
Using a Molecular Dynamics Simulation to Investigate Asphalt Nano-Cracking under External Loading Conditions


1
National Center for Materials Service Safety, University of Science and Technology Beijing, Beijing 100083, China
2
Joint USTB-Virginia Tech Lab on Multifunctional Materials, USTB, Beijing 100083, China, Virginia Tech, Blacksburg, VA 24061, USA
3
Institute of Highway Engineering, RWTH Aachen University, D52074 Aachen, Germany
*
Author to whom correspondence should be addressed.
Appl. Sci. 2017, 7(8), 770; https://doi.org/10.3390/app7080770
Submission received: 8 June 2017
/
Revised: 6 July 2017
/
Accepted: 26 July 2017
/
Published: 28 July 2017
(This article belongs to the Special Issue Advanced Asphalt Materials and Paving Technologies)
Abstract
:Recent research shows that macro-scale cracking in asphalt binder may originate from its intrinsic defects at the nano-scale. In this paper, a molecular dynamics (MD) simulation was conducted to evaluate the nucleation of natural defects in asphalt. The asphalt microstructure was modeled using an ensemble of three different types of molecules to represent a constituent species: asphaltenes, naphthene aromatics and saturates, where the weight proportion of 20:60:20 was used to create an asphalt-like ensemble of molecules. Tension force was then applied on the molecular boundaries to study the crack initiation and propagation. It was discovered that the natural distribution of atoms at microscale would affect the intrinsic defects in asphalt and further influence crack initiation and propagation in asphalt.
1. Background and Introduction
Asphalt pavement is one of the main highway forms throughout the world. With the continuous development of the world economy, there have been tremendous numbers of vehicles passing on the pavements every day, causing significant irreversible damage to pavement. It is therefore necessary to study asphalt material performance, since it is directly relevant to its service life, where structure is one of the most important factors that affects its mechanical performance. Recent studies show that the asphalt material microstructure may affect its properties at the macro-scale [1,2].
There have been many experimental approaches on the asphalt material microstructure due to the development of modern testing instruments. Mikhailenko et al. [3], used scanning electron microscopy (SEM) to observe the microstructure of asphalt materials. Fourier transform infrared spectroscopy (FTIR) can be used to study the distribution and contents of asphalt components [4,5,6]. Gel permeation chromatography (GPC) can determine the molecular weight and distribution of asphalt rapidly and reliably [7]. Differential scanning calorimetry (DSC) can be used to analyze the difference of the thermal stability of asphalt [8,9]. Atomic force microscope (AFM) can be used to observe three-dimensional (3D) spatial images of asphalt [10,11,12]. These new experimental methods can be used to further investigate the effect of modifiers on the modification of asphalt materials at nanometer scales [13,14]. For example, SEM can be used to observe and compare the microstructures of different nanometer modified asphalts [15], and AFM, FTIR, GPC and DSC can be used to further analyze the modify mechanism of nano-modifier materials.
There also have been many studies on asphalt material properties using the simulation, where molecular dynamics (MD) simulation is one of the effective methods to investigate the microstructure and micro-properties of asphalt materials [16,17,18]. In the aspect of asphalt oxidation aging, MD simulation can be employed to investigate internal chemical and molecular mechanical property changes [19]. Molecular Dynamics simulation can also be used to determine the thermodynamic properties of asphaltene before and after oxidative aging [20]. In other aspects, molecular dynamics simulation can also be used to study molecular movements and microstructure changes of epoxy modified asphalt in the process of healing [21]. Xu et al. [22], conducted AFM experiments and MD simulation to study the adhesion between asphalt and aggregate. The mechanical properties of asphalt and aggregate can be further investigated by MD simulation at the nano-scale [23], and by investigating mechanical properties of the asphalt-aggregate interface [24].
Although there has been significant progress in asphalt material microstructure and micro-properties, research on the fundamental mechanism of asphalt microstructure cracking using simulations and experiment approaches are very few. In this study, MD simulation was conducted to study micro-crack initiation, and propagation of typical asphalt molecular structures. The ultimate goal was to try to understand the fundamental mechanism of asphalt molecular structure cracking.
2. Molecular Dynamics Simulations
In this study, the MD simulation was employed to investigate the mechanical behavior of asphalt molecules.
2.1. Asphalt Molecular Structure Construction
Asphalt is often characterized chemically by its hydrocarbon class composition, e.g., three main constituent species including asphaltene, naphthene aromatics and saturates. The three-fraction asphalt model was employed to establish the molecular structure following Zhang and Greenfield [25], where three different types of molecules are used to represent the corresponding constituent species, and the three are then formed together to create an asphalt-like ensemble of molecules. In this model, 1,7-dimethylnapthalene was used to represent naphthene aromatics, and n-C22 (n-doccosane) molecules were used to represent saturates, with the mass fraction of asphaltenes, naphthene aromatics, and saturates being approximately 20:60:20, respectively.
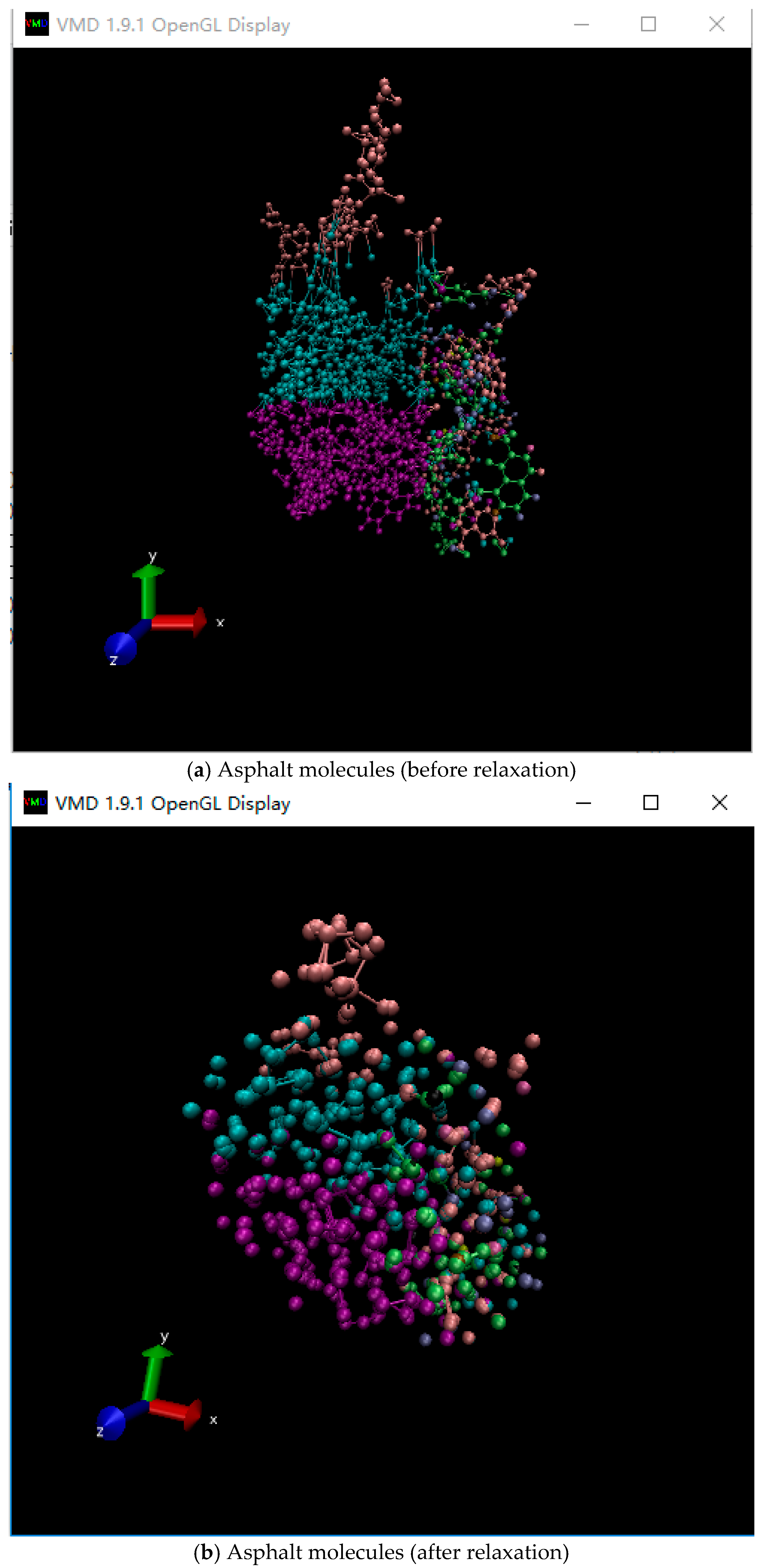
The condensed-phase optimized molecular potentials for atomistic simulation studies (COMPASS) force field was used for our simulation, considering that this force field could accurately describe the inorganic material properties. The initial molecular structure was constructed by using the commercial molecular computation software Material Studio 7.0 (BIOVIA Company, San Diego, CA, USA). The construction function in amorphous cell tools of Material Studio was used to construct the representative volume element in asphalt binder. The target density of the final configurations was set to 1 g/cm3. Table 1 shows the detailed configuration of the asphalt-like ensemble. Figure 1 shows the constructed molecular structure, where the edge length (Å) of the lattice is 50.9 × 50.9 × 50.9. The atoms in each molecule in the asphalt-like amorphous cell were then subjected to conjugate gradient energy minimization within 10,000 fs.
Our molecular dynamics simulation was split into two parts: constructing the model in Materials Studio due to convenience, and applying the loading conditions in the Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) software. After construction of the asphalt molecular model in Material Studio, it was exported in a PDB-format file and imported into the VMD (Visual Molecular Dynamics) software (University of Illinois at Urbana-Champaign, Urbana-Champaign, IL, USA). By using the “Topo writelammpsdata asphalt.data full” command, the asphalt data file can be read by the LAMMPS software.
The following potential function was used in our LAMMPS simulation for convenience:
where E is the potential, is the depth of a potential well that reflects the attraction between two atoms, is the distance between atoms when the action potential is equal to 0, r is the distance between two atoms, and rc is the cutoff.
2.2. Simulation Results
The relaxation process of asphalt molecules is shown in Figure 2. The relaxation was used to reach the energy minimization state of the asphalt molecular structure. It can be seen that the relaxation process would influence the structure of asphalt molecules, where agglomeration and densification occurred.
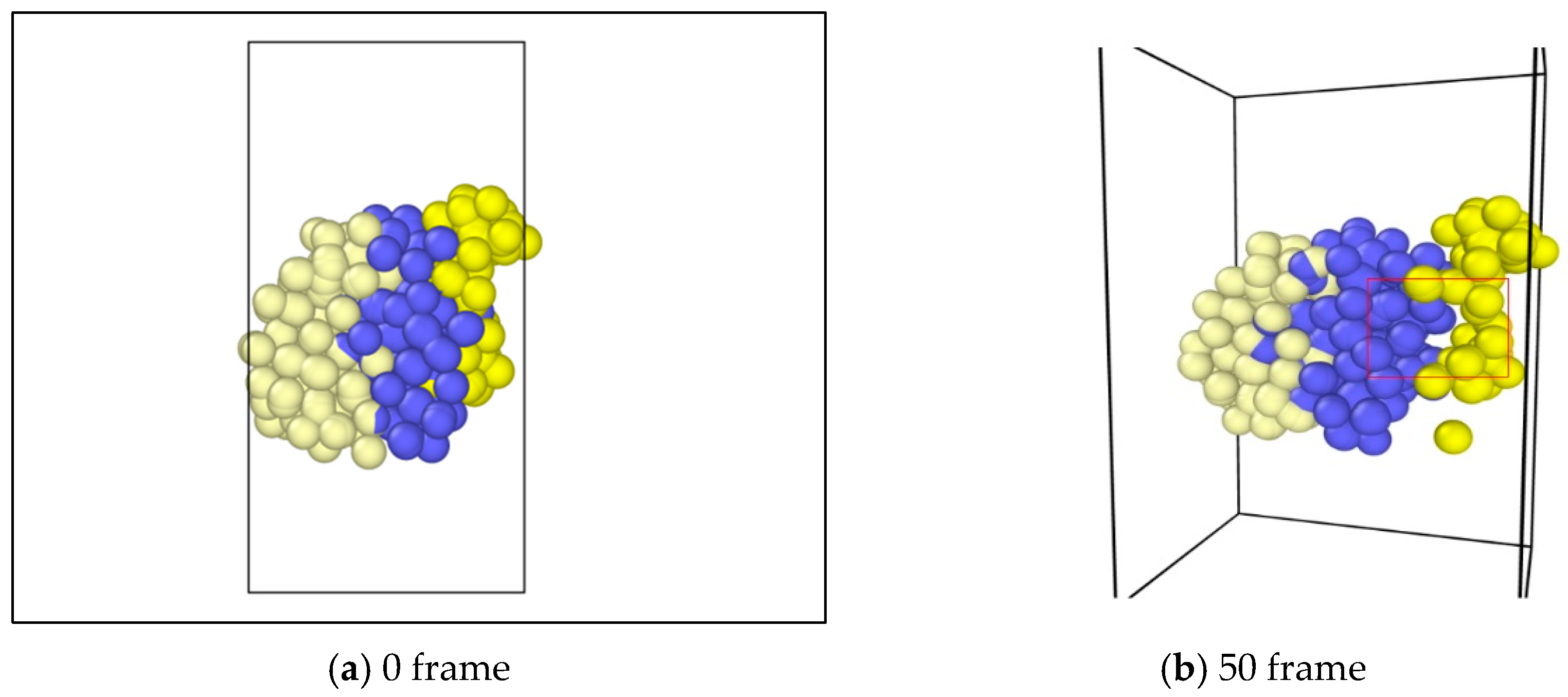
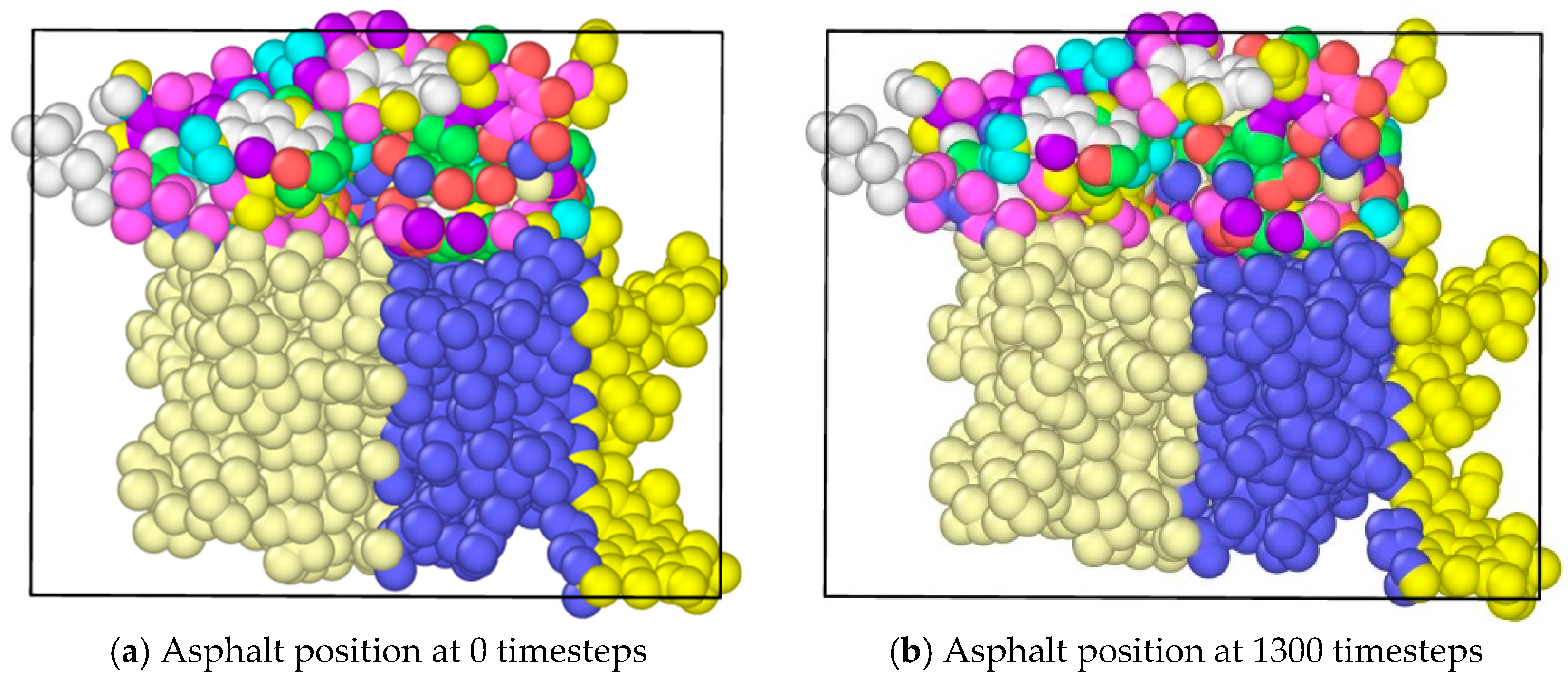
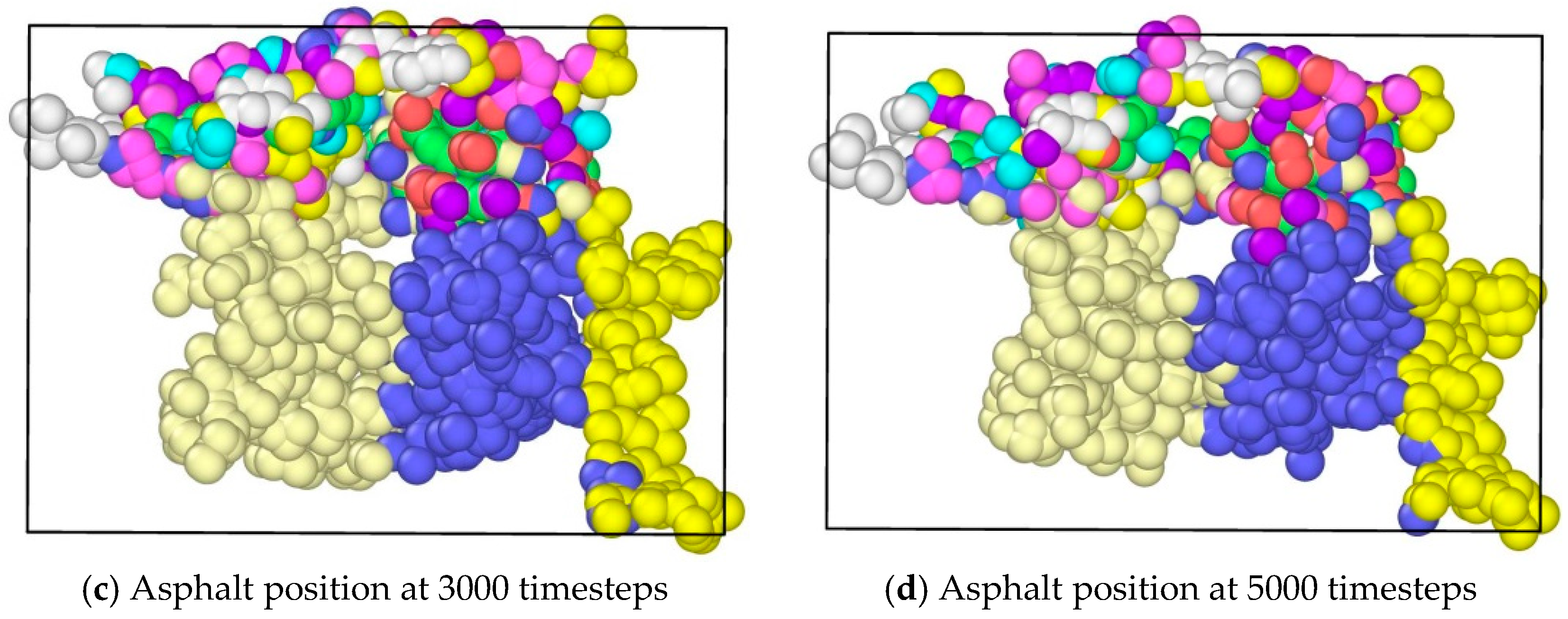
In the study, molecular dynamics simulation were exerted on the asphalt layer, where “region” and “group” commands were applied to set different regions in the asphalt molecule ensemble. The velocity command was used to set the velocities of different groups at 0.3 Å/fs along the y axis to simulate the state of tension. The ensemble temperature was set to room temperature, which is 298.15 K. For every 100 time steps, the simulation temperature was adjusted. For asphalt molecular ensemble, the energy minimization process was firstly conducted to achieve the stability of the system, and then the simulation of the stretching process was conducted. The time step was set to 0.003 fs, and a total operation time with 6000 steps was conducted. In the OVITO (Open Visualization Tool) software, asphalt molecules were sliced (Figure 3), where the internal development of asphalt molecules under tension could be observed. Figure 3a shows the complete whole microstructure and Figure 3b shows the half-sliced part. It was found that, along with the applying of continuous tension loading, obvious gaps could be found in the internal asphalt molecule structure (Figure 4), where these locations may be the weak regions and prone to easily crack.
The radial distribution function (RDF) is an important parameter to describe molecular structure. The essence of the radial distribution function gab (r) is the atomic ratio of local density and average density in the system [26]. When A and B atoms are distributed uniformly in the spherical shell, the probability is calculated as follows:
where V is the total volume of the simulation system, and are the number of A and B atoms, and are the average density of atoms in the simulation system, and PA and PB are the uniform distribution probabilities of A and B atoms.
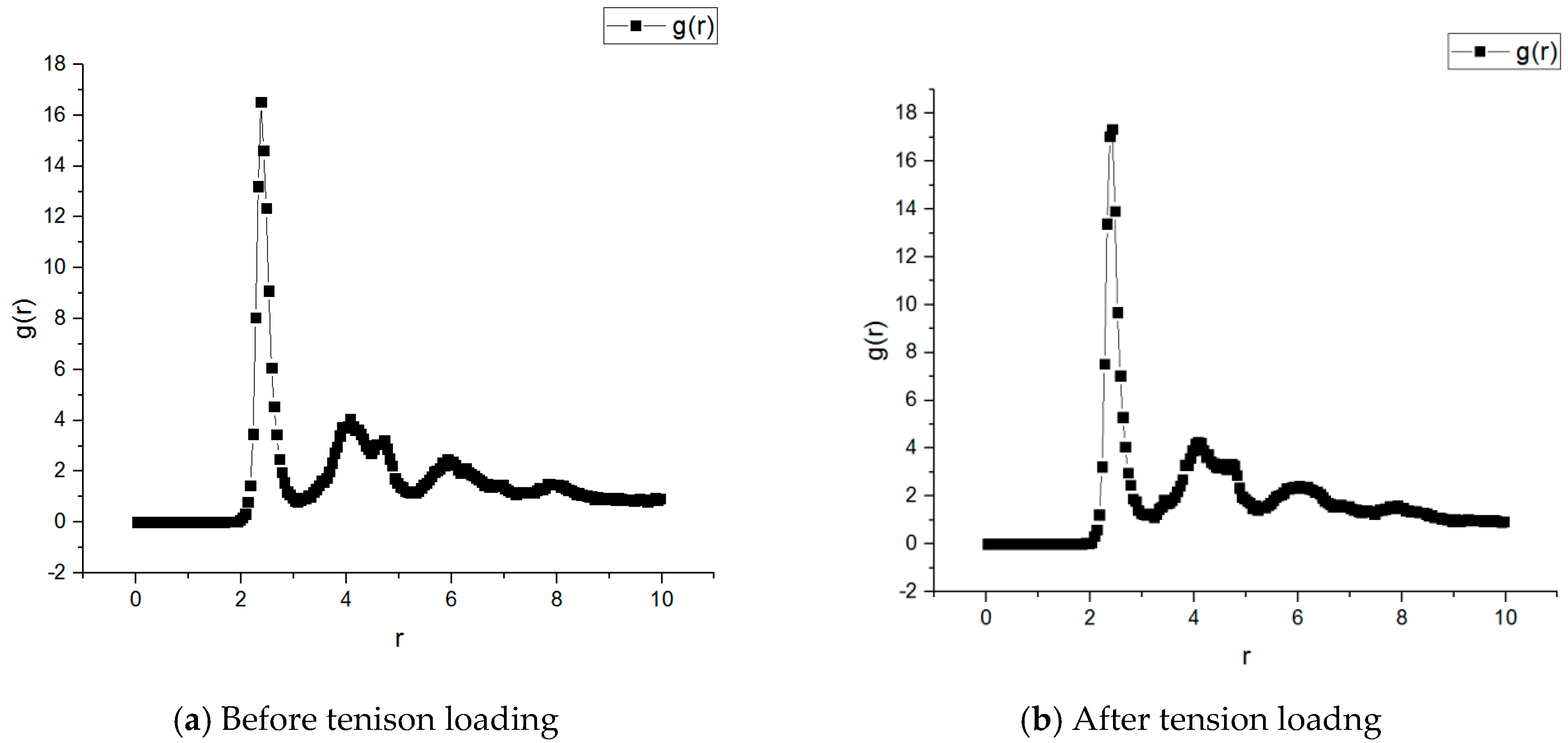
By calculating the radial distribution function between different atoms, the local distribution of different atoms could be studied, as shown in Figure 5. It is observed that there existed a small difference in RDF before and after tension loading. For the asphalt microstructure before tension loading, when the distance was 2.375 Å, a maximum value of 16.5250 occurred; for the asphalt microstructure after the tension loading, when the distance was 2.375 Å, maximum value of 17.039 occurred. The trends of the curves of the two curves were completely consistent, indicating that most of the atoms in the asphalt microstructure did not significantly change their locations while a small portion changed the locations. It was further concluded that the applied tension loading would not only result in the whole asphalt microstructure change, but also in some of the specific locations.
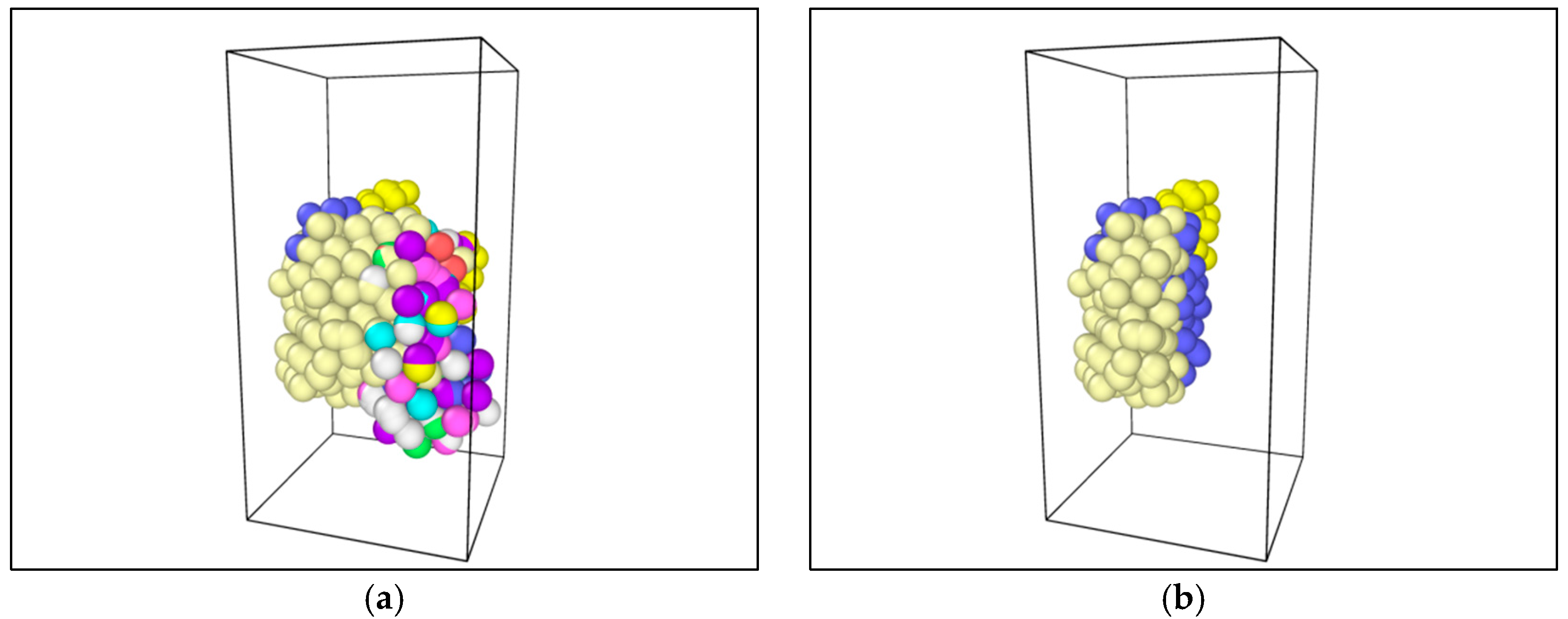
The application of tension loadings on the asphalt molecular structure was done by setting a constant speed in its y direction (vertical direction) of different regions, resulting in the stretching of the asphalt microstructure. Figure 6 shows the morphological changes of asphalt microstructures. Note that different color represents different atom types on the top and three different divisions on the bottoms. It was noticed that by applying the tension force, the original “perfect” structure generally stretched, and a “hole”, i.e., natural defect, occurred in the microstructure, which could be seen as the initiation of the microcrack. It was expected that with an increase in the tension loading, the inside small hole propagated and finally resulted in the failure of the asphalt microstructure [27].
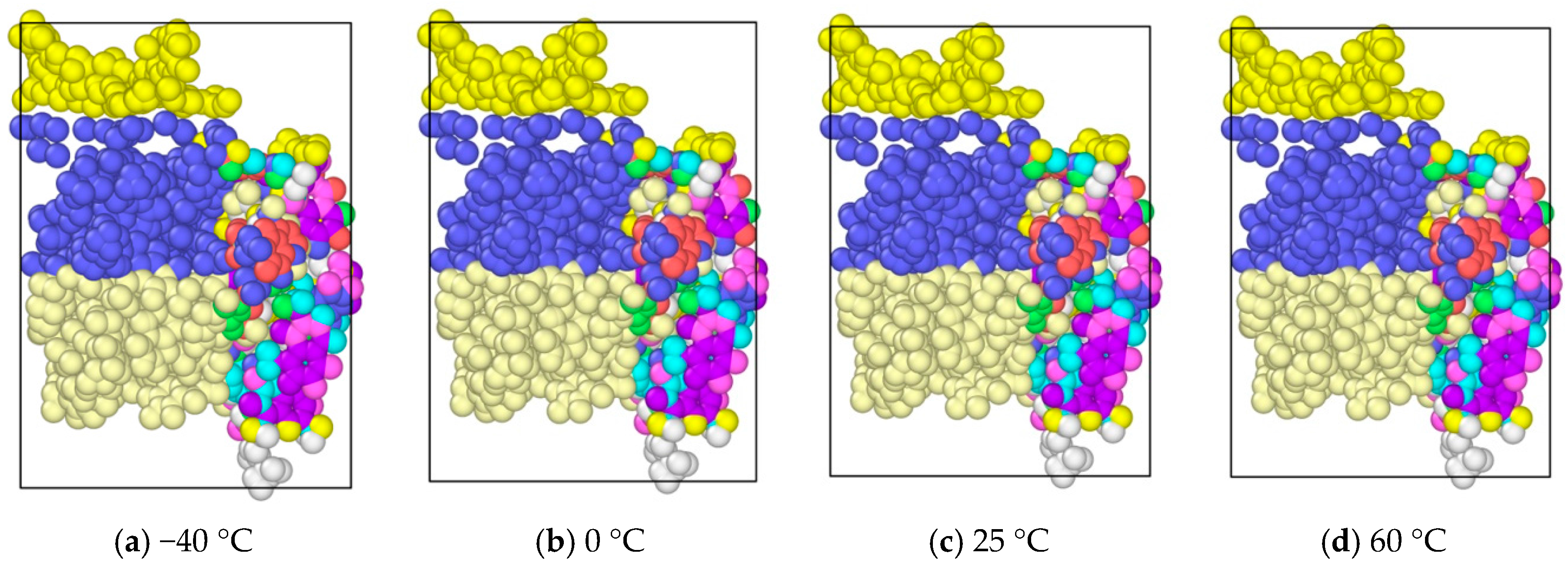
In order to analyze the temperature effects on the asphalt molecular, four kinds of different temperatures were chosen, including −40, 0, 25 and 60 °C, to analyze the temperature influence on the morphology and properties of the asphalt molecules during tension. The time step was 0.003 fs, with a total of 5000 steps. The asphalt molecular morphology with different temperture at 5000 timesteps are shown in Figure 7. It was seen that temperature seemed do not have a significant influence on the asphalt microstructure evolution. It might be caused by the small molecule amount in the model. Compared with the internal molecular component distribution, temperature does not have a significant effect on the molecular structure for this computation scale. To further study the temperature effects on asphalt microstructure evolution quantitatively and in more detail, future studies will be conducted on a supercomputer.
Through the analysis, it was found that there were “holes” in the asphalt molecules by stretching, with these holes existing due to the distribution of molecules, creating the original defects. The results showed that the original defects naturally existed in the asphalt molecules, resulting in different cracking performance. Note that since this was a preliminary study on the nucleation of natural defects in asphalt, we did not study this phenomenon quantitatively. In future studies, a parameter such as “total distance moved” by the atoms from some reference points may be proposed to describe this phenomenon.
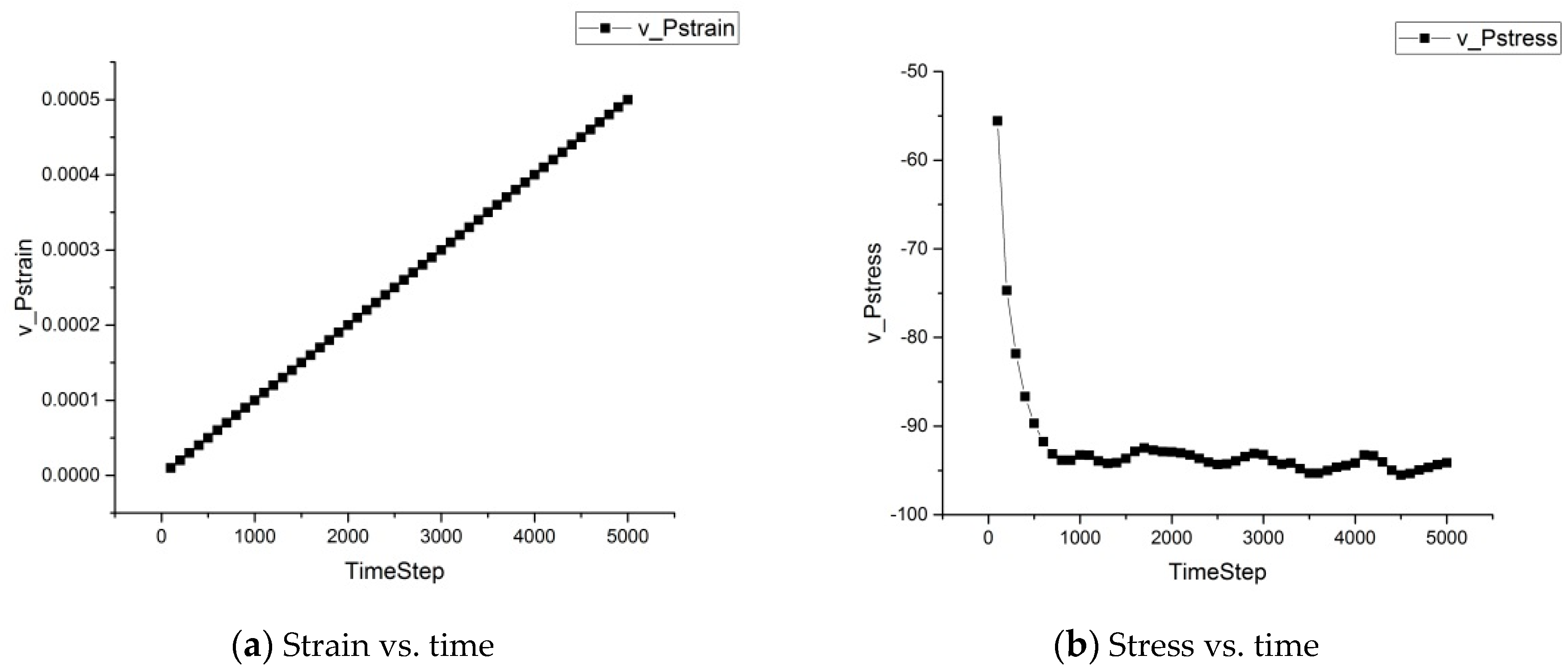
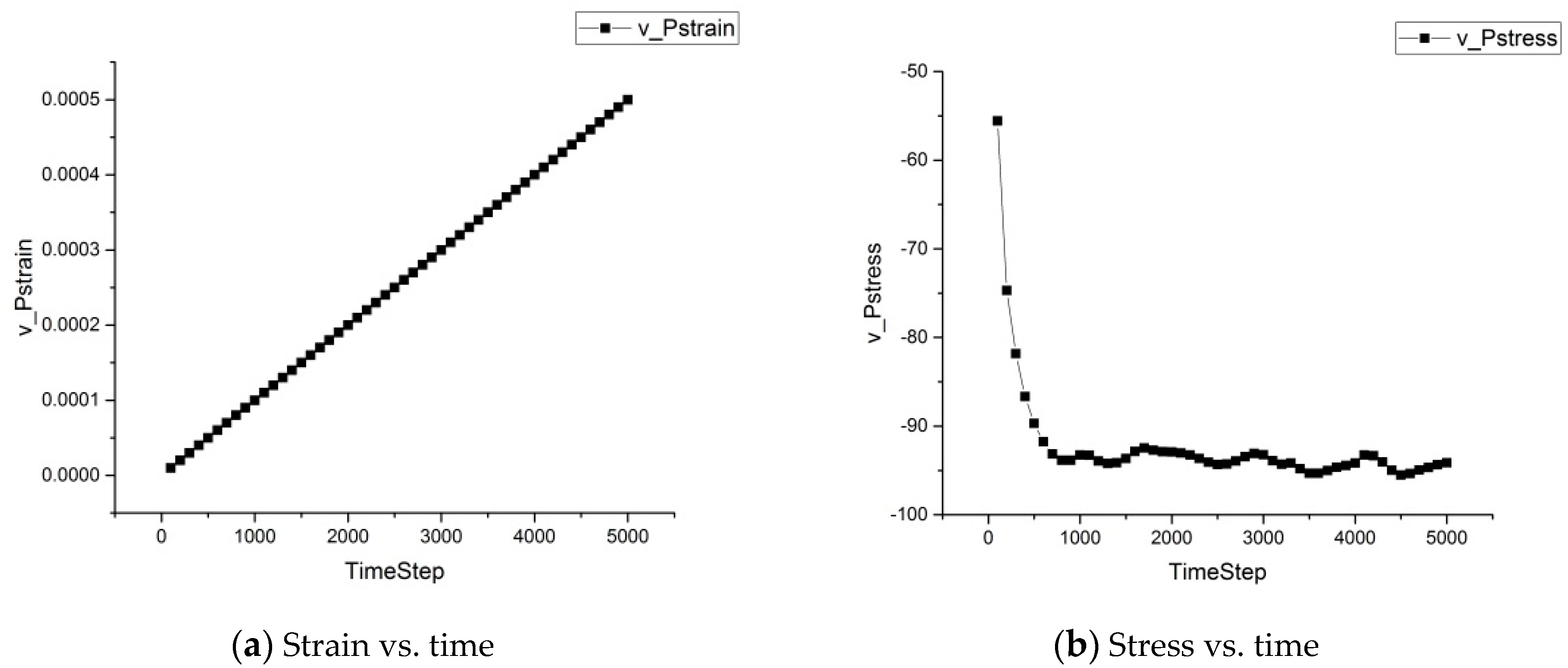
The strain and stress developments over time for the asphalt microstructure subject to external loading at 25 °C are shown in Figure 8. Figure 8a shows that the strain increased almost linearly, as we applied a constant strain deformation rate. Figure 8b shows that the stress gradually decreased and remained almost constant after 1300 timesteps, indicating that due to the occurrence of the “internal hole”, the asphalt microstructure bore less stress, and when the final structure was stabilized, the stress became stable.
It should be mentioned that our current research results seem to be a bit different from the observations in previous studies: 1. Menapace et al. (2016) discovered that temperature will significantly affect the “Bumble-bee” structure in asphalt samples at microscale [28]; 2. Jahangir et al. (2015) claimed that the tension loading would result in the change in asphalt microstructure [11]. There might be two reasons for the differences: 1. We currently only apply a very small tension loading. Therefore, compared with the internal molecule component distribution, the latter will have a larger effect on the molecular structure; 2. The current molecular model is established with relatively small molecule amount due to the limitation of our computer ability. It is expected that a much larger molecular model with millions of molecules may resemble the results by the previous research.
3. Conclusions
In this study, the MD simulation was conducted to model the asphalt molecular structure. The following conclusions were made:
- It was discovered that by applying the tension force, the original “perfect” structure generally stretched and a “hole”, i.e., natural defect, occurred in the microstructure, which can be seen as the initiation of the microcrack. It was expected that with an increase of tension loading, the inside small hole would propagate and finally result in the failure of the asphalt microstructure.
- It was observed that there existed a difference in RDF before and after tension loading. For the asphalt microstructure before the tension loading, when the distance was 2.375 Å, a maximum value of 16.5250 occurred; for the asphalt microstructure after the tension loading, when the distance was 2.375 Å, maximum value of 17.039 occurred. The trends of the curves for the two curves were completely consistent, indicating that many of the atoms in the asphalt microstructure did not change their locations, while a small portions changed.
Acknowledgments
The first author would like to express his sincerely gratitude to Amit Bhasin at the University of Texas at Austin for sharing the asphalt molecular structure. The research performed in this paper is supported by National Natural Science Foundation of China (No. 41372320), the Natural Science Foundation of Shandong Province (ZR2015EQ009), the Fundamental Research Funds for the Central Universities (06500036), and the 111 Project (Grant No. B12012).
Author Contributions
Yue Hou, Linbing Wang, Dawei Wang, Xin Qu, and Jiangfeng Wu conducted the numerical simulations; and Yue Hou wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guo, M.; Tan, Y.; Wang, L.; Hou, Y. Diffusion of asphaltene, resin, aromatic and saturate components of asphalt on mineral aggregates surface: Molecular dynamics simulation. Road Mater. Pavement 2017, 18, 149–158. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, L.; Wang, D.; Liu, P.; Guo, M.; Yu, J. Characterization of Bitumen Micro-Mechanical Behavior Using AFM, Phase Dynamics Theory and MD Simulation. Materials 2017, 10, 208. [Google Scholar] [CrossRef]
- Mikhailenko, P.; Khadim, H.; Baaj, H. Observation of Asphalt Binder Microstructure with ESEM. In Proceedings of the Transportation Association of Canada Conference, Toronto, ON, Canada, 25–28 September 2016. [Google Scholar]
- Sun, D.Q.; Zhang, L.W.; Zhang, X.L. Quantification of SBS content in SBS polymer modified asphalt by FTIR. Adv. Mater. Res. 2011, 287, 953–960. [Google Scholar] [CrossRef]
- Perez-Martinez, M.; Marsac, P.; Gabet, T.; Chailleux, E. Prediction of the Mechanical Properties of Aged Asphalt Mixes from FTIR Measurements. In Proceedings of the 8th RILEM International Symposium on Testing and Characterization of Sustainable and Innovative Bituminous Materials, Ancona, Italy, 7–9 October 2015; Springer: Haarlem, The Netherlands, 2015. [Google Scholar]
- Feng, Z.G.; Bian, H.J.; Li, X.J.; Yu, J.Y. FTIR analysis of UV aging on bitumen and its fractions. Mater. Struct. 2016, 49, 1381–1389. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, K.; Yin, J.; Zheng, M. Analysis on thermo-stability of modified asphalt using GPC (gel permeation chromatography). Railway Eng. 2013, 8, U414. [Google Scholar]
- Chen, X.L.; Sun, Y.S.; Han, Y.X.; Tian, Z.F. Microstructure and DSC of organic bentonite modified bitumen. J. Northeast. Univer. 2012, 33, 743–747. [Google Scholar]
- Chen, X.L.; Jiang, Y.Z.; Sun, Y.S.; Han, Y.X. Influence of DCCF on the properties of asphalt by DSC. Adv. Mater. Res. 2012, 454, 35–40. [Google Scholar] [CrossRef]
- Nazzal, M.D.; Abu-Qtaish, K.; Powers, D. Using atomic force microscopy to evaluate the nanostructure and nanomechanics of warm mix asphalt. J. Mater. Civ. Eng. 2015, 27, 1–9. [Google Scholar] [CrossRef]
- Jahangir, R.; Little, D.; Bhasin, A. Evolution of asphalt binder microstructure due to tensile loading determined using AFM and image analysis techniques. Int. J. Pavement Eng. 2015, 16, 337–349. [Google Scholar] [CrossRef]
- Tan, Y.; Guo, M. Micro- and nano-characteration of interaction between asphalt and filler. J. Test. Eval. 2014, 42, 1–9. [Google Scholar] [CrossRef]
- Khattak, M.J.; Khattab, A.; Zhang, P.; Rizvi, H.R.; Pesacreta, T. Microstructure and fracture morphology of carbon nano-fiber modified asphalt and hot mix asphalt mixtures. Mater. Struct. 2013, 46, 2045–2057. [Google Scholar] [CrossRef]
- Zareshahabadi, A.; Shokuhfar, A.; Ebrahiminejad, S. Microstructure and properties of nanoclay reinforced asphalt binders. Defect Diffus. Forum 2010, 579–583. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, S.; Xiao, D. Nano-Modified Asphalt and Road Performance; Science Press: Beijing, China, 2012. [Google Scholar]
- Rapaport, D.C.; Blumberg, R.L.; Mckay, S.R.; Christian, W. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Hou, Y.; Sun, W.; Huang, Y.; Ayatollahi, M.; Wang, L.; Zhang, J. Diffuse Interface Model to Investigate the Asphalt Concrete Cracking Subjected to Shear Loading at a Low Temperature. J. Cold Reg. Eng. 2017, 31, 04016009. [Google Scholar] [CrossRef]
- Hou, Y.; Guo, M.; Ge, Z.; Wang, L.; Sun, W. Mixed-Mode I-II Cracking Characterization of Mortar using Phase-Field Method. J. Eng. Mech. 2017, 143, 04017033. [Google Scholar] [CrossRef]
- Pan, J.; Tarefder, R.A. Investigation of asphalt aging behaviour due to oxidation using molecular dynamics simulation. Mol. Simul. 2016, 42, 1–12. [Google Scholar] [CrossRef]
- Tarefder, R.A.; Arisa, I.R. Molecular Dynamic Simulation of Oxidative Aging in Asphaltene. In Proceedings of the Pavements and Materials: Characterization and Modeling Symposium at Emi Conference, Los Angeles, CA, USA, 8–11 August 2010. [Google Scholar]
- Zhou, X.; Wu, S.; Liu, G.; Pan, P. Molecular simulations and experimental evaluation on the curing of epoxy bitumen. Mater. Struct. 2016, 49, 1–7. [Google Scholar] [CrossRef]
- Xu, M.; Yi, J.; Feng, D.; Huang, Y.; Wang, D. Analysis of adhesive characteristics of asphalt based on atomic force microscopy and molecular dynamics simulation. ACS Appl. Mater. Interfaces 2016, 8, 12393–12403. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, L. Nanoscale Modeling of the Mechanical Properties of Asphalt and Aggregate. In Proceedings of the Pavements and Materials: Characterization and Modeling Symposium at Emi Conference, Los Angeles, CA, USA, 8–11 August 2010. [Google Scholar]
- Lu, Y.; Wang, L. Nanoscale modelling of mechanical properties of asphalt–aggregate interface under tensile loading. Int. J. Pavement Eng. 2010, 11, 393–401. [Google Scholar] [CrossRef]
- Zhang, L.; Greenfield, M.L. Analyzing properties of model asphalts using molecular simulation. Energy Fuels 2007, 21, 1712–1716. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, B.; Zhang, Y.; Wei, J.; Cao, X. Molecular Dynamics Simulation to Investigate the Influence of SBS on Molecular Agglomeration Behavior of Asphalt. J. Mater. Civ. Eng. 2015, 27. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, L.; Yue, P.; Pauli, T.; Sun, W. Modeling Mode I: Cracking Failure in Asphalt Binder by Using Nonconserved Phase-Field Model. J. Mater. Civ. Eng. 2014, 26, 684–691. [Google Scholar] [CrossRef]
- Menapace, I.; Masad, E.; Bhasin, A. Effect of treatment temperature on the microstructure of asphalt binders: Insights on the development of dispersed domains. J. Microsc. 2016, 262, 12–27. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Constructed asphalt in amorphous cell.

Figure 2.
The relaxation process of asphalt molecules.
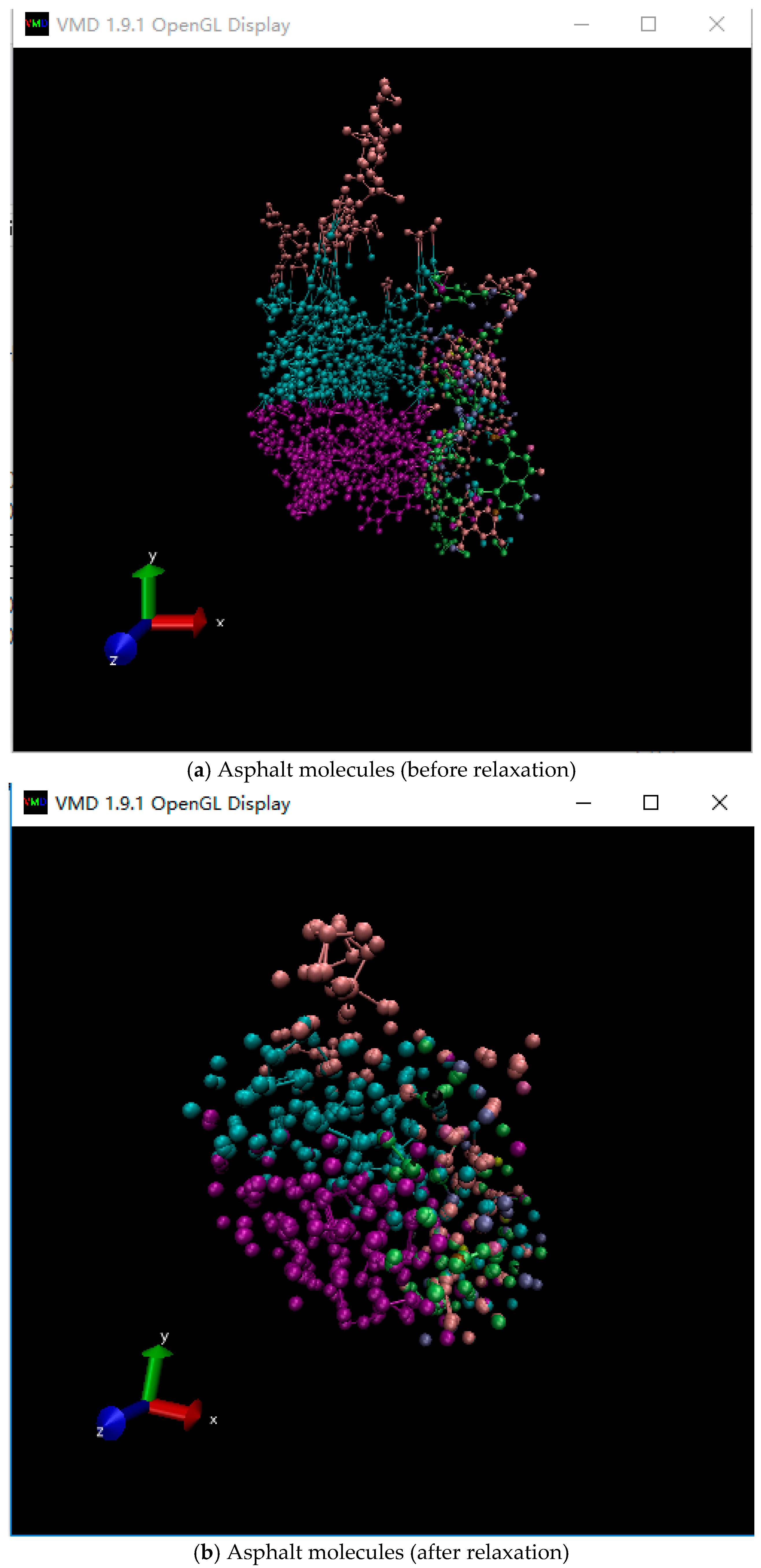
Figure 3.
Slicing of the asphalt molecules.

Figure 4.
Development of gap under tension loading.
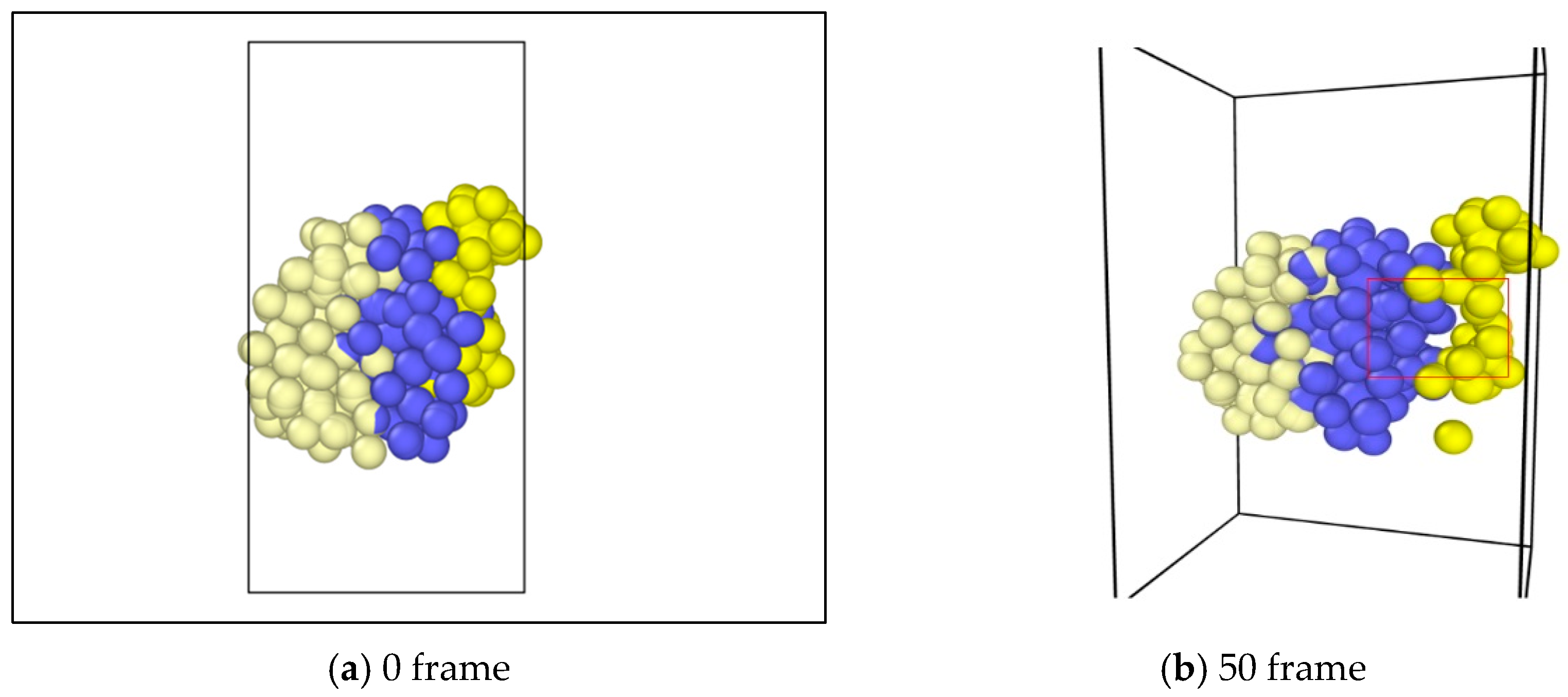
Figure 5.
Radial distribution function (RDF) of asphalt molecule before and after tension loading.
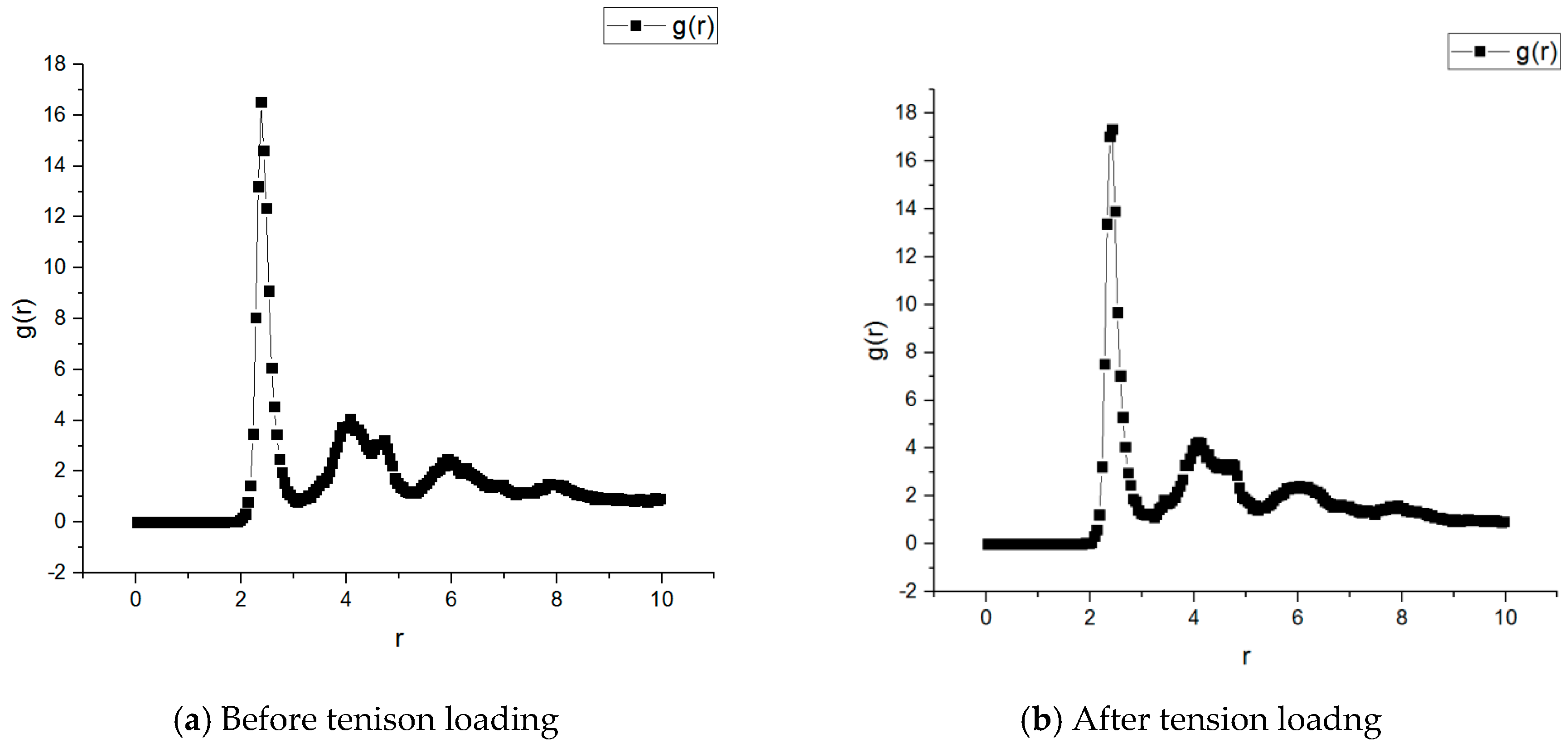
Figure 6.
Morphological changes of asphalt molecules under uniaxial tension.
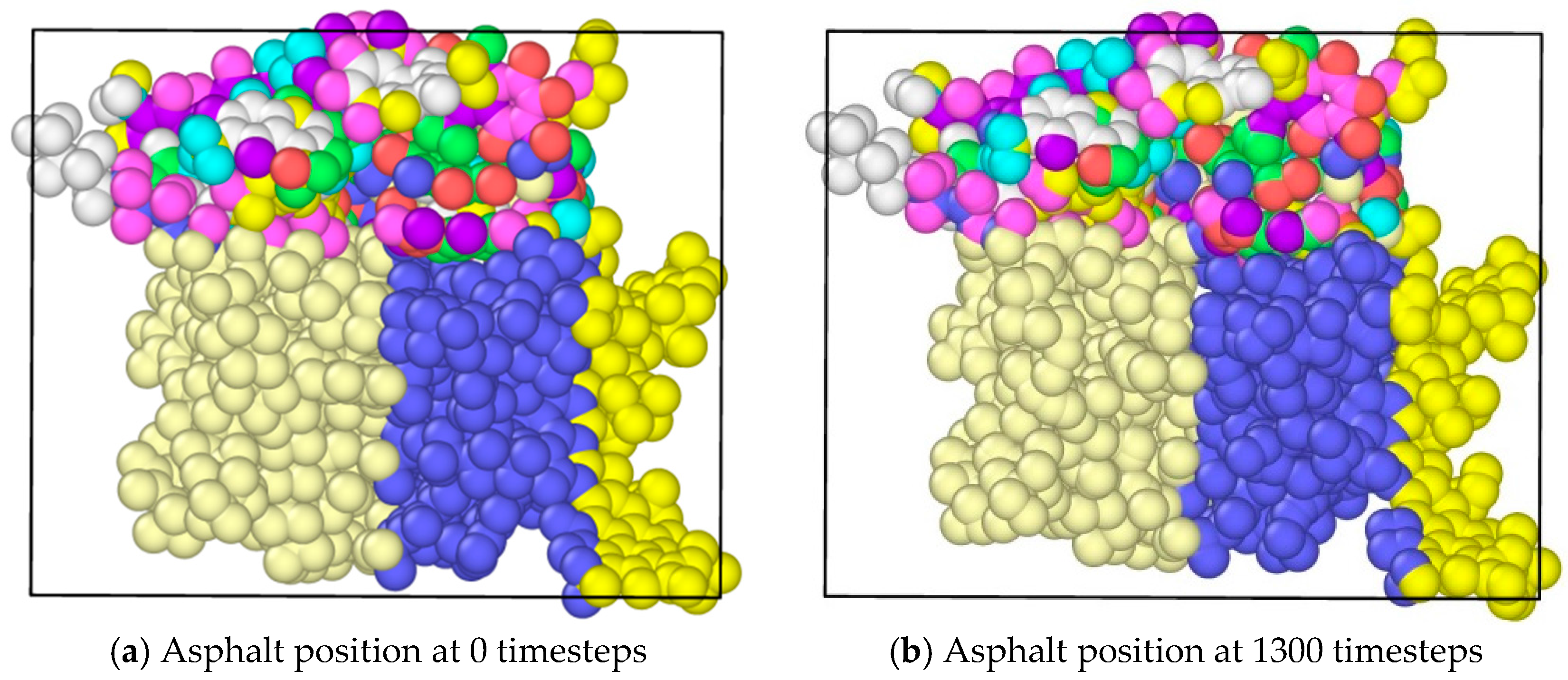
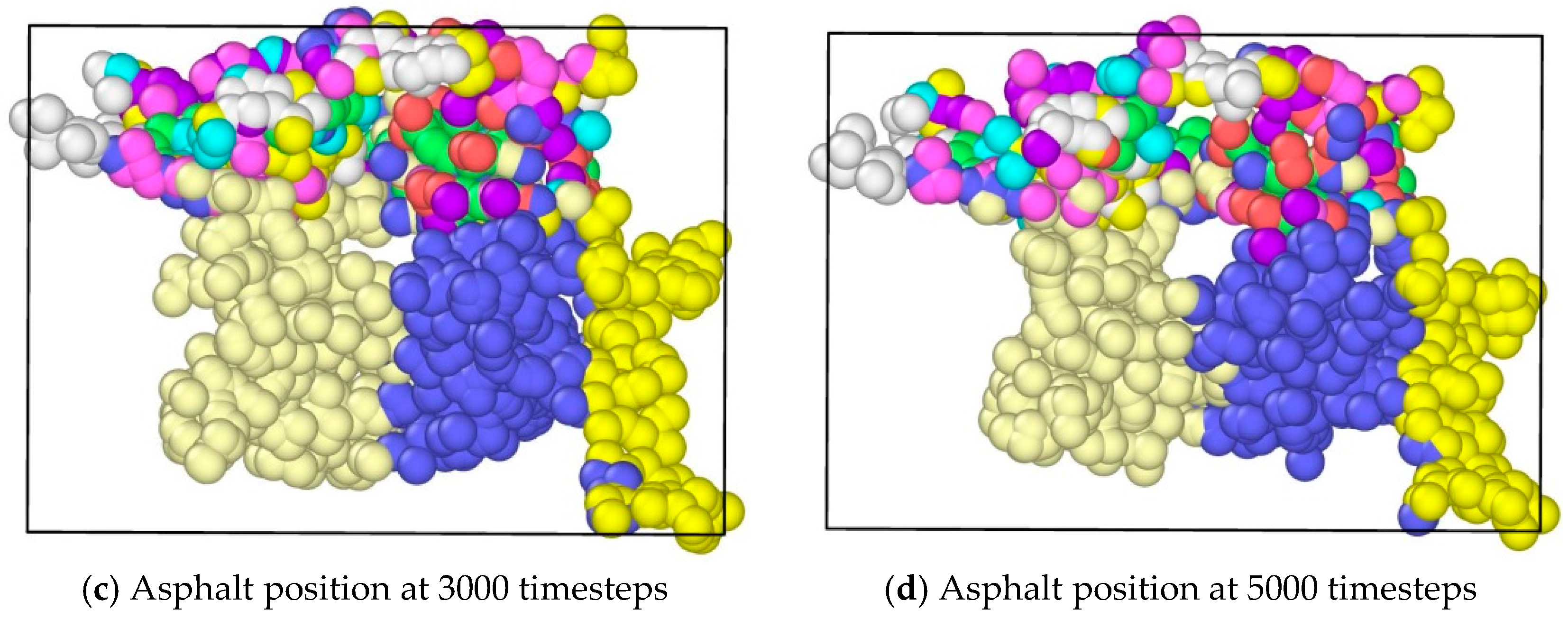
Figure 7.
Morphological changes of asphalt molecules at different temperature.
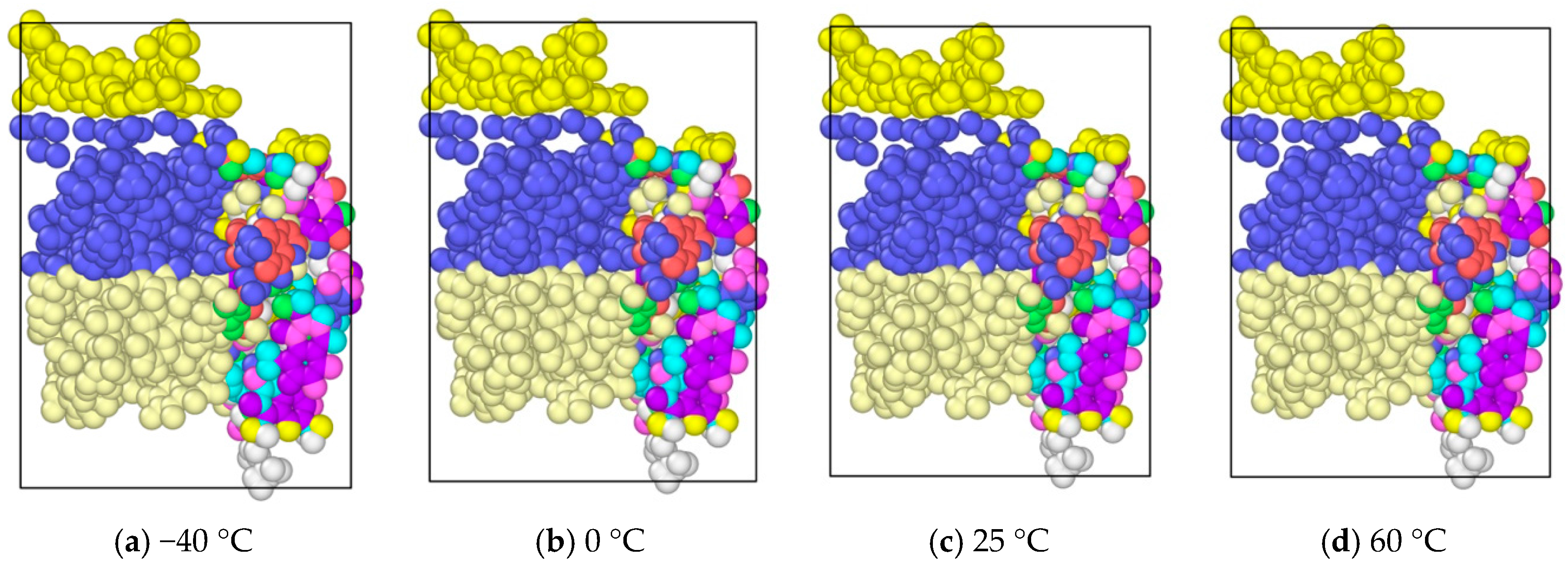
Figure 8.
Strain and stress developments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Configuration of the asphalt-like ensemble.
| Molecular Components | Asphaltene | 1,7-Dimethylnapthalene | n-Doccosane |
|---|---|---|---|
| Number | 18 | 306 | 49 |
| Approximate Mass ratio | 20 | 60 | 20 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hou, Y.; Wang, L.; Wang, D.; Qu, X.; Wu, J. Using a Molecular Dynamics Simulation to Investigate Asphalt Nano-Cracking under External Loading Conditions. Appl. Sci. 2017, 7, 770. https://doi.org/10.3390/app7080770
AMA Style
Hou Y, Wang L, Wang D, Qu X, Wu J. Using a Molecular Dynamics Simulation to Investigate Asphalt Nano-Cracking under External Loading Conditions. Applied Sciences. 2017; 7(8):770. https://doi.org/10.3390/app7080770
Chicago/Turabian StyleHou, Yue, Linbing Wang, Dawei Wang, Xin Qu, and Jiangfeng Wu. 2017. "Using a Molecular Dynamics Simulation to Investigate Asphalt Nano-Cracking under External Loading Conditions" Applied Sciences 7, no. 8: 770. https://doi.org/10.3390/app7080770
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.