Follow-Up of Peripheral IL-1β and IL-6 and Relation with Apoptotic Death in Drug-Resistant Temporal Lobe Epilepsy Patients Submitted to Surgery

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Information
2.2. Samples
2.3. Immunoenzymatic Assay for IL-1β and IL-6 in Serum and Brain Tissue
2.4. Extraction of Neocortical Tissue Proteins and Western Blot to Evaluate NF-κB
2.5. Immunohistochemistry to Annexin V and Synaptophysin
2.6. Ethical Considerations
2.7. Statistical Processing
3. Results
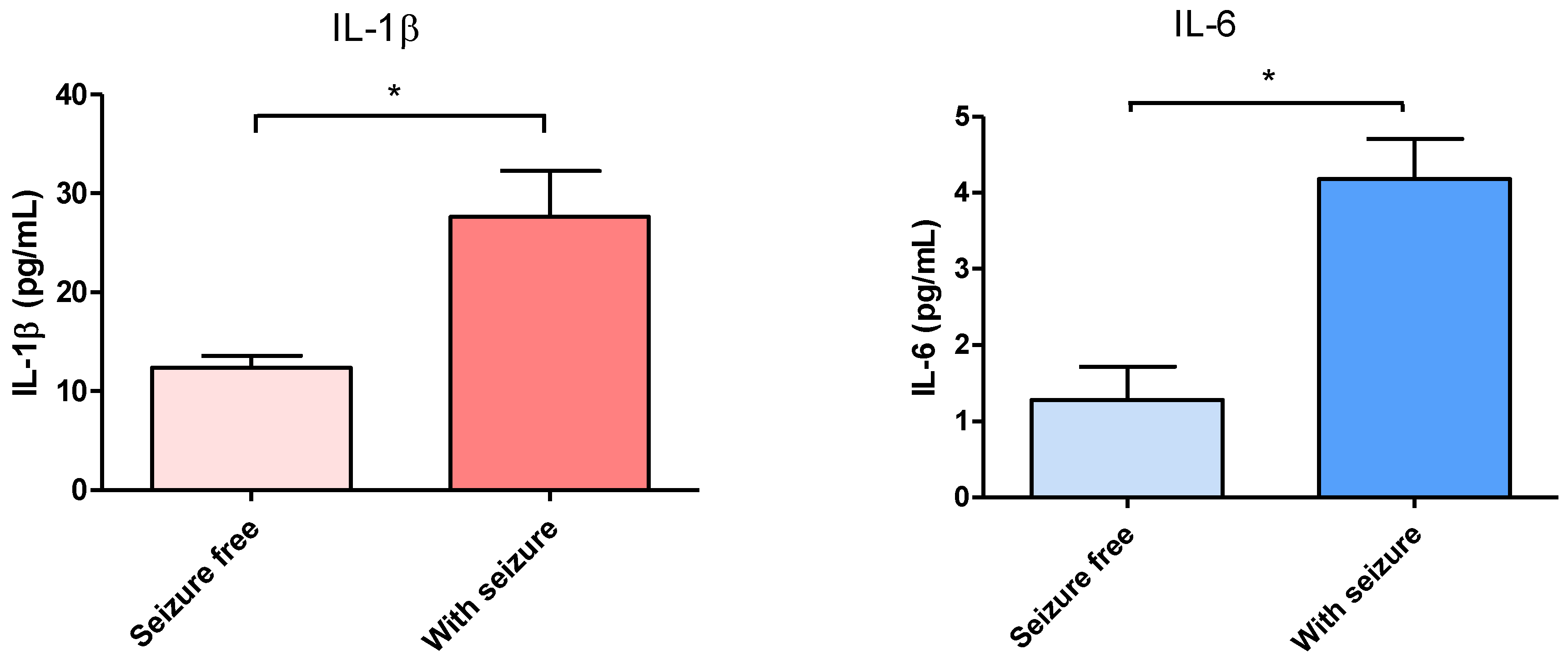
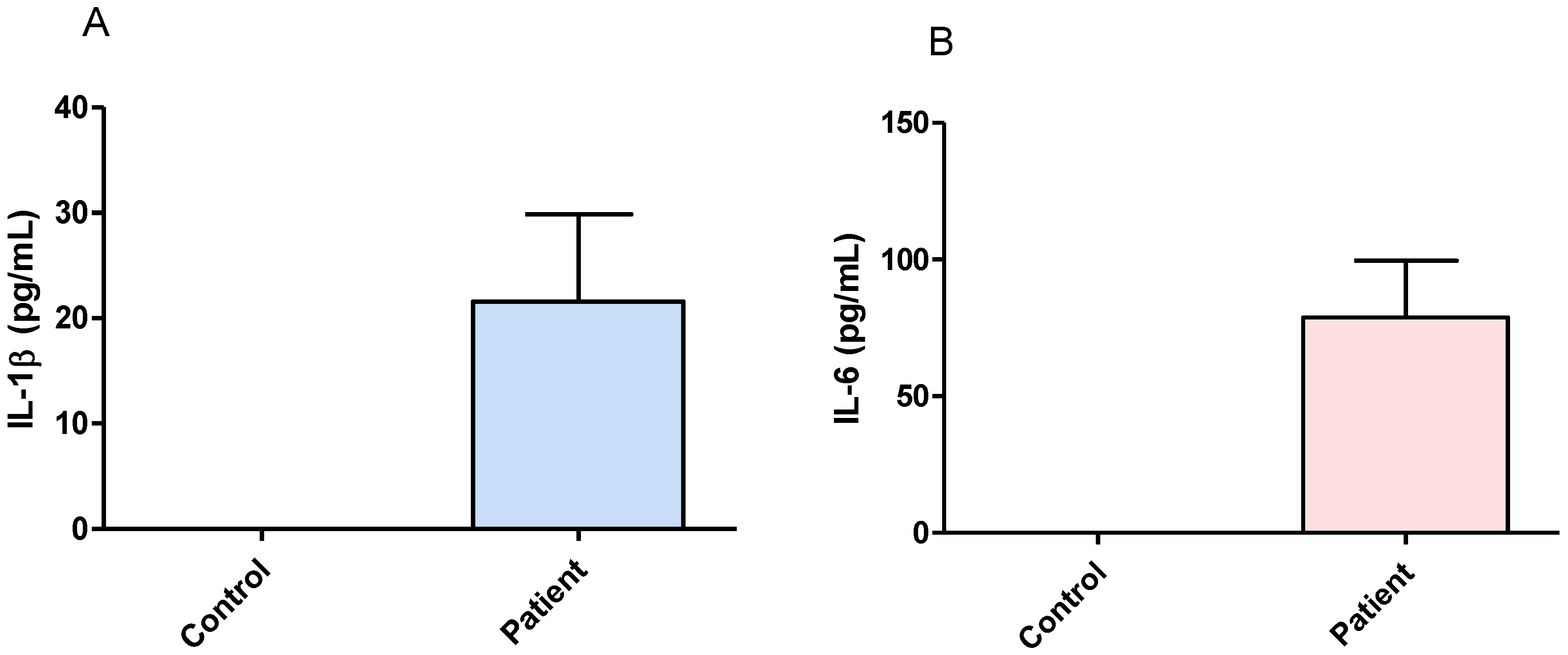
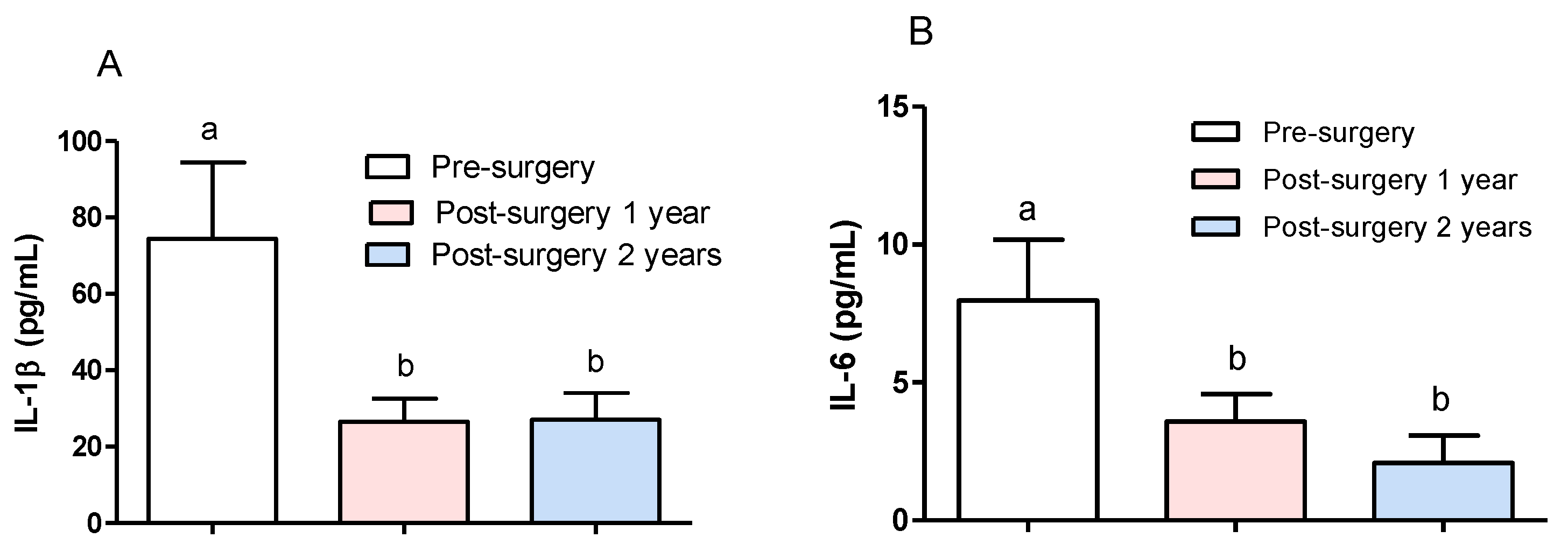
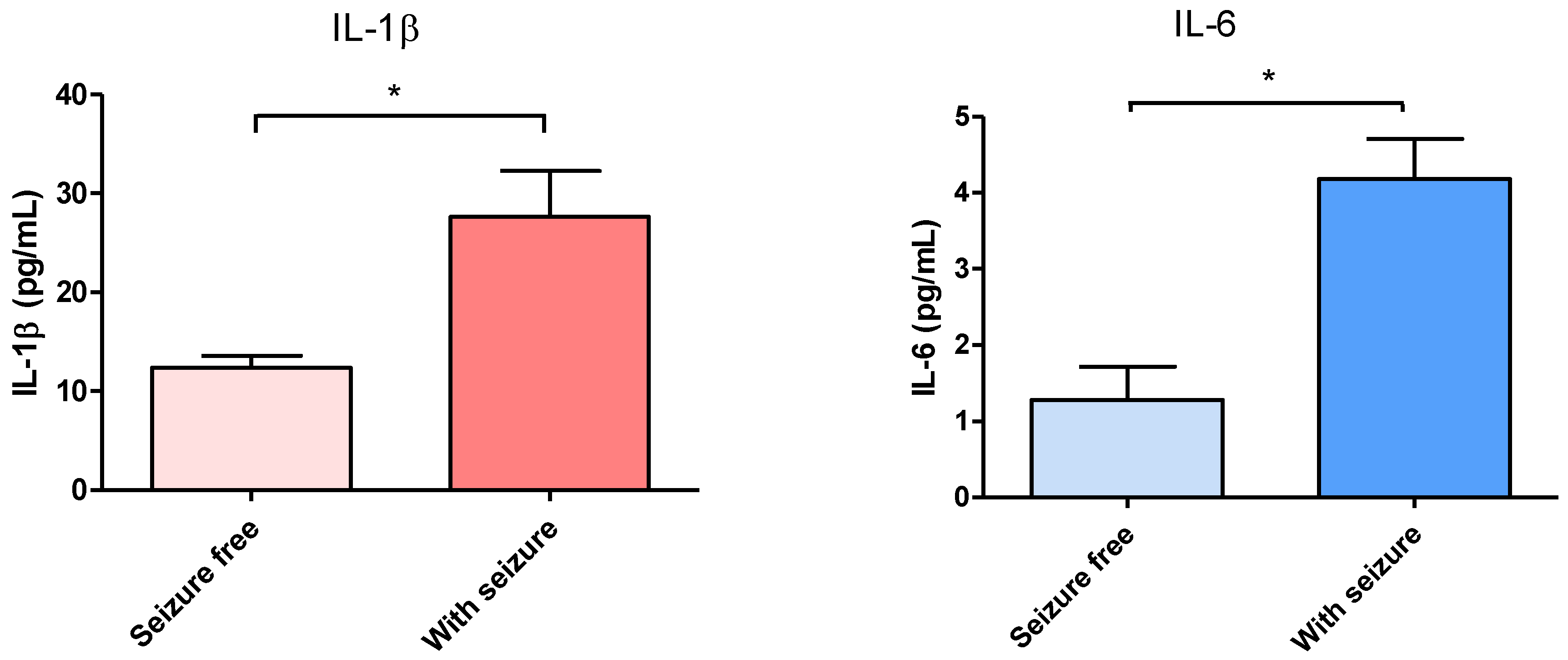
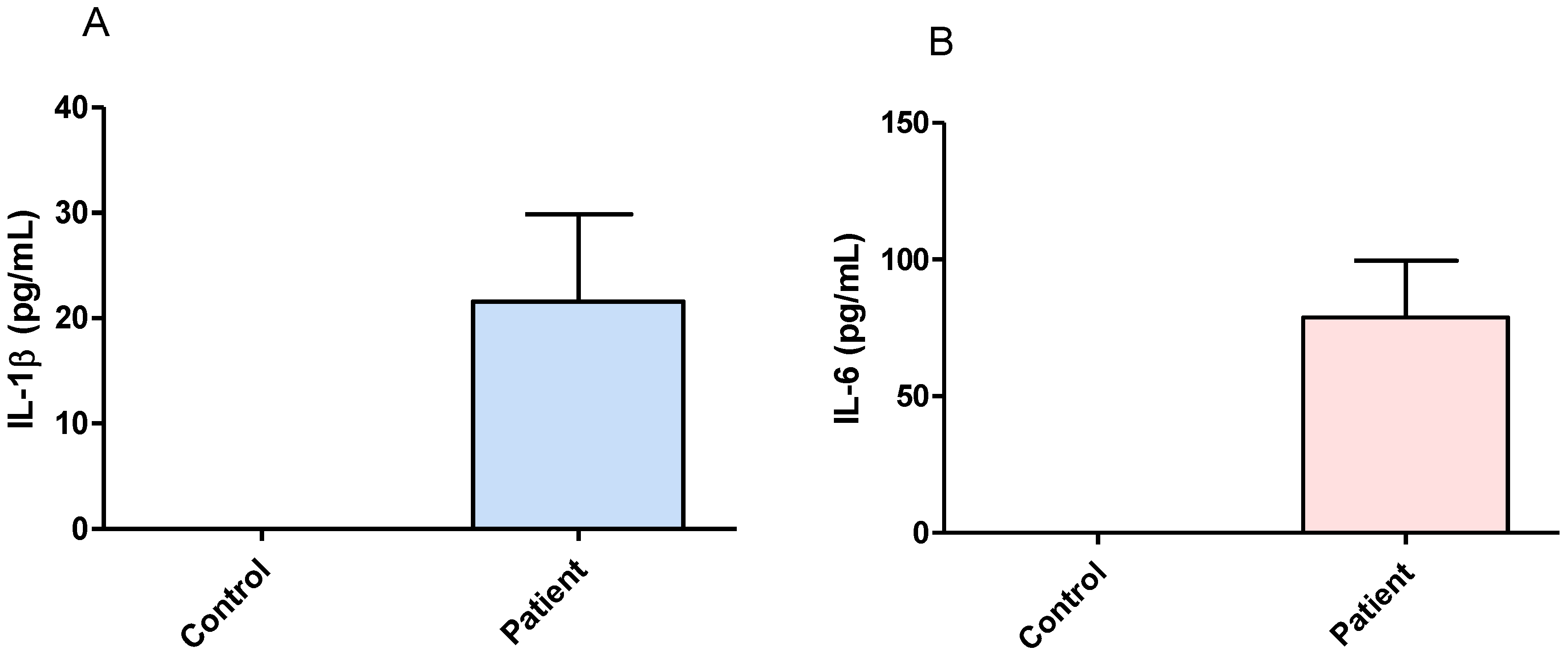
3.1. Effect of Surgical Treatment in DRTLE Patients on the Peripheral (Serum) and Central (Neocortical Tissue) Concentrationsof IL-1βand IL-6.
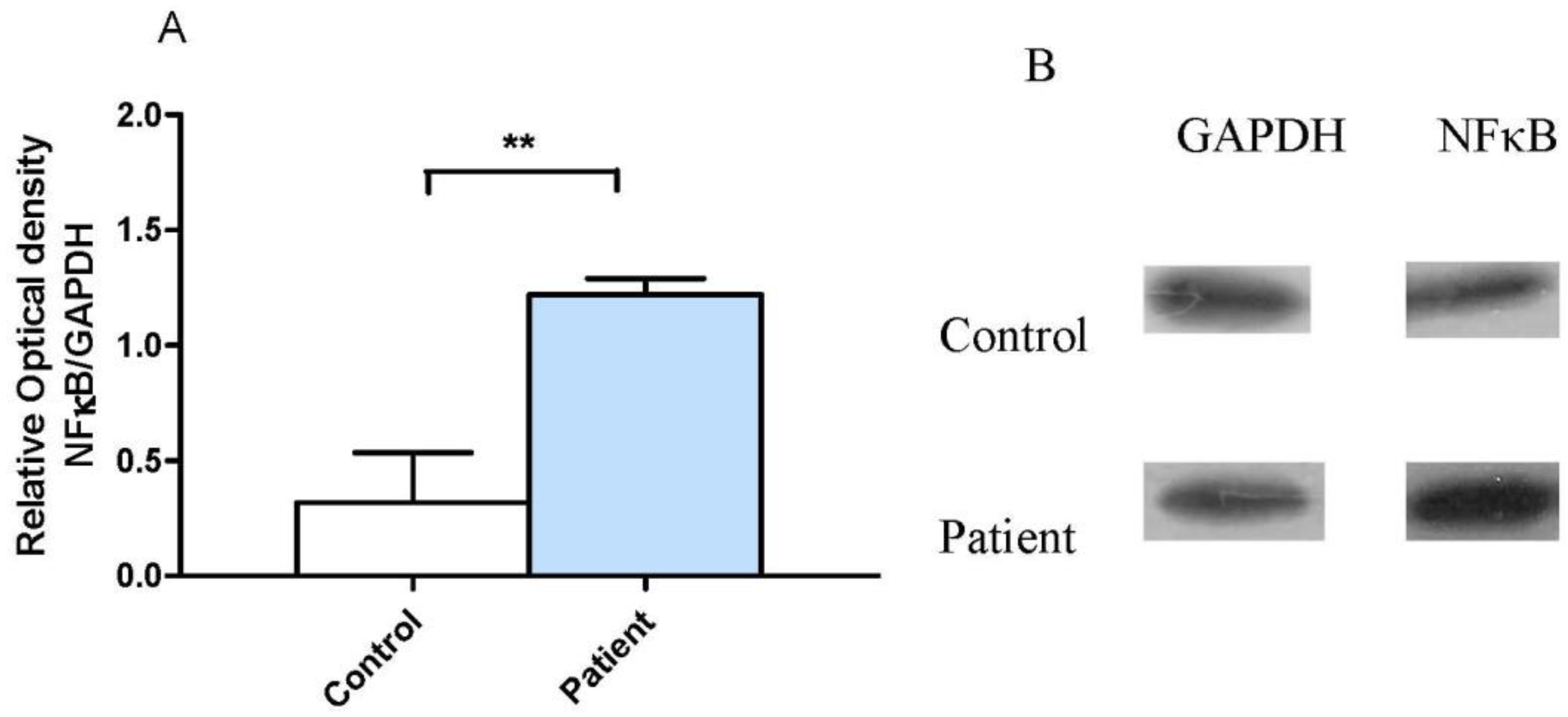
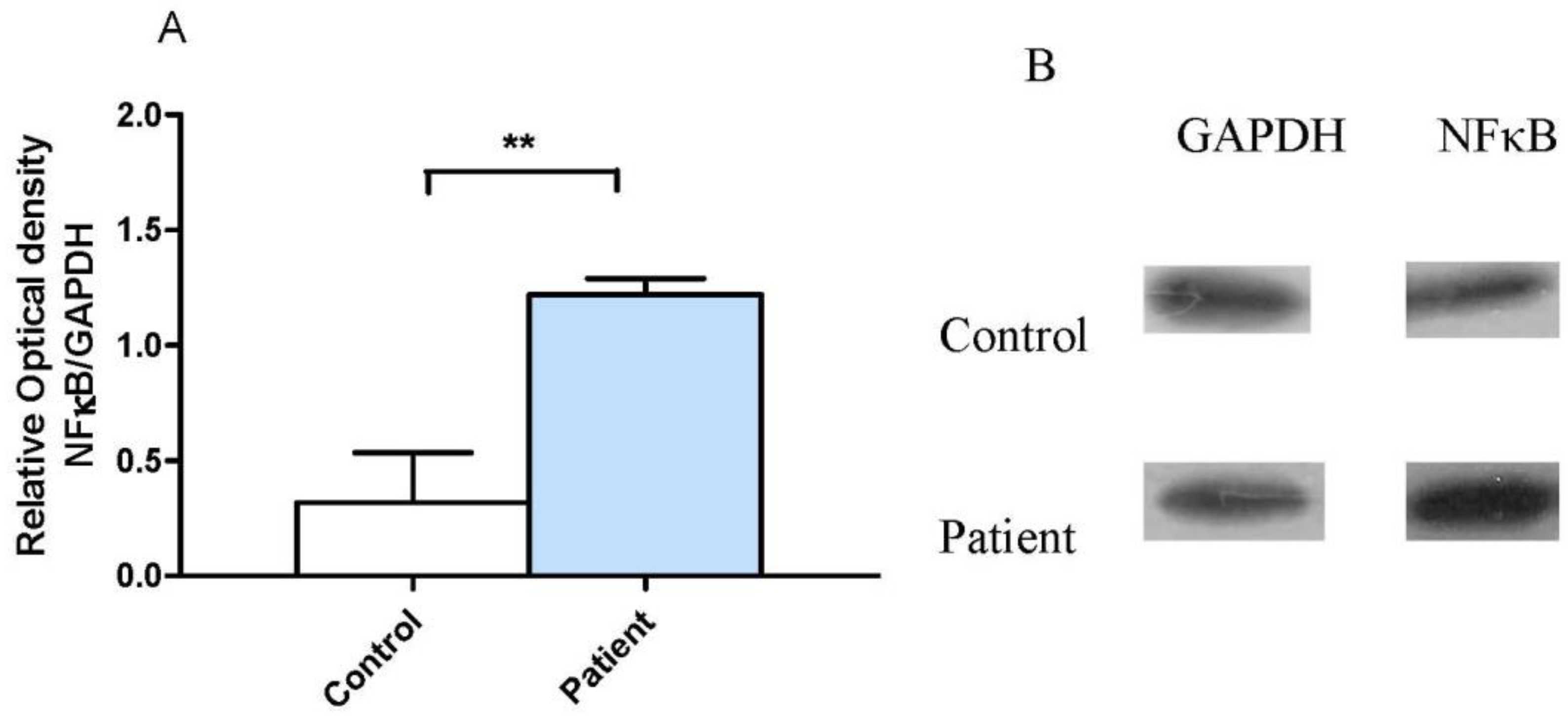
3.2. Neocortical Tissue Expression of NF-κB
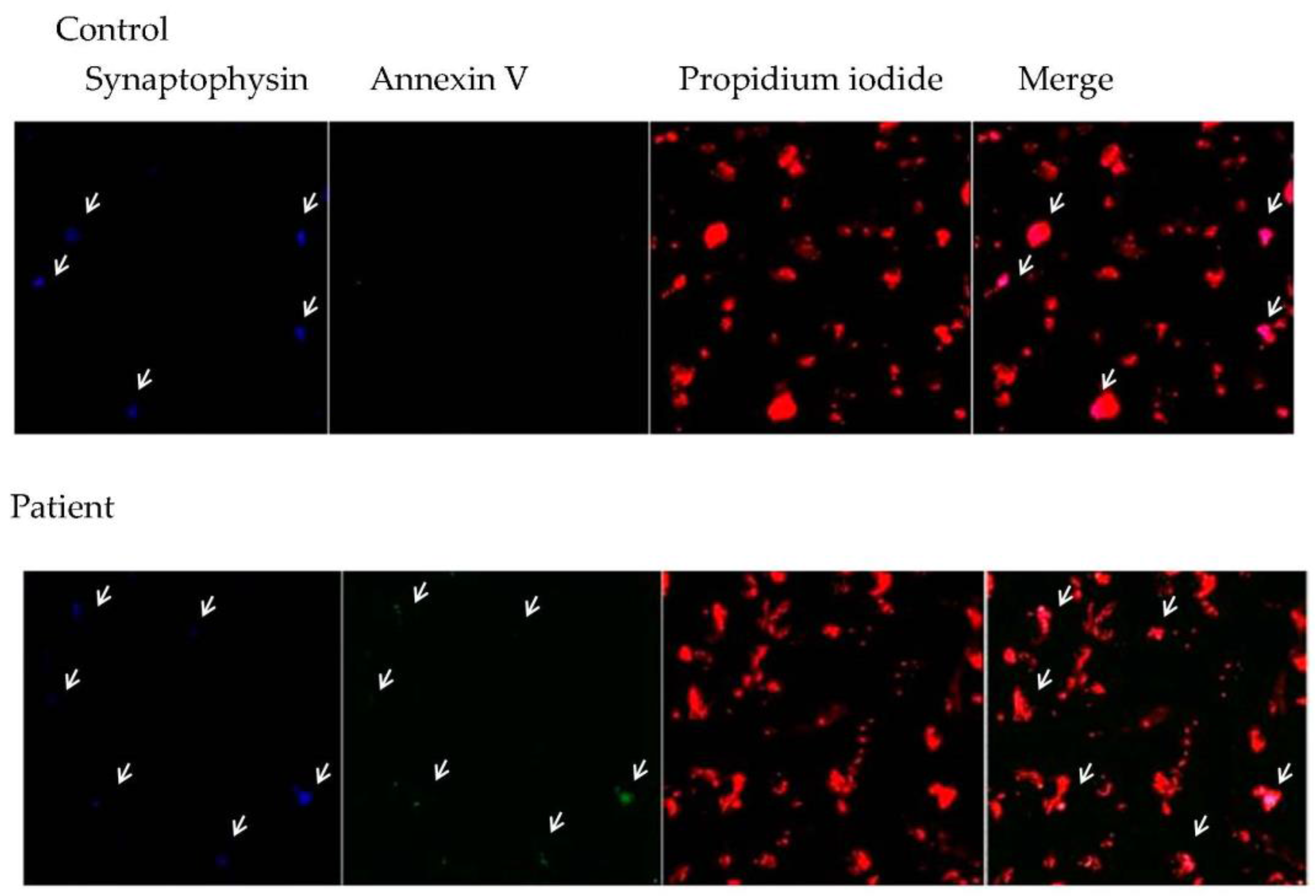
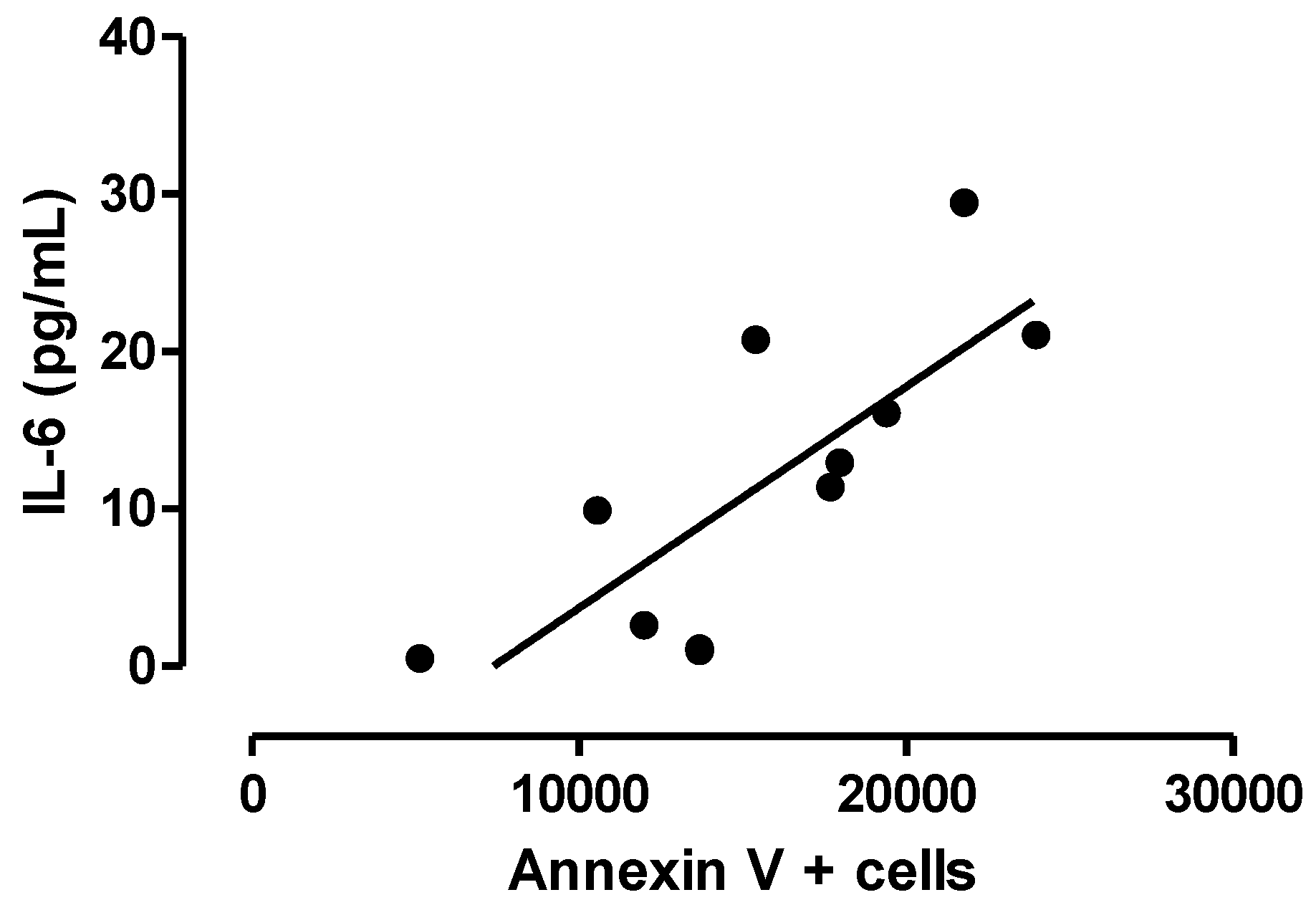
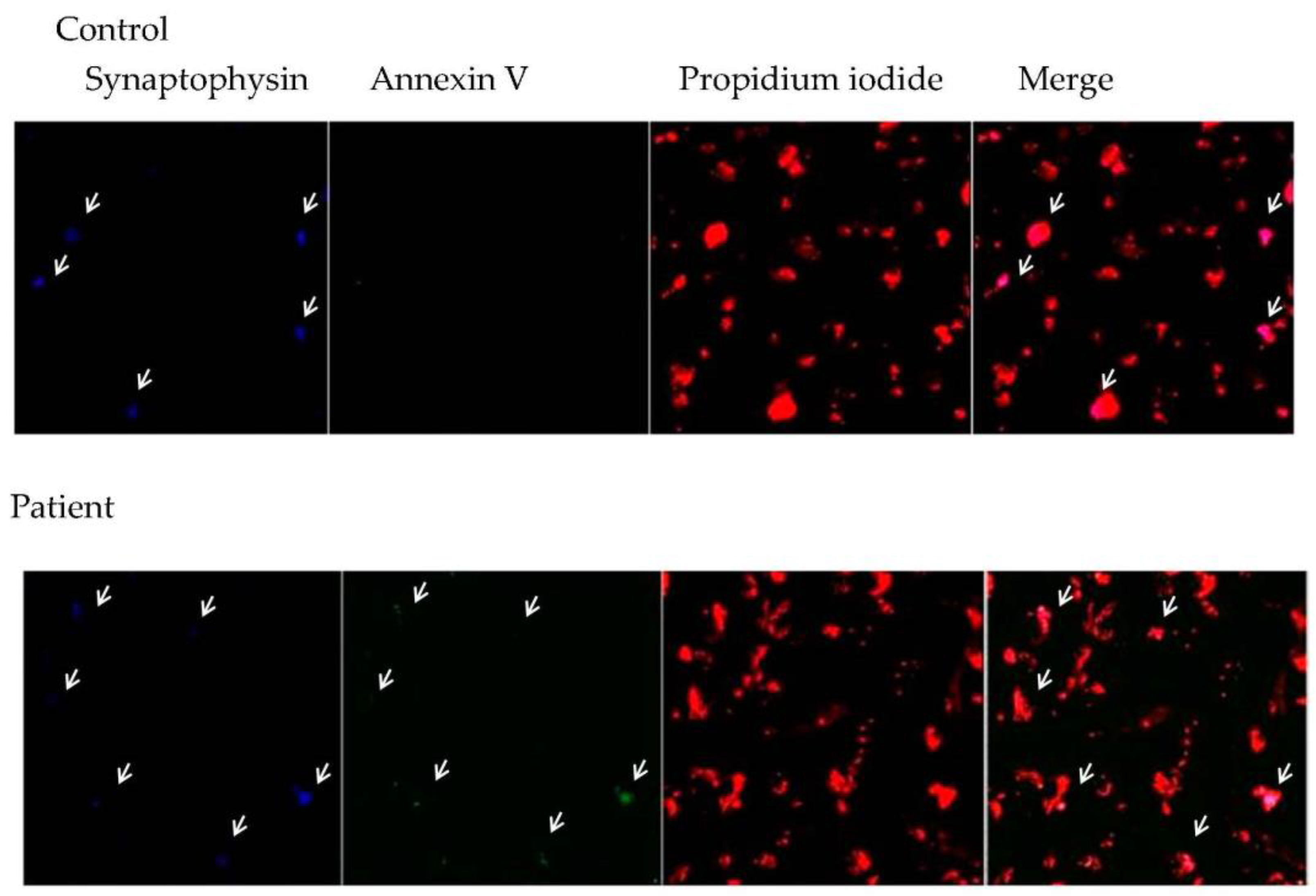
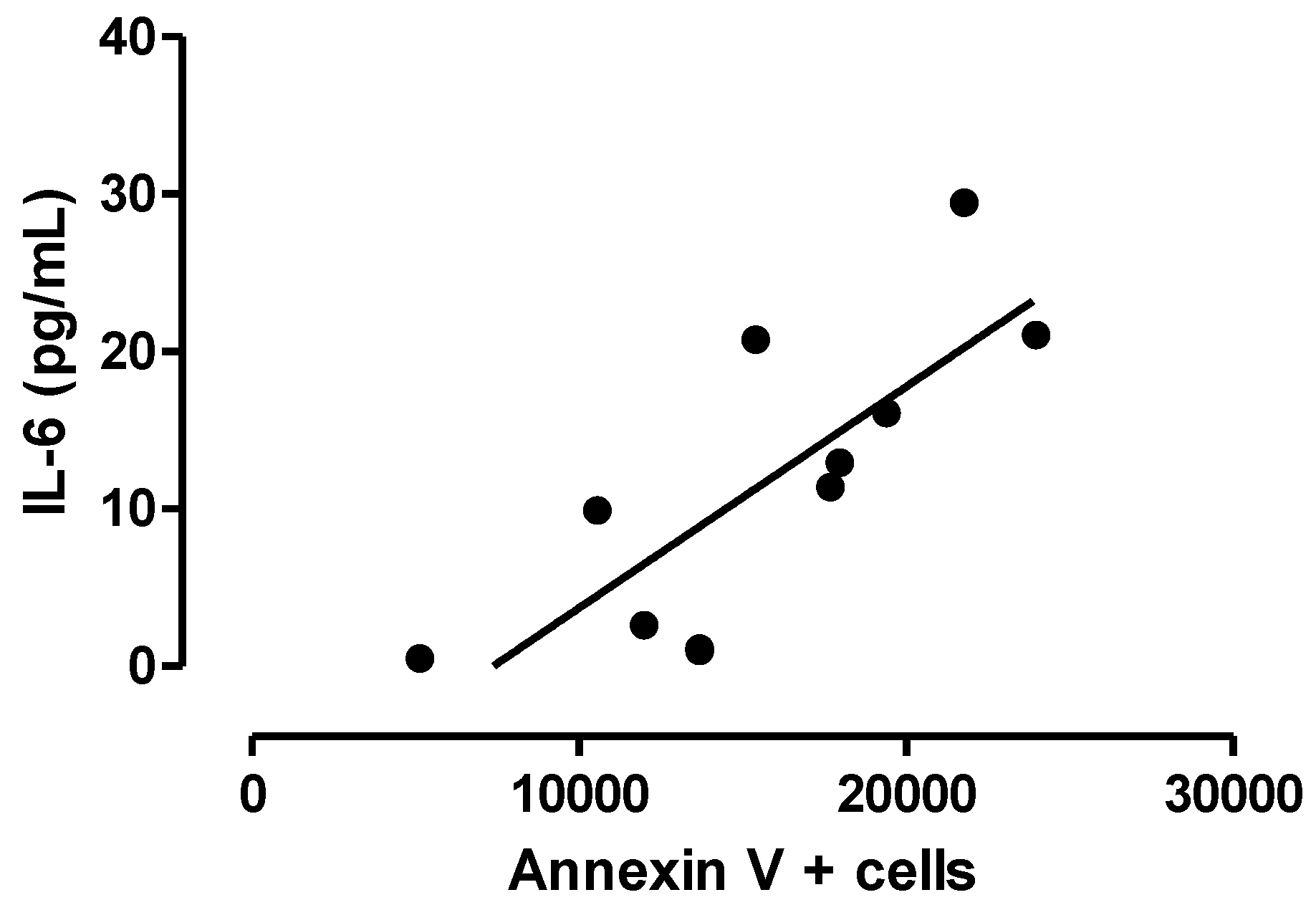
3.3. Relationship between Central Inflammation and Apoptotic Neuronal Death
4. Discussion
4.1. Inflammatory Cytokines (IL-1β and IL-6)
4.2. Expression of Molecules Associated withInflammation (NF-κB) and Apoptotic Death in Brain Tissue from DRTLE Patients
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Thurman, D.J.; Beghi, E.; Begley, C.E.; Berg, A.T.; Buchhalter, J.R.; Ding, D.; Hesdorffer, D.C.; Hauser, W.A.; Kazis, L.; Kobau, R.; et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 2011, 52 (Suppl. 7), 2–26. [Google Scholar] [CrossRef] [PubMed]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.E.; Frigerio, F.; Ravizza, T.; Ricci, E.; Tse, K.; Jenkins, R.E.; Sills, G.J.; Jorgensen, A.; Porcu, L.; Thippeswamy, T.; et al. Molecular isoforms of high-mobility group box 1 are mechanistic biomarkers for epilepsy. J. Clin. Investig. 2017, 92001. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshe, S.L.; et al. Ilae classification of the epilepsies: Position paper of the ilae commission for classification and terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Auvin, S.; Ravizza, T.; Aronica, E.; Aronica, E.; Ravizza, T.; Zurolo, E.; Vezzani, A. Glia-neuronal interactions in ictogenesis and epileptogenesis: Role of inflammatory mediators astrocyte immune responses in epilepsy. Glia 2012, 60, 1258–1268. [Google Scholar]
- Morales-Chacon, L.M.; Alfredo Sanchez, C.C.; Minou Baez, M.M.; Rodriguez, R.R.; Lorigados, P.L.; Estupinan, D.B. Multimodal imaging in nonlesional medically intractable focal epilepsy. Front Biosci. (Elite Ed.) 2015, 7, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Fois, A.; Vascotto, M. Use of intravenous immunoglobulins in drug-resistant epilepsy. Childs Nerv. Syst. 1990, 6, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Aarli, J.A. Epilepsy and the immune system. Arch. Neurol. 2000, 57, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Balosso, S.; Vezzani, A. Inflammation and prevention of epileptogenesis. Neurosci. Lett. 2011, 497, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Ozkara, C.; Vigevano, F. Immuno- and antiinflammatory therapies in epileptic disorders. Epilepsia 2011, 52 (Suppl. 3), 45–51. [Google Scholar] [CrossRef] [PubMed]
- Callenbach, P.M.; Jol-Van Der Zijde, C.M.; Geerts, A.T.; Arts, W.F.; Van Donselaar, C.A.; Peters, A.C.; Stroink, H.; Brouwer, O.F.; Van Tol, M.J. Immunoglobulins in children with epilepsy: The dutch study of epilepsy in childhood. Clin. Exp. Immunol. 2003, 132, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beghi, E.; Shorvon, S. Antiepileptic drugs and the immune system. Epilepsia 2011, 52 (Suppl. 3), 40–44. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Bartfai, T.; Bianchi, M.; Rossetti, C.; French, J. Therapeutic potential of new antiinflammatory drugs. Epilepsia 2011, 52 (Suppl. 8), 67–69. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A. Anti-inflammatory drugs in epilepsy: Does it impact epileptogenesis? Expert. Opin. Drug Saf. 2015, 14, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Lorigados-Pedre, L.; Morales-Chacon, L.; Pavon-Fuentes, N.; Serrano-Sanchez, T.; Robinson-Agramonte, M.A.; Garcia-Navarro, M.E.; der-del Busto, J.E. Immunological disorders in epileptic patients are associated to the epileptogenic focus localization. Rev. Neurol. 2004, 39, 101–104. [Google Scholar] [PubMed]
- Lorigados, P.L.; Morales Chacon, L.M.; Orozco, S.S.; Pavon, F.N.; Estupinan, D.B.; Serrano, S.T.; Garcia, M.I.; Rocha, A.L. Inflammatory mediators in epilepsy. Curr. Pharm. Des. 2013, 19, 6766–6772. [Google Scholar] [CrossRef]
- Algattas, H.; Huang, J.H. Traumatic brain injury pathophysiology and treatments: Early, intermediate, and late phases post-injury. Int. J. Mol. Sci. 2013, 15, 309–341. [Google Scholar] [CrossRef] [PubMed]
- Gales, J.M.; Jehi, L.; Nowacki, A.; Prayson, R.A. The role of histopathologic subtype in the setting of hippocampal sclerosis-associated mesial temporal lobe epilepsy. Hum. Pathol. 2017, 63, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.A.; Aronica, E.; Vezzani, A.; Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarker candidates in epilepsy: Emerging evidence from preclinical and clinical studies. Neuropathol. Appl. Neurobiol. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Liu, H.; Di, Q. Modulation of immunity and the inflammatory response: A new target for treating drug-resistant epilepsy. Curr. Neuropharmacol. 2013, 11, 114–127. [Google Scholar] [PubMed]
- Heida, J.G.; Moshe, S.L.; Pittman, Q.J. The role of interleukin-1beta in febrile seizures. Brain Dev. 2009, 31, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Kobayashi, Y.; Fujii, Y.; Kobayashi, M. Increased interleukin-6 and high-sensitivity c-reactive protein levels in pediatric epilepsy patients with frequent, refractory generalized motor seizures. Seizure 2015, 25, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Uludag, I.F.; Duksal, T.; Tiftikcioglu, B.I.; Zorlu, Y.; Ozkaya, F.; Kirkali, G. IL-1β, IL-6 and IL1Ra levels in temporal lobe epilepsy. Seizure 2015, 26, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Noe, F.; Zardoni, D.; Vaghi, V.; Sifringer, M.; Vezzani, A. Interleukin converting enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1β production. Neurobiol. Dis. 2008, 31, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.M.; Blumcke, I.; Sander, J.W.; Loscher, W. Infections, inflammation and epilepsy. Acta Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Rijkers, K.; Majoie, H.J.; Hoogland, G.; Kenis, G.; De, B.M.; Vles, J.S. The role of interleukin-1 in seizures and epilepsy: A critical review. Exp. Neurol. 2009, 216, 258–271. [Google Scholar] [CrossRef] [PubMed]
- Engel, J., Jr. Update on surgical treatment of the epilepsies. Summary of the second international palm desert conference on the surgical treatment of the epilepsies (1992). Neurology 1993, 43, 1612–1617. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Maheu, M.; Lopez, J.P.; Vaillancourt, K.; Cruceanu, C.; Gross, J.A.; Arnovitz, M.; Mechawar, N.; Turecki, G. Effects of postmortem interval on biomolecule integrity in the brain. J. Neuropathol. Exp. Neurol. 2015, 74, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.A.; Wang, C.; Hernandez, D.; Siedlak, S.L.; Rodgers, M.S.; Achar, R.K.; Fahmy, L.M.; Torres, S.L.; Petersen, R.B.; Zhu, X.; et al. Individual case analysis of postmortem interval time on brain tissue preservation. PLoS ONE 2016, 11, e0151615. [Google Scholar]
- Banuelos-Cabrera, I.; Cuellar-Herrera, M.; Velasco, A.L.; Velasco, F.; Alonso-Vanegas, M.; Carmona, F.; Guevara, R.; Arias-Montano, J.A.; Rocha, L. Pharmacoresistant temporal lobe epilepsy modifies histamine turnover and h3 receptor function in the human hippocampus and temporal neocortex. Epilepsia 2016, 57, e76–e80. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Bernardino, L.; Ferreira, R.; Cristovao, A.J.; Sales, F.; Malva, J.O. Inflammation and neurogenesis in temporal lobe epilepsy. Curr. Drug Targets. CNS Neurol. Disord. 2005, 4, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Ravizza, T.; Zurolo, E.; Vezzani, A. Astrocyte immune responses in epilepsy. Glia 2012, 60, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Ravizza, T.; Gagliardi, B.; Noe, F.; Boer, K.; Aronica, E.; Vezzani, A. Innate and adaptive immunity during epileptogenesis and spontaneous seizures: Evidence from experimental models and human temporal lobe epilepsy. Neurobiol. Dis. 2008, 29, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Viviani, B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology 2015, 96, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Fabene, P.F.; Navarro, M.G.; Martinello, M.; Rossi, B.; Merigo, F.; Ottoboni, L.; Bach, S.; Angiari, S.; Benati, D.; Chakir, A.; et al. A role for leukocyte-endothelial adhesion mechanisms in epilepsy. Nat. Med. 2008, 14, 1377–1383. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Pascente, R.; Ravizza, T. Biomarkers of epileptogenesis: The focus on glia and cognitive dysfunctions. Neurochem. Res. 2017, 42, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Di, Q.; Hu, Y.; Zhang, Y.F.; Su, L.Y.; Liu, X.H.; Li, L.C. A meta-analysis of pro-inflammatory cytokines in the plasma of epileptic patients with recent seizure. Neurosci. Lett. 2012, 514, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Crino, P.B. Inflammation in epilepsy: Clinical observations. Epilepsia 2011, 52 (Suppl. 3), 26–32. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, S. Metabolic consequences of antiepileptic drugs. Curr. Opin. Neurol. 2010, 23, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Mintzer, S.; Miller, R.; Shah, K.; Chervoneva, I.; Nei, M.; Skidmore, C.; Sperling, M.R. Long-term effect of antiepileptic drug switch on serum lipids and c-reactive protein. Epilepsy Behav. 2016, 58, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Liimatainen, S.; Fallah, M.; Kharazmi, E.; Peltola, M.; Peltola, J. Interleukin-6 levels are increased in temporal lobe epilepsy but not in extra-temporal lobe epilepsy. J. Neurol. 2009, 256, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Lorigados, L.; Morales, L.; Pav¢n, N.; Serrano, T.; Robinson, M.A.; Garc¡a, M.E.; Bender, J.E. Alteraciones inmunol¢gicas en pacientes epil‚pticos asociadas a la localizaci¢n del foco epileptog‚nico. Rev. Neurol. 2004, 39, 101–104. [Google Scholar]
- Laflamme, N.; Rivest, S. Toll-like receptor 4: The missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001, 15, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Jansson, D.; Rustenhoven, J.; Feng, S.; Hurley, D.; Oldfield, R.L.; Bergin, P.S.; Mee, E.W.; Faull, R.L.; Dragunow, M. A role for human brain pericytes in neuroinflammation. J. Neuroinflamm. 2014, 11, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Librizzi, L.; Noe, F.; Vezzani, A.; De, C.M.; Ravizza, T. Seizure-induced brain-borne inflammation sustains seizure recurrence and blood-brain barrier damage. Ann. Neurol. 2012, 72, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Louboutin, J.P.; Strayer, D.S. Relationship between the chemokine receptor CCR5 and microglia in neurological disorders: Consequences of targeting CCR5 on neuroinflammation, neuronal death and regeneration in a model of epilepsy. CNS Neurol. Disord. Drug Targets 2013, 12, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Riazi, K.; Galic, M.A.; Pittman, Q.J. Contributions of peripheral inflammation to seizure susceptibility: Cytokines and brain excitability. Epilepsy Res. 2010, 89, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Cuellar-Herrera, M.; Velasco, A.L.; Velasco, F.; Chavez, L.; Orozco-Suarez, S.; Armagan, G.; Turunc, E.; Bojnik, E.; Yalcin, A.; Benyhe, S.; et al. Mu opioid receptor mrna expression, binding, and functional coupling to g-proteins in human epileptic hippocampus. Hippocampus 2012, 22, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Nordli, D.R., Jr.; Alden, T.D.; DiPatri, A., Jr.; Laux, L.; Kelley, K.; Rosenow, J.; Schuele, S.U.; Rajaram, V.; Koh, S. Cellular injury and neuroinflammation in children with chronic intractable epilepsy. J. Neuroinflamm. 2009, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Van Gassen, K.L.; de Wit, M.; Koerkamp, M.J.; Rensen, M.G.; van Rijen, P.C.; Holstege, F.C.; Lindhout, D.; de Graan, P.N. Possible role of the innate immunity in temporal lobe epilepsy. Epilepsia 2008, 49, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Turrin, N.P.; Rivest, S. Innate immune reaction in response to seizures: Implications for the neuropathology associated with epilepsy. Neurobiol. Dis. 2004, 16, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Gahring, L.C.; Days, E.L.; Kaasch, T.; Gonzalez de, M.M.; Owen, L.; Persiyanov, K.; Rogers, S.W. Pro-inflammatory cytokines modify neuronal nicotinic acetylcholine receptor assembly. J. Neuroimmunol. 2005, 166, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, C.; Zou, L.P.; Ahlenius, S.; Winblad, B.; Schultzberg, M. Inhibition of kainic acid induced expression of interleukin-1 beta and interleukin-1 receptor antagonist mrna in the rat brain by nmda receptor antagonists. Brain Res. Mol. Brain Res. 2000, 85, 103–113. [Google Scholar] [CrossRef]
- Vezzani, A.; Conti, M.; De, L.A.; Ravizza, T.; Moneta, D.; Marchesi, F.; De Simoni, M.G. Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: Functional evidence for enhancement of electrographic seizures. J. Neurosci. 1999, 19, 5054–5065. [Google Scholar] [PubMed]
- Lorigados, P.L.; Orozco, S.S.; Morales, C.L.; Garcia, M.I.; Estupinan, D.B.; Bender del Busto, J.E.; Pavon, F.N.; Paula, P.B.; Rocha, A.L. Neuronal death in the neocortex of drug resistant temporal lobe epilepsy patients. Neurologia 2008, 23, 555–565. [Google Scholar]
- Henshall, D.C.; Engel, T. Contribution of apoptosis-associated signaling pathways to epileptogenesis: Lessons from Bcl-2 family knockouts. Front Cell Neurosci. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Crampton, S.J.; O’Keeffe, G.W. Nf-kappab: Emerging roles in hippocampal development and function. Int. J. Biochem. Cell Biol. 2013, 45, 1821–1824. [Google Scholar] [CrossRef] [PubMed]
- Teocchi, M.A.; Ferreira, A.Ç.; da Luz de Oliveira, E.P.; Tedeschi, H.; D’Souza-Li, L. Hippocampal gene expression dysregulation of klotho, nuclear factor kappa b and tumor necrosis factor in temporal lobe epilepsy patients. J. Neuroinflamm. 2013, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Crespel, A.; Coubes, P.; Rousset, M.C.; Brana, C.; Rougier, A.; Rondouin, G.; Bockaert, J.; Baldy-Moulinier, M.; Lerner-Natoli, M. Inflammatory reactions in human medial temporal lobe epilepsy with hippocampal sclerosis. Brain Res. 2002, 952, 159–169. [Google Scholar] [CrossRef]
- DeFelipe-Oroquieta, J.; Arellano, J.I.; Alonso, L.; Mu¤oz, A. Neuropatología de la epilepsia del lóbulo temporal: Alteraciones primarias y secundarias de los circuitos corticales y epileptogenicidad. Rev. Neurol. 2002, 34, 401–408. [Google Scholar] [PubMed]
- Lorigados, L.; Orozco, S.; Morales-Chacon, L.; Estupi¤an, B.; García, I.; Rocha, L. Excitotoxicity and neuronal death in epilepsy. Biotechnol. Apl. 2013, 30, 9–16. [Google Scholar]
- Liou, A.K.; Clark, R.S.; Henshall, D.C.; Yin, X.M.; Chen, J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: A review on the stress-activated signaling pathways and apoptotic pathways. Prog. Neurobiol. 2003, 69, 103–142. [Google Scholar] [CrossRef]
- Rojas, J.; Gonzalez, M.E.; Lorigados, L.; Morales, L.; Riverón, G.; Bauz, Y. Oxidative estress markers in surgically treated patients with refractory epilepsy. Clin. Biochem. 2007, 40, 292–298. [Google Scholar]
- Henshall, D.C. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Trans. 2007, 35, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D.C.; Simon, R.P. Epilepsy and apoptosis pathways. J. Cereb. Blood Flow Metab. 2005, 25, 1557–1572. [Google Scholar] [CrossRef] [PubMed]
- Narkilahti, S.; Nissinen, J.; Pitkanen, A. Administration of caspase 3 inhibitor during and after status epilepticus in rat: Effect on neuronal damage and epileptogenesis. Neuropharmacology 2003, 44, 1068–1088. [Google Scholar] [CrossRef]
- Graham, S.H.; Chen, J.; Stetler, R.A.; Zhu, R.L.; Jin, K.L.; Simon, R.P. Expression of the proto-oncogene bcl-2 is increased in the rat brain following kainate-induced seizures. Restor. Neurol. Neurosci. 1996, 9, 243–250. [Google Scholar] [PubMed]
- Roy, M.; Hom, J.J.; Sapolsky, R.M. Hsv-mediated delivery of virally derived anti-apoptotic genes protects the rat hippocampus from damage following excitotoxicity, but not metabolic disruption. Gene Ther. 2002, 9, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Naziroglu, M.; Ovey, I.S. Involvement of apoptosis and calcium accumulation through trpv1 channels in neurobiology of epilepsy. Neuroscience 2015, 293, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Teocchi, M.A.; D’Souza-Li, L. Apoptosis through death receptors in temporal lobe epilepsy-associated hippocampal sclerosis. Mediat. Inflamm. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Cause of Death | Postmortem Interval (h) |
|---|---|---|
| 1 | Pulmonary thromboembolism | 2.5 |
| 2 | Vehicle accident | 3 |
| 3 | Vehicle accident | 4 |
| 4 | Vehicle accident | 3 |
| 5 | Vehicle accident | 3.5 |
| Patient Number | Age (Year) | Gender | Personal Pathological History | Side of Focus (R, L) | Disease Duration | Pre-Surgery Number of Seizures per Month | Engel Scale | |
|---|---|---|---|---|---|---|---|---|
| One Year Post-Surgery | Two Years Post-Surgery | |||||||
| 1 | 23 | F | Bronchial asthma | L | 16 | 11 | IIIA | IIIA |
| 2 | 41 | F | No history | L | 15 | 15 | IIIA | IIIA |
| 3 | 35 | F | Febrile seizures | R | 35 | 12 | IA | IA |
| 4 | 26 | F | Cranioencephalic trauma | R | 11 | 13 | IA | IA |
| 5 | 35 | M | Meningoencephalitis | R | 34 | 12 | IA | IA |
| 6 | 31 | M | Febrile seizures | R | 30 | 10 | IA | IA |
| 7 | 41 | M | Bronchial asthma | L | 36 | 21 | IA | IA |
| 8 | 26 | F | Febrile seizures | L | 26 | 15 | IIA | IIIA |
| 9 | 36 | M | Cranioencephalic trauma | R | 32 | 12 | IA | IA |
| 10 | 37 | F | Bronchial asthma | R | 21 | 11 | IA | IA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorigados Pedre, L.; Morales Chacón, L.M.; Pavón Fuentes, N.; Robinson Agramonte, M.D.l.A.; Serrano Sánchez, T.; Cruz-Xenes, R.M.; Hung, M.-L.D.; Estupiñán Díaz, B.; Báez Martín, M.M.; Orozco-Suárez, S. Follow-Up of Peripheral IL-1β and IL-6 and Relation with Apoptotic Death in Drug-Resistant Temporal Lobe Epilepsy Patients Submitted to Surgery. Behav. Sci. 2018, 8, 21. https://doi.org/10.3390/bs8020021
Lorigados Pedre L, Morales Chacón LM, Pavón Fuentes N, Robinson Agramonte MDlA, Serrano Sánchez T, Cruz-Xenes RM, Hung M-LD, Estupiñán Díaz B, Báez Martín MM, Orozco-Suárez S. Follow-Up of Peripheral IL-1β and IL-6 and Relation with Apoptotic Death in Drug-Resistant Temporal Lobe Epilepsy Patients Submitted to Surgery. Behavioral Sciences. 2018; 8(2):21. https://doi.org/10.3390/bs8020021
Chicago/Turabian StyleLorigados Pedre, Lourdes, Lilia M. Morales Chacón, Nancy Pavón Fuentes, María De los A. Robinson Agramonte, Teresa Serrano Sánchez, Rachel M. Cruz-Xenes, Mei-Li Díaz Hung, Bárbara Estupiñán Díaz, Margarita M. Báez Martín, and Sandra Orozco-Suárez. 2018. "Follow-Up of Peripheral IL-1β and IL-6 and Relation with Apoptotic Death in Drug-Resistant Temporal Lobe Epilepsy Patients Submitted to Surgery" Behavioral Sciences 8, no. 2: 21. https://doi.org/10.3390/bs8020021