Chiral β-Amino Alcohols as Ligands for the Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation of N-Phosphinyl Ketimines

Abstract
:
1. Introduction
2. Experimental Section
2.1. General
2.2. Procedure for the Preparation of N-Phosphinylimines 1



2.3. Procedure for the Asymmetric Transfer Hydrogenation of Imines 1



3. Results and Discussion

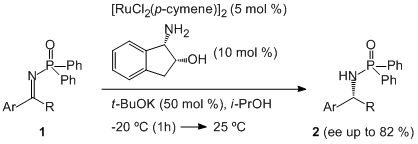{kind=link}
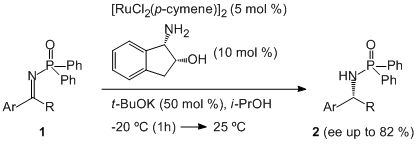{kind=link}

| Entry | Ru-dimer (mol%) | L1(mol%) | Base | T (°C) | t (h) | Product 2a | ||
|---|---|---|---|---|---|---|---|---|
| Base | mol% | Yield b (%) | ee c (%) | |||||
| 1 | 5 | 20 | KOH | 50 | 25 | 5 | 56 | 66 |
| 2 | 5 | 10 | KOH | 50 | 25 | 3 | 76 | 72 |
| 3 | 5 | 5 | KOH | 50 | 25 | 6 | 55 | 64 |
| 4 | 5 | 10 | KOH | 75 | 25 | 3 | 84 | 66 |
| 5 | 5 | 10 | KOH | 25 | 25 | 6 | 78 | 66 |
| 6 | 5 | 10 | LiOH | 50 | 25 | 5 | 63 | 70 |
| 7 | 5 | 10 | NaOH | 50 | 25 | 3 | 55 | 68 |
| 8 | 3 | 6 | KOH | 30 | 25 | 18 | 53 | 74 |
| 9 | 5 | 10 | KOH | 50 | 50 | 2 | 74 | 60 |
| 10 | 5 | 10 | KOH | 50 | 0 | 29 | 48 | 78 |
| 11 d | 5 | 10 | KOH | 50 | 0 to 25 | 19 | 70 | 78 |
| 12 d | 5 | 10 | KOH | 50 | −20 to 25 | 22 | 80 | 80 |
| 13 d | 5 | 10 | t-BuOK | 50 | −20 to 25 | 20 | 87 | 82 |
| 14 d | 5 | 10 | t-BuOK | 50 | −40 to 25 | 20 | 85 | 78 |

| Entry | Imine | Ligand | t (h) | Product 2 | ||||
|---|---|---|---|---|---|---|---|---|
| No. | R1 | R2 | No. | Yield b (%) | ee c (%) | |||
| 1 | 1a | H | Me | L1 | 20 | 2a | 87 | 82 |
| 2 | 1a | H | Me | L2 | 20 | 2a | 49 | 48 |
| 3 | 1a | H | Me | L3 | 20 | 2a | 39 | 53 |
| 4 | 1a | H | Me | L4 | 22 | 2a | — d | — |
| 5 | 1a | H | Me | L5 | 25 | 2a | — e | — |
| 6 | 1b | H | Et | L1 | 20 | 2b | 90 | 82 |
| 7 | 1c | Cl | Me | L1 | 20 | 2c | 85 | 80 |
4. Conclusions
Acknowledgements
References and Notes
- Jacques, J.; Collet, A.; Wilen, S.H. Enantiomers, Racemates and Resolution; John Wiley & Sons: New York, NY, USA, 1981. [Google Scholar]
- Juaristi, E.; Escalante, J.; León-Romo, J.L.; Reyes, A. Recent applications of α-phenylethylamine (α-PEA) in the preparation of enantiopure compounds. Part 1. Incorporation in chiral catalysts. Part 2. α-PEA and derivatives as resolving agents. Tetrahedron: Asymmetry 1998, 9, 715–740. [Google Scholar] [CrossRef]
- Fogassy, E.; Nógrádi, M.; Kozma, D.; Egri, G.; Pálovics, E.; Kiss, V. Optical resolution methods. Org. Biomol. Chem. 2006, 4, 3011–3030. [Google Scholar] [CrossRef]
- Faigl, F.; Fogassy, E.; Nógrádi, M.; Pálovics, E.; Schindler, J. Strategies in optical resolution: A practical guide. Tetrahedron: Asymmetry 2008, 19, 519–536. [Google Scholar] [CrossRef]
- Nugent, T.C. Chiral Amine Synthesis. Methods, Developments and Applications; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Whitesell, J.K. C2 Symmetry and asymmetric induction. Chem. Rev. 1989, 89, 1581–1590. [Google Scholar]
- Lucet, D.; Le Gall, T.; Mioskowski, C. The chemistry of vicinal diamines. Angew. Chem. Int. Ed. 1998, 37, 2580–2627. [Google Scholar] [CrossRef]
- Waldmann, H. C2-symmetric amines as chiral auxiliaries. In Organic Synthesis Highlights II; Waldmann, H., Ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2008. [Google Scholar]
- Braese, S.; Baumann, T.; Dahmen, S.; Vogt, H. Enantioselective catalytic syntheses of α-branched chiral amines. Chem. Commun. 2007, 1881–1890. [Google Scholar]
- Nugent, T.C.; El-Shazly, M. Chiral amine synthesis. Recent developments and trends for enamide reduction, reductive amination, and imine reduction. Adv. Synth. Catal. 2010, 352, 753–819. [Google Scholar]
- Gladiali, S.; Alberico, E. Asymmetric transfer hydrogenation: Chiral ligands and applications. Chem. Soc. Rev. 2006, 35, 226–236. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Palmer, M.J.; Wills, M. Asymmetric transfer hydrogenation of C=O and C=N bonds. Tetrahedron 1999, 10, 2045–2061. [Google Scholar] [CrossRef]
- Wills, M.; Palmer, M.; Smith, A.; Kenny, J.; Walsgrove, T. Recent developments in the area of asymmetric transfer hydrogenation. Molecules 2000, 5, 4–18. [Google Scholar]
- Clapham, S.E.; Hadzovic, A.; Morris, R.H. Mechanisms of the H2-hydrogenation and transfer hydrogenation of polar bonds catalyzed by ruthenium hydride complexes. Coord. Chem. Rev. 2004, 248, 2201–2237. [Google Scholar]
- Ikariya, T.; Blacker, A.J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef]
- Wang, C.; Wu, X.; Xiao, J. Broader, greener, and more efficient: Recent advances in asymmetric transfer hydrogenation. Chem. Asian J. 2008, 3, 1750–1770. [Google Scholar] [CrossRef]
- Wills, M. Imino reductions by transfer hydrogenation. In Modern Reduction Methods; Andersson, P.G., Munslow, I.J., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 271–296. [Google Scholar]
- Malacea, R.; Poli, R.; Manoury, E. Asymmetric hydrosilylation, transfer hydrogenation, and hydrogenation of ketones catalyzed by iridium complexe. Coord. Chem. Rev. 2010, 254, 729–752. [Google Scholar]
- Spindler, F.; Blaser, H.-U. Enantioselective hydrogenation of C=N functions and enamines. Handb. Homog. Hydrog. 2007, 3, 1193–1214. [Google Scholar]
- Fabrello, A.; Bachelier, A.; Urrutigoïty, M.; Kalck, P. Mechanistic analysis of the transition metal-catalyzed hydrogenation of imines and functionalized enamines. Coord. Chem. Rev. 2010, 254, 273–287. [Google Scholar]
- Krzyzanowska, B.; Stec, W.J. A new approach to the synthesis of primary amines, isothiocyanates, and 1-aminoalkanephosphonates via N-phosphinyl aldoximes and ketoxime. Synthesis 1978, 521–524. [Google Scholar] [CrossRef]
- Andersson, P.G.; Guijarro, D.; Tanner, D. Preparation and use of aziridino alcohols as promoters for the enantioselective addition of dialkylzinc reagents to N-(diphenylphosphinoyl) imines. J. Org. Chem. 1997, 62, 7364–7375. [Google Scholar] [CrossRef]
- For a review on the use of N-phosphinoylimines in stereoselective synthesis, see: Weinreb, S.M.; Orr, R.K. N-Phosphinoylimines: An emerging class of reactive intermediates for stereoselective organic synthesis. Synthesis 2005, 1205–1227. [Google Scholar] [CrossRef]
- Vilaivan, T.; Bhanthumnavin, W.; Sritana-Anant, Y. Recent advances in catalytic asymmetric addition to imines and related C=N systems. Curr. Org. Chem. 2005, 9, 1315–1392. [Google Scholar] [CrossRef]
- Spindler, F.; Blaser, H.-U. The highly enantioselective hydrogenation of N-diphenylphosphinylketimines with cationic Rh ferrocenyldiphosphine catalysts. Adv. Synth. Catal. 2001, 343, 68–70. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Zhou, Y.-G. Highly enantioselective Pd-catalyzed asymmetric hydrogenation of N-diphenylphosphinyl ketimines. Synlett 2006, 1189–1192. [Google Scholar]
- Wang, Y.-Q.; Lu, S.-M.; Zhou, Y.-G. Highly enantioselective Pd-catalyzed asymmetric hydrogenation of activated imines. J. Org. Chem. 2007, 72, 3729–3734. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Shimizu, H. Copper(I)-catalyzed asymmetric hydrosilylations of imines at ambient temperatures. Angew. Chem. Int. Ed. 2004, 43, 2228–2230. [Google Scholar] [CrossRef]
- Nolin, K.A.; Ahn, R.W.; Toste, F.D. Enantioselective reduction of imines catalyzed by a rhenium(V)-oxo complex. J. Am. Chem. Soc. 2005, 127, 12462–12463. [Google Scholar]
- Park, B.-M.; Mun, S.; Yun, J. Zinc-catalyzed enantioselective hydrosilylation of imines. Adv. Synth. Catal. 2006, 348, 1029–1032. [Google Scholar] [CrossRef]
- Bandini, M.; Melucci, M.; Piccinelli, F.; Sinisi, R.; Tommasi, S.; Umani-Ronchi, A. New chiral diamino-bis(tert-thiophene): An effective ligand for Pd- and Zn-catalyzed asymmetric transformations. Chem. Commun. 2007, 4519–4521. [Google Scholar]
- Nolin, K.A.; Ahn, R.W.; Kobayashi, Y.; Kennedy-Smith, J.J.; Toste, F.D. Enantioselective reduction of ketones and imines catalyzed by (CN-Box)ReV-oxo complexes. Chem. Eur. J. 2010, 16, 9555–9562. [Google Scholar]
- Graves, C.R.; Scheidt, K.A.; Nguyen, S.T. Enantioselective MSPV reduction of ketimines using 2-propanol and (BINOL)Al(III). Org. Lett. 2006, 8, 1229–1232. [Google Scholar]
- Yamada, T.; Nagata, T.; Sugi, K.D.; Yorozu, K.; Ikeno, T.; Ohtsuka, Y.; Miyazaki, D.; Mukaiyama, T. Enantioselective borohydride reduction catalyzed by optically active cobalt complexes. Chem. Eur. J. 2003, 9, 4485–4509. [Google Scholar]
- Dai-Ho, G.; Mariano, P.S. Novel photochemical-diradical cyclization methods for protoberberine alkaloid synthesis. Preparation of (±)-xylopinine and (±)-stylopine. J. Org. Chem. 1987, 52, 704–706. [Google Scholar] [CrossRef]
- Kwak, S.H.; Lee, S.A.; Lee, K.-I. Highly enantioselective Rh-catalyzed transfer hydrogenation of N-sulfonyl ketimines. Tetrahedron 2010, 21, 800–804. [Google Scholar] [CrossRef]
- Guijarro, D.; Pablo, O.; Yus, M. Ruthenium-catalysed asymmetric transfer hydrogenation of N-(tert-butanesulfinyl)imines. Tetrahedron Lett. 2009, 50, 5386–5388. [Google Scholar] [CrossRef]
- Guijarro, D.; Pablo, O.; Yus, M. Asymmetric synthesis of chiral primary amines by transfer hydrogenation of N-(tert-butanesulfinyl)ketimines. J. Org. Chem. 2010, 75, 5265–5270. [Google Scholar] [CrossRef]
- Guijarro, D.; Pablo, O.; Yus, M. Achiral β-amino alcohols as efficient ligands for the ruthenium-catalyzed asymmetric transfer hydrogenation of sulfinylimines. Tetrahedron Lett. 2011, 52, 789–791. [Google Scholar] [CrossRef]
- Pablo, O.; Guijarro, D.; Kovács, G.; Lledós, A.; Ujaque, G.; Yus, M. A versatile Ru catalyst for the asymmetric transfer hydrogenation of both aromatic and aliphatic sulfinylimines. Chem. Eur. J. 2012, in press. [Google Scholar]
- Zhou, S.; Fleischer, S.; Junge, K.; Das, S.; Addis, D.; Beller, M. Enantioselective synthesis of amines: General, efficient iron-catalyzed asymmetric transfer hydrogenation of imines. Angew. Chem. Int. Ed. 2010, 49, 8121–8125. [Google Scholar]
- Zhang, G.; Wen, X.; Wang, Y.; Mo, W.; Ding, C. Sodium nitrite catalyzed aerobic oxidative deoximation under mild conditions. J. Org. Chem. 2011, 76, 4665–4668. [Google Scholar]
- Masumoto, S.; Usuda, H.; Suzuki, M.; Kanai, M.; Shibasaki, M. Catalytic enantioselective strecker reaction of ketoimines. J. Amer. Chem. Soc. 2003, 125, 5634–5635. [Google Scholar]
- Almansa, R.; Guijarro, D.; Yus, M. Enantioselective addition of dialkylzinc reagents to N-(diphenylphosphinoyl)imines catalyzed by β-aminoalcohols with the prolinol skeleton. Tetrahedron: Asymmetry 2007, 18, 2828–2840, (retention times of the two enantiomers of compounds 2a and 2b). [Google Scholar]
- See reference 28 for the retention times of the two enantiomers of compound 2c.
- Almansa, R.; Guijarro, D.; Yus, M. N-Benzylprolinol: An efficient catalyst for the enantioselective addition of dialkylzinc reagents to N-(diphenylphosphinoyl)imines. Tetrahedron: Asymmetry 2007, 18, 896–899. [Google Scholar] [CrossRef]
- Almansa, R.; Guijarro, D.; Yus, M. Enantioselective addition of dialkylzinc reagents to N-(diphenylphosphinoyl)imines catalyzed by β-amino alcohols with the prolinol skeleton. Tetrahedron: Asymmetry 2007, 18, 2828–2840. [Google Scholar] [CrossRef]
- Almansa, R.; Guijarro, D.; Yus, M. Microwave-accelerated enantioselective addition of dialkylzinc reagents to N-(diphenylphosphinoyl)imines catalyzed by β-amino alcohols with the prolinol skeleton. Tetrahedron: Asymmetry 2008, 19, 1376–1380. [Google Scholar] [CrossRef]
- For an excellent review on the use of cis-aminoindanol in asymmetric synthesis see: Gallou, I.; Senanayake, C.H. cis-1-Amino-2-indanol in drug design and applications to asymmetric processes. Chem. Rev. 2006, 106, 2843–2874. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pablo, Ó.; Guijarro, D.; Yus, M. Chiral β-Amino Alcohols as Ligands for the Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation of N-Phosphinyl Ketimines. Appl. Sci. 2012, 2, 1-12. https://doi.org/10.3390/app2010001
Pablo Ó, Guijarro D, Yus M. Chiral β-Amino Alcohols as Ligands for the Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation of N-Phosphinyl Ketimines. Applied Sciences. 2012; 2(1):1-12. https://doi.org/10.3390/app2010001
Chicago/Turabian StylePablo, Óscar, David Guijarro, and Miguel Yus. 2012. "Chiral β-Amino Alcohols as Ligands for the Ruthenium-Catalyzed Asymmetric Transfer Hydrogenation of N-Phosphinyl Ketimines" Applied Sciences 2, no. 1: 1-12. https://doi.org/10.3390/app2010001