
Melodic Similarity and Applications Using Biologically-Inspired Techniques



1
Department of Information and Computing Sciences, Utrecht University, 3584 CC Utrecht, The Netherlands
2
David R. Cheriton School of Computer Science, University of Waterloo, Waterloo, ON N2L 3G1, Canada
*
Author to whom correspondence should be addressed.
Appl. Sci. 2017, 7(12), 1242; https://doi.org/10.3390/app7121242
Submission received: 30 September 2017
/
Revised: 23 November 2017
/
Accepted: 27 November 2017
/
Published: 1 December 2017
(This article belongs to the Special Issue Sound and Music Computing)
Abstract
:Music similarity is a complex concept that manifests itself in areas such as Music Information Retrieval (MIR), musicological analysis and music cognition. Modelling the similarity of two music items is key for a number of music-related applications, such as cover song detection and query-by-humming. Typically, similarity models are based on intuition, heuristics or small-scale cognitive experiments; thus, applicability to broader contexts cannot be guaranteed. We argue that data-driven tools and analysis methods, applied to songs known to be related, can potentially provide us with information regarding the fine-grained nature of music similarity. Interestingly, music and biological sequences share a number of parallel concepts; from the natural sequence-representation, to their mechanisms of generating variations, i.e., oral transmission and evolution respectively. As such, there is a great potential for applying scientific methods and tools from bioinformatics to music. Stripped-down from biological heuristics, certain bioinformatics approaches can be generalized to any type of sequence. Consequently, reliable and unbiased data-driven solutions to problems such as biological sequence similarity and conservation analysis can be applied to music similarity and stability analysis. Our paper relies on such an approach to tackle a number of tasks and more notably to model global melodic similarity.
1. Introduction
In 2016, digital music revenues overtook physical revenues for the first time (www.ifpi.org/downloads/GMR2016.pdf), a testament to the music industry’s adaptability to the digital age. Listeners are currently able to stream and explore massive collections of music such as Spotify’s (www.spotify.com) library of around 30 million tracks. Such a development has changed not only the way people listen to music, but also the way they interact with it. According to a 2015 survey (www.midiaresearch.com/blog/midia-chart-of-the-week-music-discovery), 35% of users of streaming services use them to discover new songs and artists, new and exciting music for their unique personal taste or listening habits. At the same time, the proliferation of digital music services has raised the listeners’ interest in the accompaniment chords (www.chordify.com), the lyrics (www.musixmatch.com), the original versions of a cover, the sample (loop) (www.whosampled.com) that a song uses and many more scenarios that service providers cannot deal with manually.
This development brings Music Information Retrieval (MIR) to the centre of attention. The field includes research about accurate and efficient computational methods, applied to various music retrieval and classification tasks such as melody retrieval, cover song detection, automatic chord extraction and of course music recommendation. Such applications require us to build representations of previously seen classes (e.g., sets of covers of the same song), which can be only compared to a query (e.g., a cover song whose original is unknown) by means of a meaningful music similarity function. A robust MIR system should model the fuzziness and uncertainty of the differences between two musical items perceived as similar. As Van Kranenburg argues specifically about folk song melodies: “knowledge about the relation between a desired melody and the way this melody is sung from memory” can increase the robustness of melody retrieval tasks [1].
However, this “knowledge”, the exact mechanics of perceived similarity, is still unknown or incomplete [2]. This is not surprising considering music’s inherently complex nature [3,4]. The perceived similarity between two musical pieces is known to be subjective: judgements of different individuals can vary significantly. Marsden [5] argues that similarity involves interpretation, which by itself is a personal creative act. Ellis et al. [6] argue that the individual perception of similarity can show variation depending on the listener’s mood or familiarity with the musical culture and can even change through time. The individual interpretation can be affected also by the multidimensionality of music, since similarity between two songs can be a function of timbre, melody, rhythm, structure or indeed any combinations of those (or other) dimensions. To make matters worse, music similarity is known to be contextual, thus depending on the circumstances of comparison. Deliège [7] argues that similarity can appear as stable only when the context, “the structure of the natural world or a specific cultural system” is quite stable itself.
To overcome, or avoid addressing the aforementioned issues, many MIR approaches to similarity rely on cognition studies, expert heuristics, music theory or formalized models in general. Cognition studies are scientifically well-founded, but often cannot capture the general consensus due to practical limitations, such as access to a sufficient number of participants that fit a certain profile for the study. Expert knowledge, on the other hand, can be a valuable source of information, but with regard to music, expert knowledge cannot fully explain its highly complex nature and the sophisticated human perception. In addition, heuristic approaches have the risk of being descriptive rather than predictive. Formalized models founded on music theory typically neglect that it is not a theory of music perception of similarity. In addition, such models have the highest risk of being solely descriptive, thus not providing us with new knowledge. To their defence, all such approaches can have a certain practical validity, but limited explanatory power, as long as they are evaluated only on a reliable ground-truth and are applied to narrow contexts. Human ratings of similarity are highly problematic with studies showing that subjects are inconsistent with each other and even with themselves [8,9]. Regarding the assessment of similarity between song-triads particularly, Tversky [10] argues that subjects are affected by the song order of appearance and even the song popularity. Regarding the context, a one-fits-all model of similarity is impossible, and as Marsden argues: “the best one can hope for is a measure which will usefully approximate human judgements of similarity in a particular situation” [5].
As long as music cognition fails to provide us a #00AEEFprint of how to develop a computational, generalizable model of music similarity, we are required to explore alternative, data-driven approaches that aim to model the knowledge extracted from the data and the data relations. Data-driven music similarity is not a new concept in MIR, but such studies [11,12] have focused on high-level similarity (genre, artist) where listeners’ opinions are fuzzy. Approaches on more fine-grained music similarity at the note or chord level, such as the work of Hu et al. [13], are scarce for a legitimate reason: in order for the data relations to be bias-free and visible, the data need to be organized in a proper-for-knowledge-extraction form. Properly annotated and disambiguated corpora of note-to-note or chord-to-chord relationships are extremely hard to find.
Fortunately, algorithms that properly organize sequential data have been widely used and are fundamental in the field of bioinformatics. One of the most notable algorithms from the vast bioinformatics toolbox, pairwise sequence alignment via dynamic programming, has been successfully adapted by MIR to compare musical items such as melodies [14] or chord sequences [15]. On closer look, musical and biological sequences are not as unrelated as one might think: even as early as the 1950s, it had been observed that they share a number of resembling concepts [16]. Krogh states that “the variation in a class of sequences can be described statistically, and this is the basis for most methods used in biological sequence analysis” [17]. By acknowledging that the variation of certain quantifiable musical features in a group of related music sequences can be described statistically, as well [18], we gain access to a number of sophisticated, data-driven approaches and bias-free tools that can be adopted from bioinformatics, allowing the modelling of music similarity.
1.1. From Bioinformatics to MIR
Bioinformatics use statistical and computational techniques to connect molecular biology and genetics. Bioinformatics deal with different types of data. DNA sequences carry most of the inherited genetic information of living organisms. These sequences can be represented as a string over a four-letter alphabet {A,C,G,T}, where each symbol represents a nucleotide base. DNA sequences can be as long as several billion symbols, depending on the organism. The instructions to form proteins, which are essential to living organisms, are encoded in the DNA in the form of subsequences or sections called genes. Through a translation process, certain genes are mapped into long chains of amino acids, which fold into three-dimensional protein structures. For computational purposes, proteins can likewise be considered as strings of characters (typically several hundred symbols) from a 20-letter alphabet (since there are 20 different common amino acids).
Music, unlike static forms of art, has a temporal nature. As such, music perception relies on temporal processing [19]. As Gurney argues regarding melodies specifically: “The elements are units succeeding one another in time; and though each in turn, by being definitely related to its neighbours, is felt as belonging to a larger whole” [20]. The same idea actually holds for other music elements, such as chords (notes sounding almost simultaneously) or rhythm. It is therefore not surprising that certain music items, such as symbolic scores, chord transcriptions and others, similarly to DNA or proteins, can be naturally represented as sequences of characters from a finite alphabet. When it comes to music applications, the importance of sequence representation has been demonstrated most notably by Casey and Slaney [21] and by numerous other works that adopted it over the years.
A core assumption of molecular biology is that of homology: related sequences diverge from a common ancestor through random processes, such as mutation, insertion, deletion, and more complex events, aided by natural selection. This process of genetic variation provides the basis for the biodiversity of organisms. Homologues might share preferentially “conserved” regions, subjected to fewer mutations compared to the rest of the sequence [22], which are considered crucial for the functionality of a protein [23]. Similarly, a fundamental observation in music is that music information passing orally, or in other form, can be subjected to noise. Due to our limited cognitive capacity, or for artistic purposes, a musical piece can change throughout a network of musical actors. A folk song that has been transmitted from mouth to mouth and from generation to generation, might differ dramatically from its original version. Even recorded songs can differ when covered by other artists or performed live. There is a strong resemblance to biological evolution since music homologues can occur by altering, inserting, deleting or duplicating music elements to a certain extent [16]. Intuitively also, certain salient parts of a melody or a chord progression are less likely to mutate, thus remaining “conserved”, in an alternative version.
Identifying similarity is crucial not only for MIR, but for bioinformatics applications, as well. Finding homologues through sequence-similarity search is key. Besides the systematic organization, homologue search can help relate certain characteristic behaviours of a poorly-known protein sequence [24]. In addition, experimental results on model species can be applied to humans. Pairwise sequence alignment is the most popular method for assessing the similarity of two sequences. The idea is to introduce gaps ‘-’ to sequences so that they share the same length, while placing “related” sequence elements in the same positions. As such, pairwise alignment aims to find the optimal alignment with respect to a scoring function that optimally captures the evolutionary relatedness between amino acids (how probable it is for one amino acid to be mutated to another). Another important bioinformatics application is finding conserved regions or patterns among multiple homologue sequences which allows for the estimation of their evolutionary distance, for phylogenetic analysis and more. This is achieved by aligning three or more sequences simultaneously, a process typically called Multiple Sequence Alignment (MSA).
1.2. Contribution
In this paper, we argue that MIR can benefit immensely by exploring the full potential of tools, methods and knowledge from the field of bioinformatics and biological sequence analysis, particularly considering melodic-similarity related applications. Despite the high resemblance of concepts (see Table 1), MIR has yet to fully adopt sophisticated solutions such as multiple sequence alignment. As Van Kranenburg suggested, there is a potential for MIR to harvest the bioinformatics’ long history of algorithm development, improvement and optimization for biological sequence analysis [1].
Our previous works on aligning polyphonic voices [25] and melody retrieval [26] more notably, briefly touched on the relationship between MIR and bioinformatics. However, their ideas and bioinformatics-inspired solutions facilitated the work presented in this paper. As such, this paper’s contribution relies first on establishing a strong connection between musical and biological sequences. This allows us to adopt analysis pipelines and algorithms from bioinformatics to: (a) gain new insights regarding music similarity by performing a stability analysis, and (b) present novel solutions for tackling melody retrieval by modelling global similarity. Most importantly, our pipelines are purely data-driven and free of heuristics, as opposed to other MIR methods. To validate the generalization-ability of our approach, we apply it to two melodic datasets of different music. As such, we diverge from previous MIR studies that focused on a specific subset of all possible music. In addition, previous work on datasets of chord sequences [27] also supports the usability of this approach to more than melodic data.
The remainder of this paper is organized as follows: Section 2 acts as an introduction the fundamental sequence comparison and analysis tools derived from bioinformatics. Section 3 describes the musical datasets used in our work. From there on, we apply the bioinformatics methods and tools to the datasets. Section 4 investigates the concept of “meaningful” alignments, while Section 5 uses the findings of 4 to present an analysis of music stability. Section 6 tackles the problems of modelling global similarity. Finally, Section 7 discusses the conclusions of this paper.
2. Methods and Tools
This section aims to describe the fundamental methods used in biological sequence analysis: pairwise alignment and multiple sequence alignment. Understanding their mechanics and limitations is crucial for successfully applying them to MIR tasks. However, the reader familiar with these methods can skip to Section 3 directly.
2.1. Pairwise Alignment
An intuitive method for DNA or protein sequence comparison is the Levenshtein (or Edit) distance, which computes the minimal number of one-symbol substitutions, insertions and deletions to transform one sequence into the other. Such operations can be naturally mapped to the biological process of mutation. Given a cost for each operation, the weighted Levenshtein distance can be computed using dynamic programming. The major drawback of the Levenshtein distance is that it captures the divergence of the two sequences rather than their relatedness or, the important to this paper, similarity. In addition, it does not allow for identifying conserved regions between the sequences, since it is a purely mathematical distance function. As such, computing the similarity of two DNA or protein sequences is typically performed using alignment, the converse to Edit distance. During alignment, gaps ‘-’ that represent symbols that were deleted from the sequences via the process of evolution [28], are introduced in the sequences, until they have the same length and the amount of “relatedness” between symbols at corresponding positions is maximized.
More formally, consider two sequences over an alphabet of symbols , and with all . An alignment A of X and Y, consists of two sequences and over , such that , where if we remove all ‘-’ from , we are left with X and Y respectively. The number of possible alignments A for a pair of sequences is exponential in n and m, so an optimal alignment should be selected given a scoring function that typically derives from a model of “relatedness” between the symbols of , where the goal is to put similar symbols at the same position. The most typical such scoring function is the alignment score:
where . The scoring function v is typically encoded as an matrix called the substitution matrix. Most pairwise alignment methods use a Dynamic Programming (DP) method, credited to Needleman and Wunsch [29], which computes the optimal (highest scoring) alignment by filling a cost matrix D recursively:
where is the gap penalty for aligning a symbol to a gap. An extension uses an affine gap penalty based on the assumption that the occurrence of consecutive deletions/insertions is more probable than the occurrence of the same amount of isolated mutations [28]: for a gap of length z, the gap penalty would be:
where d and e are the gap open and gap extension penalties respectively. To optimize an alignment that uses an affine gap penalty requires a slightly more complex DP algorithm [30]. In the simple non-affine gap case, the score of the optimal alignment is stored in , while the alignment itself can be obtained by backtracking from to . The Needleman and Wunsch approach is a global alignment method, since it aims to find the best score among alignments of full-length sequences. On the other hand, the local alignment framework, first optimized by Smith and Waterman [31], aims to find the highest scoring alignments of partial sequences by tracking back from instead of , and by forcing all to be non-negative. Local alignment allows for the identification of substrings (patterns) of high similarity.
When affine gaps are not considered, meaningful, high-quality alignments are solely dependent on the knowledge captured by the substitution matrix used [30]: optimal alignments with good scoring matrices will assign high scores to pairs of related sequences, while giving a low alignment score to unrelated sequences. More formally, given the two sequences their alignment score should represent the relative likelihood that the two sequences are related as opposed to being unrelated (aligned by chance). This is typically modelled by a ratio, denoted as odds ratio:
where M is a probabilistic model of related sequences and R is a model generating unrelated sequences. If is the frequency of a symbol a, and both X and Y have the same distribution under R, then for the random alignment case aligned pairs happen independently, which translates to:
For the matching case, where aligned pairs happen with a joint probability p, the probability for the alignment is:
In order to get an additive scoring system, it is standard practice to get the logarithm of Equation (3), which after substitution becomes:
A substitution matrix can be considered nothing more than a matrix arrangement of the values (scores) of all possible pairwise symbol combinations.
Sequence alignment via dynamic programming and its time-series counterpart, Dynamic Time Warping (DTW), have been fundamental tools for many MIR tasks since first being applied in a melody retrieval task by Mongeau and Sankoff [32]. Alignment, despite being considered an ill-posed problem for strongly deviating versions of a musical piece [33], has proven to be very useful for identification or classification tasks where strong similarities are present [1,34] and high scoring alignment has been shown to correlate well with human judgements [35,36]. It has been used for cover song detection [37], pattern mining [38], extensively for query-by-humming [14,39] and in other MIR tasks. Interestingly, DTW has been extended to align items that cannot be naturally represented as single sequences, such as polyphonic music [40] or audio [41,42]. Consequently, alignment has been also key to finding correspondences among related music items of not the same format (typically called music synchronization): it has been used for score following, the task of aligning different music representations such as audio and score or MIDI (Musical Instrument Digital Interface) [41,43]. Describing alignment’s numerous MIR applications exceeds the scope of this study. However, a complete overview of DTW in music up until 2007 can be found in the work of Müller [44].
2.2. Multiple Sequence Alignment
A multiple sequence alignment inserts gaps into more than two sequences over an alphabet so that they have the same length and the relatedness between symbols in the same columns is maximized. Formally, given k sequences over an alphabet and a gap symbol ‘-’ , and let be a mapping that removes all gaps from a sequence containing gaps. A multiple sequence alignment A consists of k sequences over such that for all i, for all p, and for all i.
Similar to pairwise alignment, there is a great number of possible MSAs for a single input of sequences [30]. We typically want to pick the most “meaningful” considering our task at hand. More formally: given an objective scoring function that maps each alignment to a real number, we are interested in . There are many such functions [28], but the most widely used is the Weighted Sum-Of-Pairs (WSOP or SOP) [45], a summing of scores of all symbol-pairs per column. Let be the i-th column j-th row of A, the SOP is defined as such:
where is a weight assigned to the pair of sequences and corresponds to an iteration over all pairs of rows in the column. Naturally, the objective function can be adapted to accommodate affine gaps. Computing the optimal MSA is unfortunately NP-complete [46] and cannot be used in realistic scenarios that include numerous and long sequences. Therefore in the field of bioinformatics, heuristic approaches that give good alignments, though not guaranteed to be optimal, have been developed. According to Kemena and Notredame [47], more than 100 different MSA algorithms have been proposed over the last 30 years but discussing them in detail exceeds the scope of this paper.
MSA algorithms have found a rather small application in MIR. Liu [48] uses the progressive alignment algorithm to compare different music performances represented as strings derived from chroma features (distribution of the signal’s energy across a set of pitch classes). In a similar manner Wang et al. [49] showed that progressive alignment of multiple versions can stabilize the comparison for hard-to-align recordings that can lead to an increase in alignment accuracy and robustness. Finally in a tangential task, Knees et al. [50] use a progressive alignment approach to align multiple lyrics gathered from various online sources.
3. Melodic Sequence Data
Music comprises sound events that can be pitched or unpitched (percussive) with either stable or unstable pitch. In the context of this paper we consider the tone, a fixed frequency sound (pitch), to be the most important musical element. In music notation (scores), tones are represented as notes with accompanying duration values. A series of notes arranged in time and perceived as a distinct group or idea, is what we roughly define as a melody, although years of musicological studies have failed to agree on a consensus definition. Poliner et al. [51] define it as “the single (monophonic) pitch sequence that a listener might reproduce if asked to whistle or hum a piece of polyphonic music, and that a listener would recognize as being the essence of that music when heard in comparison.” As Kim et al. [52] also mention, one can recognize a song (out of all known songs) just by its melody even though it might have been corrupted with noise or cut short. This observation is a testament to melody’s importance to music perception. As such, melodies have been at the centre of musicological research [53] and music cognition [54]. In MIR, melody extraction from audio has been an active research topic, since melodies can act as robust and efficient features for song retrieval [55]. Query-by-humming, i.e., retrieving similar items using a sung melody as a query, has been also an important, on-going MIR task [56,57].
When it comes to comparing melodies in terms of their similarity, sequence representation is key; we need to carefully select the music features that we will represent as sequences [47]. As Volk et al. [2] argue based on relevant studies, music similarity works on many dimensions, such as melodic, rhythmic or harmonic, but the musicological insights regarding the relative importance of each dimension are insufficient. The works of Van Kranenburg [1] and Hillewaere et al. [58] revealed the importance of the pitch dimension, so our work considers melodies as pitch-contours, meaning series of relative pitch transitions constrained to the region between and semitones (folded to one octave so that a jump of an octave is treated as unison and therefore ignored). Besides their simplicity and key-invariance, pitch contours have been found to be more significant to listeners for assessing melodic similarity than alternative representations [59]. In our work, all sequences of pitch transitions are mapped to an extension of the 20-letter alphabet that is used to represent the naturally occurring amino acid for ease of adaptation.
3.1. Datasets
Reliable analysis and modelling of similarity requires first and foremost datasets of unambiguous relationships between music items. Marsden [5] among others, makes a strong case regarding the validity of similarity ranking annotations, considering the paradigm differences of the listening experiments that generated them. However, he is more supportive to binary or definite annotations of similarity, such as songs known to be covers, or songs known to be related from musicological studies. Such data can be used to verify a computational model with regard to its retrieval or classification performance, since the distance for music items within a category should be less than the distance of items belonging to different categories. As such, this paper uses two datasets of symbolically represented melodies of varying size and nature, containing melodies that are considered related (e.g., covers of the same song) grouped into definite groups called either families, classes or cliques. Summary statistics for both sets are presented in Table 2.
The Annotated Corpus of the Meertens Tune Collections [60], or TuneFam-26, is a set of 360 Dutch folk songs grouped into 26 “tune families” by Meertens Institute experts. Each contains a group of melody renditions related through an oral transmission process. For this dataset, expert annotators assessed the perceived similarity of every melody over a set of dimensions (contour, rhythm, lyrics, etc.) to a set of 26 prototype “reference melodies”. In addition, the dataset contains 1426 annotated motif occurrences grouped into 104 classes, where “motifs” correspond to recurring patterns inside the melodies of a tune family. The Cover Song Variation dataset [61], or Csv-60, is a set of expert-annotated, symbolically-represented vocal melodies derived from matching structural segments (such as verses and choruses) of different renditions of sixty pop and rock songs. Csv-60 is inherently different from TuneFam-26 in two ways. First, the grouping of melodies into classes is certain: the songs were pre-chosen as known covers of songs of interest. Secondly, cover songs are typically not a by-product of an oral transmission process since cover artists have access to the original version.
4. Multiple Sequence Alignment Quality for Melodic Sequences
This paper’s main approach on modelling melodic similarity relies on capturing the variation among two or more perceived-as-similar melodies. For that we need trustworthy, “meaningful” alignments of related music sequences, such that the statistical properties of the alignment can inform us about the note-to-note relationships. Since such data can be hard to find, we are required to align related sequences using alignment algorithms. Alignment, pairwise or otherwise, with notable exceptions [63,64] has been typically used as an out-of-the-box tool to align instances of music sequences, with the sole purpose of using its score output further in a retrieval pipeline. The quality or musical appropriateness of the alignment of symbols themselves has always been evaluated via a proxy, i.e., some kind of music retrieval scenario. As long as the alignment-pipeline outperformed other approaches, its utility was considered significant. The major problem however, is that outside the proxy strategy, there are no studies or musical intuition to prefer one alignment over the other.
Identifying the features that make a “good”, meaningful alignment is an intricate task, not only for musical but biological sequences as well. Interestingly, proteins are folded into diverse and complex three-dimensional structures. Structure motifs (not to be confused with the homonym musical concept) diverge slower in the evolutionary time scale than sequences, and consequently homology detection among highly divergent sequences is easier in the structural than the sequence domain, though the actual algorithms for three-dimensional shape alignment are complex. As such, structure motifs have been used to aid the alignment of highly-divergent sequences [65]. In addition, reference alignments produced from biological information, such as a conserved structure, have been frequently used to assess the quality of an MSA [66].
We argue that similar to biological sequences, a “good” meaningful alignment of musical significance, can be only evaluated via a trustworthy reference alignment. Previous related work [67] generated “trustworthy” alignments of the Csv-60 set by using a progressive alignment algorithm extended on three musical dimensions (pitch, onset, duration). Bountouridis and Van Balen’s choice was based largely on intuition, since there is no literature supporting those three dimensions. Prätzlich and Müller [64] investigated the evaluation of music alignment by using solely triplets of recordings of the same piece and made clear that there are theoretical considerations of alignment quality-assessment without a reference alignment. Therefore, the question becomes whether there exists a musical analogy to the protein structure motifs.
In musicology shared, transformed but yet recognizable musical patterns are called “variations” and according to musicological and cognitive studies, variations are essential to the human perception of music similarity [2]. Specifically when it comes to classifying folk songs into tune families, i.e., groups of songs with a common ancestor, Cowdery [68] considers the shared patterns to be a key criterion. An annotation study on Dutch folk songs by Volk and Van Kranenuburg [4] also supported this claim by proving that shared, stable musical patterns, called motifs were important for the expert assessment of music similarity. Consequently, we can theoretically use the motif alignment as reference for evaluating the quality of musical sequence alignment. For example, consider the following sequences with expert annotated motifs “AB” (red) and “AFF” (#00AEEF): AFFGABBBBC, ABDDBBC and AFFABB. Two possible alignments with equal SOP scores are:
AFFGABB-BBC AFFGABB-BBC ––––ABDDBBC A––––BDDBBC AFF-ABB–––– AFF-ABB––––
From a musicological perspective though, the first alignment is considered of higher quality, since it aligns perfectly those subsequences that are annotated as same-label motifs. It is of high importance to investigate which MSA algorithms and settings are optimal with regard to motif alignment (for example, which algorithm would be more likely to generate the first alignment rather than the second). The following paragraphs describe the appropriate experiments to answer such question.
Our experiment pipeline comprises aligning a group of related sequences (that include motifs) using different motif-agnostic MSA strategies, and then comparing the resulting alignment of motifs to a reference optimal motif alignment. The comparison is not based on a distance function between the alignments, but rather on assigning a score to both of them. Besides the different MSA strategies (to be discussed in Section 4.3), the pipeline requires the following: first, a motif alignment scoring function that is well-founded (see Section 4.1). Secondly, it requires a dataset of musical sequences that contain annotated motifs for each clique, combined with trustworthy alignments of these motifs that would act as a reference (see Section 4.2).
4.1. Motif Alignment Scoring
The only information available to compute a meaningful motif-based MSA score is the motifs’ position in the sequence, length and notes they contain. Due to the lack of knowledge regarding which pairs of pitches should be aligned together, the motif alignment scoring method cannot be founded on the pitch dimension. We are confident for only one thing: the notes belonging to same-labelled motifs should be somehow aligned. As a consequence, we focus on an intuitive scoring function that is maximized when same-labelled motifs are maximally overlapped. Given a function that returns the motif label of the i-th note of a sequence X, the WSOP score (denoted motif-SOP) of an MSA is based on the following scoring function:
In other words, we only penalize those alignments that align notes belonging to different motifs. Alignment between notes not belonging to any motif (), and labelled notes are considered neutral since no studies or intuition suggests otherwise. The particular scoring function would assign the same motif-SOP score for both the following alignments, since only the alignment of motif labels (represented as colours) is taken into consideration:
AFFGABB-BBC –AFFGABB-BBC ––––ABDDBBC –––––ABDDBBC –AFFABB–––– A-FF-ABB––––
4.2. Dataset and Reference Motif Alignments
The TuneFam-26 dataset is the best benchmark for our experiment, since it contains related melodies (grouped into tune families) with a number of subsequences annotated by experts and uniquely labelled as motifs (see Figure 1). It is however, not the optimal benchmark since the expert annotated motifs of the same label, which can be of different lengths, do not come pre-aligned; we know which sub-sequences in the family’s melodies are motifs, but we do not know their note-to-note alignment. Since there are no trustworthy motif alignments, the optimal alignment should be a by-product of the motif-SOP function and the intuition behind it, i.e., the reference alignment should be the one that maximizes the motif-SOP score. In order to acquire that for each family, through visual inspection, we manually align the motif variations. At the same time, we consider the motif-SOP score of the original unaligned sequences as the lower bound , i.e., the worst possible scenario. The and scores allow us to normalize any motif-SOP score to a meaningful range.
4.3. Multiple Sequence Alignment Algorithms and Settings
From the numerous MSA algorithms, we selected three based on many factors including simplicity, popularity or quality of results on several bioinformatic benchmarks. One of the simplest approaches to MSA, named “star” alignment, aims at employing only pairwise alignments for building the final MSA. The idea is to first find the most “central” among the sequences, pairwise align it to each one of the rest and then combine the pairwise alignments into a single MSA. This method does not necessarily attempt to optimize the objective function (see Section 2.2) and as such is rarely used. In our case, star alignment can act as a naive baseline for the more sophisticated algorithms to be compared against.
Progressive Alignment (PA) [69] is one of the most popular and intuitive approaches, and it comprises three fundamental steps. At first, all pairwise alignments between sequences are computed to determine the similarity between each pair. At the second step, a similarity tree (guide tree) is constructed using a hierarchical clustering method, which in biological sequences is sometimes used to attempt to identify evolutionary relationships between taxa. Finally, working from the leaves of the tree to the root, one aligns alignments, until reaching the root of the tree, where a single MSA is built. The drawback of PA, is that incorrect gaps (especially those at early stages) are retained throughout the process since the moment they are first inserted (the “once a gap, always a gap” rule). Iterative refinement methods [70,71] aim to tackle this problem by iteratively removing each sequence and realigning it with a profile created from the MSA of the rest, until an objective function has been maximized. Our experiments use the PA-based T-Coffee software (Tree-based consistency objective function for alignment evaluation) [72]. T-Coffee aims to tackle the problem by making better use of information in the early stages . It uses an objective function (called COFFEE [73]) that first builds a library of all optimal pairwise alignments and secondly, scores a multiple sequence alignment by measuring its consistency with the library: how many of the aligned pairs in the MSA appear in the library.
Locating very similar short and shared sub-regions between large sequences has been in important task in bioinformatics. Such segments can efficiently reduce MSA runtimes and as a consequence, MSA solutions that incorporate some of form of segmentation, such as Dialign [74] and Mafft [75], have found successful application. Mafft in particular, is a progressive alignment method at its core, but incorporates the Fast Fourier Transform (FFT) for biological sequences. In addition, Mafft allows the usage of the iterative refinement method. For non-biological sequences, Mafft offers a “text” alignment option that excludes biological and chemically-inspired heuristics from its pipeline. In such a case, segmenting the sequences becomes a by-product of Mafft’s objective function that incorporates both a WSOP and a COFFEE-like scoring. According to Mafft’s website (mafft.cbrc.jp/alignment/software), “the use of the WSOP score has the merit that a pattern of gaps can be incorporated into the objective function”.
Mafft offers three different strategies for the initial pairwise alignment, that behave differently with regard to the structure of the sequences. Local alignment with affine gap costs localpair is appropriate for unaligned sequences centred around a conserved region. The genafpair strategy uses local alignment with generalized affine gap costs [76] and is appropriate for sequences with several conserved sub-sequences in long unalignable regions. Global alignment with affine gap costs globalpair is appropriate for throughout alignable sequences. A lesser known option, which can be applied on top of localpair and globalpair strategies, is allowshift which is appropriate for sequences that are largely similar but contaminated by small dissimilar regions.
Each MSA algorithm aims to find the alignment that maximizes the SOP score on the Identity (ID) scoring scheme, i.e., if and if . As a matrix, the ID scheme has +1 in the diagonal and −1 otherwise. The effect and importance of gap penalties, or gap settings (see Equation (3)), is well known for biological sequences [77] and for musical sequences as well [78]. Understanding their behaviour with regard to the MSA is crucial, especially when different matrices are used. Since literature suggests setting them empirically [77] and the ID matrix is used on each MSA algorithm in our case, we experiment with a only a small variety of gap settings. At the same time, we keep in mind that there is no guarantee that these settings optimize the performance of all MSA algorithms. Regarding T-Coffee, such penalties are not essential when building the MSA, since in theory the penalties are estimated from the library of pairwise alignments. In practice, it is suggested to experiment with different settings while keeping in mind that the penalties are not related to the substitution matrix. Gap open can be in the range of and gap extension in the range of .
4.4. Results
For each clique of sequences we generated a reference motif alignment manually, and computed its motif-SOP score (called ). At the same time, for each motif-agnostic configuration (MSA algorithm, gap settings), we aligned the melodic sequences. Each resulting alignment was also assigned a motif-SOP score (called ). In order to identify the best MSA configuration with respect to motif alignment, we compute its normalized motif-SOP score .
Before proceeding into the quantitative results, it is worth visually examining the alignments created by the MSA algorithms. Figure 2 presents different alignments of the tune family “Daar_ging_een_heer_1” for a number of configurations. Regarding quantitative results, Figure 3 and Table 3 present the normalized motif-SOP score for different configurations. There are a number of observations that become immediately apparent: first, the normalized motif-SOP score can be less than zero, since the original unaligned sequences, that act as the lower bound, may include correctly aligned motifs by random chance (see Figure 1). Secondly as expected, star alignment is the worst performing algorithm across all gap settings. Regarding the relative performance of the configurations themselves (excluding star alignment), a Friedman significance test showed that there was a statistically significant difference in motif-SOP depending on the configuration with . However, post hoc analysis with Wilcoxon signed-rank tests and Bonferroni correction, revealed that there were no significant differences among any pair of configurations.
Regarding the overall performance of the algorithms themselves, a Friedman significance test showed that there was a statistically significant difference in normalized motif-SOP depending on the algorithm with . Post hoc analysis with Wilcoxon signed-rank tests and a Bonferroni correction resulted in a significance level set at . p values for all possible pairs are presented in Table 4. It is clear that Mafft, run with the globalpair strategy, outperforms T-Coffee and that of the three Mafft strategies, globalpair performs the best. A Wilcoxon signed-rank test between all the Mafft algorithms using and not using the allowshift option, revealed that the allowshift option does not have a significant impact on the results, .
Finally, regarding the gap settings, significance tests showed that for Mafft and T-Coffee in general, there is a significant difference depending on the gap penalties used. For T-Coffee in particular, large gap settings such as (−60,−3) or (−40,−2) are not recommended. For MAFFT on the other hand, small gap penalties, such as (−0.8,−0.5) should be avoided. However it should be noted that for each particular Mafft strategy, gap settings have no significant effect.
4.5. Discussion
In this section, we first established a measure of MSA quality based on motifs. Secondly, we evaluated different MSA algorithms and gap settings on a dataset of folk song melodies. Despite the small dataset of 26 tune families, the results offer strong proof about the benefits of the MSA algorithms, and Mafft in particular. Regarding Mafft’s success, we hypothesize that it can be attributed to its objective function that results to gap-free segments. According to Margulis [79], the phrase structure of a melody is of major importance for the human perception of variation patterns. By treating the located sub-regions as gap-free segments, Mafft can be the closest to partitioning melodies into perceptually meaningful units without using heuristics or expert knowledge.
In general, by establishing a reliable strategy to align multiple instances of melodies, we eliminate the prerequisite to invent a retrieval/classification proxy to assess the quality of an alignment. We can also now benefit from both the alignment score and the alignment’s structure itself. Particularly regarding the latter, since the alignment of notes is musically significant, we can now extract knowledge about their relationships. For example, we can perform reliable analysis on notions such as stability (as we do in the following Section 5) or generate models of similarity (as we do in Section 6).
5. Analysis of Melodic Stability
It has been theorized that our perception and memorization of melodies is dynamic, meaning that certain musical events throughout a melody’s length, can be perceived as more stable (resistant to change) than others depending on the context [80]. Klusen et al. [81] showed that every note in a melody can be altered in an oral transmission scenario, but some notes are more stable than others. Numerous studies from cognition [80,82,83] to corpus-based analysis points-of-view [84], have also evaluated the importance of certain musical factors with regard to their influence on the perceived music stability. Since the alteration of stable elements can affect the process of recognition, stability is also a key component for understanding music similarity. Unsurprisingly, stability and music variation (stability’s counterpart ) have been at the core of both musicology and MIR. From a musicological perspective, knowledge of the mechanics of those concepts would allow researchers to trace, classify or possibly even pinpoint in time variations of songs. Similarly, scientists from computational disciplines may use knowledge of stable musical elements to improve the automatic classification and retrieval of musical objects, such as the work of Van Balen et al. [85]. Therefore, before proceeding into modelling music similarity (see Section 6), it is worth investigating the complementary concept of music stability.
Interestingly, conservation is at the centre of biological sequence analysis, in the same way that stability is at the core of musicology or MIR. As Valdar [23] nicely describes, a multiple sequence alignment of protein homologue sequences (together with the phylogeny) is a historical record that tells a story about the evolutionary processes applied and how they shaped a protein through time. Useful and important regions of a protein sequence often appear as “conserved” columns in the MSA, and major sequence events that appear on a phylogenetic tree often correspond to epochal moments in natural history.
In this section we argue that, much as an MSA of protein homologues can inform us about the statistical properties of the evolutionary processes, an MSA of related melodies can provide us with valuable information regarding the processes of musical variation. We aim to determine and analyse regions of less variation inside a selection of related melodies, or in other words, regions of melodic stability. Analysing stability requires trustworthy MSAs such that the assignment of corresponding notes across different versions can be directly observed by looking at the MSA’s columns. The findings of Section 4 allows us to be confident regarding the results of a stability analysis since it can be conducted on high-quality, musically meaningful alignments.
5.1. Setup
We are interested in applying the best alignment configuration (as established on Section 4) to the TuneFam-26 and Csv-60 melodic datasets. We can later perform an analysis on the aligned cliques (tune families or cover song melodies) by using an appropriate measure of stability applied on each column of the MSA. The results from Section 4 have indicated that the best MSA algorithm for melodic sequences is Mafft, while its globalpair and localpair strategies are indistinguishable in terms of alignment quality. Gap settings have little or no effect per strategy, Mafft options and gap penalties had minimal effect on alignment quality, so we explored several parameterizations: Mafft-globalpair with (−4,−2) gap penalties, Mafft-globalpair-allowshift with (−4,−2) gap penalties and Mafft-localpair-allowshift with (−2,−1) gap penalties.
A quantitative measure of stability, suitable for music sequences, does not exist as a result of the lack of supporting literature and research. Nevertheless, Bountouridis and Van Balen [67] use a probabilistic interpretation of the WSOP measure that aims to answer the following question: given that we observe a single, randomly chosen melodic element, what is the probability for this element to appear unchanged when we observe a new, unseen a variation of it. In practice, given a set of k aligned sequences of length m such as , the stability of the non-gap symbol e in position j is defined as:
while the stability of the j-th MSA column is simply over all unique e.
It is worth examining the related bioinformatics literature regarding the equivalent concept of conservation scores. Valdar [23] mentions that “there is no rigorous mathematical test for judging a conservation measure”. A scoring method can be only judged with respect to biochemical intuition, and therefore a number of conservation scores have been proposed through the years [22]. The same authors list a number of intuitive prerequisites that a conservation score should fulfil, including sequence weighting (to avoid bias due to near-duplicate sequences) or the consideration of prior amino acid frequencies. However, applying the same prerequisites to music sequences is not supported by any musical literature. Consequently, our analysis adopts two widely used and interpretable conservation scores from bioinformatics: the WSOP score (already discussed thoroughly) and Information Content (IC). Based on Shannon’s entropy, the IC score of the j-th column is defined as such:
where is alphabet size, is the frequency of a particular symbol e in the j-th column, while is the expected frequency of symbol e in the dataset (prior). It should be noted that symbols in a column with zero frequency are not taken into account.
5.2. Analysis
The next paragraphs present a brief analysis on stability and variation with regard to two music dimensions, position and pitch intervals. However, it is possible to extend the analysis to dimensions such as note durations [67] or interval n-grams. Janssen et al. [84] on their corpus-based analysis on the TuneFam-26 dataset, investigated stability with regard to global features related to memorability, i.e., a phrase length, its position in the melody, its repetitiveness and others.
We hypothesize that certain parts of a melody, such as the beginning or end, are more robust to variations. We are therefore interested in the stability with regard to a note’s relative position in the melody. Each column j of an MSA has a computed stability score. Each i-th index of a sequence in the MSA is assigned the stability score of its corresponding column. It should be noted that due to gaps, the i-th index of two different sequences may not correspond to the same j column. For each dataset (TuneFam-26 and Csv-60) we accumulate all the position versus stability data, where position corresponds the i-th index normalized to the range. Figure 4 and Figure 5 present the stability scores using different scoring methods (computed over three different alignment configurations) versus the relative position of a note (interval in our case) for the TuneFam-26 and Csv-60 datasets respectively. The corresponding gap ratio of the MSA versus the note position is also presented as a reference, since all conservation scores are affected by the amount of gaps per column.
For both datasets there are a number of observations (trends) that become immediately apparent: first, there is a strong indication that roughly the first half of a melody (up until 60% of its length) is more stable than the remaining. The downward slope after position 0.6 is prominent in both datasets and on all different stability scoring methods. This observation seems to agree with findings of Janssen et al. [84]; stable phrases occur relatively early in the melody. Secondly, the stability towards the final notes of a melody seems to be increasing. For the TuneFam-26 dataset in particular, the final 20% of the melody is very stable. The trend is less obvious on the Csv-60 dataset. However, it should be reminded that TuneFam-26 contains whole folk tune melodies, while Csv-60 contains melodies corresponding to structural segments of pop/rock songs; we cannot expect certain trends to be completely shared by both sets.
A potential explanation for this trend would be that artists interpreting a song creatively start out with an emphasis on the familiar material and take more liberty as the melody or segment progresses, introducing more variation along the way. But in contrast to the findings of Bountouridis and Van Balen, our results indicate that artists end with a familiar set of notes (for folk tunes more notably). This can be potentially attributed to the capacity of our short-memory; after a considerable part of varied material, our brain requires familiarity as to identify the whole piece as a variation of the original. For the Csv-60 dataset, since the melodies are shorter, the effect of short-term memory’s capacity is weaker thus explaining the less obvious trend.
We now turn our focus to pitch intervals. We hypothesize that certain pitch intervals are more stable than others, i.e., certain transitions are less likely to be varied in a variation of the melody. To test our hypothesis, we need to measure the overall stability for each interval, while avoiding biases related to their likelihood of appearing in a sequence or column. We use the Information Content measure, computed for each symbol (note interval) e in the j-th index of the MSA as such:
where is the frequency of a particular symbol e in the jth column, while is the expected frequency of symbol e (prior).
Figure 6 presents the overall stability scores per interval for the whole TuneFam-26 and Csv-60 datasets, in addition to their interval distribution. We show the results for the 13 most frequent intervals, since the remaining are too scarce for reliable analysis. Starting our analysis from the interval distribution profiles, we observe that they agree with Schoenberg’s “singableness” hypothesis, that posits (among others) that a melody consists of more stepwise than leap intervals as a result of the human voice’s nature [86]. The scarcity of chromatic jumps can be explained if we consider them as short excursions from the scale, which offer difficulties as well according to Schoenberg.
Regarding the stability-per-interval profiles, on first look, they are quite similar for the two datasets. Interestingly, the variance seems proportional to the interval’s frequency despite the fact that our stability measure IC is normalized for the expected frequency per interval. On closer look and regarding the TuneFam-26 dataset, the and intervals are significantly more stable than the , intervals of similar frequency of appearance. In addition, the +7 interval is as stable as the very frequent intervals. Therefore, we conclude that there is something inherently salient about the and intervals (at least in the TuneFam-26 dataset), but it is unsafe to make hypothesis regarding why this is the case. It should be noted that the findings of Janssen et al. [84] indicated that stable phrases are likely to comprise (among others) small pitch intervals and little surprising melodic material. However, their analysis approach is focused on stable phrases’ global features, while ours on note-level features. Therefore, a direct comparison of findings, at least for pitch intervals, cannot be performed.
6. Data-Driven Modelling of Global Similarity
The findings of our stability analysis validated the intuitive hypothesis that some notes are more likely to be altered in a melodic variation than others. As such, any fine-grained melodic similarity function needs to accommodate for that fact by integrating meaningful scores for any pair of notes. In pairwise alignment via dynamic programming, integrating domain knowledge is only possible through the substitution matrix, which constitutes a model of global similarity, since it identifies notes commonly changed into other notes. Van Kranenburg [1] extended the DTW scoring function to include multiple musical dimensions, such as inner-metric analysis or phrase boundaries. On a melody classification task, he showed that expert-based heuristics could achieve almost perfect results. De Haas [87] showed that with regard to chord sequence similarity, local alignment with a substitution matrix based on simple heuristics [15], significantly outperforms his more sophisticated geometric model that takes into consideration the temporal relations between chords. Despite their success, the major concern with such approaches is their reliance on heuristics with known issues, such as limited generalization (see Section 1).
Interestingly in bioinformatics, the problem of meaningful substitution matrices, has been addressed following a data-driven approach. The major difficulty of the scoring matrix calculation is the computation of the joint probability (see Equation (7)) that expresses the likelihood of the two symbols at homologous sites. In bioinformatics, the key idea for solving this problem is that trusted alignments of related sequences can provide information regarding the mutability of symbols. One of the most widely-used matrices for protein comparison, BLOSUM [88], is actually derived from a large number of manually constructed, expert-aligned amino-acid sequences by counting how often certain amino-acids are substituted (mutated).
It follows naturally to investigate the potential of data-driven approaches in the MIR domain as well. Hirjee and Brown [89,90] generated a data-driven phoneme substitution matrix from misheard lyrics, gathered from online sources, and successfully applied it on a lyrics retrieval task. Similarly, Bountouridis et al. [27] used online sources to generate a chord similarity matrix for the task of cover song detection. Hu et al. [13] on the other hand, based their approach on pairs of aligned sung and reference melodies for the task of query-by-humming, but failed to significantly outperform a simple heuristic matrix. This might be attributed to the lack of experimentation with gap penalties or the noisy frame-based instead of note-based representation. Another major drawback for them was the amount of data, which consisted of only 40 sung melodies. We argue that expert-based alignments are generally problematic due to their limited quantity. Online sources have been shown to be potential solutions for lyrics or chords, but their existence cannot be guaranteed for all possible musical items such as melodies.
To eliminate the need for trustworthy pre-aligned melodic variations, in this section we propose the usage of trusted alignment algorithms as discussed in Section 4. Alignments generated by such algorithms can provide us with the appropriate information to generate a substitution matrix by computing log odds ratios for any pairs of symbols. While trusted alignment algorithms reduce the need for expert or crowd-sourced alignments, they still require melodies grouped (by experts preferably) into related cliques or tune families. These are still hard to find and as such, the applicability of our approach in real-life scenarios can be limited. Interestingly, in the same way that melody cliques contain melodic variants, melodies themselves may contain short recurring fragments, intra-song motifs. Such motifs may appear in variations throughout the melody. It is therefore also possible to generate a model of similarity among intra-song motifs if properly aligned. We hypothesize that intra-song motivic similarity can approximate the melodic similarity, or in other words, independent melodies contain enough information to explain variations in melodic cliques.
In the following paragraphs we present two data-driven approaches for capturing global similarity realized as substitution matrices for the TuneFam-26 and Csv-60 datasets. First, a matrix generated by alignments of melodic variations belonging to a clique (denoted simply melodic similarity). Secondly, matrices generated from different alignments of individual melodies with themselves (denoted intra-song motivic similarity). In order to assess their quality, we later perform an experiment to evaluate their retrieval performance.
6.1. Generating Substitution Matrices
Before discussing the alignments used, we explain the general process of converting them into a scoring system (a substitution matrix). The SubsMat package from the bioinformatics library Biopython provides routines for creating the typical log-odds substitution matrices. For our data, we firstly create the Accepted Replacement Matrix (ARM), meaning the counted number of replacements (confusions) according to the alignments. In order to avoid matrix entries of value zero, we apply pseudo-counts, meaning we add one to each entry. We generate the log-odds matrix M by applying a function that builds the observed frequency matrix from the ARM. We use the default settings: log base b = 10 and a multiplication factor f of 10. For two symbols x and y, their corresponding log-odds score is:
with rounded to the nearest integer. We normalize the matrix by dividing each of its elements with , so that the maximum score assigned to a pair of symbols is one.
6.2. Computing the Alignments for Melodic and Intra-Song Motivic Similarity
For the modelling of melodic similarity, the results from Section 4 have indicated that, although Mafft is the best alignment strategy, the differences between various configurations are rather insignificant. Therefore, instead of generating a substitution matrix from clique alignments of one configuration only, we decided to use the following: Mafft-globalpair with (−4,−2) gap penalties, Mafft-globalpair-allowshift with (−4,−2) gap penalties and Mafft-localpair-allowshift with (−2,−1) gap penalties. The melodic similarity matrices generated for the TuneFam-26 and Csv-60 datasets are denoted TFam-matrix and Csv-matrix respectively.
For the modelling of intra-song motivic similarity, the idea is to align each sequence with artificial versions of itself, such that all possible instances of intra-song motifs are aligned. In such a context, a useful and informative version of a sequence is one that when aligned to the original, maximizes the overlap between different instances of perceived-as-similar motifs. This informativeness criterion partially agrees with Hertz’s and Stormo’s definition of interesting alignments: those whose symbol frequencies most differ from the a priori probabilities of the symbols [91]. However, since informativeness can be erroneously biased, we are interested in alignments that at the same time minimize the overlap between perceptually different motifs.
Let us consider an example sequence with two known motif instances “ABF” (#00AEEF), “AGG” (green) of label and one motif instance “KLM” (red) of label : XXABFXXXAGGXXXKLM. Furthermore, consider three versions of the sequence based on arbitrary splitting in segments and further duplication or shuffling: KLXXXXABFXXXAGGXF, XAGGXXABFXAGGX and AGGXXKLMXXXABFXXX. Three possible pairwise alignments of the versions with the original are:
––––XXABFXXXAGGXXXKLM XXABFXXXAGGXXXKLM– XXABFXXX–––––AGGXXXKLM LMXXXXABFXXXAGG––XK–– X–AGGXX–ABFX––AGGX ––AGGXXXKLMXXABFXXX–––
The first example contains alignment of same-label motif instances with themselves (e.g., ABF to ABF), which provide no new information regarding their variation and therefore is of no value. The second alignment matches different instances of same-label motifs (e.g., ABF to AGG) but incorrectly aligns different-label motifs (e.g., AGG to KLM). It is only the third case that satisfies our criteria of a useful version of a sequence.
In order to identify the method that can be better used in practice to align any intra-song motifs (where the actual motifs are unknown), we design a simple experiment: we select all single sequences from the TuneFam-26 dataset that contain annotated motifs with two instances and devise three version-creation methods based on intuition. We then pairwise-align each original sequence to its different versions using different configurations of motif-agnostic alignment algorithms. In our experiment, the usefulness criteria are formulated as such: we are given the set L of all motif labels in a sequence S and , the set of all instances of intra-song motifs of label . We are interested in generating and pairwise-aligning different sequence versions with S, such that average relative likelihood that the different instances , are aligned as opposed to be aligned by chance, is greater than one and maximal:
At the same time the average relative likelihood that any instances of different-labels motifs are aligned as opposed to be aligned by chance should be less than one and minimal:
In practice, we are interested in the setup (version method plus alignment configuration) that maximizes . We experiment with three different automatic methods for version creation. Each method generates versions of the original sequence which is then pairwise-aligned to the original. We experiment with . The automatic methods for version creation are as follows:
- Permutations: The original sequence is first split into n same-size segments. Each version is one of the rearrangements of the segments. In our case n is arbitrarily set to four. Although automatic melody segmentation algorithms could have been used, we decided to used a fixed number of segments for the sake of simplicity.
- Halves: The original sequence is iteratively split in subsequences of half size until their length is equal to four or their number is equal to . Each version is a sequence of length equal to the original, created by the concatenation of one of the subsequences.
- Halves and shifts: A set of versions created by shifting the sequence by of its length to the right k times, resulting to k versions. The idea is to fuse the current set with the halves. We do that by randomly selecting versions from the halves method and versions from the current set.
The different versions are pairwise-aligned to the original using the following alignment configurations: Mafft-globalpair with (−4,−2) gap penalties, Mafft-globalpair-allowshift with (−4,−2) gap penalties and Mafft-localpair-allowshift with (−2,−1) gap penalties.
The , and figures for each version-creation method over all and for each alignment configuration, are presented in Figure 7. We notice that is greater than one and is less than 1 for most setups, meaning that useful alignments are indeed generated. However, the versions created with the halves method () and aligned to the original with localpair-allowshift with (−2,−1) gap penalties, achieve the highest (see the third column, second row in Figure 7). As such, we generate matrices (denoted halves-:4 and halves-:6 for both datasets) based on this configurations.
6.3. Experimental Setup
We are interested in evaluating whether the scoring matrices generated from alignments using the methods of the previous Section 6.2, outperform the the standard ±1 scoring matrix on the TuneFam-26 and Csv-60 datasets. In the retrieval case, we want to rank higher those melodies belonging to the same tune family or clique as the query. In the classification task, we want the tune family or clique of the highest ranked melody to correspond to the query’s (that is, we are doing a k-Nearest Neighbour (kNN) classification experiment with ).
Regarding the gap settings for this experiment, we should be extremely careful: the significant variation among the distribution of scores in between the matrices, renders the effect of the gap settings unpredictable, which can be problematic when aiming for a fair matrix comparison. Intuitively, there are two possible solutions: either compute the optimal gap settings per matrix, e.g., via a training process that optimizes the sensitivity (true positive rate) and selectivity (true negative rate) [92], or present their performance across a set of different penalties. The first approach is suitable for large datasets but is prone to over-fitting, and lacks a proper theoretical framework [93]. The second approach resembles the task of systematically comparing classifiers , which allows for a more complete view of each matrix by exploring the effect of the gap settings. Such an approach follows an intuitive classifier quality principle that agrees with our goal to develop generalizable solutions: “if a good classification is achieved only for a very small range in the parameter space, then for many applications it will be very difficult to achieve the best accuracy rate provided by the classifier” [94].
Picking a range of gap settings for each matrix that fairly represent its quality is not trivial. To solve the problem of fair matrix comparison, we need a meaningful intermediate mapping between two gap spaces and that work on matrices A and B respectively; or a single function under which (,A) and (,B) have the same image (are equivalent). Given two sequences to be aligned, we argue that two settings and are equivalent and comparable only when they are of same flexibility, meaning they result to alignments of equal length relative to the original sequences (which translates to equal ratio of gaps to non-gap symbols for both settings). This idea is based on the observation that for two settings that result to the same amount of gaps, the alignment quality is solely dependent on the matrices used; as such, the matrices can be compared fairly. To compute the flexibility values for each of the TuneFam-26 and Csv-60 datasets, we randomly selected a subset of 50 sequences and pairwise aligned them using a range of different gap settings per matrix (d, e with 0.1 intervals and ). We used subsets instead of whole datasets for efficiency reasons, while the gap boundaries 0.1 and 2.0 are considered typical. For each alignment of sequences and of length and respectively, we computed the gap to non-gap ratio , where corresponds to the amount of gaps in the alignment. The average r over all pairwise alignments using a gap setting on the matrix is what we consider the setting’s flexibility for that particular dataset. Given the mapping of each gap setting to the flexibility space, we can now fairly compare matrices by investigating their retrieval performance across different flexibility values.
6.4. Results
Figure 8a,b present the average precision and classification accuracy per substitution matrix over a range of flexibility values for the TuneFam-26 and Csv-60 datasets respectively. For the TuneFam-26 dataset and starting from the performance of the TFam-matrix, we observe that it significantly increases the retrieval performance across all gap settings. In average, the TFam-matrix increases the mean average precision from ID’s 0.65 to 0.69, indicating that some meaningful similarity/variation knowledge has been indeed captured. The Csv-matrix presents a higher retrieval performance than the ID matrix, but the significance is not constant across all flexibilities. The same holds for the intra-song motivic matrices halves-:4 and halves-:6. If we concatenate the average precision scores over all flexibilities per matrix, besides the TFam-matrix (see Figure 8a (top-right)) and perform a Friedman test, we discover that there is a significant difference between the four matrices. Post hoc analysis shows that the difference is due to the difference in between all pairs of matrices except halves-:4 and halves-:6. With regard to the classification accuracy, we do not observe a significant difference among the matrices.
For the Csv-60 dataset, the differences between matrices are more accentuated even through visual inspection. The Csv-matrix and learned matrix from the folk tunes collection TFam-matrix, significantly outperform ID across almost all flexibilities. The implication of their similar performance in average will be discussed in the next section. Regarding the intra-song motivic matrices, both present significantly better performance than ID. Excluding Csv-matrix, a Friedman test with post hoc analysis on the concatenated average precision, reveals significant difference between all pairs of matrices except for the halves-:4 (0.74) and halves-:6 (0.75).
6.5. Discussion
The results offer a number of interesting findings that are secondary to our main question, e.g., the insignificant difference among matrices for the classification task implies the existence of almost-duplicates for each query. Or the inverse relation between the retrieval performance of each matrix to the flexibility value, indicates that real-life retrieval systems should aim for gap settings of low flexibility. However, most importantly, our results strongly suggest that data-driven matrices, learned from either melody variations or intra-song motif variations, capture some meaningful relationships between notes that can find application in melody retrieval. In the case of TuneFam-26, the results are obviously not impressive despite their statistical significance. Van Kranenburg’s heuristics on the same dataset and task, pushed the MAP and classification accuracy to 0.85 and 0.98 respectively [1]. However, Van Kranenburg used only one arbitrarily selected gap setting (−0.8, −0.5), thus leaving the effect of gap settings uninvestigated. In our case however, we established a fairer framework for comparing matrices. In addition compared to our data-driven approach, Van Kranenburg had to experiment with a large number of heuristics to find the optimal. For the Csv-60 dataset, and in contrast to TuneFam-26, learning note relationships from folk tune variations or intra-song motifs seems to have a much more very positive effect in the overall retrieval performance. The reason behind this difference is unclear, but we can speculate based on intuition. In general, we observe that the vertical variation, i.e., among melodies belonging to the same family/clique, in the TuneFam-26 is more informative than the vertical variation in Csv-60. This explains why the TFam-matrix is successful on both datasets, while Csv-matrix is only successful on Csv-60. Probably, tune families contain an adequate amount of melodic variations that allows for the generation of an informative matrix. At the same time the horizontal variation, i.e., among intra-song motifs, is similarly informative in both datasets. This explains why the performance of halves-:4 and halves-:6 matrices lies in between that of the ID and the best performing matrix for each dataset.
In summary, the results indicate that vertical variation models are more beneficial in a retrieval scenario. At the same time, the captured relationships of the horizontal models seem inadequate to approximate their performance. This implies that the way a song varies across its length does not follow the same principles as its variation through time, but further confirmation with note-to-note alignments of intra-song motifs and melodic variations is required. Nevertheless, the modelling of horizontal variation can be considered highly appropriate for practical scenarios of melody retrieval and classification where clique information is unavailable.
7. Conclusions
Modelling music similarity is a fundamental, but intricate task in MIR. Most previous works on music similarity, practical or theoretical, relied heavily on heuristics. In contrast, our work focused on acquiring knowledge on music and melodic similarity in particular from the data itself. Since data-driven methods and tools have been under development for years in bioinformatics, and since biological and music sequence share resembling concepts, we investigated their applicability inside a musical context.
First, we tackled the concept of meaningful and musically significant alignments of related melodies, by applying the bioinformatics structural alignment metaphor to music motifs. Our results revealed that the Mafft multiple alignment algorithm, which uses gap-free sections as anchor points, is a natural fit for multiple melodic sequences; a strong indication of the importance of musical patterns for melodic similarity. Trusted MSA techniques made it possible to organize melodic variations such that melodic stability/variation can be analysed. We argue that our stability analysis findings are free of heuristics or biases that might have been introduced following other approaches.
Secondly, we investigated the modelling of global melodic similarity. We captured the probability of one note to be changed to another in a variation and created musically appropriate note-substitution scoring matrices for melodic alignment. We then put these matrices successfully to the test by designing retrieval and classification tasks. Our data-driven modelling of music similarity outperforms the naive ±1 matrix, indicating that indeed some novel knowledge was captured. Additionally, we showed that variations inside a melody can be an alternative source for modelling the similarity of variations among tune families or cliques of covers.
In general, we showed that bioinformatics tools and methods can find successful application in music, to answer in a reliable, data-driven way a number of important, on-going questions in MIR. We argue data-driven approaches, such as ours, constitute an ideal balance between the two occasionally contradicting goals of MIR, problem solving and knowledge acquisition. Unfortunately, in the current age of big data, the potential in exploring musical relationships that can aid both the digital music services and our understanding of music itself remains largely idle. We hope that our work will stimulate future research to focus on a more constructive direction.
Author Contributions
Dimitrios Bountouridis and Daniel G. Brown both developed the relationship to bioinformatics applications and designed the experiments. Dimitrios Bountouridis performed the experiments, analysed the data and wrote the paper. Daniel G. Brown, Frans Wiering and Remco C. Veltkamp contributed to the writing of the paper.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; nor in the decision to publish the results.
References
- Van Kranenburg, P. A Computational Approach to Content-Based Retrieval of Folk Song Melodies. Ph.D. Thesis, Utrecht University, Utrecht, The Netherlands, 2010. [Google Scholar]
- Volk, A.; Haas, W.; Kranenburg, P. Towards modelling variation in music as foundation for similarity. In Proceedings of the International Conference on Music Perception and Cognition, Thessaloniki, Greece, 23–28 July 2012; pp. 1085–1094. [Google Scholar]
- Pampalk, E. Computational Models of Music Similarity and Their Application to Music Information Retrieval. Ph.D. Thesis, Vienna University of Technology, Vienna, Austria, 2006. [Google Scholar]
- Volk, A.; Van Kranenburg, P. Melodic similarity among folk songs: An annotation study on similarity-based categorization in music. Music. Sci. 2012, 16, 317–339. [Google Scholar] [CrossRef]
- Marsden, A. Interrogating melodic similarity: A definitive phenomenon or the product of interpretation? J. New Music Res. 2012, 41, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Ellis, D.P.; Whitman, B.; Berenzweig, A.; Lawrence, S. The quest for ground truth in musical artist similarity. In Proceedings of the International Society of Music Information Retrieval Conference, Paris, France, 13–17 October 2002; pp. 170–177. [Google Scholar]
- Deliège, I. Similarity perception categorization cue abstraction. Music Percept. 2001, 18, 233–244. [Google Scholar] [CrossRef]
- Novello, A.; McKinney, M.F.; Kohlrausch, A. Perceptual evaluation of music similarity. In Proceedings of the International Society of Music Information Retrieval, Victoria, BC, Canada, 8–12 October 2006; pp. 246–249. [Google Scholar]
- Jones, M.C.; Downie, J.S.; Ehmann, A.F. Human similarity judgements: Implications for the design of formal evaluations. In Proceedings of the International Society of Music Information Retrieval, Vienna, Austria, 23–30 September 2007; pp. 539–542. [Google Scholar]
- Tversky, A. Features of similarity. Psychol. Rev. 1977, 84, 327–352. [Google Scholar] [CrossRef]
- Lamere, P. Social tagging and music information retrieval. J. New Music Res. 2008, 37, 101–114. [Google Scholar] [CrossRef]
- Berenzweig, A.; Logan, B.; Ellis, D.P.; Whitman, B. A large-scale evaluation of acoustic and subjective music-similarity measures. Comput. Music J. 2004, 28, 63–76. [Google Scholar] [CrossRef]
- Hu, N.; Dannenberg, R.B.; Lewis, A.L. A probabilistic model of melodic similarity. In Proceedings of the International Computer Music Conference, Göteborg, Sweden, 16–21 September 2002. [Google Scholar]
- Hu, N.; Dannenberg, R.B. A comparison of melodic database retrieval techniques using sung queries. In Proceedings of the 2nd ACM/IEEE-Cs Joint Conference on Digital Libraries, Portland, OR, USA, 13–17 July 2002; pp. 301–307. [Google Scholar]
- Hanna, P.; Robine, M.; Rocher, T. An alignment based system for chord sequence retrieval. In Proceedings of the 9th ACM/IEEE-Cs Joint Conference on Digital Libraries, Austin, TX, USA, 14–19 June 2009; pp. 101–104. [Google Scholar]
- Bronson, B.H. Melodic stability in oral transmission. J. Int. Folk Music Counc. 1951, 3, 50–55. [Google Scholar] [CrossRef]
- Krogh, A. An introduction to hidden markov models for biological sequences. New Compr. Biochem. 1998, 32, 45–63. [Google Scholar]
- Bascom, W. The main problems of stability and change in tradition. J. Int. Folk Music Counc. 1959, 11, 7–12. [Google Scholar] [CrossRef]
- Drake, C.; Bertrand, D. The quest for universals in temporal processing in music. Ann. N. Y. Acad. Sci. 2001, 930, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Gurney, E. The Power of Sound; Cambridge University Press: Cambridge, UK, 2011. [Google Scholar]
- Casey, M.; Slaney, M. The importance of sequences in musical similarity. In Proceedings of the International Conference On Acoustics, Speech and Signal Processing, Toulouse, France, 14–19 May 2006; pp. 5–8. [Google Scholar]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Valdar, W.S. Scoring residue conservation. Proteins Struct. Funct. Bioinform. 2002, 48, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Luscombe, N.M.; Greenbaum, D.; Gerstein, M. What is bioinformatics? A proposed definition and overview of the field. Methods Inf. Med. 2001, 40, 346–358. [Google Scholar] [PubMed]
- Bountouridis, D.; Wiering, F.; Brown, D.; Veltkamp, R.C. Towards polyphony reconstruction using multidimensional multiple sequence alignment. In Proceedings of the International Conference on Evolutionary and Biologically Inspired Music and Art, Amsterdam, The Netherlands, 19–21 April 2017; pp. 33–48. [Google Scholar]
- Bountouridis, D.; Brown, D.; Koops, H.V.; Wiering, F.; Veltkamp, R. Melody retrieval and classification using biologically-inspired techniques. In Proceedings of the International Conference on Evolutionary and Biologically Inspired Music and Art, Amsterdam, The Netherlands, 19–21 April 2017; pp. 49–64. [Google Scholar]
- Bountouridis, D.; Koops, H.V.; Wiering, F.; Veltkamp, R. A data-driven approach to chord similarity and chord mutability. In Proceedings of the International Conference on Multimedia Big Data, Taipei, Taiwan, 20–22 April 2016; pp. 275–278. [Google Scholar]
- Nguyen, K.; Guo, X.; Pan, Y. Multiple Biological Sequence Alignment: Scoring Functions, Algorithms and Evaluation; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Durbin, R.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Mongeau, M.; Sankoff, D. Comparison of musical sequences. Comput. Humanit. 1990, 24, 161–175. [Google Scholar] [CrossRef]
- Ewert, S.; Müller, M.; Dannenberg, R.B. Towards reliable partial music alignments using multiple synchronization strategies. In Proceedings of the International Workshop on Adaptive Multimedia Retrieval, Madrid, Spain, 24–25 September 2009; pp. 35–48. [Google Scholar]
- Serra, J.; Gómez, E.; Herrera, P.; Serra, X. Chroma binary similarity and local alignment applied to cover song identification. Audio Speech Lang. Process. 2008, 16, 1138–1151. [Google Scholar] [CrossRef]
- Müllensiefen, D.; Frieler, K. Optimizing measures of melodic similarity for the exploration of a large folk song database. In Proceedings of the International Society of Music Information Retrieval, Barcelona, Spain, 10–15 October 2004; pp. 1–7. [Google Scholar]
- Müllensiefen, D.; Frieler, K. Cognitive adequacy in the measurement of melodic similarity: Algorithmic vs. human judgements. Comput. Musicol. 2004, 13, 147–176. [Google Scholar]
- Sailer, C.; Dressler, K. Finding cover songs by melodic similarity. In Proceedings of the Annual Music Information Retrieval Evaluation Exchange, Victoria, BC, Canada, 8–12 September 2006; Available online: www.music-ir.org/mirex/abstracts/2006/CS_sailer.pdf (accessed on 28 November 2017).
- Ross, J.C.; Vinutha, T.; Rao, P. Detecting melodic motifs from audio for hindustani classical music. In Proceedings of the International Society of Music Information Retrieval, Porto, Portugal, 8–12 October 2012; pp. 193–198. [Google Scholar]
- Salamon, J.; Rohrmeier, M. A quantitative evaluation of a two stage retrieval approach for a melodic query by example system. In Proceedings of the International Society of Music Information Retrieval, Kobe, Japan, 26–30 October 2009; pp. 255–260. [Google Scholar]
- Hu, N.; Dannenberg, R.B.; Tzanetakis, G. Polyphonic audio matching and alignment for music retrieval. In Proceedings of the Workshop in Applications of Signal Processing to Audio and Acoustics, New Paltz, NY, USA, 19–22 October 2003; pp. 185–188. [Google Scholar]
- Ewert, S.; Müller, M.; Grosche, P. High resolution audio synchronization using chroma onset features. In Proceedings of the International Conference On Acoustics, Speech and Signal Processing, Taipei, Taiwan, 19–24 April 2009; pp. 1869–1872. [Google Scholar]
- Balke, S.; Arifi-Müller, V.; Lamprecht, L.; Müller, M. Retrieving audio recordings using musical themes. In Proceedings of the International Conference on Acoustics, Speech and Signal Processing, Shanghai, China, 20–25 March 2016; pp. 281–285. [Google Scholar]
- Raffel, C.; Ellis, D.P. Large-scale content-based matching of midi and audio files. In Proceedings of the International Society of Music Information Retrieval, Malaga, Spain, 26–30 October 2015; pp. 234–240. [Google Scholar]
- Müller, M. Information Retrieval for Music and Motion; Springer: Berlin, Germany, 2007. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. Clustal W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiang, T. On the complexity of multiple sequence alignment. J. Comput. Biol. 1994, 1, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Kemena, C.; Notredame, C. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 2009, 25, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C. Towards automatic music performance comparison with the multiple sequence alignment technique. In Proceedings of the International Conference on Multimedia Modelling, Huangshan, China, 7–9 January 2013; pp. 391–402. [Google Scholar]
- Wang, S.; Ewert, S.; Dixon, S. Robust joint alignment of multiple versions of a piece of music. In Proceedings of the International Society of Music Information Retrieval, Taipei, Taiwan, 27–31 October 2014; pp. 83–88. [Google Scholar]
- Knees, P.; Schedl, M.; Widmer, G. Multiple lyrics alignment: Automatic retrieval of song lyrics. In Proceedings of the International Society of Music Information Retrieval, London, UK, 11–15 October 2005; pp. 564–569. [Google Scholar]
- Poliner, G.E.; Ellis, D.P.; Ehmann, A.F.; Gómez, E.; Streich, S.; Ong, B. Melody transcription from music audio: Approaches and evaluation. IEEE Trans. Audio Speech Lang. Process. 2007, 15, 1247–1256. [Google Scholar] [CrossRef]
- Kim, Y.E.; Chai, W.; Garcia, R.; Vercoe, B. Analysis of a contour-based representation for melody. In Proceedings of the International Society of Music Information Retrieval, Plymouth, MA, USA, 23–25 October 2000. [Google Scholar]
- Huron, D. The melodic arch in western folksongs. Comput. Musicol. 1996, 10, 3–23. [Google Scholar]
- Margulis, E.H. A model of melodic expectation. Music Percept. Interdiscip. J. 2005, 22, 663–714. [Google Scholar] [CrossRef]
- Salamon, J.; Gómez, E.; Ellis, D.P.; Richard, G. Melody extraction from polyphonic music signals: Approaches, applications, and challenges. IEEE Signal Process. Mag. 2014, 31, 118–134. [Google Scholar] [CrossRef]
- Suyoto, I.S.; Uitdenbogerd, A.L. Simple efficient n-gram indexing for effective melody retrieval. In Proceedings of the Annual Music Information Retrieval Evaluation Exchange, London, UK, 14 September 2005; Available online: pdfs.semanticscholar.org/4103/07d4f5398b1588b04d2916f0f592813a3d0a.pdf (accessed on 28 November 2017).
- Ryynanen, M.; Klapuri, A. Query by humming of midi and audio using locality sensitive hashing. In Proceedings of the International Conderence on Acoustics, Speech and Signal Processing, Las Vegas, NV, USA, 31 March–4 April 2008; pp. 2249–2252. [Google Scholar]
- Hillewaere, R.; Manderick, B.; Conklin, D. Alignment methods for folk tune classification. In Proceedings of the Annual Conference of the German Classification Society on Data Analysis, Machine Learning and Knowledge Discovery, Hildesheim, Germany, 1–3 August 2014; pp. 369–377. [Google Scholar]
- Gómez, E.; Klapuri, A.; Meudic, B. Melody description and extraction in the context of music content processing. J. New Music Res. 2003, 32, 23–40. [Google Scholar] [CrossRef]
- Van Kranenburg, P.; de Bruin, M.; Grijp, L.; Wiering, F. The Meertens Tune Collections; Meertens Online Reports; Meertens Institute: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Bountouridis, D.; Van Balen, J. The cover song variation dataset. In Proceedings of the International Workshop on Folk Music Analysis, Istanbul, Turkey, 12–13 June 2014. [Google Scholar]
- Raghava, G.; Barton, G. Quantification of the variation in percentage identity for protein sequence alignments. BMC Bioinform. 2006, 7, 415–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewert, S.; Müller, M.; Müllensiefen, D.; Clausen, M.; Wiggins, G.A. Case study “Beatles songs” what can be learned from unreliable music alignments? In Proceedings of the Dagstuhl Seminar, Dagstuhl, Germany, 15–20 March 2009. [Google Scholar]
- Prätzlich, T.; Müller, M. Triple-based analysis of music alignments without the need of ground-truth annotations. In Proceedings of the International Conference on Acoustics, Speech and Signal Processing, Shanghai, China, 20–25 March 2016; pp. 266–270. [Google Scholar]
- Pei, J.; Grishin, N.V. Promals: Towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 2007, 23, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Blackburne, B.P.; Whelan, S. Measuring the distance between multiple sequence alignments. Bioinformatics 2012, 28, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Bountouridis, D.; Van Balen, J. Towards capturing melodic stability. In Proceedings of the Interdisciplinary Musicology Conference, Berlin, Germany, 4–6 December 2014. [Google Scholar]
- Cowdery, J.R. A fresh look at the concept of tune family. Ethnomusicology 1984, 28, 495–504. [Google Scholar] [CrossRef]
- Hogeweg, P.; Hesper, B. The alignment of sets of sequences and the construction of phyletic trees: An integrated method. J. Mol. Evol. 1984, 20, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Munson, P.J. A novel randomized iterative strategy for aligning multiple protein sequences. Comput. Appl. Biosci. Cabios 1991, 7, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, O. Optimal alignment between groups of sequences and its application to multiple sequence alignment. Comput. Appl. Biosci. Cabios 1993, 9, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Holm, L.; Higgins, D.G. Coffee: An objective function for multiple sequence alignments. Bioinformatics 1998, 14, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Morgenstern, B.; Dress, A.; Werner, T. Multiple dna and protein sequence alignment based on segment-to-segment comparison. Proc. Natl. Acad. Sci. USA 1996, 93, 12098–12103. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. Mafft: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F. Generalized affine gap costs for protein sequence alignment. Proteins Struct. Funct. Genet. 1998, 32, 88–96. [Google Scholar] [CrossRef]
- Carroll, H.; Clement, M.J.; Ridge, P.; Snell, Q.O. Effects of gap open and gap extension penalties. In Proceedings of the Biotechnology and Bioinformatics Symposium, Provo, Utah, 20–21 October 2006; pp. 19–23. [Google Scholar]
- Dannenberg, R.B.; Hu, N. Understanding search performance in query-by-humming systems. In Proceedings of the Conference of the International Society of Music Information Retrieval, Barcelona, Spain, 10–15 October 2004. [Google Scholar]
- Margulis, E.H. Musical repetition detection across multiple exposures. Music Percept. Interdiscip. J. 2012, 29, 377–385. [Google Scholar] [CrossRef]
- Bigand, E.; Pineau, M. Context effects on melody recognition: A dynamic interpretation. Curr. Psychol. Cogn. 1996, 15, 121–134. [Google Scholar]
- Klusen, E.; Moog, H.; Piel, W. Experimente zur mündlichen Tradition von Melodien. Jahrbuch Fur Volksliedforschung 1978, 23, 11–32. [Google Scholar]
- Bigand, E. Perceiving musical stability: The effect of tonal structure, rhythm, and musical expertise. J. Exp. Psychol. Hum. Percept. Perform. 1997, 23, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Schmuckler, M.A.; Boltz, M.G. Harmonic and rhythmic influences on musical expectancy. Atten. Percept. Psychophys. 1994, 56, 313–325. [Google Scholar] [CrossRef]
- Janssen, B.; Burgoyne, J.A.; Honing, H. Predicting variation of folk songs: A corpus analysis study on the memorability of melodies. Front. Psychol. 2017, 8, 621. [Google Scholar] [CrossRef] [PubMed]
- Van Balen, J.; Bountouridis, D.; Wiering, F.; Veltkamp, R. Cognition-inspired descriptors for scalable cover song retrieval. In Proceedings of the International Society of Music Information Retrieval, Taipei, Taiwan, 27–31 October 2014. [Google Scholar]
- Schoenberg, A.; Stein, L. Fundamentals of Musical Composition; Faber: London, UK, 1967. [Google Scholar]
- De Haas, W.B.; Wiering, F.; Veltkamp, R. A geometrical distance measure for determining the similarity of musical harmony. Int. J. Multimed. Inf. Retr. 2013, 2, 189–202. [Google Scholar] [CrossRef]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [PubMed]
- Hirjee, H.; Brown, D.G. Rhyme analyser: An analysis tool for rap lyrics. In Proceedings of the International Society of Music Information Retrieval, Utrecht, The Netherlands, 9–13 August 2010. [Google Scholar]
- Hirjee, H.; Brown, D.G. Solving misheard lyric search queries using a probabilistic model of speech sounds. In Proceedings of the International Society of Music Information Retrieval, Utrecht, The Netherlands, 9–13 August 2010; pp. 147–152. [Google Scholar]
- Hertz, G.Z.; Stormo, G.D. Identifying dna and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 1999, 15, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Tomii, K. Revisiting amino acid substitution matrices for identifying distantly related proteins. Bioinformatics 2013, 30, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Li, M.; Fu, H. Determination of optimal parameters of MAFFT program based on BAliBASE3.0 database. SpringerPlus 2016, 5, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Amancio, D.R.; Comin, C.H.; Casanova, D.; Travieso, G.; Bruno, O.M.; Rodrigues, F.A.; da Fontoura Costa, L. A systematic comparison of supervised classifiers. PLoS ONE 2014, 9, e94137. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The 15 unaligned sequences of the tune family “Daar ging een heer”. Colours correspond to motif labels. White colour indicates no motif label.
Figure 1.
The 15 unaligned sequences of the tune family “Daar ging een heer”. Colours correspond to motif labels. White colour indicates no motif label.
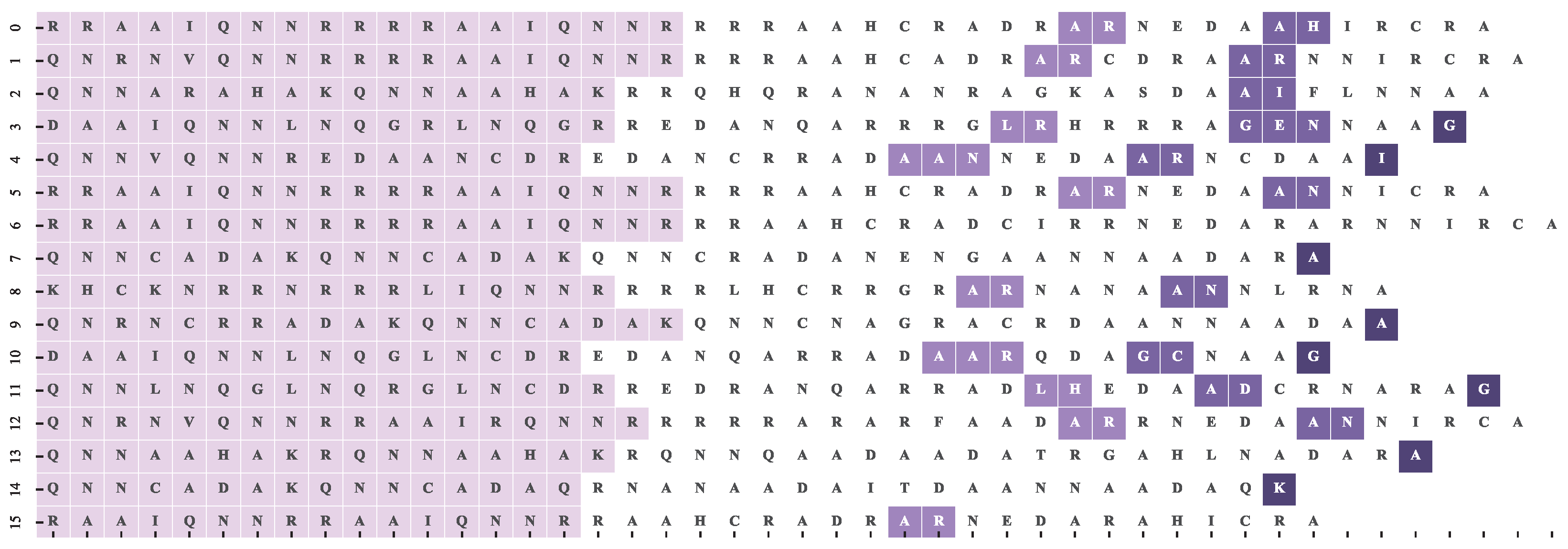
Figure 2.
Automatically aligned melodic sequences of the tune family “Daar ging een heer” using the following configurations (top–bottom): Mafft-genafpair-4.-2., Mafft-globalpair-allowshift-4.-2., Mafft-loacalpair-2.-1.5, Mafft-localpair-allowshift-3.-1.5 and T-Coffee-8-0.5. Colours correspond to motif labels. White colour indicates no motif label.
Figure 2.
Automatically aligned melodic sequences of the tune family “Daar ging een heer” using the following configurations (top–bottom): Mafft-genafpair-4.-2., Mafft-globalpair-allowshift-4.-2., Mafft-loacalpair-2.-1.5, Mafft-localpair-allowshift-3.-1.5 and T-Coffee-8-0.5. Colours correspond to motif labels. White colour indicates no motif label.
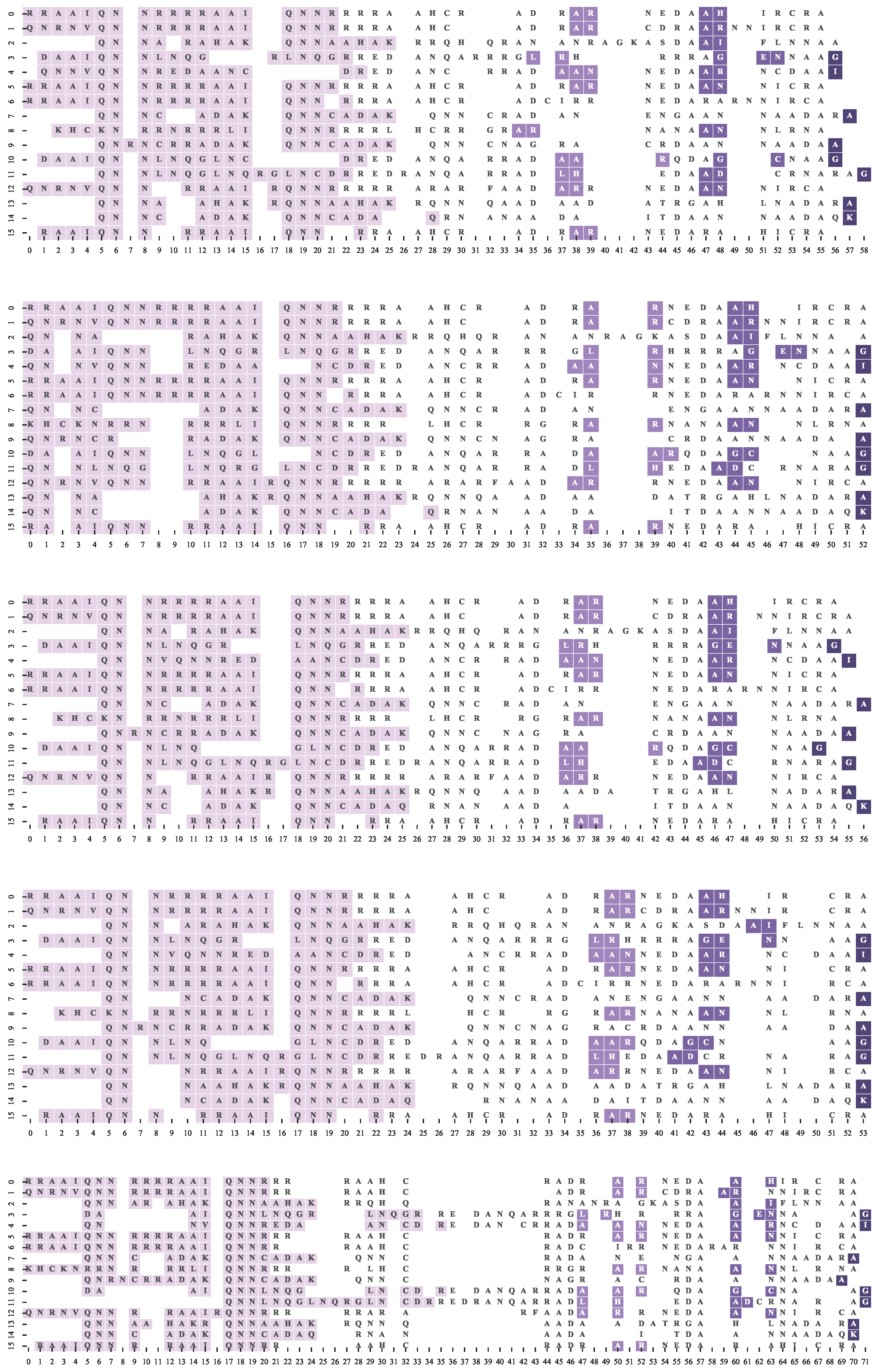
Figure 3.
Normalized motif-Sum-Of-Pairs (SOP) score (y-axis) for different gap settings (x-axis) and Multiple Sequence Alignment (MSA) algorithms.
Figure 3.
Normalized motif-Sum-Of-Pairs (SOP) score (y-axis) for different gap settings (x-axis) and Multiple Sequence Alignment (MSA) algorithms.
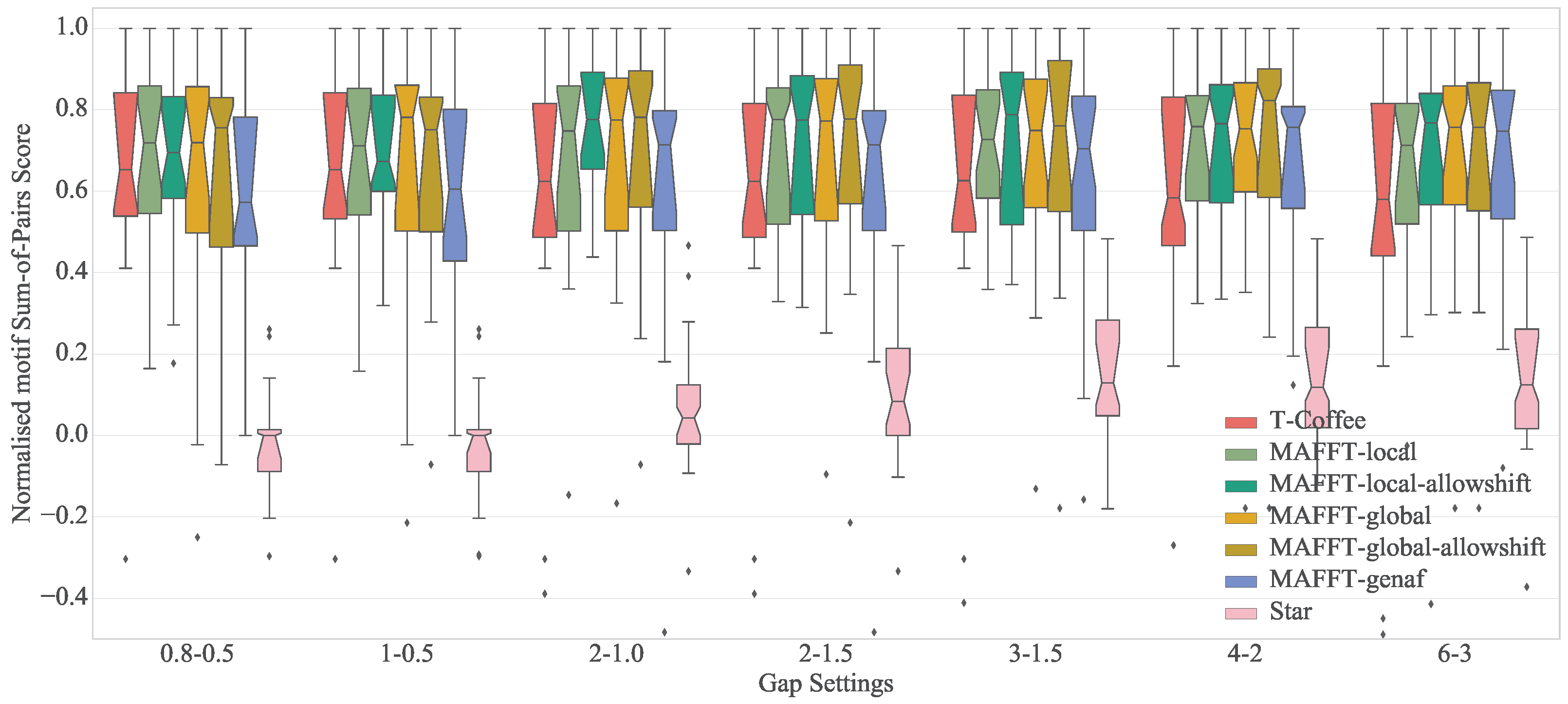
Figure 4.
Position versus various stability scores (Information Content (IC), Weighted Sum-Of-Pairs (WSOP) and PS) for the TuneFam-26 dataset using three different alignment configurations. Position versus gap ratio is also presented (first to the left). Points are quantised to 10 bins.
Figure 4.
Position versus various stability scores (Information Content (IC), Weighted Sum-Of-Pairs (WSOP) and PS) for the TuneFam-26 dataset using three different alignment configurations. Position versus gap ratio is also presented (first to the left). Points are quantised to 10 bins.
Figure 5.
Position versus various stability scores (IC, WSOP and PS) for the Csv-60 dataset using three different alignment configurations. Position versus gap ratio is also presented (first to the left). Points are quantised to 10 bins.
Figure 5.
Position versus various stability scores (IC, WSOP and PS) for the Csv-60 dataset using three different alignment configurations. Position versus gap ratio is also presented (first to the left). Points are quantised to 10 bins.
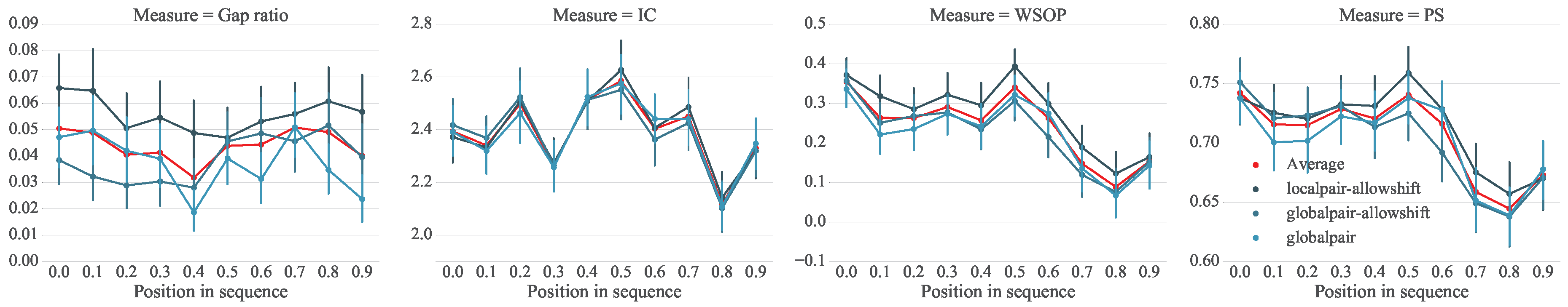
Figure 6.
Pitch interval versus IC stability for the TuneFam-26 (left) and Csv-60 (right) datasets using three different alignment configurations. Interval frequencies per dataset are also presented. Results for only the 13 most frequent intervals are presented.
Figure 6.
Pitch interval versus IC stability for the TuneFam-26 (left) and Csv-60 (right) datasets using three different alignment configurations. Interval frequencies per dataset are also presented. Results for only the 13 most frequent intervals are presented.

Figure 7.
The average relative likelihood (first column) that the different motif instances are aligned as opposed to be aligned by chance, the average relative likelihood (middle column) that any instances of different-labels motifs are aligned as opposed to be aligned by chance, and (last column) figures for each version-creation method (per row) over all and for each alignment configuration.
Figure 7.
The average relative likelihood (first column) that the different motif instances are aligned as opposed to be aligned by chance, the average relative likelihood (middle column) that any instances of different-labels motifs are aligned as opposed to be aligned by chance, and (last column) figures for each version-creation method (per row) over all and for each alignment configuration.
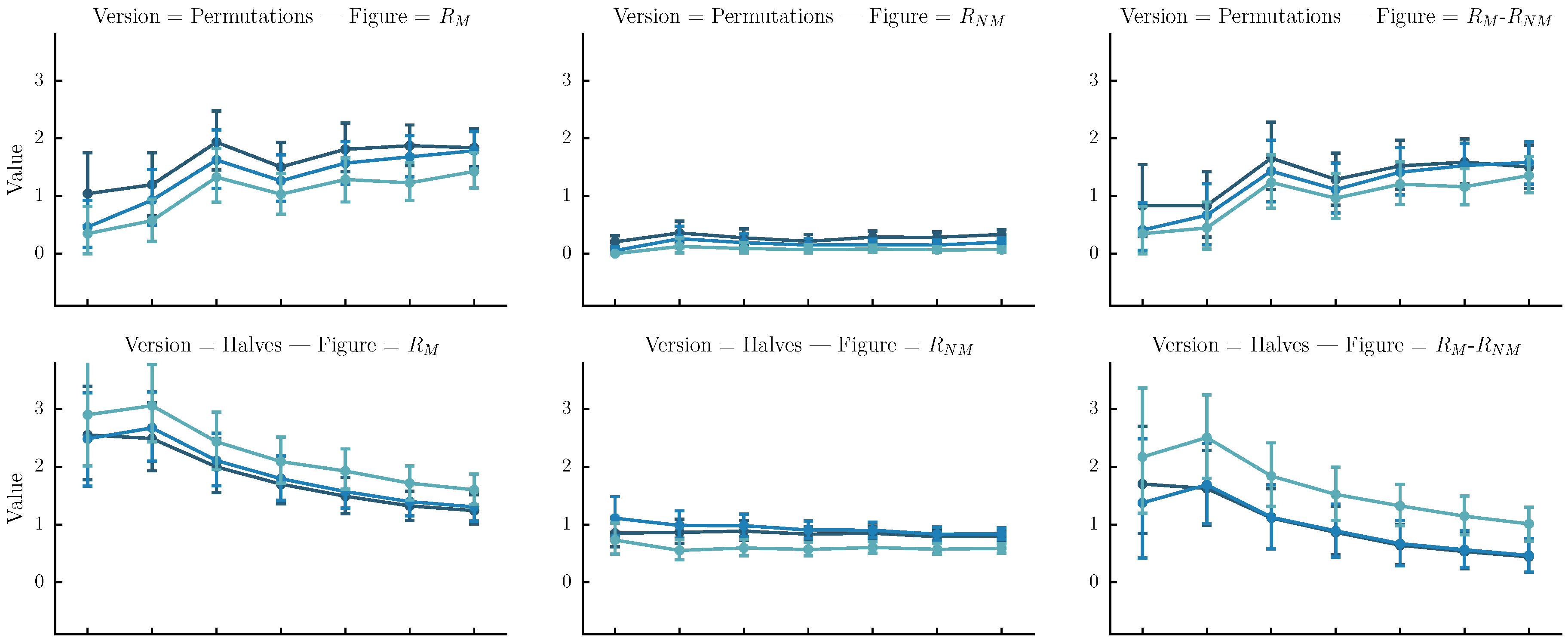
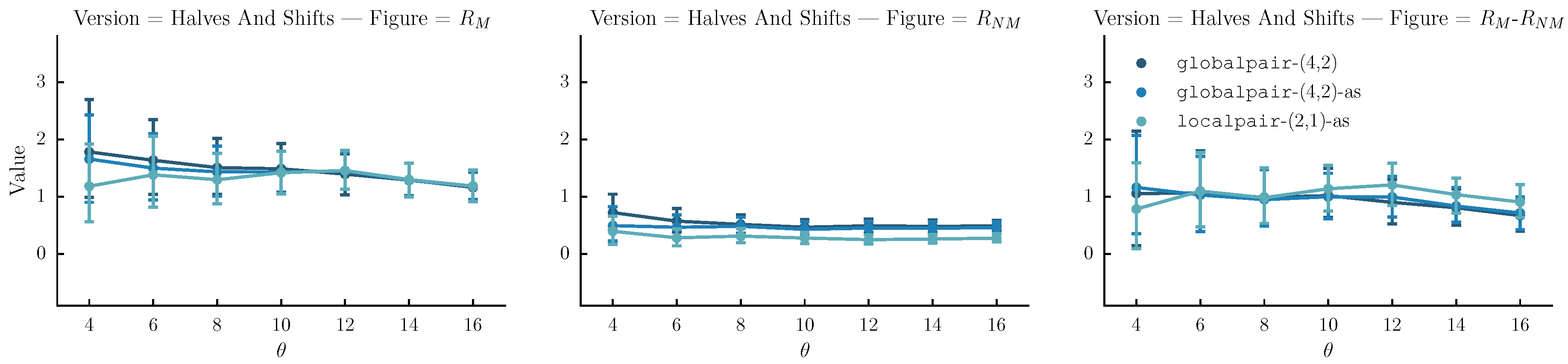
Figure 8.
Average precision and classification accuracy for each matrix over a range of flexibility values (left) for the TuneFam-26 (a) and Csv-60 (b) datasets. The average precision and accuracy over all flexibilities per matrix are also presented on the right.
Figure 8.
Average precision and classification accuracy for each matrix over a range of flexibility values (left) for the TuneFam-26 (a) and Csv-60 (b) datasets. The average precision and accuracy over all flexibilities per matrix are also presented on the right.
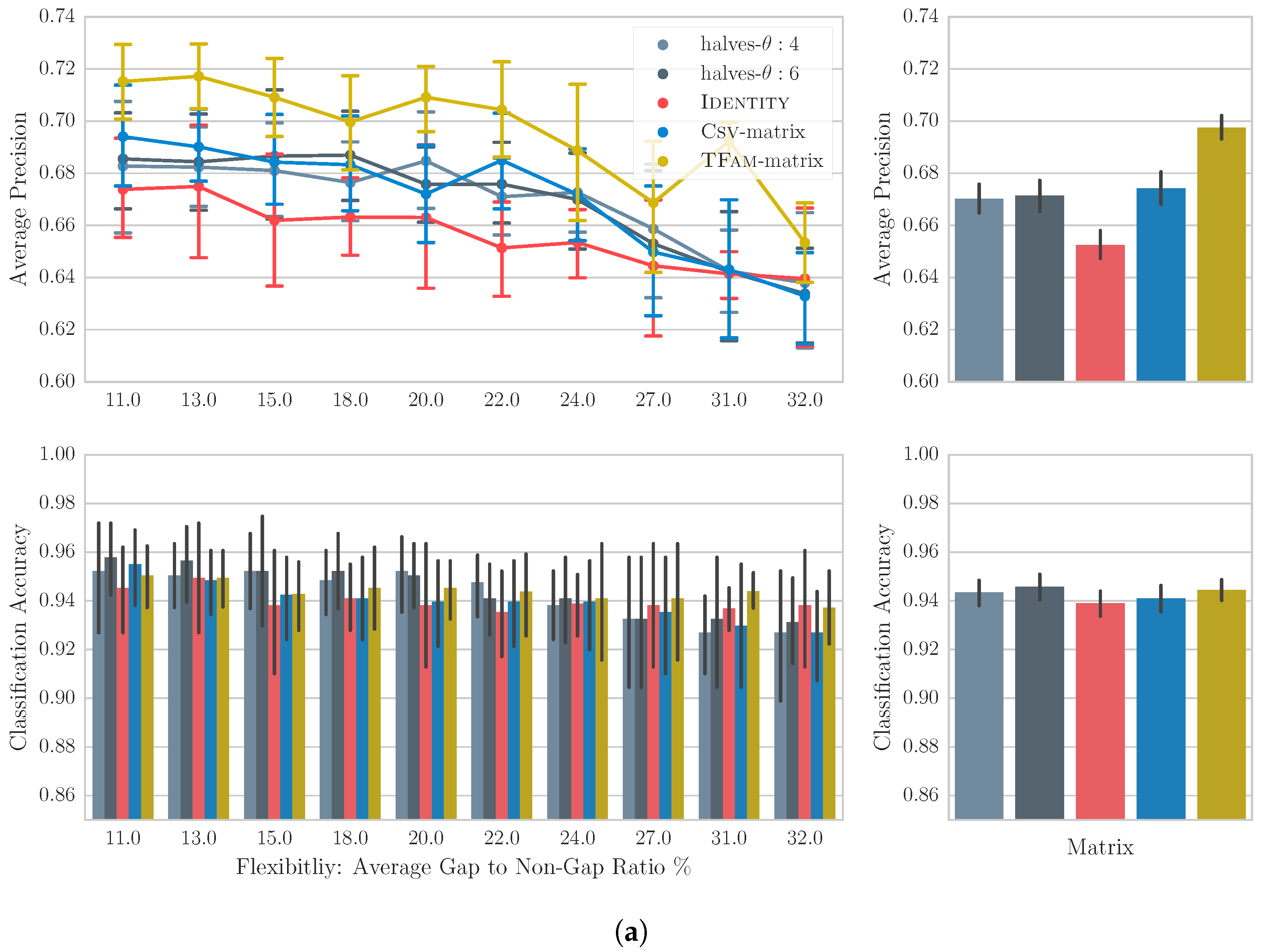
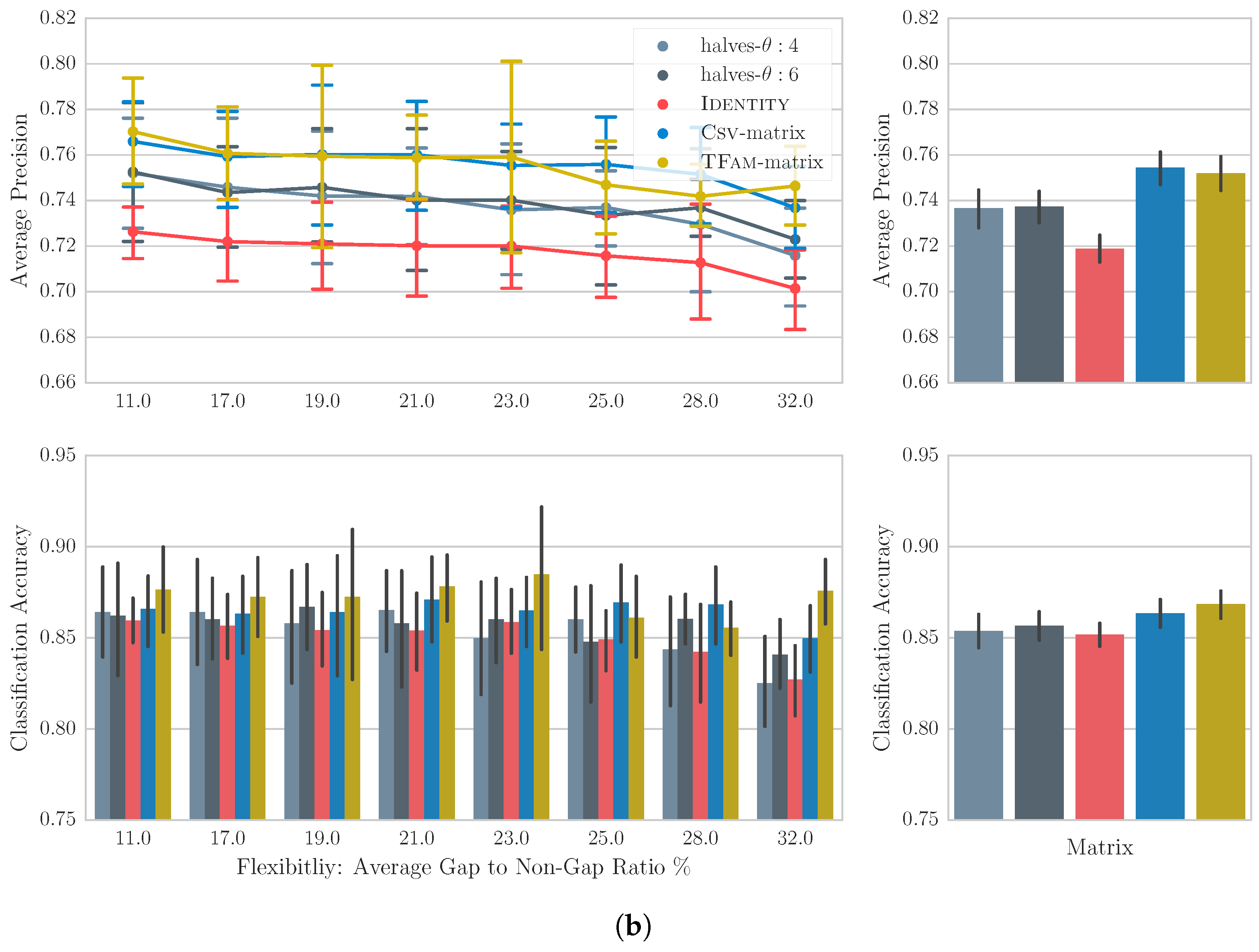

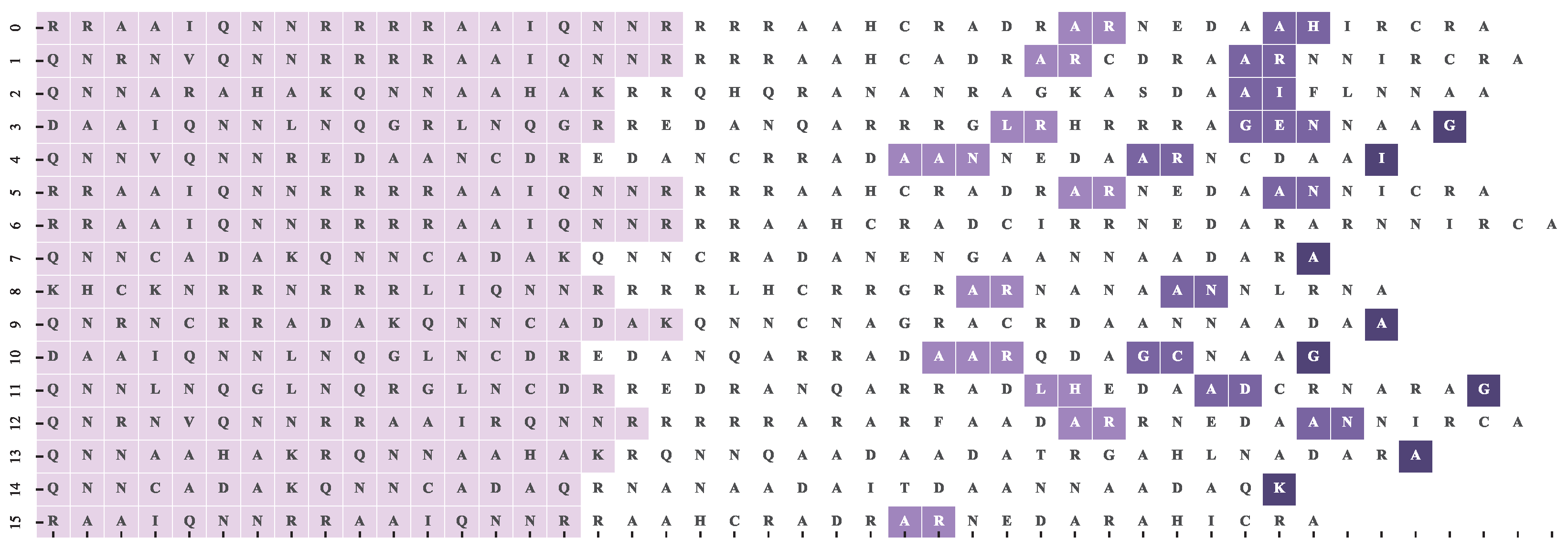{kind=link}
{kind=link}
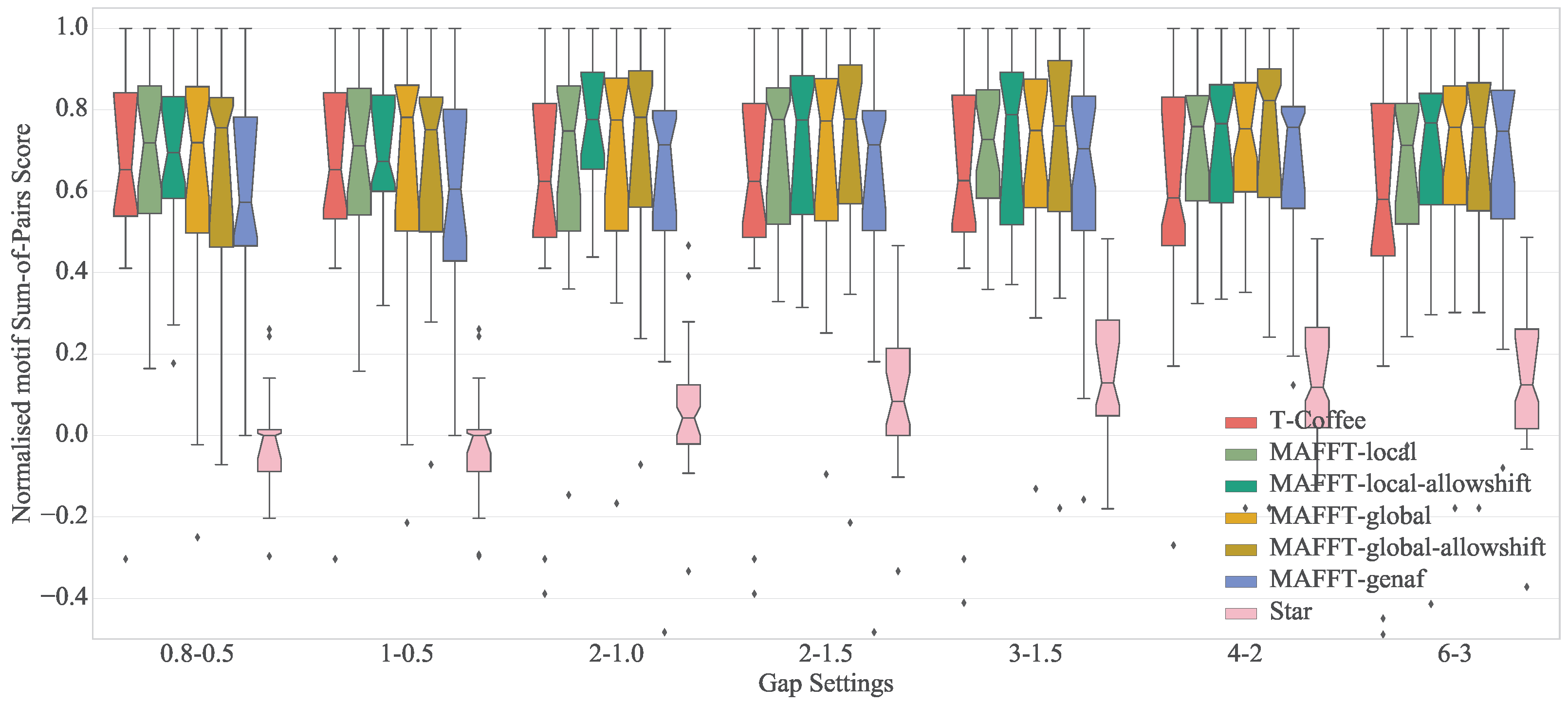{kind=link}
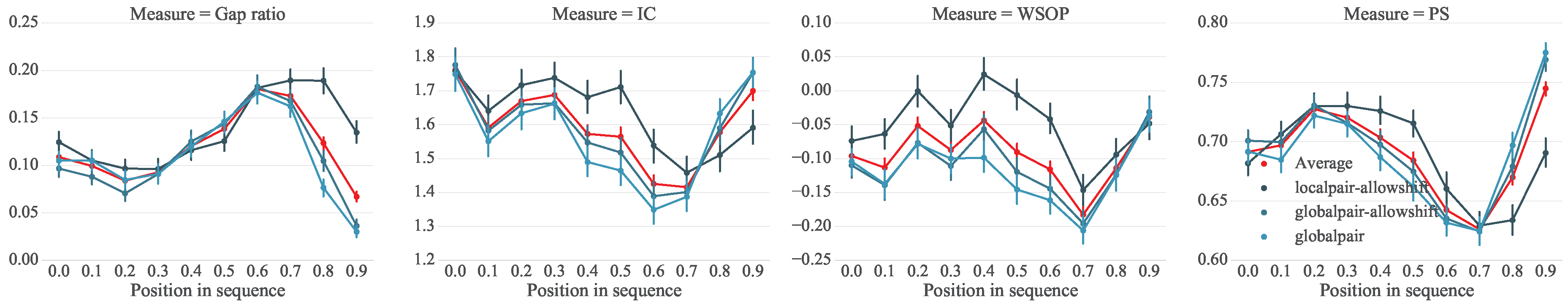{kind=link}
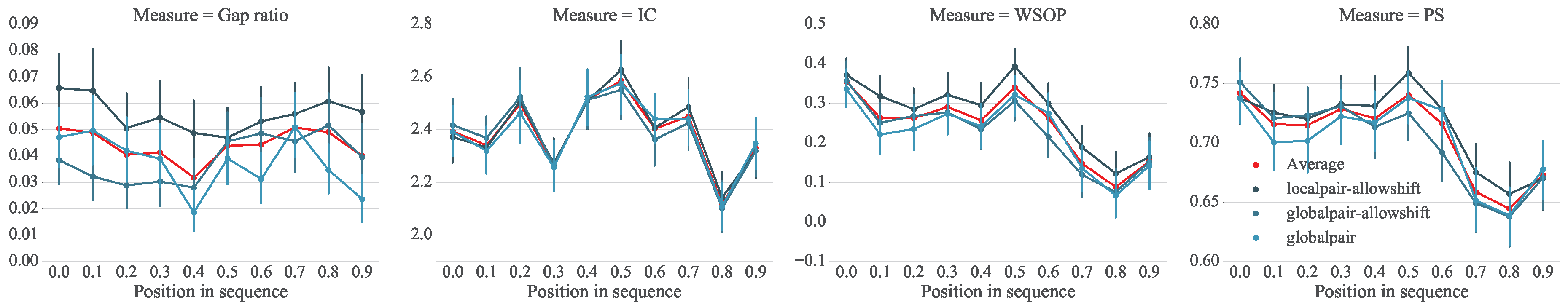{kind=link}
{kind=link}
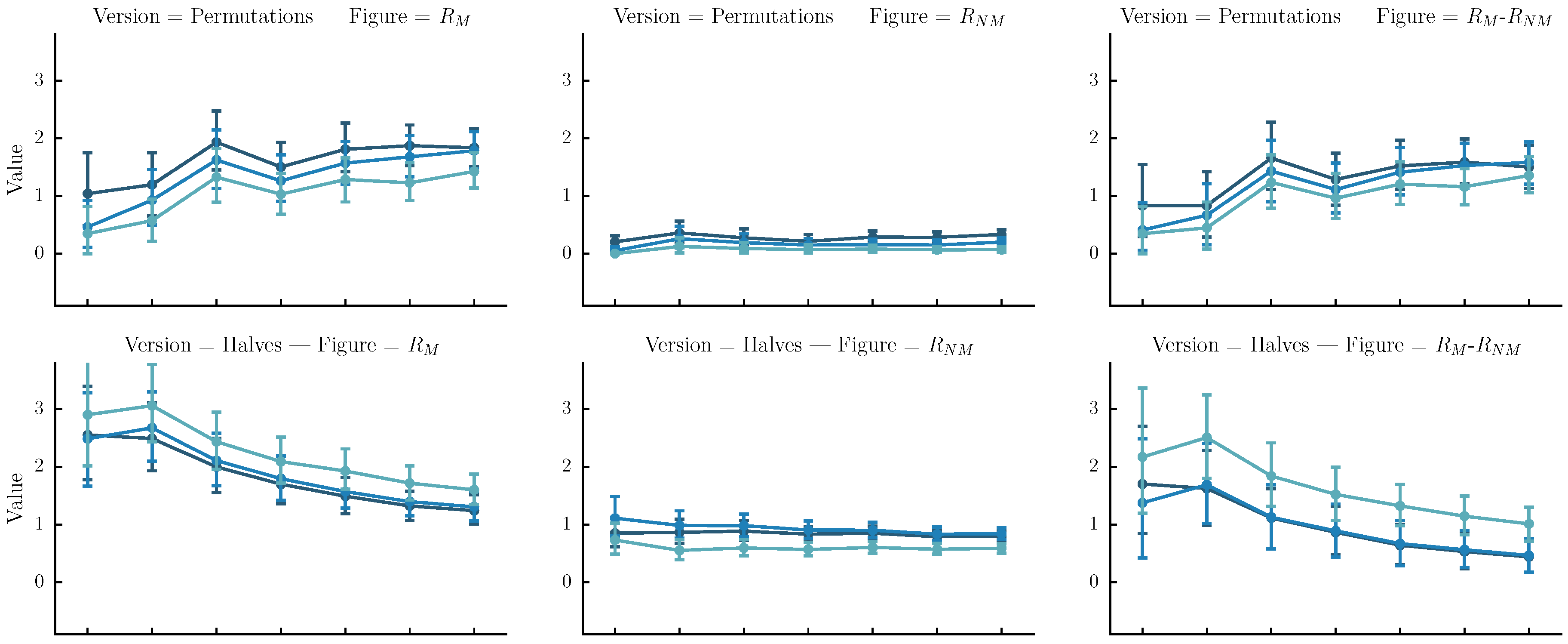{kind=link}
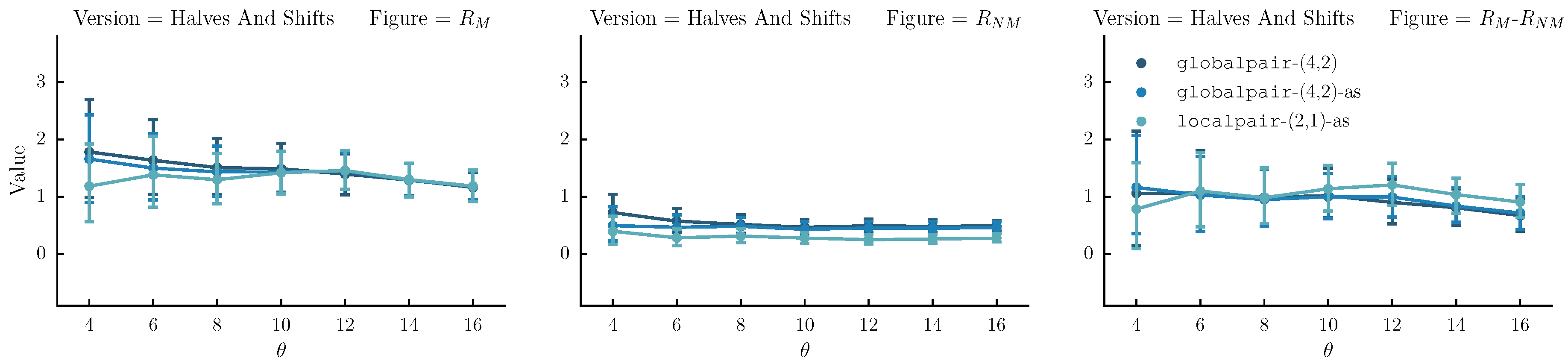{kind=link}
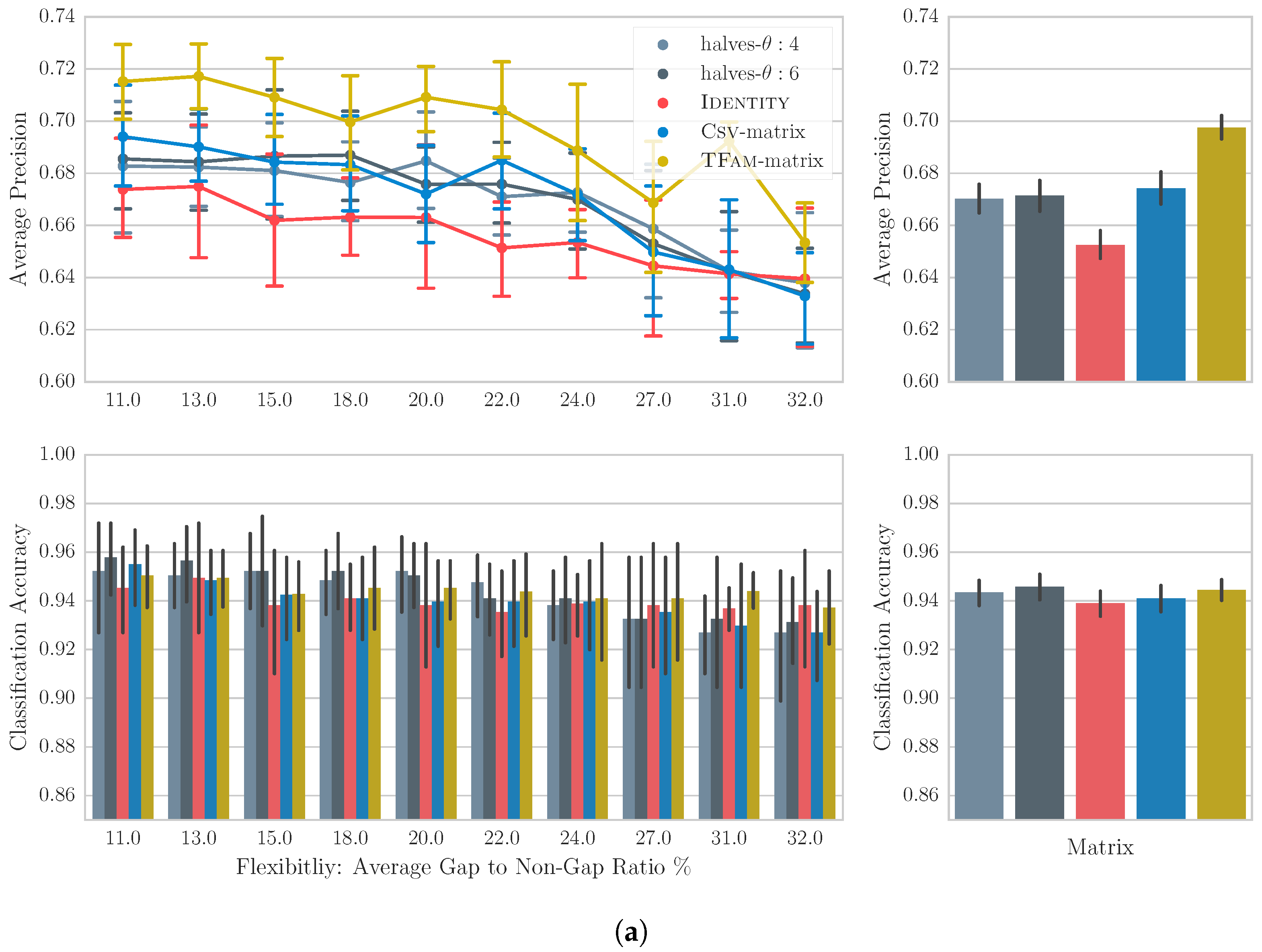{kind=link}
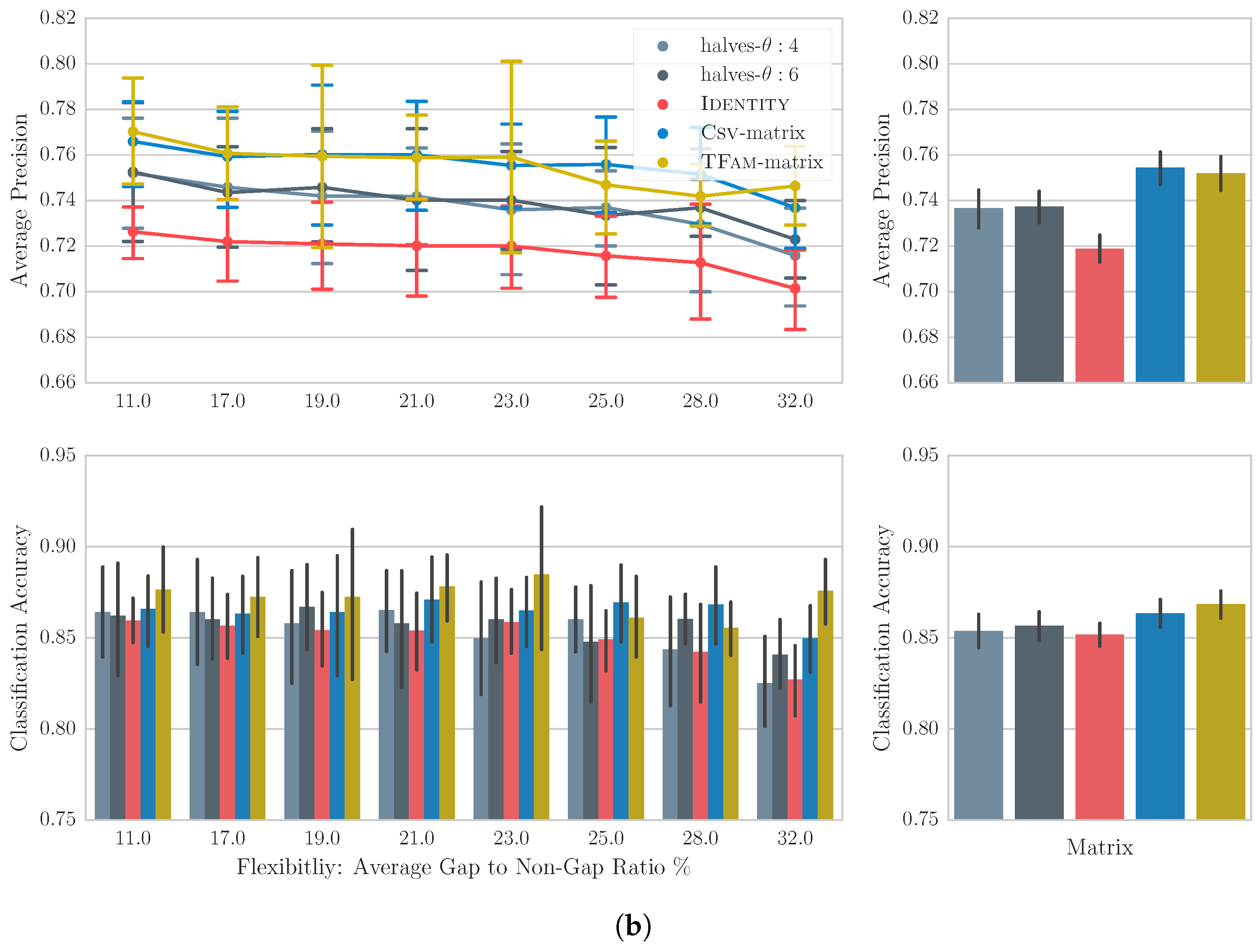{kind=link}
Table 1.
Shared concepts and terms between music and bioinformatics.
| Music | Bioinformatics |
|---|---|
| Melodies, chord progressions | DNA, proteins |
| Oral transmission, cover songs | Evolution |
| Variations, covers | Homologues |
| Tune family, clique | Homology, family |
| Cover song identification, melody retrieval | Homologue detection |
| Stability | Conservation |
Table 2.
Summary statistics for the datasets considered in our work. We also present the Area Under the Curve (AUC) value for the Receiver Operating Characteristic curve (ROC) on the Percentage Sequence Identity (PID). Given two aligned sequences, the PID score is simply the number of identical positions in the alignment divided by the number of aligned positions [62]. The higher the AUC PID the more similar the sequences are in a clique compared to the whole dataset. It should be noted that the alphabet size presented corresponds to the number of unique symbols appearing in the dataset.
Table 2.
Summary statistics for the datasets considered in our work. We also present the Area Under the Curve (AUC) value for the Receiver Operating Characteristic curve (ROC) on the Percentage Sequence Identity (PID). Given two aligned sequences, the PID score is simply the number of identical positions in the alignment divided by the number of aligned positions [62]. The higher the AUC PID the more similar the sequences are in a clique compared to the whole dataset. It should be noted that the alphabet size presented corresponds to the number of unique symbols appearing in the dataset.
| Summary statistics | TuneFam-26 | Csv-60 |
|---|---|---|
| Number of cliques | 26 | 60 |
| Clique Size median (var) | 13.0 (4.016) | 4.0 (1.146) |
| Sequence Length median (var) | 43.0 (15.003) | 26.0 (10.736) |
| AUC PID | 0.84 | 0.94 |
| Alphabet Size | 22 | 22 |
Table 3.
Median (standard deviation) normalized motif-SOP scores for different MSA algorithms and gap settings. For T-Coffee, the gap open values are multiplied by 10.
Table 3.
Median (standard deviation) normalized motif-SOP scores for different MSA algorithms and gap settings. For T-Coffee, the gap open values are multiplied by 10.
| Algorithm | 0.8–0.5 | 1–0.5 | 2–1.0 | 2–1.5 | 3–1.5 | 4–2 | 6–3 |
|---|---|---|---|---|---|---|---|
| Mafft-genaf | 0.57 (0.95) | 0.61 (0.83) | 0.71 (0.68) | 0.71 (0.68) | 0.70 (0.61) | 0.76 (0.53) | 0.75 (0.80) |
| Mafft-global | 0.72 (0.60) | 0.78 (0.60) | 0.77 (0.49) | 0.77 (0.48) | 0.75 (0.50) | 0.75 (0.41) | 0.76 (0.25) |
| Mafft-global-allowshift | 0.76 (0.83) | 0.75 (0.70) | 0.78 (0.49) | 0.78 (0.47) | 0.76 (0.46) | 0.82 (0.40) | 0.76 (0.26) |
| Mafft-local | 0.72 (0.58) | 0.71 (0.57) | 0.75 (0.50) | 0.78 (0.45) | 0.73 (0.46) | 0.76 (0.38) | 0.71 (0.24) |
| Mafft-local-allowshift | 0.69 (0.68) | 0.67 (0.72) | 0.78 (0.60) | 0.77 (0.45) | 0.79 (0.45) | 0.77 (0.35) | 0.77 (0.29) |
| T-Coffee | 0.65 (0.72) | 0.65 (0.72) | 0.62 (0.78) | 0.62 (0.78) | 0.63 (0.80) | 0.58 (0.95) | 0.58 (1.04) |
| Star | 0.00 (0.49) | 0.00 (0.48) | 0.04 (0.33) | 0.08 (0.37) | 0.13 (0.37) | 0.12 (0.29) | 0.12 (0.24) |
Table 4.
p values of the Wilcoxon signed-rank tests for pairs of algorithms with regard to the normalized motif-SOP score. “-a” indicates the allowshift option. p-values larger than 0.05 are not presented.
Table 4.
p values of the Wilcoxon signed-rank tests for pairs of algorithms with regard to the normalized motif-SOP score. “-a” indicates the allowshift option. p-values larger than 0.05 are not presented.
| Algorithm | Mafft-genaf | Mafft-global | Mafft-global-a | Mafft-local | Mafft-local-a |
|---|---|---|---|---|---|
| Mafft-global | |||||
| Mafft-global-a | |||||
| Mafft-local | |||||
| Mafft-local-a | |||||
| T-Coffee |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bountouridis, D.; Brown, D.G.; Wiering, F.; Veltkamp, R.C. Melodic Similarity and Applications Using Biologically-Inspired Techniques. Appl. Sci. 2017, 7, 1242. https://doi.org/10.3390/app7121242
AMA Style
Bountouridis D, Brown DG, Wiering F, Veltkamp RC. Melodic Similarity and Applications Using Biologically-Inspired Techniques. Applied Sciences. 2017; 7(12):1242. https://doi.org/10.3390/app7121242
Chicago/Turabian StyleBountouridis, Dimitrios, Daniel G. Brown, Frans Wiering, and Remco C. Veltkamp. 2017. "Melodic Similarity and Applications Using Biologically-Inspired Techniques" Applied Sciences 7, no. 12: 1242. https://doi.org/10.3390/app7121242
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.