
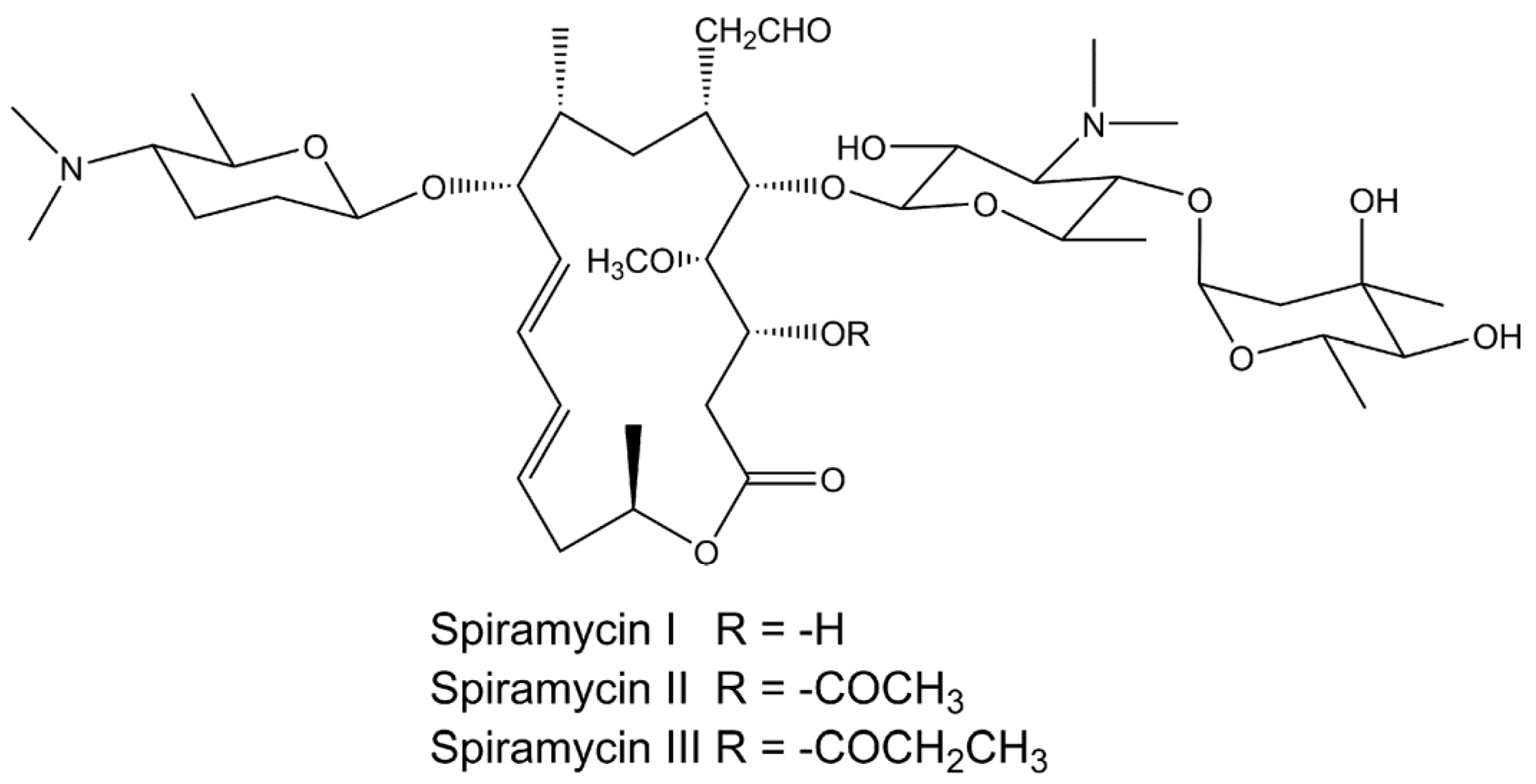
Physicochemical Properties and In Vitro Dissolution of Spiramycin Microparticles Using the Homogenate-Antisolvent Precipitation Process


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
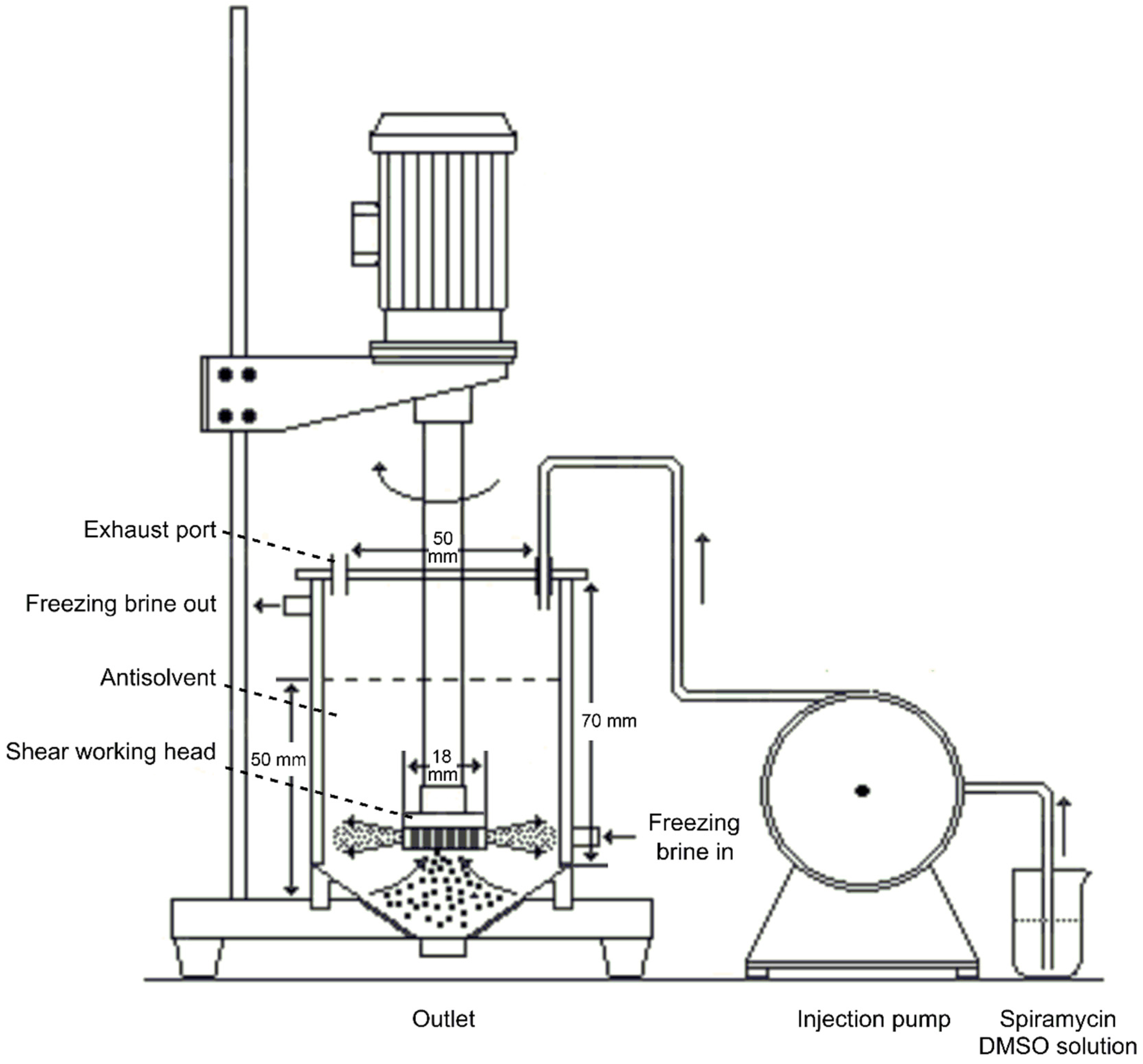
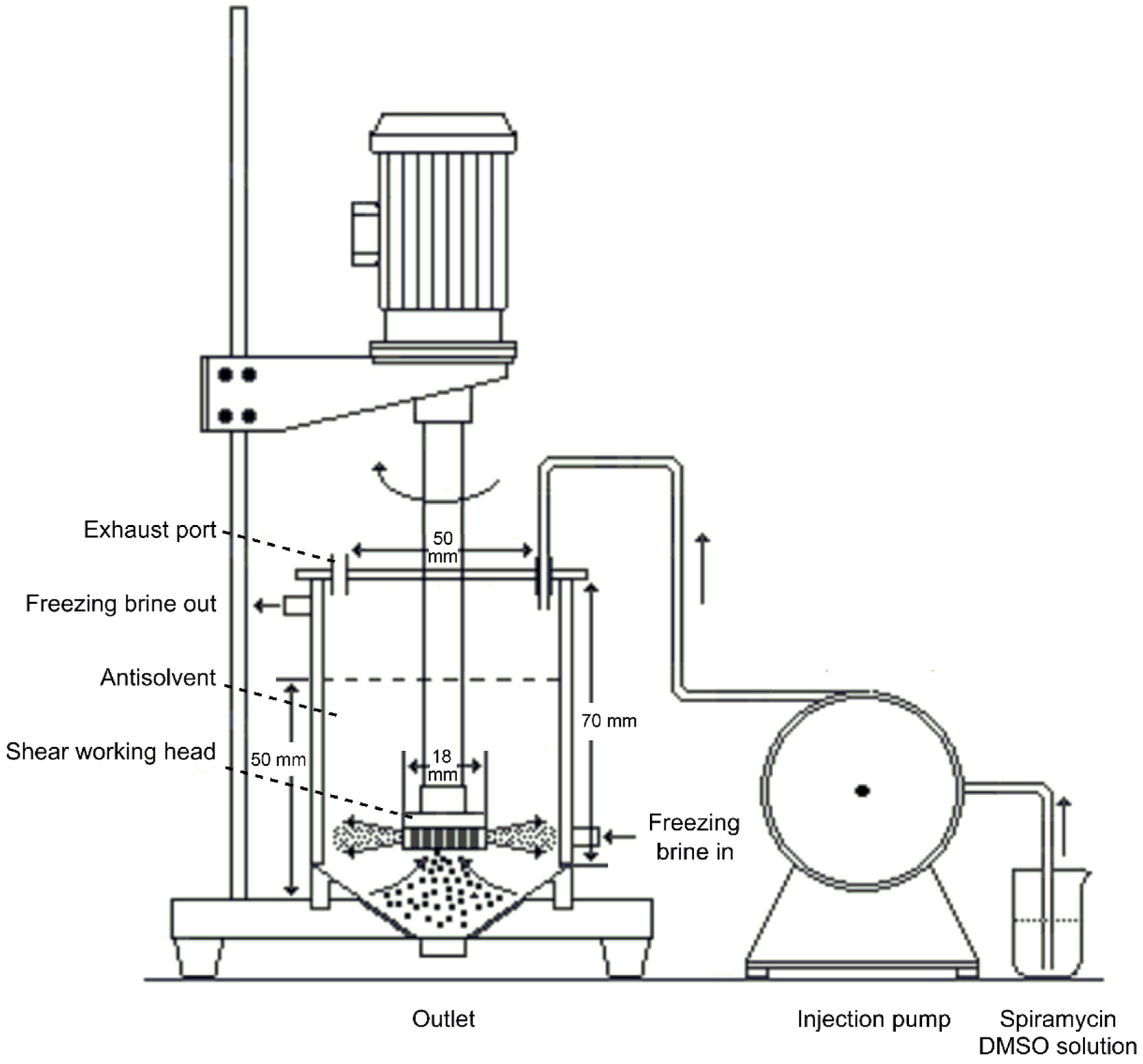
2.2. Homogenate-Antisolvent Precipitation Procedure
2.3. Determination of Spiramycin Content by High-Performance Liquid Chromatography (HPLC)
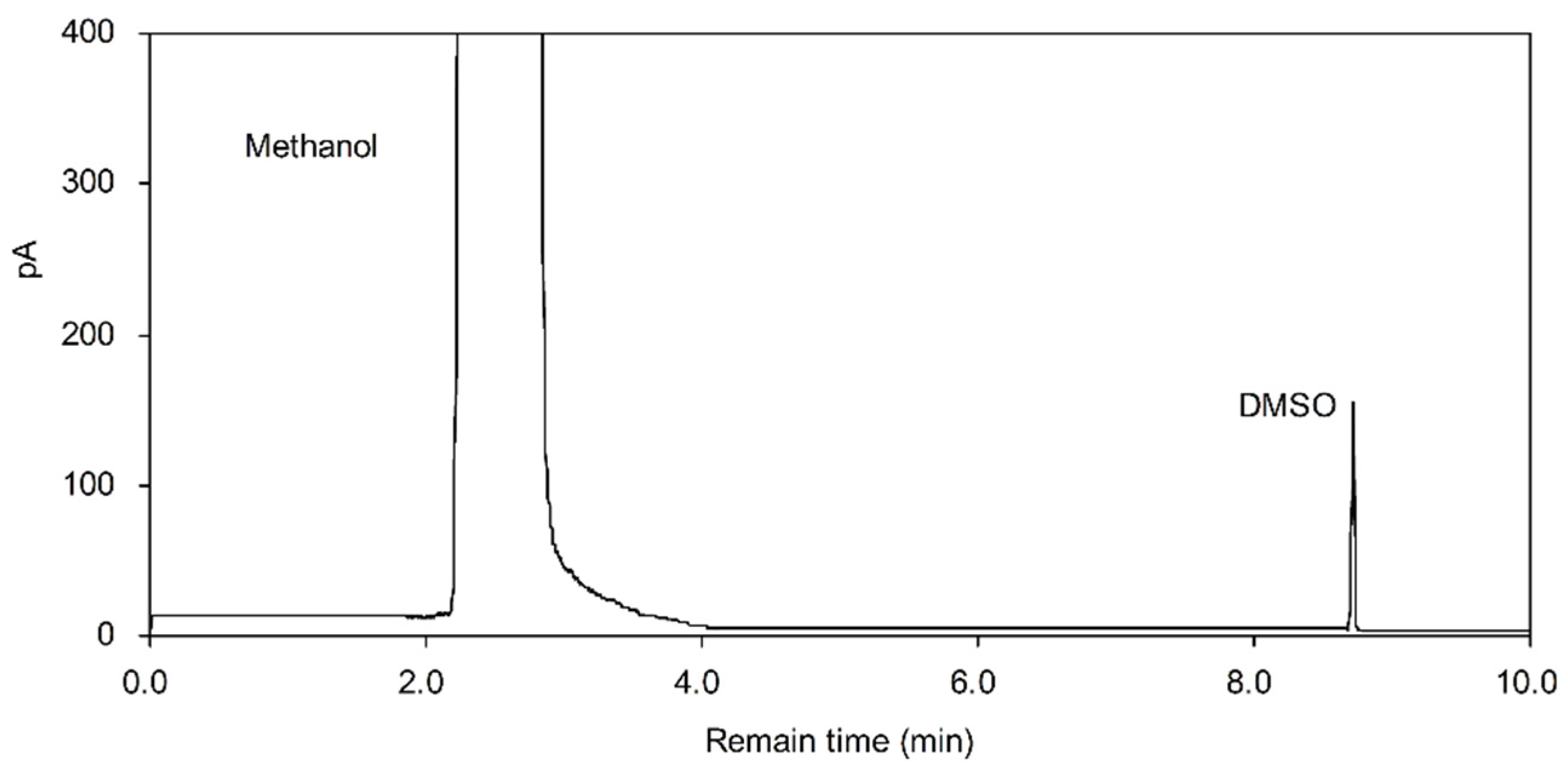
2.4. Residual Solvent Determination
2.5. Physicochemical Evaluation of Spiramycin
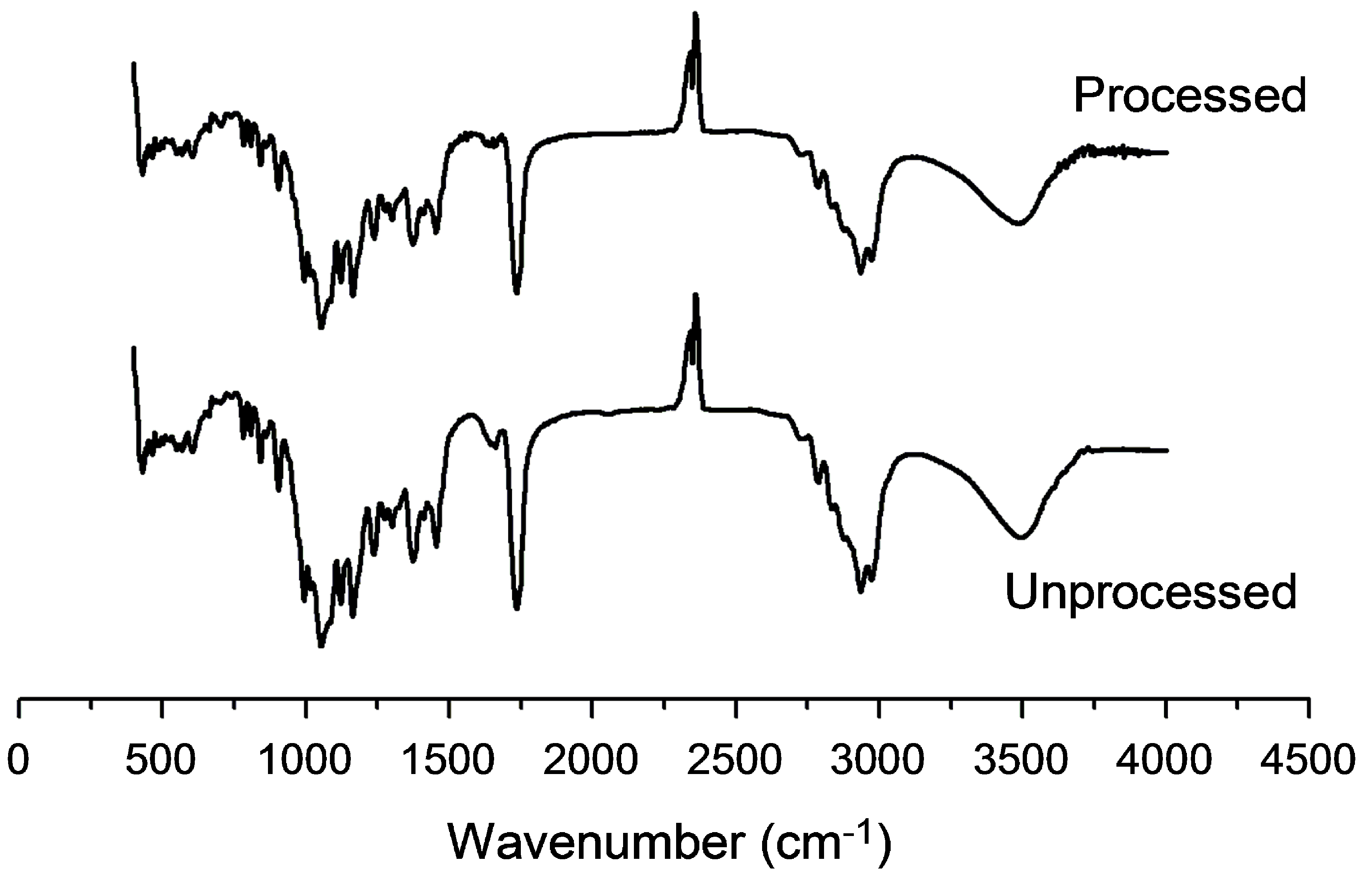
2.5.1. Fourier Transform Infrared Spectroscopy
2.5.2. Powder X-ray Diffraction
2.5.3. Scanning Electron Microscope
2.5.4. Particle Size Analysis
2.6. Dissolution Test
3. Results and Discussion
3.1. Optimization of Single HAP Conditions
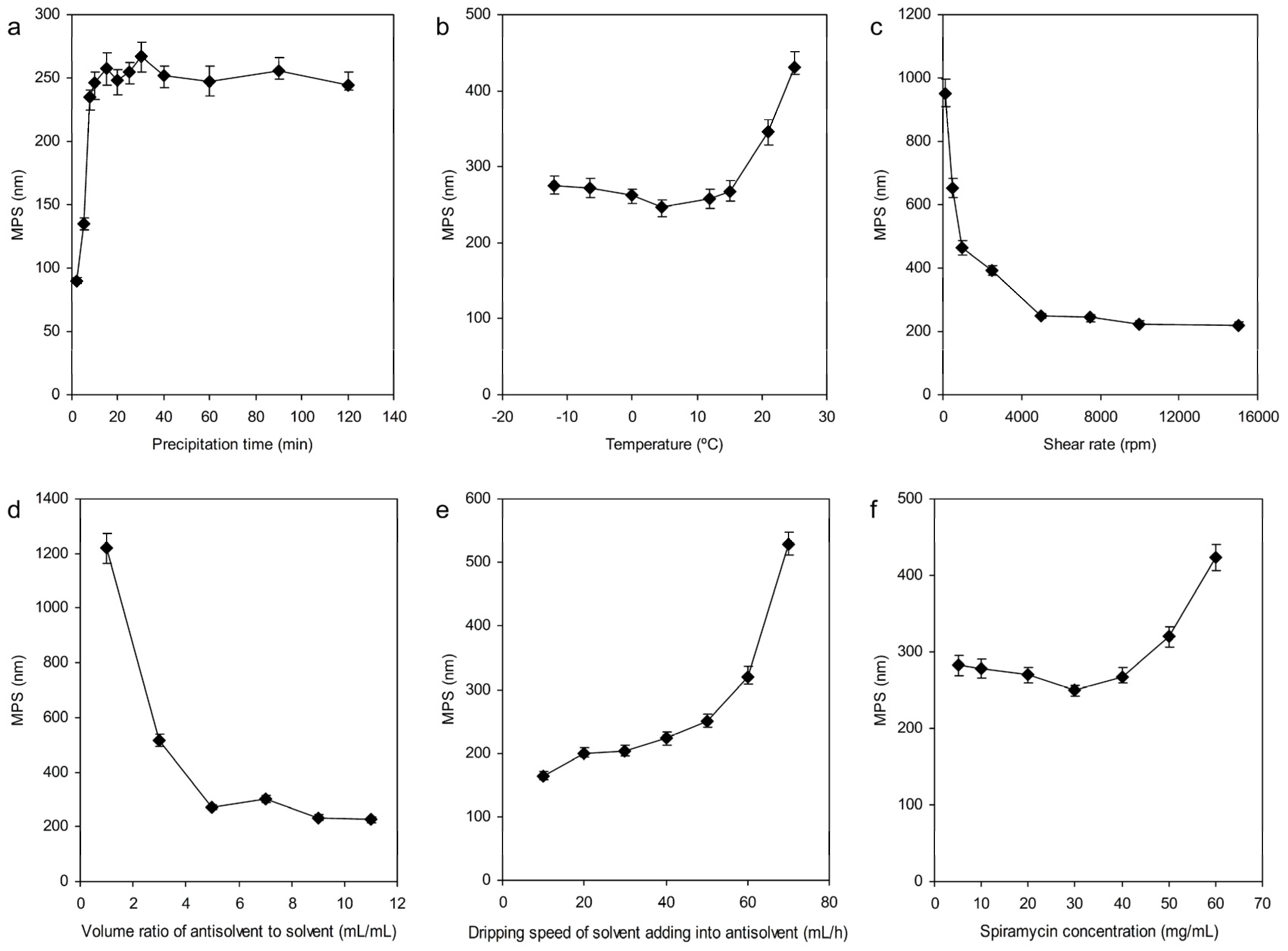
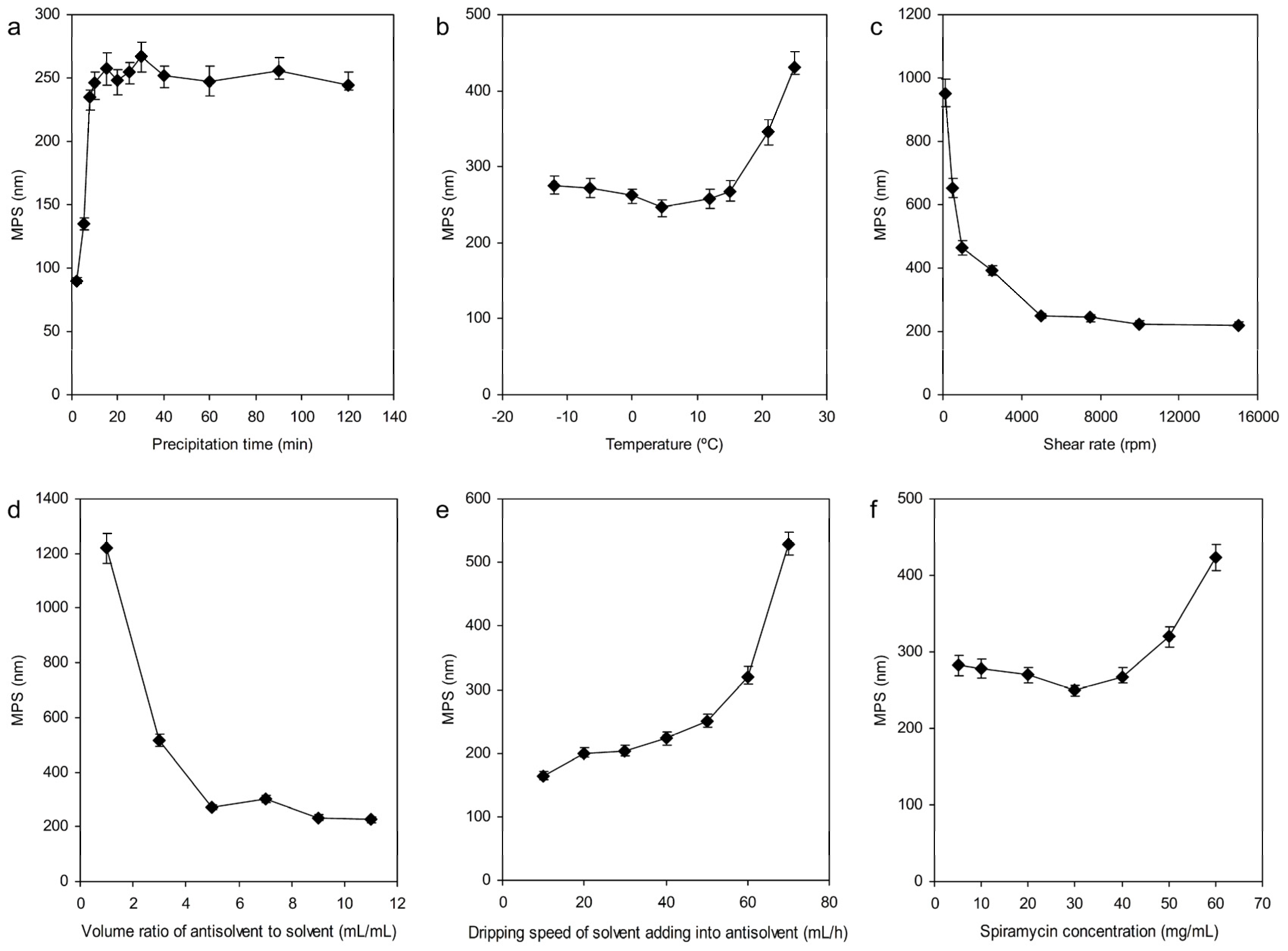
3.1.1. Effect of Precipitation Time
3.1.2. Precipitation Temperature
3.1.3. Shear Rate
3.1.4. The Volume Ratio of Antisolvent to Solvent
3.1.5. The Dripping Speed of Solvent Adding into Antisolvent
3.1.6. Concentration of Spiramycin
3.2. Optimization Parameters by Response Surface Methodology
3.3. Physicochemical Evaluation of Spiramycin
3.3.1. Fourier Transform Infrared Spectroscopy Analysis
3.3.2. Powder X-ray Diffraction Analysis
3.3.3. Scanning Electron Microscope (SEM)
3.4. Solvent Residues Analysis
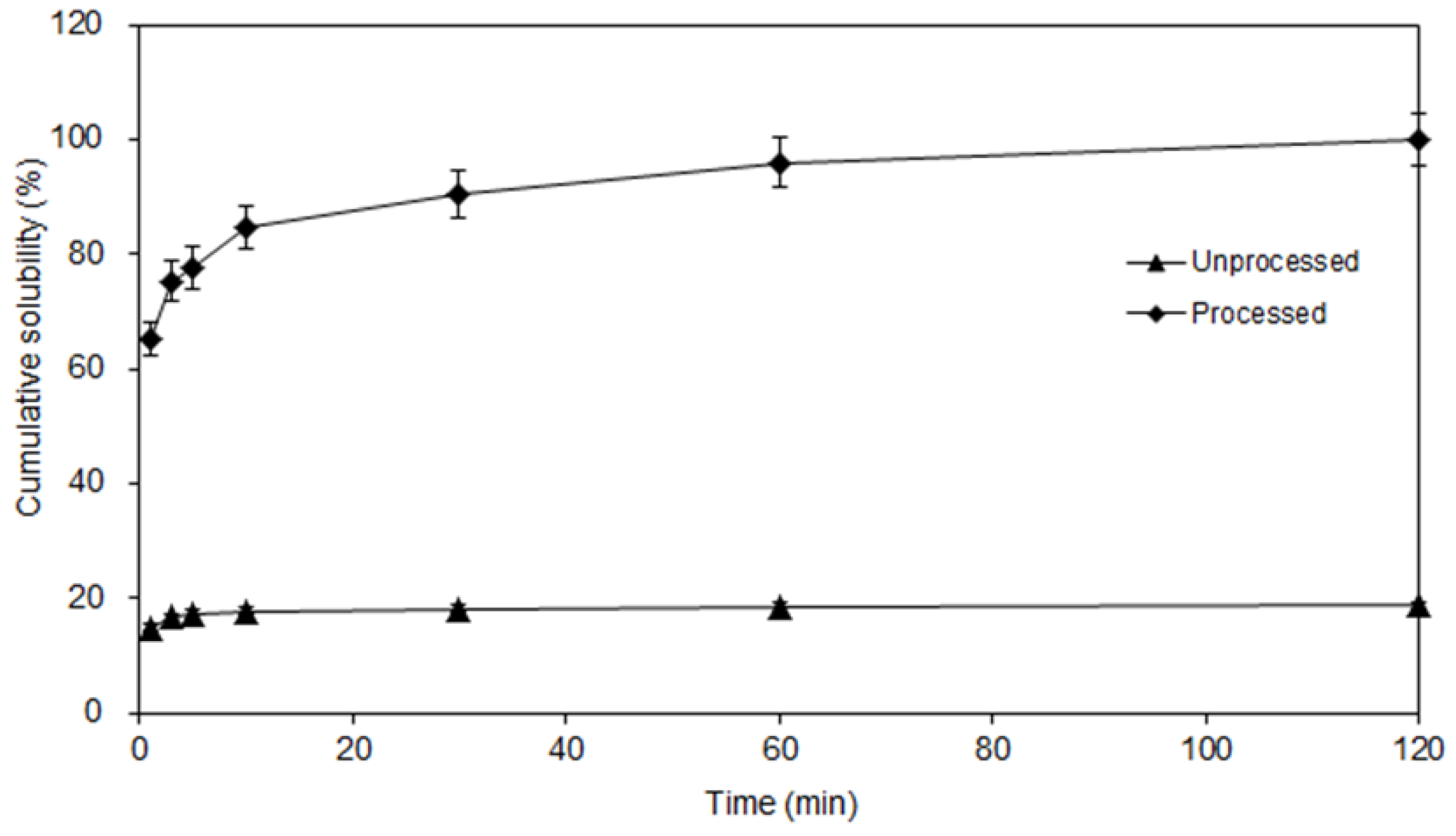
3.5. Dissolution Studies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kernbaum, S. Susceptibility of mycoplasmas and chlamydiae to macrolides. J. Antimicrob. Chemother. 1985, 16, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, E.; Keller, N. Spiramycin renaissance. J. Antimicrob. Chemother. 1998, 42, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.T.; Awad, C.E.; Ali, A.; Matyas, R.; Vital, A.C.; Silva, C.O. Comparison of spiramycin and clarithromycin for community-acquired lower respiratory tract infections. Int. J. Clin. Pract. 1996, 53, 433–436. [Google Scholar]
- Bunetel, L.; Guerin, J.; Agnani, G.; Piel, S.; Pinsard, H.; Corbel, J.C.; Bonnaure-Mallet, M. In vitro study of the effect of titanium on porphyromonas gingivalis in the presence of metronidazole and spiramycin. Biomaterials 2001, 22, 3067–3072. [Google Scholar] [CrossRef]
- Mourier, P.; Brun, A. Study of the metabolism of spiramycin in pig liver. J. Chromatogr. B 1997, 704, 197–205. [Google Scholar] [CrossRef]
- Engel, G.; Farid, N.; Faul, M.; Richardson, L.; Winneroski, L. Salt form selection and characterization of LY333531 mesylate monohydrate. Int. J. Pharm. 2000, 198, 239–247. [Google Scholar] [CrossRef]
- Perng, C.; Kearney, A.; Palepu, N.; Smith, B.; Azzarano, L. Assessment of oral bioavailability enhancing approaches for SB-247083 using flow-through cell dissolution testing as one of the screens. Int. J. Pharm. 2003, 250, 147–156. [Google Scholar] [CrossRef]
- Ahmed, A. Mechanism of inhibition of protein synthesis by spiramycin. Biochim. Biophys. Acta 1968, 166, 205–217. [Google Scholar] [CrossRef]
- Brisson-Noel, A.; Trieu-Cuot, P.; Courvalin, P. Mechanism of action of spiramycin and other macrolides. J. Antimicrob. Chemother. 1988, 22, 13–23. [Google Scholar] [PubMed]
- Choi, W.S.; Kim, H.I.; Kwak, S.S.; Chung, H.Y.; Chung, H.Y.; Yamamoto, K.; Oguchi, T.; Tozuka, Y.; Yonemochi, E.; Terada, K. Amorphous ultrafine particle preparation for improvement of bioavailability of insoluble drugs: Grinding characteristics of fine grinding mills. Int. J. Miner. Process. 2004, 74, S165–S172. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, G.; Neilly, J.; Marsh, K.; Mawhinney, D.; Sanzgiri, Y. Enhancing the bioavailability of ABT-963 using solid dispersion containing Pluronic F-68. Int. J. Pharm. 2004, 286, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, A.; Ambike, A.; Jadhav, B.; Mahadik, K. Characterization of curcumin-PVP solid dispersion obtained by spray drying. Int. J. Pharm. 2004, 271, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Farinha, A.; Bica, A.; Tavares, P. Improved bioavailability of amicronizedmege strol acetate tablet formulation in humans. Drug Dev. Ind. Pharm. 2000, 26, 567–570. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Haruna, Y.; Otsuka, M. Dissolution process analysis using model-free Noyes-Whitney integral equation. Colloids Surf. B 2013, 102, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Li, D.Y.; Zhang, Y.Z.; Kang, Z.T. Preparation of magnesium ferrite nanoparticles by ultrasonic wave-assisted aqueous solution ball milling. Ultrason. Sonochem. 2013, 20, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.P.; Müller, R.H. Production and characterization of highly concentrated nanosuspensions by high pressure homogenization. Int. J. Pharm. 2001, 214, 21–24. [Google Scholar] [CrossRef]
- Aoki, M.; Nishimura, H.; Mimura, A.; Kita, S.; Yasuzawa, T.; Terada, K. Identification of the degradation products of the steroid sulfatase inhibitor KW-2581 in jet mill-micronized powder. J. Pharm. Sci. 2013, 102, 1760–1772. [Google Scholar] [CrossRef] [PubMed]
- Chan, L.W.; Lee, C.C.; Heng, P.W. Ultrafine grinding using a fluidized bed opposed jet mill: Effects of feed load and rotational speed of classifier wheel on particle shape. Drug Dev. Ind. Pharm. 2002, 28, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Huang, J.; Zu, Y.; Ma, C.; Wang, H.; Sun, X.; Sun, Z. Preparation and radical scavenging activities of polymeric procyanidins nanoparticles by a supercritical antisolvent (SAS) process. Food Chem. 2011, 128, 1152–1159. [Google Scholar] [CrossRef]
- Yang, L.; Sun, Z.; Zu, Y.; Zhao, C.; Sun, X.; Zhang, Z.; Zhang, L. Physicochemical properties and oral bioavailability of ursolic acid nanoparticles using supercritical anti-solvent (SAS) process. Food Chem. 2012, 132, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Prosapio, V.; de Marco, I.; Scognamiglio, M.; Reverchon, E. Folic acid-PVP nanostructured composite microparticles by supercritical antisolvent precipitation. Chem. Eng. J. 2015, 277, 286–294. [Google Scholar] [CrossRef]
- Majerik, V.; Charbit, G.; Badens, E.; Horváth, G.; Szokonya, L.; Bosc, N.; Teillaud, E. Bioavailability enhancement of an active substance by supercritical antisolvent precipitation. J. Supercrit. Fluids 2007, 40, 101–110. [Google Scholar] [CrossRef]
- Chen, F.; Li, T.; Li, S.; Hou, K.; Liu, Z.; Li, L.; Cui, G.; Zu, Y.; Yang, L. Preparation and characterization of Tripterygium wilfordii multi-glycoside nanoparticle using supercritical anti-solvent process. Int. J. Mol. Sci. 2014, 15, 2695–2711. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, X.; Zu, Y.; Zhang, Y. Preparation and characterization of paclitaxel nanosuspension using novel emulsification method by combining high speed homogenizer and high pressure homogenization. Int. J. Pharm. 2015, 490, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Homayouni, A.; Sadeghi, F.; Varshosaz, J.; Afrasiabi, G.H.; Nokhodchi, A. Promising dissolution enhancement effect of soluplus on crystallized celecoxib obtained through antisolvent precipitation and high pressure homogenization techniques. Colloids Surf. B 2014, 122, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Wang, Y.; Yin, Q.; Glennon, B.; Zhong, J.; Ouyang, J.; Huang, X.; Hao, H. Crystallization methods for preparation of nanocrystals for drug delivery system. Curr. Pharm. Des. 2015, 21, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Varghese, S.; Kurian, T. Preparation of ultra-fine dispersions of zinc oxide by simple ball-milling: Optimization of process parameters. Powder Technol. 2015, 71, 187–192. [Google Scholar] [CrossRef]
- De Paiva, L.B.; de Oliveira, A.M.; Gavioli, R.R. Preparation and properties of rubber powder from modified-SBR latex by spray drying process. Powder Technol. 2014, 264, 507–513. [Google Scholar] [CrossRef]
- Tenorio, A.; Gordillo, M.D.; Pereyra, C.; de la Ossa, E.J.M. Controlled submicro particle formation of ampicillin by supercritical antisolvent precipitation. J. Supercrit. Fluids 2007, 40, 308–316. [Google Scholar] [CrossRef]
- Fodor-Kardos, A.; Toth, J.; Gyenis, J. Preparation of protein loaded chitosan microparticles by combined precipitation and spherical agglomeration. Powder Technol. 2013, 244, 16–25. [Google Scholar] [CrossRef]
- Ober, C.A.; Kalombo, L.; Swai, H.; Gupta, R.B. Preparation of rifampicin/lactose microparticle composites by a supercritical antisolvent-drug excipient mixing technique for inhalation delivery. Powder Technol. 2013, 236, 132–138. [Google Scholar] [CrossRef]
- Nielsen, P.; Gyrd-hansen, N. Bioavailability of spiramycin and lincomycin after oral administration to fed and fasted pigs. J. Vet. Pharmacol. Ther. 1998, 21, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Pharmacopoeia of the People’s Republic of China; China Medical Science and Technology Press: Beijing, China, 2015.
- Prieto, G.; Meeldijk, J.D.; de Jong, K.P.; de Jongh, P.E. Interplay between pore size an d nanoparticle spatial distribution: Consequences for the stability of CuZn/SiO2 methanol synthesis catalysts. J. Catal. 2013, 303, 31–40. [Google Scholar] [CrossRef]
- Moura, A.P.; Cavalcante, L.S.; Sczancoski, J.C.; Stroppa, D.G.; Paris, E.C.; Ramirez, A.J.; Varela, J.A.; Longo, E. Structure and growth mechanism of CuO plates obtained by microwave-hydrothermal without surfactants. Adv. Powder Technol. 2010, 21, 197–202. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Zhang, Z.; Le, Y.; Shen, Z.; Chen, J. Micronization of atorvastatin calcium by antisolvent precipitation process. Int. J. Pharm. 2009, 374, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Le, Y.; Shen, Z.; Yun, J. Preparation of ultrafine beclomethasone dipropionate drug powder by antisolvent precipitation. Ind. Eng. Chem. Res. 2007, 46, 4839–4845. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, C.; Chen, G. Interaction of macro- and micromixing on particle size distribution in reactive precipitation. Chem. Eng. Sci. 1996, 51, 1957–1966. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, M.; Shao, L.; Wang, Y.; Yun, J.; Chew, N.; Chan, H. Feasibility of preparing nanodrugs by high-gravity reactive precipitation. Int. J. Pharm. 2004, 269, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, M.E.; Hotze, M.A.; Johnston, K.P.; Williams, R.O. Drug nanoparticles by antisolvent precipitation: Mixing energy versus surfactant stabilization. Langmuir 2006, 22, 8951–8959. [Google Scholar] [CrossRef] [PubMed]
- Kakran, M.; Sahoo, N.; Li, L.; Judeh, Z.; Wang, Y.; Chong, K.; Loh, L. Fabrication of drug nanoparticles by evaporative precipitation of nanosuspension. Int. J. Pharm. 2010, 383, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shen, Z.; Zhong, J.; Hu, T.; Chen, J.; Ma, Z.; Yun, J. Preparation of amorphous cefuroxime axetil nanoparticles by controlled nanoprecipitation method without surfactants. Int. J. Pharm. 2006, 323, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zu, S.; Chen, F.; Liu, Z.; Li, X.; Yang, L.; Zu, Y.; Zhao, X.; Zhang, L. Preparation and characterization of cefquinome sulfate microparticles for transdermal delivery by negative-pressure cavitation antisolvent precipitation. Powder Technol. 2016, 294, 429–436. [Google Scholar] [CrossRef]
- Nelson, K.G. The Kelvin equation and solubility of small particles. J. Pharm. Sci. 1972, 61, 479–480. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | BBD Experiments | ANOVA | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| X1 b | X2 | X3 | Y1 | Y2 | Source | Sum of Squares | Degree of Freedom | Mean Square | F | p | |||||||
| 1 | 10 (−1) | 3 (−1) | 50 (0) | 294 | 297 | Model | 36,570 | 9 | 36,570 | 113 | <0.0001 c | ||||||
| 2 | 30 (+1) | 3 (−1) | 50 (0) | 349 | 346 | X1 | 903 | 1 | 903 | 25 | 0.0016 c | ||||||
| 3 | 10 (−1) | 7 (+1) | 50 (0) | 280 | 283 | X2 | 3445 | 1 | 3445 | 96 | <0.0001 c | ||||||
| 4 | 30 (+1) | 7 (+1) | 50 (0) | 280 | 277 | X3 | 13,695 | 1 | 13,695 | 380 | <0.0001 c | ||||||
| 5 | 10 (−1) | 5 (0) | 40 (−1) | 279 | 274 | X1X2 | 756 | 1 | 756 | 21 | 0.0025 c | ||||||
| 6 | 30 (+1) | 5 (0) | 40 (−1) | 305 | 306 | X1X3 | 121 | 1 | 121 | 3 | 0.1097 | ||||||
| 7 | 10 (−1) | 5 (0) | 60 (+1) | 368 | 367 | X2X3 | 930 | 1 | 930 | 26 | 0.0014 c | ||||||
| 8 | 30 (+1) | 5 (0) | 60 (+1) | 372 | 378 | X12 | 5984 | 1 | 5984 | 166 | <0.0001 c | ||||||
| 9 | 20 (0) | 3 (−1) | 40 (−1) | 300 | 302 | X22 | 879 | 1 | 879 | 24 | 0.0017 c | ||||||
| 10 | 20 (0) | 7 (+1) | 40 (−1) | 228 | 230 | X32 | 8413 | 1 | 8413 | 233 | <0.0001 c | ||||||
| 11 | 20 (0) | 3 (−1) | 60 (+1) | 357 | 355 | Residual | 252 | 7 | 36 | ||||||||
| 12 | 20 (0) | 7 (+1) | 60 (+1) | 346 | 344 | Lack of fit | 123 | 3 | 41 | 1 | 0.3970 | ||||||
| 13 | 20 (0) | 5 (0) | 50 (0) | 240 | 249 | Pure error | 129 | 4 | 32 | ||||||||
| 14 | 20 (0) | 5 (0) | 50 (0) | 247 | 249 | Corrected total | 36,822 | 16 | |||||||||
| 15 | 20 (0) | 5 (0) | 50 (0) | 255 | 249 | Credibility analysis of the regression equations | |||||||||||
| 16 | 20 (0) | 5 (0) | 50 (0) | 249 | 249 | Standard deviation | Mean | CV (%) | R2 | Adjust R2 | Predicted R2 | Adequacy precision | |||||
| 17 | 20 (0) | 5 (0) | 50 (0) | 252 | 249 | 6.01 | 294.18 | 2.04 | 0.9931 | 0.9843 | 0.9410 | 31.94 | |||||
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wu, X.; Xie, F.; Wang, Z.; Zhang, X.; Jiang, L. Physicochemical Properties and In Vitro Dissolution of Spiramycin Microparticles Using the Homogenate-Antisolvent Precipitation Process. Appl. Sci. 2017, 7, 10. https://doi.org/10.3390/app7010010
Zhang X, Wu X, Xie F, Wang Z, Zhang X, Jiang L. Physicochemical Properties and In Vitro Dissolution of Spiramycin Microparticles Using the Homogenate-Antisolvent Precipitation Process. Applied Sciences. 2017; 7(1):10. https://doi.org/10.3390/app7010010
Chicago/Turabian StyleZhang, Xiaonan, Xia Wu, Fengying Xie, Zhongjiang Wang, Xiuling Zhang, and Lianzhou Jiang. 2017. "Physicochemical Properties and In Vitro Dissolution of Spiramycin Microparticles Using the Homogenate-Antisolvent Precipitation Process" Applied Sciences 7, no. 1: 10. https://doi.org/10.3390/app7010010