Self-Assembled Fluorinated Organogelators for Surface Modification

Abstract
:1. Introduction


2. Experimental Section
2.1. General
2.2. Synthesis of 2H,2H,3H,3H-Perfluorononoyl Chloride and 1H,1H,2H,2H-Perfluorooctyl Isocyanate
2.3. Synthesis of 2H,2H,3H,3H-Perfluoroheptanoyl Chloride and 1H,1H,2H,2H-Perfluorohexyl Isocyanate
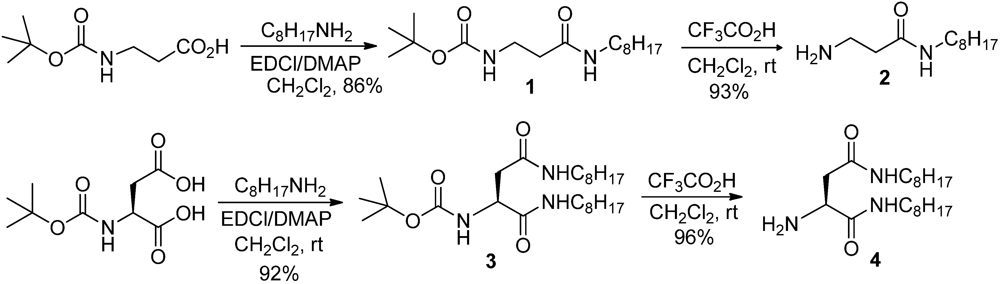
2.4. Synthesis of 1
2.5. Synthesis of 2
2.6. Synthesis of 3
2.7. Synthesis of 4
2.8. Synthesis of 5a
2.9. Synthesis of 5b
2.10. Synthesis of 5c
2.11. Synthesis of 6a
2.12. Synthesis of 6b
2.13. Synthesis of 6c
2.14. Synthesis of 7a
2.15. Synthesis of 7b
2.16. Synthesis of 7c
2.17. Synthesis of 8a
2.18. Synthesis of 8b
2.19. Synthesis of 8c
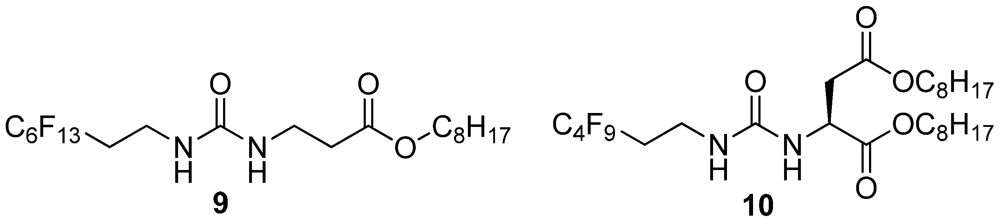
2.20. Synthesis of 9
2.21. Synthesis of 10
2.22. Gelation Experiments in Organic Solvents
2.23. Gel-Impregnation of Nonwoven Supports in Organic Solvents
3. Results and Discussion
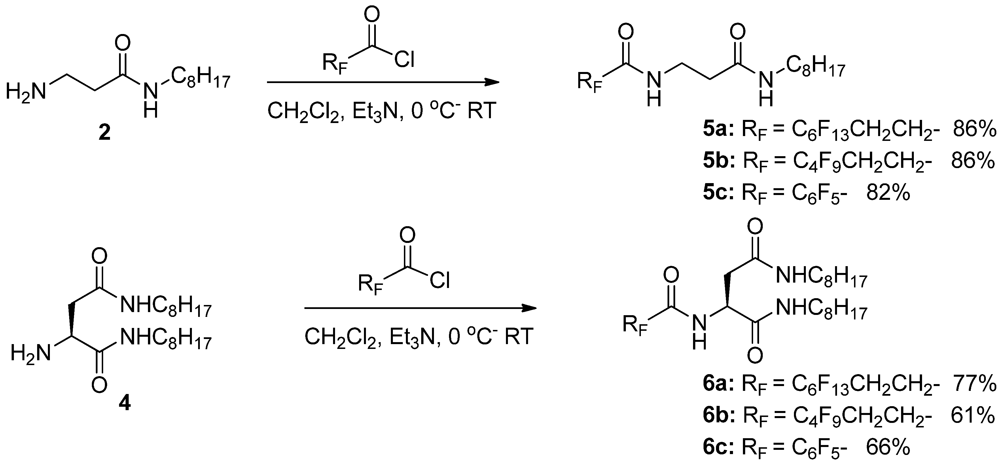
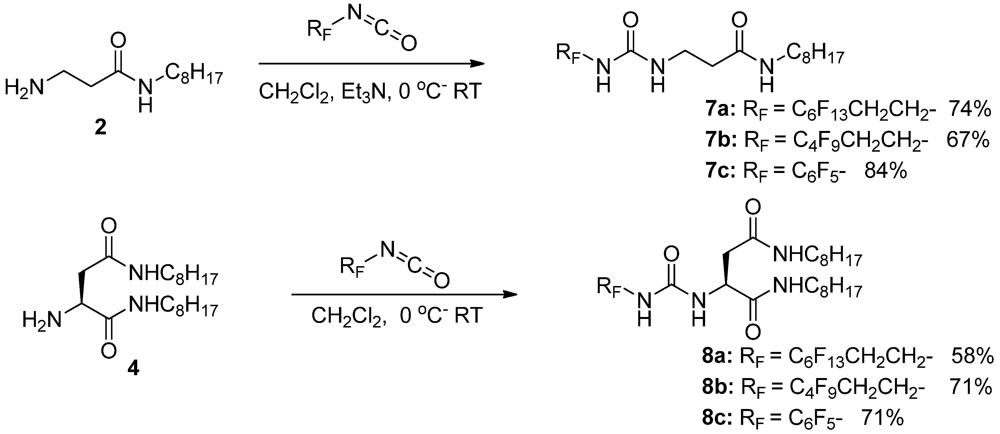
3.1. Organogelators: Design and Synthesis

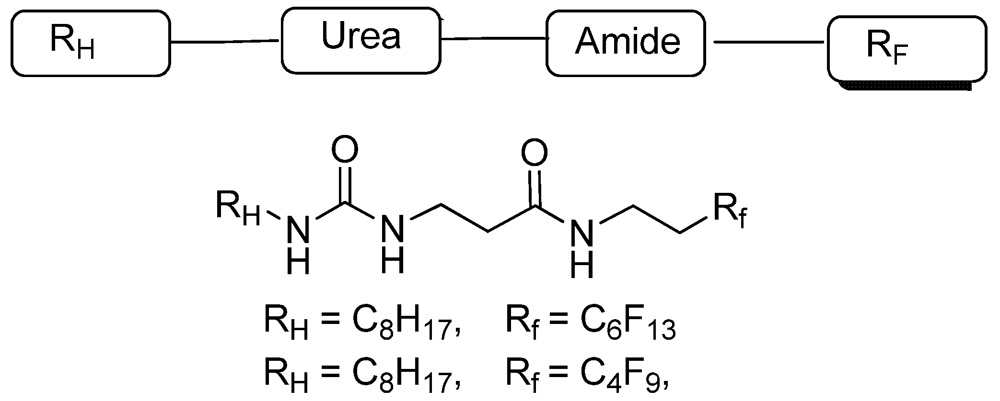
3.1.1. Partially Fluorinated-bis- and Tris-Amides

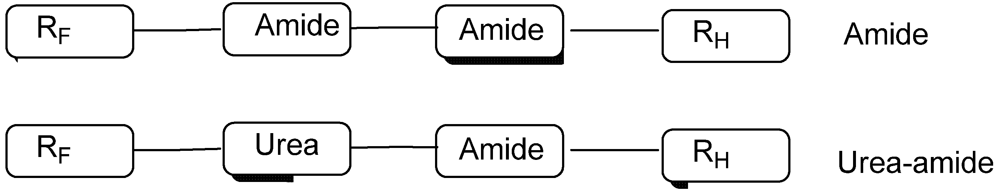
3.1.2. Partially Fluorinated Urea-Amides

3.2. Gelation in Organic Solvents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Gelation Conditions | Appearance b | |
|---|---|---|---|
| Gelator Concentration mg/mL | Solvent | ||
| 5a | 2,3 | Acetone | Partial Gel |
| 3–4 | n -Butanol | Partial Gel | |
| 5b | 3 | Acetone | Partial Gel |
| 5c | 0.5–4 | Acetone, THF, CH2Cl2, | Clear Solution or |
| n -Butanol, hexane, methanol | Precipitate | ||
| 6a | 3 a | Acetone | Opaque Gel |
| 4 a | n -Butanol | Opaque Gel | |
| 6b | 2 a | CH2Cl2 | Hazy Gel |
| 3 a | Acetone | Hazy Gel | |
| 6c | 2 a | Acetone, methanol | Opaque Gel |
| 7a | 2 a | THF or acetone | Opaque Gel |
| 3 | n -Butanol | Partial Gel | |
| 7b | 3 a | Acetone or THF | Opaque Gel |
| 7c | 2 | CH2Cl2 | Partial Gel |
| 8a | 2 a | Acetone | Opaque Gel |
| 3 a | CH2Cl2 | Hazy Gel | |
| 2–4 | THF, n -butanol | Partial Gel | |
| 8b | 2 a | CH2Cl2 | Opaque Gel |
| 3 a | Acetone | Hazy Gel | |
| 8c | 0.5 a, 1, 2, 3 | Acetone | Hazy Gel |
| 0.5 a | CH2Cl2 | Transparent Gel | |
| 2 | THF | Opaque Gel | |

3.3. Surface Modification of Nonwoven Supports in Organic Solvents

| Composite | Average Amount of Dry Gel Impregnated per Unit Area, g/cm2 | Contact angle † | |||
|---|---|---|---|---|---|
| Water | Hexadecane | ||||
| Adv CA | Rec CA | Adv CA | Rec CA | ||
| 6a on Tyvek® | 0.0014 | 141 ± 4 | 134 ± 1 | 89 ± 2 | 64 ± 1 |
| 6a on Kolon® | 0.0027 | 157 ± 6 | 144 ± 3 | 94 ± 5 | 61 ± 3 |
| 6b on Tyvek® | 0.0041 | 128 ± 4 | 112 ± 3 | 60 ± 2 | 30 ± 3 |
| 6b on Kolon® | 0.0012 | 134 ± 1 | 118 ± 2 | 59 ± 4 | 29 ± 5 |
| 7a on Tyvek® | 0.0015 | 140 ± 4 | 126 ± 1 | 84 ± 4 | 43± 1 |
| 7a on Kolon® | 0.0030 | 154 ± 3 | 138 ± 2 | 87± 4 | 42± 2 |
| 8c on Tyvek® | 0.0013 | 161 ± 1 | 148 ± 1 | Absorbed in 25 s | |
| 8c on Kolon® | 0.0037 | 158 ± 2 | 145 ± 1 | Absorbed in 30 s | |
| Untreated Tyvek® | --- | 108 ± 1 | 78 ± 1 | Absorbed on contact | |
| Untreated Kolon® | --- | 115 ± 4 | 85 ± 4 | Absorbed on contact | |
4. Conclusions
Acknowledgments
References
- Terech, P.; Weiss, R.G. Low-molecular mass gelators of organic liquids and the properties of their gels. Chem. Rev. 1997, 97, 3133–3160. [Google Scholar]
- Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Weiss, R.G.; Terech, P. (Eds.) Springer: Dordrecht, The Netherlands, 2006.
- George, M.; Weiss, R.G. Molecular organogels. Soft matter comprised of low-molecular-mass organic gelators and organic liquids. Acc. Chem. Res. 2006, 39, 489–497. [Google Scholar] [CrossRef]
- van Esch, J.H.; Feringa, B.L. New functional materials based on self-assembling organogels: From serendipity towards design. Angew. Chem. Int. Ed. 2000, 39, 2263–2266. [Google Scholar]
- Srinivasan, S.; Babu, P.A.; Mahesh, S.; Ajayaghosh, A. Reversible self-assembly of entrapped fluorescent gelators in polymerized styrene gel matrix: Erasable thermal imaging via recreation of supramolecular architectures. J. Am. Chem. Soc. 2009, 131, 15122–15123. [Google Scholar]
- Sahoo, S.; Kumar, N.; Bhattacharya, C.; Sagiri, S.S.; Jain, K.; Pal, K.; Ray, S.S.; Nayak, B. Organogels: Properties and applications in drug delivery. Des. Monomers Polym. 2011, 14, 95–108. [Google Scholar]
- Debnath, S.; Shome, A.; Dutta, S.; Das, Prasanta, K. Dipeptide-based low-molecular-weight efficient organogelators and their application in water purification. Chem.-A Eur. J. 2008, 14, 6870–6881. [Google Scholar]
- Smith, D.K. Molecular gels: Nanostructured soft materials. In Organic Nanostructures; Atwood, J.L., Steed, J.W., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 111–154. [Google Scholar]
- Hughes, N.E.; Marangoni, A.G.; Wright, A.J.; Rogers, M.A.; Rush, J.W.E. Potential food applications of edible oil organogels. Trends Food Sci. Technol. 2009, 20, 470–480. [Google Scholar]
- Fages, F.; Voegtle, F.; Zinic, M. Systematic design of amide- and urea-type gelators with tailored properties. Top. Curr. Chem. 2005, 256, 77–131. [Google Scholar]
- Allix, F.; Curcio, P.; Pham, Q.; Pickaert, G.; Jamart-Grégoire, G. Evidence of intercolumnar π-π stacking interactions in amino-acid-based low-molecular-weight organogels. Langmuir 2010, 26, 16818–16827. [Google Scholar]
- Song, B.; Wei, H.; Wang, Z.; Zhang, X.; Smet, M.; Dehae, W. Supramolecular nanofibers by self-organization of bola-amphiphiles through a combination of hydrogen bonding and π–π stacking interactions. Adv. Mater. 2007, 19, 416–420. [Google Scholar]
- Suzuki, M.; Hanabusa, K. Polymer organogelators that make supramolecular organogels through physical cross-linking and self-assembly. Chem. Soc. Rev. 2010, 39, 455–463. [Google Scholar]
- de Loos, M.; Feringa, B.L.; van Esch, J.H. Design and application of self-assembled low molecular weight hydrogels. Eur. J. Org. Chem. 2005, 3615–3631. [Google Scholar]
- Hirst, A.R.; Coates, I.A.; Boucheteau, T.R.; Miravet, J.F.; Escuder, B.; Castelletto, V.; Hamley, I.W.; Smith, D.K. Low-molecular-weight gelators: Elucidating the principles of gelation based on gelator solubility and a cooperative self-assembly model. J. Am. Chem. Soc. 2008, 130, 9113–9121. [Google Scholar]
- Liu, J.; Ma, J.; Chen, C. Structure-property relationship of a class of efficient organogelators and their multistimuli responsiveness. Tetrahedron 2011, 67, 85–91. [Google Scholar]
- Mohmeyer, N.; Schmidt, H. Synthesis and structure-property relationships of amphiphilic organogelators. Chem. Eur. J. 2007, 13, 4499–4509. [Google Scholar]
- Raynal, M.; Laurent, B. Organogel formation rationalized by Hansen solubility parameters. Chem. Commun. 2011, 47, 8271–8273. [Google Scholar]
- Srinivasan, S.; Praveen, V.K.; Philip, R.; Ajayaghosh, A. Bioinspired superhydrophobic coatings of carbon nanotubes and linear π systems based on the “bottom-up” self-assembly approach. Angew. Chem. Int. Ed. 2008, 47, 5750–5754. [Google Scholar]
- Ren, C.L.; Xu, S.Y.; Xu, J.; Chen, H.Y.; Zeng, H.Q. Planar macrocyclic fluoropentamers as supramolecular organogelators. Org. Lett. 2011, 13, 3840–3843. [Google Scholar]
- Hanabusa, K. Development of organogelators based on supramolecular chemistry. In Macromolecular Nanostructured Materials; Ueyama, N., Harada, A., Eds.; Springer Series in Materials Science: Dordrecht, The Netherlands, 2004; Volume 78, pp. 118–137. [Google Scholar]
- Shi, C.; Huang, Z.; Kilic, S.; Xu, J.; Enick, R.M.; Beckman, E.J.; Carr, A.J.; Melendez, R.E.; Hamilton, A.D. The gelation of CO2: A sustainable route to the creation of microcellular materials. Science 1999, 286, 1540–1543. [Google Scholar]
- Zhou, Y.; Yi, T.; Li, T.; Zhou, Z.; Li, F.; Huang, W.; Huyang, C. Morphology and wettability tunable two-dimensional superstructure assembled by hydrogen bonds and hydrophobic interactions. Chem. Mater. 2006, 18, 2974. [Google Scholar]
- Yamanaka, M.; Sada, K.; Miyata, M.; Hanabusa, K.; Nakano, K. Construction of superhydrophobic surfaces by fibrous aggregation of perfluoroalkyl chain-containing organogelators. Chem. Commun. 2006, 21, 2248–2250. [Google Scholar]
- Nakajima, A.; Hashimoto, K.; Watanabe, T. Some Recent studies on super-hydrophobic films. Monateshefte Fur Chemie 2001, 132, 31–41. [Google Scholar]
- Feng, L.; Li, S.; Li, Y.; Li, H.; Zhang, L.; Zhai, J.; Yanlin, S.; Liu, B.; Jiang, L.; Zhu, D. Super-hydrophobic surfaces: from natural to artificial. Adv. Mater. 2002, 14, 1857–1864. [Google Scholar]
- Chen, W.; Fadeev, A.Y.; Che Hsieh, M.; Oner, D.; Youngblood, J.; McCarthy, T. Ultrahydrophobic and ultralyophobic surfaces: Some comments and examples. Langmuir 1999, 15, 3395–3399. [Google Scholar]
- Barthlott, W.; Neinhuis, C. Purity of the sacred lotus, or escape from contamination in biological surfaces. Planta 1997, 202, 1–8. [Google Scholar]
- Nun, E.; Oles, M.; Schleich, B. Lotus-effect-surfaces. Macromol. Symp. 2002, 187, 677–682. [Google Scholar]
- Gould, P. Smart clean surfaces. Mater. Today 2003, 6, 44–48. [Google Scholar]
- Raghavanpillai, A.; Reinartz, S.; Hutchenson, K.W. Hydrophobic and oleophobic surface modification using fluorinated bis-urea and bis-amide gelators. J. Fluor. Chem. 2009, 130, 410–417. [Google Scholar]
- Schoonbeek, F.S.; Van Esch, J.H.; Hulst, R.; Kellogg, R.M.; Feringa, B.L. Geminal Bis-ureas as gelators for organic solvents: Gelation properties and structural studies in solution and in the gel state. Chem. Eur. J. 2000, 6, 2633–2643. [Google Scholar]
- Raghavanpillai, A.; Franco, V.A.; Meredith, W.E. Hydrophobic and oleophobic surface modification using gelling agents derived from amino acids. J. Fluor. Chem. 2012, 135, 187–194. [Google Scholar]
- Jouani, A.M.; Szonyi, F.; Cambon, A. Synthesis of 2-F-alkylethyl isocyanates. J. Fluor. Chem. 1992, 56, 85–92. [Google Scholar]
- Honda, K.; Morita, M.; Otsuka, H.; Takahara, A. Molecular aggregation structure and surface properties of poly(fluoroalkyl acrylate) thin films. Macromolecules 2005, 38, 5699–5705. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raghavanpillai, A.; Franco, V.A. Self-Assembled Fluorinated Organogelators for Surface Modification. Appl. Sci. 2012, 2, 175-191. https://doi.org/10.3390/app2010175
Raghavanpillai A, Franco VA. Self-Assembled Fluorinated Organogelators for Surface Modification. Applied Sciences. 2012; 2(1):175-191. https://doi.org/10.3390/app2010175
Chicago/Turabian StyleRaghavanpillai, Anilkumar, and Vincent A. Franco. 2012. "Self-Assembled Fluorinated Organogelators for Surface Modification" Applied Sciences 2, no. 1: 175-191. https://doi.org/10.3390/app2010175