Therapeutic Advances for Huntington’s Disease
by
 ,
,
Ashok Kumar
1,†, 
Vijay Kumar
2,*,† ,
,
Kritanjali Singh
3,
Sukesh Kumar
4,
You-Sam Kim
2,
Yun-Mi Lee
2 and
Jong-Joo Kim
2,* 1
Department of Genetics, Sanjay Gandhi Post-Graduate Institute of Medical Sciences, Lucknow 226014, UP, India
2
Department of Biotechnology, Yeungnam University, Gyeongsan, Gyeongbuk 38541, Korea
3
Central Research Station, Subharti Medical College, Swami Vivekanand Subharti University, Meerut 250002, India
4
PG Department of Botany, Nalanda College, Bihar Sharif, Magadh University, Bihar 824234, India
*
Authors to whom correspondence should be addressed.
†
Contributed equally as co-first authors.
Brain Sci. 2020, 10(1), 43; https://doi.org/10.3390/brainsci10010043
Submission received: 5 November 2019
/
Revised: 9 January 2020
/
Accepted: 10 January 2020
/
Published: 12 January 2020
(This article belongs to the Special Issue Juvenile Onset Huntington's Disease)
Abstract
:Huntington’s disease (HD) is a progressive neurological disease that is inherited in an autosomal fashion. The cause of disease pathology is an expansion of cytosine-adenine-guanine (CAG) repeats within the huntingtin gene (HTT) on chromosome 4 (4p16.3), which codes the huntingtin protein (mHTT). The common symptoms of HD include motor and cognitive impairment of psychiatric functions. Patients exhibit a representative phenotype of involuntary movement (chorea) of limbs, impaired cognition, and severe psychiatric disturbances (mood swings, depression, and personality changes). A variety of symptomatic treatments (which target glutamate and dopamine pathways, caspases, inhibition of aggregation, mitochondrial dysfunction, transcriptional dysregulation, and fetal neural transplants, etc.) are available and some are in the pipeline. Advancement in novel therapeutic approaches include targeting the mutant huntingtin (mHTT) protein and the HTT gene. New gene editing techniques will reduce the CAG repeats. More appropriate and readily tractable treatment goals, coupled with advances in analytical tools will help to assess the clinical outcomes of HD treatments. This will not only improve the quality of life and life span of HD patients, but it will also provide a beneficial role in other inherited and neurological disorders. In this review, we aim to discuss current therapeutic research approaches and their possible uses for HD.
1. Introduction
Huntington’s disease (HD) is genetically inherited in an autosomal dominant fashion. It is a fatal neurodegenerative disease, caused by an abnormal triplet repeat expansion of CAG (cytosine-adenine-guanine) within the huntingtin (HTT) gene on chromosome 4p16.3, causing a mutated huntingtin protein (mHTT) [1,2,3,4,5]. HD is predominantly characterized by adult-onset, progressive motor dysfunction, cognitive impairment and psychiatric symptoms (depression, anxiety, obsessive-compulsive disorder, and psychosis). Chorea, incoordination, and rigidity are common motor symptoms due to neurotoxicity of mHTT, leading to brain atrophy of the striatum, thalamus, cerebellum, brain stem and cortex [6,7,8,9]. Clinically, HD includes juvenile HD (onset less than 21 years, and marked clinical symptoms), and late-onset HD (after the age of 60 years) [10,11,12]. Alcohol, drug, and tobacco abuse were associated with earlier onset of HD, and hasten motor onset in women. These abuses have more significant associations in females than in males [13,14]. Children with CAG repeats ≥39, had significantly lower measures of head circumference, weight, and body mass index [15,16,17]. Disrupted sleep, tics, pain, itching, and psychosis are the common symptoms of juvenile HD [18].
Presently, there is no remedy for HD, and the disease progresses manifests with a presumed continuation of 15–20 years after the appearance of the first symptom [12,19]. The identification of novel biomarkers involves the development of new treatment strategies. The current therapy is palliative and does not change the course of the disease. Tetrabenazine (TBZ; Xenazine™) was approved for the remedy of chorea in HD by the U.S. food and drug administration (FDA). Additionally, the deuterated version of TBZ, deutetrabenazine (AUSTEDO™), has an improved pharmacokinetic profile and was recently approved by the FDA for the treatment of Huntington chorea. In the last review [20], we discussed different promising agents in the treatment of HD, and their phases under clinical trial. Here we describe updates related to these promising agents which will cure HD.
2. Pathogenesis of the HD
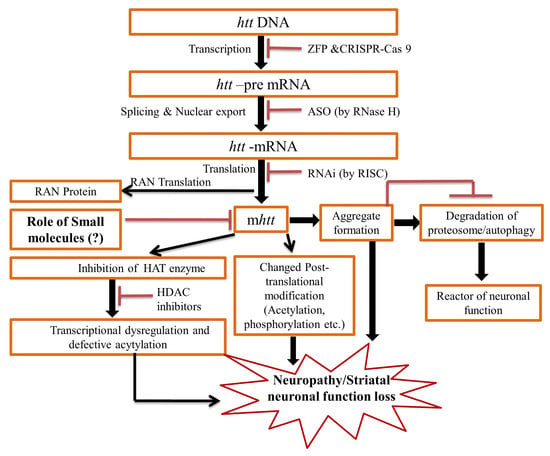
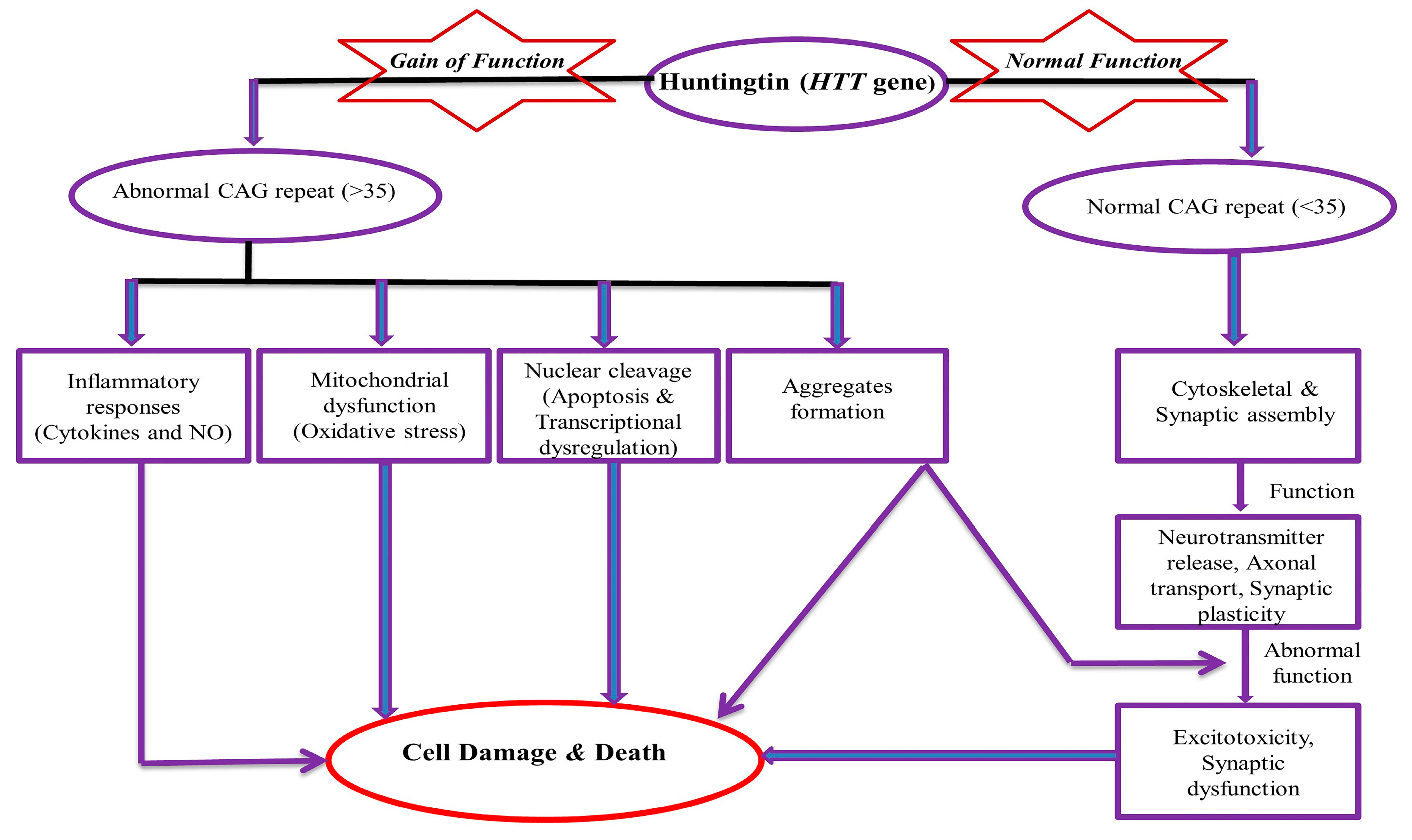
HD is a monogenic disease with prevalence of about 1 in 7,500 individuals in the general population [21,22]. The normal allele has less than 27 CAG repeats and intermediate alleles have 27–35 repeats. CAG repeats of 36–39 will develop HD with less penetrance. Individuals who have 40 or more CAG repeats will develop HD with full penetrance. It is also reported that the higher the CAG expansion, the earlier the onset and the greater the disease severity [12,23]. Kremer et al. reported the largest expansion of 121 trinucleotides [24]. CAG codon encodes glutamine α-amino acid (symbol Gln or Q). Glutamine (C5H10N2O3) is synthesized from glutamate and ammonia by the enzyme glutamine synthetase. It is mainly produced in muscle, the lungs, and the brain and acts as a precursor to the neurotransmitter glutamate [25]. CAG has glutamine amino acids within the HTT gene and it is not toxic in itself. However, the polyglutamine expansion involves the formation of aggregate and ultimately becomes toxic. It is the principal factor for the manifestation of HD because aggregates are never a remarkable feature in the brain of normal subjects [26,27]. Aggregate formations are accountable for secondary problems, like inflammatory responses (altered cytokine and nitric oxide level), mitochondrial dysfunction (imbalanced level of free radicals and oxidative stress markers), nuclear cleavage, apoptosis, excitotoxicity, transcriptional altered regulation, and lastly, are responsible for the altered neuropathological feature (cause of cell death/damage) (Figure 1). Approximately 70% of the variation of the disease is due to expanded CAG repeats, while 13% of the variation is due to polymorphisms in the GRIK2 gene [28]. These depict the importance of secondary factors that affect disease onset, its severity, and possible output.
3. Therapeutic Update
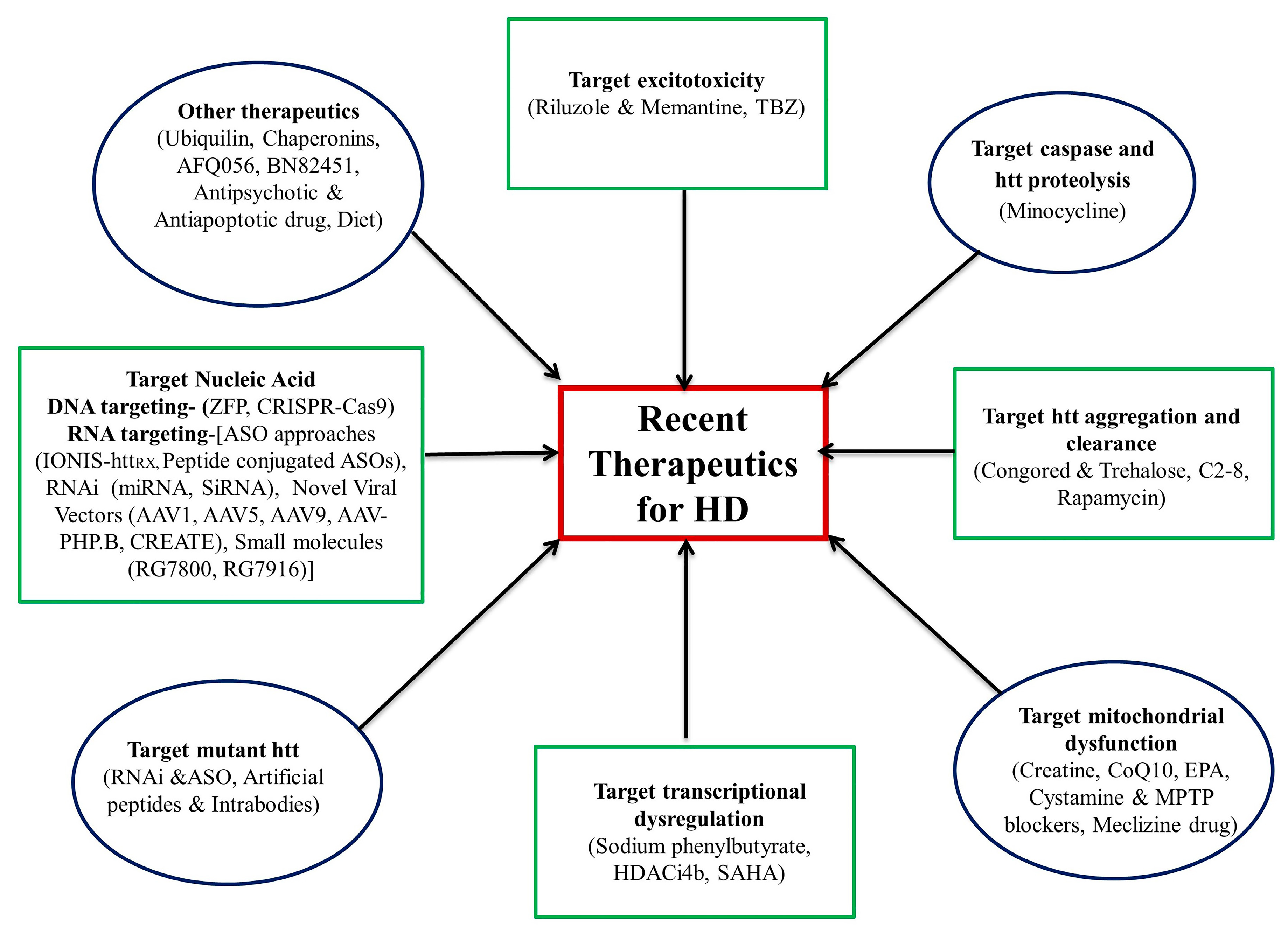
Currently, many drugs are under clinical trial. In the following subsection, we discuss their therapeutic status and their potential role in treatment. These drugs are summarized in Table 1 and Figure 2.
3.1. Drugs against Excitotoxicity
3.1.1. Riluzole and Memantine Drug
Riluzole is a glutamate inhibitor that reduces abnormal movement in amyotrophic lateral sclerosis (ALS) patients [29,30]. In a double-blinded trial, riluzole did not decrease symptoms of HD, nor was it neuroprotective [31].
Memantine is an antagonist of extrasynaptic N-methyl-D-aspartate (NMDA) receptors and is used for the treatment of moderate-severe dementia in Alzheimer’s disease (AD). It diminishes striatal cell death, hinders disease progression and improves cognitive function related to HD [32,33]. The combination of memantine and risperidone prevented the expected progression of motor symptoms, cognitive decline, and psychosis over a 6-month study period [34]. However, memantine dosing may be critical, as rodents on low-dose memantine had decreased pathology, while a high-dose of memantine worsened rodent outcomes and possibly promoted cell death [35,36,37].
3.1.2. Tetrabenazine (TBZ) and Deutetrabenazine
TBZ inhibits the dopamine pathway by inhibiting vesicular monoamine transporter (VMAT) type 2 and consequently decreases available dopamine in the synapse and its interaction with postsynaptic dopamine receptors [38,39,40]. Deutetrabenazine contains a deuterium atom and is a novel inhibitor of VMAT2. In indirect treatment comparison studies, deutetrabenazine was found to have a favorable tolerability profile compared to tetrabenazine [41]. In mouse models, TBZ ameliorated chorea and other motor symptoms, and reduced striatal neuronal cell loss [38].
3.2. Targeting Caspase Activities and Huntingtin Proteolysis
Minocycline
Minocycline is a tetracycline analog and can cross the blood–brain barrier (BBB) and inhibits the expression of caspase-3 and caspase-1 [42,43]. Treatment with minocycline proved to be neuroprotective, and to improve the disease phenotype [30,42,44]. A human trial study observed motor (unified HD rating scale (UHDRS)), and cognitive (mini-mental state examination (MMSE)) improvement in 14 HD patients who took 100 mg of minocycline for 6 months [43]. This study was continued for another 18 months, and it was found that MMSE, TMS, total functional capacity (TFC) and independence scale were all stabilized after treatment, reducing the expected decline in these measures. There was also a decrease in psychiatric symptoms at 24 months, which was not apparent after 6 months of treatment [44]. In a pilot study, Thomas et al. found improvement in MMSE, UHDRS, and abnormal involuntary movements scale (AIMS), in 30 patients with HD who were given minocycline for 6 months [45].
3.3. Targeting HTT Aggregation and Clearance
3.3.1. Congo Red and Trehalose
Congo red dye binds preferably to β-sheets with amyloid fibrils. When injected into HD mice, it preserved normal protein synthesis and degradation, and improved motor functions. This dye promotes clearance of expanded polyQ repeats and inhibits polyglutamine oligomer formation through the disruption of preformed oligomers. Congo red dye also prevented ATP depletion and caspase activation [46,47,48].
3.3.2. Compound C2–8
Compound C2–8 inhibits polyglutamine aggregates in brain slices and cell cultures. It has improved motor function, decreased the amount of neuronal atrophy, and decreased the size of the mHTT aggregates in R6/2 mice [52,53]. There is no ongoing human trial using this compound currently listed on clinicaltrials.gov.
3.3.3. Rapamycin
mTOR is a protein kinase that phosphorylates many proteins and plays a key role in various cellular functions (like autophagy and transcription). mTOR interacts with mHTT and localizes to these polyglutamine aggregates, and thus sequestration of mTOR reduces the activity of mTOR, resulting in a decrease in autophagy and a decrease in the clearance of mHTT. mTOR phosphorylates S6K1 (a key regulator of cell volume), therefore mHTT-related impairment of mTOR may account for the brain atrophy in HD. Rapamycin (which inhibits mTOR and consequently induces autophagy) decreased mHTT aggregates and improved neuronal survival in the drosophila HD model. Rapamycin also improved motor performance and decreased striatal neuropathology in mouse models of HD [54,55,56].
3.4. Targeting Mitochondrial Dysfunction
3.4.1. Creatine
Creatine (with antioxidant properties) reduced serum levels of 8-hydroxy-2′-deoxyguanosine (8-OH-2′-dG) in HD patients [85,86] and is safe and tolerable at a dose of 15 g twice daily [87]. In a trial study (with HD patients), receiving a dose of 8 g/day of creatine was secure and well-tolerated but produced no marked change on the UHDRS scale [86]. A randomized double-blind study trial measuring TFC (up to 40 g daily) was terminated early due to futility criteria being reached. The use of creatine fails to delay functional decline in an early manifestation of HD [57]. In another controlled study, creatine (5 g/day; 1 year) treatment resulted in better muscle function capacity in patients with neuromuscular disease but did not show improvements in neuromuscular function and the cognitive status of stage I–III HD patients [88].
3.4.2. Coenzyme Q10
Coenzyme Q10 cofactor is involved in ATP production in the electron transport chain (ETC) of mitochondria, and its supplementation in HD patients may improve mitochondrial function [89]. It was neuroprotective in R6/2 mice, delaying motor deficit, atrophy, and inclusion, and extending survival [90,91]. In a phase III randomized clinical trial, coenzyme Q10 was not effective and the trial was stopped as the futility criteria were reached (http://hdsa.org/wp-content/uploads/2015/01) [58].
3.4.3. Eicosapentaenoic Acid (EPA)
Ethyl-EPA is a derivative of the n-3 polyunsaturated fatty acid EPA, which binds to the peroxisome proliferator-activated receptor of mitochondria [92]. Ethyl-EPA improves the neuronal function by inhibiting caspase and reducing mitochondrial damage by reducing the activity of the c-Jun N-terminal kinase (JNK) pathway [93,94]. Treatment with ethyl-EPA (2 g/day) showed a stable/improved motor function. However, intent-to-treat analysis showed no significant change between ethyl-EPA and placebo for total motor score 4 (TMS–4) subscale in HD patients (stage III) [95]. Patients with fewer CAG repeats showed significant improvement in TMS-4.
In a phase III, double-blind randomized control trial, ethyl-EPA did not show improvement in TMS, cognition or global impression over 6 months. After 6 months, all participants (both those in the treatment and placebo group) were given ethyl-EPA. Those in the original treatment group showed a better motor function (indicated by TMS scores). This suggests that ethyl-EPA needs a longer period before improvement can be observed which might possibly reflect a disease modification [96]. In a recent study, no significant improvement of the treatment group over placebo group was found in measures of TMS or UHDRS scores [59].
3.4.4. Cystamine and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) Blockers
Both cystamine and MPTP increase the survival effects of HD cells and inhibit oxidative damage [60].
3.4.5. Meclizine
Meclizine, an antihistamine drug, inhibits oxidative metabolism and apoptosis, and is neuroprotective in drosophila model. Energy metabolism deficits and neuronal degeneration are hallmarks of HD, so treatment with meclizine is a potential strategy, especially since it crosses the BBB [20,61] (Table 1 and Figure 2). There is no human clinical trial for this drug listed in clinicaltrials.gov.
3.5. Targeting Transcriptional Dysregulation
3.5.1. Sodium Phenylbutyrate
Administration of sodium phenylbutyrate (an HDAC inhibitor) to N171-82Q symptomatic mice showed less brain atrophy and extended survival rates. Further, it increased and decreased histone acetylation and methylation, respectively, in the rodent brain. It also downregulated caspases involved in apoptosis [62]. A dose-response study highlighted that sodium phenylbutyrate was safe, secure, effective and well-tolerable in HD patients [97].
3.5.2. HDACi4b
HDACi4b (a pimelic diphenylamide HDAC inhibitor) improves motor impairment as well as decreases neurodegeneration in mouse models of HD. Oral administration of HDACi4b to mice showed improvement in these motor deficits. These mice also showed less striatal atrophy and brain-size reduction. HDACi4b reversed hypoacetylation of the H3 histone subunit that occurs in the presence of mHTT, and mRNA expression was returned to normal levels [63].
3.5.3. Suberoylanilide Hydroxamic Acid (SAHA)
Histone acetylation in the brain is increased by SAHA (by inhibiting HDAC) which improves motor impairments in transgenic R6/2 mice. SAHA can be taken orally because it crosses the BBB, however, this has not been tested in humans [64].
3.5.4. Mithramycin and Chromomycin
3.6. Agents Targeting Mutant Huntingtin
3.6.1. RNA Interference (RNAi) and Antisense Oligonucleotide (ASO)
ASO and RNAi execute their knockdown function by allele and nonallele-selective manners [100,101,102]. As an example, modified ASO (peptide nucleic acid i.e., PNA) enables the selective recognition of the mutant allele and selective inhibition of mHTT expression in human fibroblasts [103]. RNAi reduced neuropathology, improved motor behavior and extended viability in HD [102,104,105].
3.6.2. Intrabodies and Artificial Peptides
3.7. Nucleic Acid-Targeting Therapies
3.7.1. Therapies Targeting DNA
Currently, zinc finger proteins (ZFPs) and CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR-associated system) are under investigation.
ZFPs
ZFPs are one of the most abundant protein groups and have various functions, including regulation of DNA, RNA, and protein function. They can bind to specific sequences of DNA and can be used as therapeutic compounds. ZFPs reduce mHTT expression without affecting the expression of other genes/wild-type HTT [67,102,107].
CRISPR-Cas9
CRISPR-Cas9 is involved in viral defense mechanisms of bacteria which recognize and destroy foreign DNA. CRISPR-Cas9 is involved in the excision of CAG repeats to make harmless alleles and silence the mHTT expression by insertion of stop codon/missense mutations [68,108,109,110]. In HD140Q-knockin mice, it was demonstrated that CRISPR-Cas9 can be used to reduce mHTT and improve motor function, but not to increase the lifespan of these mice [69]. Ekman et al. showed that CRISPR-Cas9 can be used for mHTT editing, which can extend survival and improve motor function in the R6 mice following intrastriatal delivery [111].
3.7.2. RNA Targeting Therapies
The four major methods to inhibit the function of mHTT mRNA are: ASOs, RNAi compounds, novel viral vectors, and small-molecule splicing modulators.
ASO Approaches
ASO are single-stranded DNA (ssDNA) molecules that primarily bind to a specific sequence on RNA and regulate post-transcriptional gene expression [112]. The ssDNA diffuses well in the CNS and is taken up by neurons. Therefore, the injection of ASOs into the cerebrospinal fluid (CSF) results in ubiquitous delivery of drugs and suppresses the production of mHTT [70] (Table 1). However, ASO delivery has some side effects, like thrombocytopenia which was observed in some human trials of ASOs [113]. ASO can ameliorate transcriptional dysregulation and reduce the level of mHTT and improve behavior in the YAC128, YAC18, and BACHD mouse models of HD [114,115].
IONIS-HTTRx is an important ASO. It has 12–25 nucleotides and transforms phosphodiester linkages to phosphorothioate. IONIS-HTTRx caused a remarkable reduction in HTT mRNA and protein expression [71]. The injection of ASOs (conjugated with peptides), produced wide CNS distribution and longer life span in the spinal muscular atrophy (SMA) mouse model [116]. In a recent study of phase I–IIa trial, HTTRx lessened the concentration of mutant HTT in CSF of HD patients. Therefore, ASO compounds not only suppress the expression of HTT mRNA and the huntingtin protein in CNS, but also in CSF [117].
RNAi Approaches
RNA interference is a gene-silencing process that uses short interfering RNA (siRNA), short hairpin RNA (shRNA), bi-functional shRNA and microRNA (miRNA). The combination of neural progenitor stem cell therapy and RNAi therapy can ameliorate symptoms in mouse models of HD [118]. In the animal models (R6/1, R62, N171-82Q, RAT AAV-HD70d) of HD, siRNA, shRNA, and miRNA treatments have been used to reduce neuropathology and improve motor function [6,72,104]. AMT-130 (adeno-associated virus vector) contains an artificial miRNA which produces a huntingtin-lowering molecule. Side effects include peripheral neuropathy observed in clinical trials of siRNA. RNAi has been tested in rodents and its delivery system has been tested in nonhuman primates [119].
Small Molecule Approach
Small molecules like RG7800 showed ocular complications in the Δ7 mouse model of spinal muscular atrophy (SMA) [120], while the phase I trial of the molecule RG7916 (risdiplam) was recently completed (NCT02633709). RG7800 and RG7916 are splicing modifiers, which change the way the pre-mRNA is spliced so that it contains all the information necessary to make a functional protein. They promote the production of a full-length and functional protein from the gene. RG7800 increases the survival motor neuron (SMN) protein level by modifying the splicing of the SMN2 mRNA. RG7800 is shown to promote the inclusion of exon 7 in SMN2 mRNA, generating full-length mRNA, using fibroblasts from an SMA type I patient. In the SMA mouse model, the treatment of RG7800 showed a dose-dependent increase in SMN protein levels [121]. Oral administration of RG7800 in SMA patients increased the functional SMN protein level up to two-fold from baseline [122]. New work is now underway to identify these molecules and their possible role in the lowering of mutant HTT gene and protein expression [102,123].
3.8. Other Therapeutics Advancements
3.8.1. Ubiquilin
3.8.2. Chaperonins
3.8.3. AFQ056
AFQ056 did not improve chorea in a randomized double-blind clinical trial [79].
3.8.4. BN82451
BN82451 inhibits cyclooxygenases and provides antioxidant, anti-inflammatory and neuroprotective effects [80]. It also improved motor function and survival, decreased brain atrophy, neuronal atrophy, and neuronal mHTT inclusions in the R6/2 mice [81]. Recently, a phase II clinical trial has been completed in male HD patients. As per clinicaltrials.gov (NCT02231580), no results have been published.
3.8.5. Antipsychotic Drugs
Antipsychotic drugs are used to treat chorea associated symptoms because they block or modulate dopamine receptors. Many antipsychotic drugs (especially typical antipsychotics) produce motor dysfunction resembling Parkinson’s disease (PD). Currently, a phase III trial comparing TBZ with olanzapine and tiapridal is under evaluation [39].
3.8.6. Antiapoptotic Drugs
Caspase cleavage (mainly caspase-3 and -6) occur in mHTT [83]. Mutating the caspase cleavage sites on mHTT leads to neuroprotection and prevents neurodegeneration in yeast artificial chromosome (YAC) mice that express mHTT. Caspase-3 and -6 resistant mice did not develop HD neurodegeneration, indicating that cleavage at these caspase sites plays an important role in neurodegeneration of HD [82,123,124].
3.8.7. Diet
Various studies indicate that a Mediterranean-type diet may delay the onset of other neurodegenerative diseases, like AD, PD, dementia and cognitive impairment [125]. Recently a study highlighted that a Mediterranean-type diet affects the time to HD phenoconversion. In fact, eating high amounts of dairy products was associated with an increased risk of phenoconversion. This may be due to a lower level of urate, which leads to a faster progression and manifestation of HD. These types of diet-related studies need further investigation [84,107,126]. Intermittent fasting promotes autophagy and cleared the mHTT [127].
3.9. Some Promising Clinical Trials
3.9.1. Cysteamine (CYST)
Cysteamine controls oxidation levels through increasing concentration of glutathione, activation of protein catabolism through the hindrance of transglutaminase and induction of heat shock proteins (HSPs) effects [128]. Inhibition of transglutaminase is the putative mode of action and is observed in R6/2 and zQ175 mouse models of HD [129]. The Cysteamine-HD phase II/III trials indicated a delay in the release of cysteamine in HD patients.
3.9.2. Pridopidine
Pridopidine is a modulator of the dopamine 2 receptor [130] and activates the sigma-1 receptor [131]. In the most recent trials of pridopidine, no improvement in motor symptoms (unified Huntington’s Disease rating scale - total motor score (UHDRS-TMS)) was observed with placebo [132,133,134]. High level of pridopidine is found in brain-derived neurotrophic factor (BDNF) and diminishes mHTT aggregate size and improved motor performance in R6/2 mice [135,136]. In an early-stage HD, improvement in the total motor score (TMS) was observed in treated patients [136,137].
3.9.3. Triheptanoin
Triheptanoin is a triglyceride that reverses the metabolic defects in HD by supplying substrates to the Krebs cycle [138]. Recently, a phase II study using triheptanoin was conducted in the early phase of HD patients (listed at clincialtrials.gov under #NCT02453061).
3.9.4. Latrepirdine (Dimebon)
Latrepirdine stabilize and enhance mitochondrial membranes and functions. In a short duration trial, latrepirdine promoted cognitive improvement (MMSE) in mild to moderate HD patients [11,139]. At the time of writing this, a total of 13 clinical trials using latrepirdine have been reported on clinicaltrials.gov (NCT00497159, NCT01085266, NCT00920946, NCT00387270, NCT00988624, NCT00827034, NCT00990613, NCT00824590, NCT00931073, NCT00831506, NCT00788047, NCT00825084).
3.9.5. Amantadine
3.9.6. Lamotrigine
3.9.7. Selisistat
3.9.8. Tauroursodeoxycholic Acid/Ursodiol
Tauroursodeoxycholic acid (TUDCA) is a bile acid and has antiapoptotic properties in a mouse model of HD. Mice given TUDCA showed less striatal atrophy, apoptosis, and reduced locomotor and sensory-motor defects [148]. A commercially available uroursodeoxycholic acid precursor, ursodiol, has been examined in a phase I trial, but to date, not reported.
3.9.9. Laquinimod
Laquinimod reduces the expression of Bax, responsible for the release of cytochrome C from mitochondria and activation of caspases, causing apoptosis and production of toxic mHTT fragments. This drug improves motor function and reduces depressive behaviors in mice. It is recently undergoing a phase II clinical trial in human HD patients [149]. Laquinimod ameliorates myelination deficiency and behavioral deficits in the YAC128 mouse model of HD [150,151].
3.9.10. Kynurenine Inhibitors
The enzyme indoleamine 2,3 dioxygenase (IDO1) catalyzes the conversion of tryptophan into kynurenine. Kynurenine is then metabolized into 3-hydroxykynurenine (3-HK) and quinolinic acid, both of which are neurotoxic and are increased in HD. In contrast, kynurenine is also metabolized into kynurenic acid, which is neuroprotective. In HD, an imbalance exists between the neurotoxic products and neuroprotective products and targeting the rate-limiting step of IDO1 could effectively shift the balance toward neuroprotective [152]. Kynurenine 3-monooxygenase is the enzyme that catalyzes the conversion of kynurenine into 3-HK. Treating microglial cells from R6/2 mice with a kynurenine 3-monooxygenase inhibitor (Ro 61–8048) showed dramatically reduced 3-HK levels compared to the vector containing cells [153].
4. Conclusions and Future perspectives
The current therapeutic investigations of HD mainly focus on excitotoxicity, dopamine pathway, caspase inhibitors, mHTT aggregation, mitochondrial dysfunction, transcriptional dysregulation, and diet. The application of robust molecular imaging and digital biomarkers may provide a valuable therapeutic boost to the design of clinical trials. Additionally, the increased openness of regulatory agencies for effectiveness will also promote the development of clinical trials. The advancement of modern technologies, and the availability of various promising agents/molecules enable the development of therapies which will further improve the quality of research and outcomes in HD patients. The most promising drugs are those that target the production of mHTT protein and block its actions.
Author Contributions
Conceptualization, A.K., V.K. J.-J.K.; methodology, A.K., V.K.; writing—original draft preparation, A.K., V.K.; writing—review and editing J.-J.K., S.K., V.K., Y.-M.L., Y.-S.K., A.K., K.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| Abnormal involuntary movements scale | AIMS |
| Alzheimer’s disease | AD |
| Antisense Oligonucleotide | ASO |
| Blood-brain barrier | BBB |
| Brain-derived neurotrophic factor | BDNF |
| Clustered regularly interspaced short palindromic repeats-CRISPR-associated system | CRISPR-Cas9 |
| Cerebrospinal fluid | CSF |
| Cytosine-adenine-guanine | CAG |
| Eicosapentaenoic acid | EPA |
| Electron transport chain | ETC |
| Food and Drug Administration | FDA |
| Huntington disease | HD |
| Huntingtin gene | HTT |
| micro RNA | miRNA |
| Mini-mental state examination | MMSE |
| Mutant huntingtin protein | mHTT |
| Mammalian target of rapamycin | mTOR |
| 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine | MPTP |
| N-methyl-D-aspartate | NMDA |
| Parkinson’s disease | PD |
| RNA Interference | RNAi |
| small interfering RNA | siRNA |
| Spinal muscular atrophy | SMA |
| Suberoylanilide hydroxamic acid | SAHA |
| Tauroursodeoxycholic acid | TUDCA |
| Tetrabenazine | TBZ |
| Total functional capacity | TFC |
| Total motor score | TMS |
| Unified HD rating scale | UHDRS |
| Vesicular monoamine transporter 2 | VMAT2 |
| Zinc finger proteins | ZFPs |
References
- Kim, S.D.; Fung, V.S. An update on Huntington’s disease: From the gene to the clinic. Curr. Opin. Neurol. 2014, 27, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Conrad, A.; Epping, E.; Mathews, K.; Magnotta, V.; Dawson, J.D.; Nopoulos, P. Effect of Trinucleotide Repeats in the Huntington’s Gene on Intelligence. EBioMedicine 2018, 31, 47–53. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.M.; Zhang, Y.B.; Wu, Z.Y. Huntington′s Disease: Relationship Between Phenotype and Genotype. Mol. Neurobiol. 2017, 54, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Moss, D.J.H.; Pardinas, A.F.; Langbehn, D.; Lo, K.; Leavitt, B.R.; Roos, R.; Durr, A.; Mead, S.; TRACK-HD investigators; REGISTRY investigators; et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. 2017, 16, 701–711. [Google Scholar] [CrossRef]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L.; et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassel, B.; Tessler, S.; Faull, R.L.; Emson, P.C. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 2008, 33, 232–237. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. Huntington’s disease: Underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013, 38, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Chao, T.K.; Hu, J.; Pringsheim, T. Risk factors for the onset and progression of Huntington disease. Neurotoxicology 2017, 61, 79–99. [Google Scholar] [CrossRef]
- Fusilli, C.; Migliore, S.; Mazza, T.; Consoli, F.; De Luca, A.; Barbagallo, G.; Ciammola, A.; Gatto, E.M.; Cesarini, M.; Etcheverry, J.L.; et al. Biological and clinical manifestations of juvenile Huntington’s disease: A retrospective analysis. Lancet Neurol. 2018, 17, 986–993. [Google Scholar] [CrossRef]
- Horizon Investigators Of The Huntington Study Group; European Huntington’s Disease Network. A randomized, double-blind, placebo-controlled study of latrepirdine in patients with mild to moderate Huntington disease. JAMA Neurol. 2013, 70, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Byars, J.A.; Beglinger, L.J.; Moser, D.J.; Gonzalez-Alegre, P.; Nopoulos, P. Substance abuse may be a risk factor for earlier onset of Huntington disease. J. Neurol. 2012, 259, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.L.; Kamholz, J.A.; Moser, D.J.; Feely, S.M.; Paulsen, J.S.; Nopoulos, P.C. Substance abuse may hasten motor onset of Huntington disease: Evaluating the Enroll-HD database. Neurology 2017, 88, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Mathews, K.; Schlaggar, B.; Perlmutter, J.; Paulsen, J.S.; Epping, E.; Burmeister, L.; Nopoulos, P. Measures of growth in children at risk for Huntington disease. Neurology 2012, 79, 668–674. [Google Scholar] [CrossRef] [Green Version]
- Aylward, E.H.; Liu, D.; Nopoulos, P.C.; Ross, C.A.; Pierson, R.K.; Mills, J.A.; Long, J.D.; Paulsen, J.S.; PREDICT-HD Investigators; Coordinators of the Huntington Study Group. Striatal volume contributes to the prediction of onset of Huntington disease in incident cases. Biol. Psychiatry 2012, 71, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Tereshchenko, A.; Magnotta, V.; Epping, E.; Mathews, K.; Espe-Pfeifer, P.; Martin, E.; Dawson, J.; Duan, W.; Nopoulos, P. Brain structure in juvenile-onset Huntington disease. Neurology 2019, 92, e1939–e1947. [Google Scholar] [CrossRef] [Green Version]
- Moser, A.D.; Epping, E.; Espe-Pfeifer, P.; Martin, E.; Zhorne, L.; Mathews, K.; Nance, M.; Hudgell, D.; Quarrell, O.; Nopoulos, P.; et al. A survey-based study identifies common but unrecognized symptoms in a large series of juvenile Huntington’s disease. Neurodegener. Dis. Manag. 2017, 7, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar Singh, S.; Kumar, V.; Kumar, D.; Agarwal, S.; Rana, M.K. Huntington’s disease: An update of therapeutic strategies. Gene 2015, 556, 91–97. [Google Scholar] [CrossRef]
- Evans, S.J.; Douglas, I.; Rawlins, M.D.; Wexler, N.S.; Tabrizi, S.J.; Smeeth, L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Fisher, E.R.; Hayden, M.R. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk. Mov. Disord. 2014, 29, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.A. Genetics of Huntington disease. Handb. Clin. Neurol. 2017, 144, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef]
- Newsholme, P.; Lima, M.M.; Procopio, J.; Pithon-Curi, T.C.; Doi, S.Q.; Bazotte, R.B.; Curi, R. Glutamine and glutamate as vital metabolites. Braz. J. Med. Biol. Res. 2003, 36, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Becher, M.W.; Kotzuk, J.A.; Sharp, A.H.; Davies, S.W.; Bates, G.P.; Price, D.L.; Ross, C.A. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: Correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 1998, 4, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Lutz, R.E. Trinucleotide repeat disorders. Semin. Pediatr. Neurol. 2007, 14, 26–33. [Google Scholar] [CrossRef]
- Palfi, S.; Riche, D.; Brouillet, E.; Guyot, M.C.; Mary, V.; Wahl, F.; Peschanski, M.; Stutzmann, J.M.; Hantraye, P. Riluzole reduces incidence of abnormal movements but not striatal cell death in a primate model of progressive striatal degeneration. Exp. Neurol. 1997, 146, 135–141. [Google Scholar] [CrossRef]
- Turck, P.; Frizzo, M.E. Riluzole stimulates BDNF release from human platelets. Biomed. Res. Int. 2015, 2015, 189307. [Google Scholar] [CrossRef]
- Landwehrmeyer, G.B.; Dubois, B.; de Yebenes, J.G.; Kremer, B.; Gaus, W.; Kraus, P.H.; Przuntek, H.; Dib, M.; Doble, A.; Fischer, W.; et al. Riluzole in Huntington’s disease: A 3-year, randomized controlled study. Ann. Neurol. 2007, 62, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Beister, A.; Kraus, P.; Kuhn, W.; Dose, M.; Weindl, A.; Gerlach, M. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. In Focus on Extrapyramidal Dysfunction; Müller, T., Riederer, P., Eds.; Springer: Vienna, Austria, 2004; pp. 117–122. [Google Scholar]
- Lee, S.T.; Chu, K.; Park, J.E.; Kang, L.; Ko, S.Y.; Jung, K.H.; Kim, M. Memantine reduces striatal cell death with decreasing calpain level in 3-nitropropionic model of Huntington’s disease. Brain Res. 2006, 1118, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Cankurtaran, E.S.; Ozalp, E.; Soygur, H.; Cakir, A. Clinical experience with risperidone and memantine in the treatment of Huntington’s disease. J. Natl. Med. Assoc. 2006, 98, 1353–1355. [Google Scholar] [PubMed]
- Dau, A.; Gladding, C.M.; Sepers, M.D.; Raymond, L.A. Chronic blockade of extrasynaptic NMDA receptors ameliorates synaptic dysfunction and pro-death signaling in Huntington disease transgenic mice. Neurobiol. Dis. 2014, 62, 533–542. [Google Scholar] [CrossRef]
- Okamoto, S.; Pouladi, M.A.; Talantova, M.; Yao, D.; Xia, P.; Ehrnhoefer, D.E.; Zaidi, R.; Clemente, A.; Kaul, M.; Graham, R.K.; et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat. Med. 2009, 15, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- Milnerwood, A.J.; Gladding, C.M.; Pouladi, M.A.; Kaufman, A.M.; Hines, R.M.; Boyd, J.D.; Ko, R.W.; Vasuta, O.C.; Graham, R.K.; Hayden, M.R.; et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010, 65, 178–190. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, X.; Li, Y.; Tang, T.S.; Bezprozvanny, I. Tetrabenazine is neuroprotective in Huntington’s disease mice. Mol. Neurodegener. 2010, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Coppen, E.M.; Roos, R.A. Current Pharmacological Approaches to Reduce Chorea in Huntington’s Disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef] [Green Version]
- de Tommaso, M.; Serpino, C.; Sciruicchio, V. Management of Huntington’s disease: Role of tetrabenazine. Ther. Clin. Risk Manag. 2011, 7, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Claassen, D.O.; Carroll, B.; De Boer, L.M.; Wu, E.; Ayyagari, R.; Gandhi, S.; Stamler, D. Indirect tolerability comparison of Deutetrabenazine and Tetrabenazine for Huntington disease. J. Clin. Mov. Disord. 2017, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M.; et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Heuberger, C.; Reisecker, F. Minocycline for Huntington’s disease: An open label study. Neurology 2003, 60, 883–884. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, R.M.; Hodl, A.K.; Hofmann, P.; Kapfhammer, H.P. Neuroprotection in Huntington’s disease: A 2-year study on minocycline. Int. Clin. Psychopharmacol. 2004, 19, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Ashizawa, T.; Jankovic, J. Minocycline in Huntington’s disease: A pilot study. Mov. Disord. 2004, 19, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef]
- Frid, P.; Anisimov, S.V.; Popovic, N. Congo red and protein aggregation in neurodegenerative diseases. Brain Res. Rev. 2007, 53, 135–160. [Google Scholar] [CrossRef]
- McGowan, D.P.; van Roon-Mom, W.; Holloway, H.; Bates, G.P.; Mangiarini, L.; Cooper, G.J.; Faull, R.L.; Snell, R.G. Amyloid-like inclusions in Huntington’s disease. Neuroscience 2000, 100, 677–680. [Google Scholar] [CrossRef]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef]
- Lee, H.J.; Yoon, Y.S.; Lee, S.J. Mechanism of neuroprotection by trehalose: Controversy surrounding autophagy induction. Cell Death Dis. 2018, 9, 712. [Google Scholar] [CrossRef]
- Fernandez-Estevez, M.A.; Casarejos, M.J.; Lopez Sendon, J.; Garcia Caldentey, J.; Ruiz, C.; Gomez, A.; Perucho, J.; de Yebenes, J.G.; Mena, M.A. Trehalose reverses cell malfunction in fibroblasts from normal and Huntington’s disease patients caused by proteosome inhibition. PLoS ONE 2014, 9, e90202. [Google Scholar] [CrossRef]
- Wang, N.; Lu, X.H.; Sandoval, S.V.; Yang, X.W. An independent study of the preclinical efficacy of C2-8 in the R6/2 transgenic mouse model of Huntington’s disease. J. Huntington’s Dis. 2013, 2, 443–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, V.; Fox, J.H.; Lieberman, G.; Dorsey, K.; Matson, W.; Waldmeier, P.; Housman, D.E.; Kazantsev, A.; Young, A.B.; Hersch, S.; et al. A small-molecule therapeutic lead for Huntington’s disease: Preclinical pharmacology and efficacy of C2-8 in the R6/2 transgenic mouse. Proc. Natl. Acad. Sci. USA 2007, 104, 16685–16689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Pryor, W.M.; Biagioli, M.; Shahani, N.; Swarnkar, S.; Huang, W.C.; Page, D.T.; MacDonald, M.E.; Subramaniam, S. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci. Signal. 2014, 7, ra103. [Google Scholar] [CrossRef]
- Hersch, S.M.; Schifitto, G.; Oakes, D.; Bredlau, A.L.; Meyers, C.M.; Nahin, R.; Rosas, H.D.; Huntington Study Group CREST-E Investigators and Coordinators. The CREST-E study of creatine for Huntington disease: A randomized controlled trial. Neurology 2017, 89, 594–601. [Google Scholar] [CrossRef] [Green Version]
- McGarry, A.; McDermott, M.; Kieburtz, K.; de Blieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M.; et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, J.J.; Rosser, A.; Craufurd, D.; Squitieri, F.; Mallard, N.; Landwehrmeyer, B. Ethyl-eicosapentaenoic acid treatment in Huntington’s disease: A placebo-controlled clinical trial. Mov. Disord. 2015, 30, 1426–1429. [Google Scholar] [CrossRef]
- Mao, Z.; Choo, Y.S.; Lesort, M. Cystamine and cysteamine prevent 3-NP-induced mitochondrial depolarization of Huntington’s disease knock-in striatal cells. Eur. J. Neurosci. 2006, 23, 1701–1710. [Google Scholar] [CrossRef]
- Gohil, V.M.; Offner, N.; Walker, J.A.; Sheth, S.A.; Fossale, E.; Gusella, J.F.; MacDonald, M.E.; Neri, C.; Mootha, V.K. Meclizine is neuroprotective in models of Huntington’s disease. Hum. Mol. Genet. 2011, 20, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Gardian, G.; Browne, S.E.; Choi, D.K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [Green Version]
- Southwell, A.L.; Ko, J.; Patterson, P.H. Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease. J. Neurosci. 2009, 29, 13589–13602. [Google Scholar] [CrossRef] [Green Version]
- Garriga-Canut, M.; Agustin-Pavon, C.; Herrmann, F.; Sanchez, A.; Dierssen, M.; Fillat, C.; Isalan, M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. USA 2012, 109, E3136–E3145. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investg. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Franich, N.R.; Fitzsimons, H.L.; Fong, D.M.; Klugmann, M.; During, M.J.; Young, D. AAV vector-mediated RNAi of mutant huntingtin expression is neuroprotective in a novel genetic rat model of Huntington’s disease. Mol. Ther. 2008, 16, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Stanek, L.M.; Sardi, S.P.; Mastis, B.; Richards, A.R.; Treleaven, C.M.; Taksir, T.; Misra, K.; Cheng, S.H.; Shihabuddin, L.S. Silencing mutant huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum. Gene Ther. 2014, 25, 461–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaranch, L.; Blits, B.; San Sebastian, W.; Hadaczek, P.; Bringas, J.; Sudhakar, V.; Macayan, M.; Pivirotto, P.J.; Petry, H.; Bankiewicz, K.S.; et al. MR-guided parenchymal delivery of adeno-associated viral vector serotype 5 in non-human primate brain. Gene Ther. 2017, 24, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lim, P.J.; Yin, C.; Rieckher, M.; Vogel, B.E.; Monteiro, M.J. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum. Mol. Genet. 2006, 15, 1025–1041. [Google Scholar] [CrossRef] [Green Version]
- Safren, N.; El Ayadi, A.; Chang, L.; Terrillion, C.E.; Gould, T.D.; Boehning, D.F.; Monteiro, M.J. Ubiquilin-1 overexpression increases the lifespan and delays accumulation of Huntingtin aggregates in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2014, 9, e87513. [Google Scholar] [CrossRef] [Green Version]
- Thulasiraman, V.; Yang, C.F.; Frydman, J. In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J. 1999, 18, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Kalisman, N.; Adams, C.M.; Levitt, M. Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. USA 2012, 109, 2884–2889. [Google Scholar] [CrossRef] [Green Version]
- Reilmann, R.; Rouzade-Dominguez, M.L.; Saft, C.; Sussmuth, S.D.; Priller, J.; Rosser, A.; Rickards, H.; Schols, L.; Pezous, N.; Gasparini, F.; et al. A randomized, placebo-controlled trial of AFQ056 for the treatment of chorea in Huntington’s disease. Mov. Disord. 2015, 30, 427–431. [Google Scholar] [CrossRef]
- Chabrier, P.E.; Auguet, M. Pharmacological properties of BN82451: A novel multitargeting neuroprotective agent. CNS Drug Rev. 2007, 13, 317–332. [Google Scholar] [CrossRef]
- Klivenyi, P.; Ferrante, R.J.; Gardian, G.; Browne, S.; Chabrier, P.E.; Beal, M.F. Increased survival and neuroprotective effects of BN82451 in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2003, 86, 267–272. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellington, C.L.; Ellerby, L.M.; Hackam, A.S.; Margolis, R.L.; Trifiro, M.A.; Singaraja, R.; McCutcheon, K.; Salvesen, G.S.; Propp, S.S.; Bromm, M.; et al. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J. Biol. Chem. 1998, 273, 9158–9167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marder, K.; Gu, Y.; Eberly, S.; Tanner, C.M.; Scarmeas, N.; Oakes, D.; Shoulson, I.; Huntington Study Group, P.I. Relationship of Mediterranean diet and caloric intake to phenoconversion in Huntington disease. JAMA Neurol. 2013, 70, 1382–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.; Rosas, H.D.; Hersch, S.M.; Ferrante, R.J. The therapeutic role of creatine in Huntington’s disease. Pharmacol. Ther. 2005, 108, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Hersch, S.M.; Gevorkian, S.; Marder, K.; Moskowitz, C.; Feigin, A.; Cox, M.; Como, P.; Zimmerman, C.; Lin, M.; Zhang, L.; et al. Creatine in Huntington disease is safe, tolerable, bioavailable in brain and reduces serum 8OH2’dG. Neurology 2006, 66, 250–252. [Google Scholar] [CrossRef]
- Rosas, H.D.; Doros, G.; Gevorkian, S.; Malarick, K.; Reuter, M.; Coutu, J.P.; Triggs, T.D.; Wilkens, P.J.; Matson, W.; Salat, D.H.; et al. PRECREST: A phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology 2014, 82, 850–857. [Google Scholar] [CrossRef] [Green Version]
- Verbessem, P.; Lemiere, J.; Eijnde, B.O.; Swinnen, S.; Vanhees, L.; Van Leemputte, M.; Hespel, P.; Dom, R. Creatine supplementation in Huntington’s disease: A placebo-controlled pilot trial. Neurology 2003, 61, 925–930. [Google Scholar] [CrossRef]
- Andrich, J.; Saft, C.; Gerlach, M.; Schneider, B.; Arz, A.; Kuhn, W.; Muller, T. Coenzyme Q10 serum levels in Huntington’s disease. J. Neural Transm. Suppl. 2004, 68, 111–116. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Calingasan, N.Y.; Wille, E.J.; Cormier, K.; Smith, K.; Ferrante, R.J.; Beal, M.F. Combination therapy with coenzyme Q10 and creatine produces additive neuroprotective effects in models of Parkinson’s and Huntington’s diseases. J. Neurochem. 2009, 109, 1427–1439. [Google Scholar] [CrossRef] [Green Version]
- Jump, D.B. The biochemistry of n-3 polyunsaturated fatty acids. J. Biol. Chem. 2002, 277, 8755–8758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonergan, P.E.; Martin, D.S.; Horrobin, D.F.; Lynch, M.A. Neuroprotective effect of eicosapentaenoic acid in hippocampus of rats exposed to gamma-irradiation. J. Biol. Chem. 2002, 277, 20804–20811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.S.; Lonergan, P.E.; Boland, B.; Fogarty, M.P.; Brady, M.; Horrobin, D.F.; Campbell, V.A.; Lynch, M.A. Apoptotic changes in the aged brain are triggered by interleukin-1beta-induced activation of p38 and reversed by treatment with eicosapentaenoic acid. J. Biol. Chem. 2002, 277, 34239–34246. [Google Scholar] [CrossRef] [Green Version]
- Puri, B.K.; Leavitt, B.R.; Hayden, M.R.; Ross, C.A.; Rosenblatt, A.; Greenamyre, J.T.; Hersch, S.; Vaddadi, K.S.; Sword, A.; Horrobin, D.F.; et al. Ethyl-EPA in Huntington disease: A double-blind, randomized, placebo-controlled trial. Neurology 2005, 65, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Huntington Study Group TREND-HD Investigators. Randomized controlled trial of ethyl-eicosapentaenoic acid in Huntington disease: The TREND-HD study. Arch. Neurol. 2008, 65, 1582–1589. [Google Scholar] [CrossRef]
- Hogarth, P.; Lovrecic, L.; Krainc, D. Sodium phenylbutyrate in Huntington’s disease: A dose-finding study. Mov. Disord. 2007, 22, 1962–1964. [Google Scholar] [CrossRef] [PubMed]
- Stack, E.C.; Del Signore, S.J.; Luthi-Carter, R.; Soh, B.Y.; Goldstein, D.R.; Matson, S.; Goodrich, S.; Markey, A.L.; Cormier, K.; Hagerty, S.W.; et al. Modulation of nucleosome dynamics in Huntington’s disease. Hum. Mol. Genet. 2007, 16, 1164–1175. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Hwang, Y.J.; Kim, K.Y.; Kowall, N.W.; Ryu, H. Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef]
- Sah, D.W.; Aronin, N. Oligonucleotide therapeutic approaches for Huntington disease. J. Clin. Investg. 2011, 121, 500–507. [Google Scholar] [CrossRef]
- Miniarikova, J.; Evers, M.M.; Konstantinova, P. Translation of MicroRNA-Based Huntingtin-Lowering Therapies from Preclinical Studies to the Clinic. Mol. Ther. 2018, 26, 947–962. [Google Scholar] [CrossRef] [Green Version]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Matsui, M.; Corey, D.R. Allele-selective inhibition of mutant huntingtin by peptide nucleic acid-peptide conjugates, locked nucleic acid, and small interfering RNA. Ann. N. Y. Acad. Sci. 2009, 1175, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, R.L.; McBride, J.L.; Martins, I.; Shen, S.; Xing, Y.; Carter, B.J.; Davidson, B.L. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 2009, 17, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P.; et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009, 65, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; Maschat, F. The P42 peptide and Peptide-based therapies for Huntington’s disease. Orphanet J. Rare Dis. 2016, 11, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malankhanova, T.B.; Malakhova, A.A.; Medvedev, S.P.; Zakian, S.M. Modern Genome Editing Technologies in Huntington’s Disease Research. J. Huntingt. Dis. 2017, 6, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vachey, G.; Deglon, N. CRISPR/Cas9-Mediated Genome Editing for Huntington’s Disease. Methods Mol. Biol. 2018, 1780, 463–481. [Google Scholar] [CrossRef]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Dabrowska, M.; Olejniczak, M. Gene Therapy for Huntington’s Disease Using Targeted Endonucleases. Methods Mol. Biol. 2020, 2056, 269–284. [Google Scholar] [CrossRef]
- Ekman, F.K.; Ojala, D.S.; Adil, M.M.; Lopez, P.A.; Schaffer, D.V.; Gaj, T. CRISPR-Cas9-Mediated Genome Editing Increases Lifespan and Improves Motor Deficits in a Huntington’s Disease Mouse Model. Mol. Ther. Nucleic Acids 2019, 17, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Larrouy, B.; Blonski, C.; Boiziau, C.; Stuer, M.; Moreau, S.; Shire, D.; Toulme, J.J. RNase H-mediated inhibition of translation by antisense oligodeoxyribonucleotides: Use of backbone modification to improve specificity. Gene 1992, 121, 189–194. [Google Scholar] [CrossRef]
- Crooke, S.T.; Baker, B.F.; Kwoh, T.J.; Cheng, W.; Schulz, D.J.; Xia, S.; Salgado, N.; Bui, H.H.; Hart, C.E.; Burel, S.A.; et al. Integrated Safety Assessment of 2’-O-Methoxyethyl Chimeric Antisense Oligonucleotides in NonHuman Primates and Healthy Human Volunteers. Mol. Ther. 2016, 24, 1771–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanek, L.M.; Yang, W.; Angus, S.; Sardi, P.S.; Hayden, M.R.; Hung, G.H.; Bennett, C.F.; Cheng, S.H.; Shihabuddin, L.S. Antisense oligonucleotide-mediated correction of transcriptional dysregulation is correlated with behavioral benefits in the YAC128 mouse model of Huntington’s disease. J. Huntingt. Dis. 2013, 2, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.B.; Warby, S.C.; Southwell, A.L.; Doty, C.N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C.F.; Freier, S.M.; Hayden, M.R.; et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene / allele-specific silencing of mutant huntingtin. Mol. Ther. 2011, 19, 2178–2185. [Google Scholar] [CrossRef] [Green Version]
- Hammond, S.M.; Hazell, G.; Shabanpoor, F.; Saleh, A.F.; Bowerman, M.; Sleigh, J.N.; Meijboom, K.E.; Zhou, H.; Muntoni, F.; Talbot, K.; et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 2016, 113, 10962–10967. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Cho, I.K.; Hunter, C.E.; Ye, S.; Pongos, A.L.; Chan, A.W.S. Combination of stem cell and gene therapy ameliorates symptoms in Huntington’s disease mice. NPJ Regen. Med. 2019, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Ratni, H.; Karp, G.M.; Weetall, M.; Naryshkin, N.A.; Paushkin, S.V.; Chen, K.S.; McCarthy, K.D.; Qi, H.; Turpoff, A.; Woll, M.G.; et al. Specific Correction of Alternative Survival Motor Neuron 2 Splicing by Small Molecules: Discovery of a Potential Novel Medicine to Treat Spinal Muscular Atrophy. J. Med. Chem. 2016, 59, 6086–6100. [Google Scholar] [CrossRef]
- Kletzl, H.; Marquet, A.; Gunther, A.; Tang, W.; Heuberger, J.; Groeneveld, G.J.; Birkhoff, W.; Mercuri, E.; Lochmuller, H.; Wood, C.; et al. The oral splicing modifier RG7800 increases full length survival of motor neuron 2 mRNA and survival of motor neuron protein: Results from trials in healthy adults and patients with spinal muscular atrophy. Neuromuscul. Disord. 2019, 29, 21–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, N.S.; Dorsey, E.R.; Hayden, M.R. Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat. Rev. Drug Discov. 2018, 17, 729–750. [Google Scholar] [CrossRef]
- Pattison, L.R.; Kotter, M.R.; Fraga, D.; Bonelli, R.M. Apoptotic cascades as possible targets for inhibiting cell death in Huntington’s disease. J. Neurol. 2006, 253, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Sofi, F.; Macchi, C.; Casini, A. Mediterranean Diet and Minimizing Neurodegeneration. Curr. Nutr. Rep. 2013, 2, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Tangney, C.C.; Wang, Y.; Sacks, F.M.; Bennett, D.A.; Aggarwal, N.T. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 1007–1014. [Google Scholar] [CrossRef] [Green Version]
- Ehrnhoefer, D.E.; Martin, D.D.O.; Schmidt, M.E.; Qiu, X.; Ladha, S.; Caron, N.S.; Skotte, N.H.; Nguyen, Y.T.N.; Vaid, K.; Southwell, A.L.; et al. Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol. Commun. 2018, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Borrell-Pages, M.; Canals, J.M.; Cordelieres, F.P.; Parker, J.A.; Pineda, J.R.; Grange, G.; Bryson, E.A.; Guillermier, M.; Hirsch, E.; Hantraye, P.; et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J. Clin. Investg. 2006, 116, 1410–1424. [Google Scholar] [CrossRef] [Green Version]
- Menalled, L.B.; Kudwa, A.E.; Oakeshott, S.; Farrar, A.; Paterson, N.; Filippov, I.; Miller, S.; Kwan, M.; Olsen, M.; Beltran, J.; et al. Genetic deletion of transglutaminase 2 does not rescue the phenotypic deficits observed in R6/2 and zQ175 mouse models of Huntington’s disease. PLoS ONE 2014, 9, e99520. [Google Scholar] [CrossRef] [Green Version]
- Waters, S.; Tedroff, J.; Ponten, H.; Klamer, D.; Sonesson, C.; Waters, N. Pridopidine: Overview of Pharmacology and Rationale for its Use in Huntington’s Disease. J. Hunt. Dis. 2018, 7, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ryskamp, D.; Wu, L.; Wu, J.; Kim, D.; Rammes, G.; Geva, M.; Hayden, M.; Bezprozvanny, I. Pridopidine stabilizes mushroom spines in mouse models of Alzheimer’s disease by acting on the sigma-1 receptor. Neurobiol. Dis. 2019, 124, 489–504. [Google Scholar] [CrossRef]
- de Yebenes, J.G.; Landwehrmeyer, B.; Squitieri, F.; Reilmann, R.; Rosser, A.; Barker, R.A.; Saft, C.; Magnet, M.K.; Sword, A.; Rembratt, A.; et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2011, 10, 1049–1057. [Google Scholar] [CrossRef]
- Reilmann, R.; McGarry, A.; Grachev, I.D.; Savola, J.M.; Borowsky, B.; Eyal, E.; Gross, N.; Langbehn, D.; Schubert, R.; Wickenberg, A.T.; et al. Safety and efficacy of pridopidine in patients with Huntington’s disease (PRIDE-HD): A phase 2, randomised, placebo-controlled, multicentre, dose-ranging study. Lancet Neurol. 2019, 18, 165–176. [Google Scholar] [CrossRef]
- Geva, M.; Kusko, R.; Soares, H.; Fowler, K.D.; Birnberg, T.; Barash, S.; Wagner, A.M.; Fine, T.; Lysaght, A.; Weiner, B.; et al. Pridopidine activates neuroprotective pathways impaired in Huntington Disease. Hum. Mol. Genet. 2016, 25, 3975–3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda, J.R.; Canals, J.M.; Bosch, M.; Adell, A.; Mengod, G.; Artigas, F.; Ernfors, P.; Alberch, J. Brain-derived neurotrophic factor modulates dopaminergic deficits in a transgenic mouse model of Huntington’s disease. J. Neurochem. 2005, 93, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Squitieri, F.; Di Pardo, A.; Favellato, M.; Amico, E.; Maglione, V.; Frati, L. Pridopidine, a dopamine stabilizer, improves motor performance and shows neuroprotective effects in Huntington disease R6/2 mouse model. J. Cell. Mol. Med. 2015, 19, 2540–2548. [Google Scholar] [CrossRef]
- Huntington Study Group HART Investigators. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov. Disord. 2013, 28, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Adanyeguh, I.M.; Rinaldi, D.; Henry, P.G.; Caillet, S.; Valabregue, R.; Durr, A.; Mochel, F. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015, 84, 490–495. [Google Scholar] [CrossRef]
- Kieburtz, K.; McDermott, M.P.; Voss, T.S.; Corey-Bloom, J.; Deuel, L.M.; Dorsey, E.R.; Factor, S.; Geschwind, M.D.; Hodgeman, K.; Kayson, E.; et al. A randomized, placebo-controlled trial of latrepirdine in Huntington disease. Arch. Neurol. 2010, 67, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Blanpied, T.A.; Clarke, R.J.; Johnson, J.W. Amantadine inhibits NMDA receptors by accelerating channel closure during channel block. J. Neurosci 2005, 25, 3312–3322. [Google Scholar] [CrossRef] [Green Version]
- Hosenbocus, S.; Chahal, R. Amantadine: A review of use in child and adolescent psychiatry. J. Can. Acad Child. Adolesc. Psychiatry 2013, 22, 55–60. [Google Scholar]
- Verhagen Metman, L.; Morris, M.J.; Farmer, C.; Gillespie, M.; Mosby, K.; Wuu, J.; Chase, T.N. Huntington’s disease: A randomized, controlled trial using the NMDA-antagonist amantadine. Neurology 2002, 59, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Wagstaff, A.J.; Ibbotson, T.; Perry, C.M. Lamotrigine: A review of its use in bipolar disorder. Drugs 2003, 63, 2029–2050. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Liu, Y.C.; Lee, C.T.; Lin, Y.C.; Wang, M.L.; Yang, Y.P.; Chang, K.Y.; Chiou, S.H. Revisiting the Lamotrigine-Mediated Effect on Hippocampal GABAergic Transmission. Int. J. Mol. Sci. 2016, 17, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.C. Lamotrigine in motor and mood symptoms of Huntington’s disease. World J. Biol. Psychiatry 2008, 9, 147–149. [Google Scholar] [CrossRef] [PubMed]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a SirT1 inhibitor, in patients with Huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilmann, R.; Squitieri, F.; Priller, J.; Saft, C.; Mariotti, C.; Suessmuth, S.; Nemeth, A.; Tabrizi, S.; Quarrell, O.; Craufurd, D.; et al. Safety and Tolerability of Selisistat for the Treatment of Huntington’s Disease: Results from a Randomized, Double-Blind, Placebo-Controlled Phase II Trial (S47.004). Neurology 2014, 82, S47.004. [Google Scholar] [CrossRef]
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [Green Version]
- Ehrnhoefer, D.E.; Caron, N.S.; Deng, Y.; Qiu, X.; Tsang, M.; Hayden, M.R. Laquinimod decreases Bax expression and reduces caspase-6 activation in neurons. Exp. Neurol. 2016, 283, 121–128. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Yusof, N.; Tan, J.Y.; Radulescu, C.I.; Sidik, H.; Tan, L.J.; Belinson, H.; Zach, N.; Hayden, M.R.; Pouladi, M.A.; et al. Laquinimod Treatment Improves Myelination Deficits at the Transcriptional and Ultrastructural Levels in the YAC128 Mouse Model of Huntington Disease. Mol. Neurobiol. 2019, 56, 4464–4478. [Google Scholar] [CrossRef]
- Garcia-Miralles, M.; Hong, X.; Tan, L.J.; Caron, N.S.; Huang, Y.; To, X.V.; Lin, R.Y.; Franciosi, S.; Papapetropoulos, S.; Hayardeny, L.; et al. Laquinimod rescues striatal, cortical and white matter pathology and results in modest behavioural improvements in the YAC128 model of Huntington disease. Sci. Rep. 2016, 6, 31652. [Google Scholar] [CrossRef]
- Mazarei, G.; Leavitt, B.R. Indoleamine 2,3 Dioxygenase as a Potential Therapeutic Target in Huntington’s Disease. J. Huntingt. Dis. 2015, 4, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgini, F.; Moller, T.; Kwan, W.; Zwilling, D.; Wacker, J.L.; Hong, S.; Tsai, L.C.; Cheah, C.S.; Schwarcz, R.; Guidetti, P.; et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J. Biol. Chem. 2008, 283, 7390–7400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Mechanism of Toxicity of Huntingtin (HTT) gene. NO (Nitric Oxide), CAG (cytosine-adenine-guanine).
Figure 1.
Mechanism of Toxicity of Huntingtin (HTT) gene. NO (Nitric Oxide), CAG (cytosine-adenine-guanine).

Figure 2.
Recent advancement in the therapeutics for Huntington’s disease. TBZ (Tetrabenazine); EPA (eicosapentaenoic acid); MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine); SAHA (suberoylanilide hydroxamic acid); HDACi4b (histone deacetylase inhibitors); RNAi (RNA interference); ASO (antisense oligonucleotide); ZFP (zinc finger protein); CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats); miRNA (micro RNA); siRNA (small interfering RNA).
Figure 2.
Recent advancement in the therapeutics for Huntington’s disease. TBZ (Tetrabenazine); EPA (eicosapentaenoic acid); MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine); SAHA (suberoylanilide hydroxamic acid); HDACi4b (histone deacetylase inhibitors); RNAi (RNA interference); ASO (antisense oligonucleotide); ZFP (zinc finger protein); CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats); miRNA (micro RNA); siRNA (small interfering RNA).


{kind=link}
{kind=link}
{kind=link}
Table 1.
Recent status of Huntington’s disease (HD) drug therapy.
| Drug/Reagent | Primary Target (Mechanism of Action) | Status and Principal Result | Ref. |
|---|---|---|---|
| (1) Drugs against excitotoxicity | |||
| Riluzole | Glutamate release inhibitor | Does not show efficacy in human trails | [31] |
| Memantine | N-methyl-D-aspartate (NMDA) receptor inhibitor | Demonstrated efficacy in human trial | [32,33] |
| Tetrabenazine (TBZ) | Dopamine pathway (Vesicular monoamine transporter 2 inhibitor) | Approved by food and drug administration (FDA) for treatment of chorea in HD | [38,39,40] |
| (2) Targeting Caspase and huntingtin (HTT) proteolysis | |||
| Minocycline | Caspase-dependent and independent neurodegenerative pathway | Inhibits caspase-1 and -3 mRNA upregulation, and decreases inducible nitric oxide synthetase activity | [42,44,45] |
| (3) Targeting HTT aggregation and clearance | |||
| Congo red and Trehalose | Aggregation | Showed efficacy in a rodent model | [46,49] |
| Compound C2–8 | Aggregation | Showed efficacy in a rodent model | [53] |
| Rapamycin | Aggregation mammalian target of rapamycin (mTOR) inhibitor | Showed efficacy in a rodent model | [54,55] |
| (4) Targeting mitochondrial dysfunction | |||
| Creatine | Mitochondrial dysfunction | Attained futility in human trial | [57] |
| CoQ10 | Mitochondrial dysfunction | Attained futility in human trial | [58] |
| Eicosapentaenoic acid (EPA) | Mitochondria dysfunction | A mixed scenario of positive and negative trial | [59] |
| Cystamine and mitochondrial permeability transition pore blockers | Mitochondrial dysfunction | Showed efficacy in a rodent model | [60] |
| Meclizine drug | Mitochondrial dysfunction | Showed efficacy in the fly model | [61] |
| (5) Targeting transcriptional dysregulation | |||
| Sodium phenylbutyrate | Transcriptional deregulation histone deacetylase inhibitor | Showed efficacy in a rodent model | [62] |
| HDACi4b (a pimelic diphenylamide HDAC inhibitor) | Transcriptional deregulation histone deacetylase inhibitor | Showed efficacy in a rodent model | [63] |
| Suberoylanilide hydroxamic acid | Transcriptional deregulation histone deacetylase inhibitor | Showed efficacy in a rodent model | [64] |
| Mithramycin and chromomycin | Transcriptional deregulation G-C-rich DNA binding antibiotic | Showed efficacy in a rodent model | [65] |
| (6) Targetting mutant huntingtin (mHTT) | |||
| RNA interference and antisense oligonucleotide (ASO) | Blocks transcription of mHTT | Showed efficacy in a rodent model | [6] |
| Artificial peptides and intrabodies | Targeting proline-rich domain of HTT | Showed efficacy in a rodent model | [66] |
| (7) Therapies targeting nucleic acid | |||
| Zinc finger protein | Reduced mutant protein expression | Showed efficacy in a rodent model | [67] |
| CRISPR-Cas9 | Excision of CAG repeat and, reduction of mutant HTT | Showed efficacy in a rodent model | [68,69] |
| ASO approach (IONIS-HTTRX, Peptide conjugated ASOs) | Reduction in HTT mRNA and protein | Showed efficacy in a rodent model | [70,71] |
| RNAi approach (siRNA, shRNA, and miRNA) | Improves motor and neuropathological abnormalities, silencing of HTT | Showed efficacy in a rodent model | [6,72] |
| Novel Viral Vectors (AAV1, AAV5, AAV9, AAV-PHP.B, CREATE) | Widespread transduction of cells | Showed efficacy in primate and rodent models | [73,74] |
| (8) Other therapeutics | |||
| Ubiquilin | Reduces mHTT aggregation | Showed efficacy in a rodent model | [75,76] |
| miRNA | Silence HTT | Testing in rodent and nonhuman primates | [6,72] |
| Chaperonins | Decrease mHTT aggregation | Showed efficacy in a rodent model | [77,78] |
| AFQ056 | The antagonist for glutamate receptor 5 | Showed no improvement in chorea in a clinical trial | [79] |
| BN82451 | Decreases glutamate release by blocking Na+ channels | Showed efficacy in a rodent model | [80,81] |
| Antipsychotic drug | Block or modulate dopamine receptors | Under phase III trial | - |
| Antiapoptotic drug | Cleave mHTT | Effective in mice model | [82,83] |
| Diet | Delay onset of disease | Effective result but requires further evaluation | [84] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kumar, A.; Kumar, V.; Singh, K.; Kumar, S.; Kim, Y.-S.; Lee, Y.-M.; Kim, J.-J. Therapeutic Advances for Huntington’s Disease. Brain Sci. 2020, 10, 43. https://doi.org/10.3390/brainsci10010043
AMA Style
Kumar A, Kumar V, Singh K, Kumar S, Kim Y-S, Lee Y-M, Kim J-J. Therapeutic Advances for Huntington’s Disease. Brain Sciences. 2020; 10(1):43. https://doi.org/10.3390/brainsci10010043
Chicago/Turabian StyleKumar, Ashok, Vijay Kumar, Kritanjali Singh, Sukesh Kumar, You-Sam Kim, Yun-Mi Lee, and Jong-Joo Kim. 2020. "Therapeutic Advances for Huntington’s Disease" Brain Sciences 10, no. 1: 43. https://doi.org/10.3390/brainsci10010043
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.