-50% from O2 in Ch4 [19]. Using these proportions makes a theoretical estimation of sulfate Δ17O [20] possible. Δ33S and Δ36S ≠ 0 are generated mainly by SO2 photolysis and photoexcitation by UV radiation [12,21,22], process that can most likely take place in the stratosphere above the O3 layer, where UV radiations are not filtered. Note that in the troposphere Δ33S and Δ36S = 0 is expected but some mass dependent processes can generate small isotopic anomalies as discussed in the text (Section 3.2) and in Figure 3.
Figure 1.
Volcanic SO2 main oxidation channels in the troposphere and stratosphere (gas phase reactions in red and aqueous reactions in blue). X can be two monovalents or one bivalent cation (e.g., H+, K+, Na+, or Ca2+). The average isotopic values of volcanic SO2, atmospheric oxidants and expected sulfates are indicated in grey values for Δ17O and purple for Δ33S and Δ36S. The detailed oxidation reactions are discussed in the text (Ch1, Ch2, Ch3 and Ch4). Note that sulfate oxygen atoms come partially from the atmospheric oxidants, at 25% from OH in Ch1, 50% from H2O2 in Ch2, 25% from O3 in for Ch3, and 25-50% from O2 in Ch4 [19]. Using these proportions makes a theoretical estimation of sulfate Δ17O [20] possible. Δ33S and Δ36S ≠ 0 are generated mainly by SO2 photolysis and photoexcitation by UV radiation [12,21,22], process that can most likely take place in the stratosphere above the O3 layer, where UV radiations are not filtered. Note that in the troposphere Δ33S and Δ36S = 0 is expected but some mass dependent processes can generate small isotopic anomalies as discussed in the text (Section 3.2) and in Figure 3.

Figure 2.
Multi O- and S-isotopic composition measured in volcanic sulfates. Red symbols represent primary sulfate samples collected at volcanic vents (aerosols or sulfate extracted from ashes). Blue symbols are sulfate samples extracted from volcanic deposits from tropospheric eruptions gathered at distances up to 100 km from the vent. Green symbols represent volcanic sulfates collected in deposits of super-eruptions at distances between 500 km and 5000 km from the volcanic systems. Pale green symbols correspond to ∆33S and ∆36S recalculated from the values represented by the dark green symbols, assuming dilution by non-volcanic sulfates [33]. Purple symbols represent sulfate samples from Antarctica ice cores. Mass dependent (non-MIF) compositions are emphasized by grey areas (taking into account the analytical uncertainties in 2σ) and bulk magmatic S-bearing gases composition by orange areas or dotted lines (see the Section 3.1 isotopic composition of S-bearing gases for further discussion). The theoretical δ18O and Δ17O composition of secondary sulfate generated in the atmosphere by different oxidation pathways are reported in grey areas. The different oxidation pathways via OH, H2O2, O3, and O2-TMI are detailed in the introduction of this paper and in Figure 1. The effect of UV photolysis and photoexcitation of SO2 on the sulfate Δ33S and Δ36S are shown by grey arrows. References of the figure: sulfate aerosols (1): [31]; (2): [30]; (3): [25] and references therein; sulfate extracted from volcanic ash (4): [30]; (5): [20]; (6): [35]; (7): [75]; (8): [76]; (9): [32]; (10): [77]; (11): [34]; (12): [33]; (13): [35]; (14): Unpublished data; (15): [78]; (16): [79,80,81]; (17): [82]. For the (16) and (17) dataset and due to significant background corrections, only samples with high volcanic fraction (>65%) are considered here (see [82] for further discussion); (18): [83]. Note that for stratospheric aerosols collected in ice cores, even if only high volcanic fraction samples are considered, the uncertainties are typically of about: Δ17O ± 0.5‰, Δ33S ± 0.1‰ and Δ36S ± 0.8‰ (in 2σ; see Figure 3).
Figure 2.
Multi O- and S-isotopic composition measured in volcanic sulfates. Red symbols represent primary sulfate samples collected at volcanic vents (aerosols or sulfate extracted from ashes). Blue symbols are sulfate samples extracted from volcanic deposits from tropospheric eruptions gathered at distances up to 100 km from the vent. Green symbols represent volcanic sulfates collected in deposits of super-eruptions at distances between 500 km and 5000 km from the volcanic systems. Pale green symbols correspond to ∆33S and ∆36S recalculated from the values represented by the dark green symbols, assuming dilution by non-volcanic sulfates [33]. Purple symbols represent sulfate samples from Antarctica ice cores. Mass dependent (non-MIF) compositions are emphasized by grey areas (taking into account the analytical uncertainties in 2σ) and bulk magmatic S-bearing gases composition by orange areas or dotted lines (see the Section 3.1 isotopic composition of S-bearing gases for further discussion). The theoretical δ18O and Δ17O composition of secondary sulfate generated in the atmosphere by different oxidation pathways are reported in grey areas. The different oxidation pathways via OH, H2O2, O3, and O2-TMI are detailed in the introduction of this paper and in Figure 1. The effect of UV photolysis and photoexcitation of SO2 on the sulfate Δ33S and Δ36S are shown by grey arrows. References of the figure: sulfate aerosols (1): [31]; (2): [30]; (3): [25] and references therein; sulfate extracted from volcanic ash (4): [30]; (5): [20]; (6): [35]; (7): [75]; (8): [76]; (9): [32]; (10): [77]; (11): [34]; (12): [33]; (13): [35]; (14): Unpublished data; (15): [78]; (16): [79,80,81]; (17): [82]. For the (16) and (17) dataset and due to significant background corrections, only samples with high volcanic fraction (>65%) are considered here (see [82] for further discussion); (18): [83]. Note that for stratospheric aerosols collected in ice cores, even if only high volcanic fraction samples are considered, the uncertainties are typically of about: Δ17O ± 0.5‰, Δ33S ± 0.1‰ and Δ36S ± 0.8‰ (in 2σ; see Figure 3).


Figure 3.
Δ33S versus Δ36S relationship for volcanic sulfates collected close the vents (in red), from volcanic deposits of tropospheric eruptions (in blue), super-eruptions (in green), and from ice cores recording stratospheric eruptions (in purple). All the dataset is the same as in the Figure 2. MDL: mass dependent fractionation line which has a slope between −10.0 and −5.0 (−6.58 in average) and Archean (yellow zone), corresponds to reference array defined by Archean rock samples, and have a slope between −1.5 and −0.9. For more details, the reader is invited to read the recent review on the subject by Ono [94]. Despite the analytical uncertainties, specifically on Δ36S from ice core sulfates samples (see the error bars in 2σ), which induce possible large uncertainties on the Δ33S/Δ36S ratios (lines) defined by volcanic samples, some general conclusion can still be addressed. Volcanic sulfates produced in volcanic vent at high temperatures and in the troposphere define a line with a slope of about −8.0 (R2 = 0.61), which is comparable to the MDL. However, volcanic sulfate from stratospheric and super-eruptions define a line with a slope of about −1.7 (R2 = 0.65), which is clearly not comparable to the MDL but very close to the Archean array.
Figure 3.
Δ33S versus Δ36S relationship for volcanic sulfates collected close the vents (in red), from volcanic deposits of tropospheric eruptions (in blue), super-eruptions (in green), and from ice cores recording stratospheric eruptions (in purple). All the dataset is the same as in the Figure 2. MDL: mass dependent fractionation line which has a slope between −10.0 and −5.0 (−6.58 in average) and Archean (yellow zone), corresponds to reference array defined by Archean rock samples, and have a slope between −1.5 and −0.9. For more details, the reader is invited to read the recent review on the subject by Ono [94]. Despite the analytical uncertainties, specifically on Δ36S from ice core sulfates samples (see the error bars in 2σ), which induce possible large uncertainties on the Δ33S/Δ36S ratios (lines) defined by volcanic samples, some general conclusion can still be addressed. Volcanic sulfates produced in volcanic vent at high temperatures and in the troposphere define a line with a slope of about −8.0 (R2 = 0.61), which is comparable to the MDL. However, volcanic sulfate from stratospheric and super-eruptions define a line with a slope of about −1.7 (R2 = 0.65), which is clearly not comparable to the MDL but very close to the Archean array.

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Martin, E. Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation. Geosciences 2018, 8, 198. https://doi.org/10.3390/geosciences8060198
AMA Style
Martin E. Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation. Geosciences. 2018; 8(6):198. https://doi.org/10.3390/geosciences8060198
Chicago/Turabian StyleMartin, Erwan. 2018. "Volcanic Plume Impact on the Atmosphere and Climate: O- and S-Isotope Insight into Sulfate Aerosol Formation" Geosciences 8, no. 6: 198. https://doi.org/10.3390/geosciences8060198
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.