Stroke Neuroprotection: Targeting Mitochondria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Stroke: A Brief Overview
2. Pathological Mechanisms Associated with Stroke
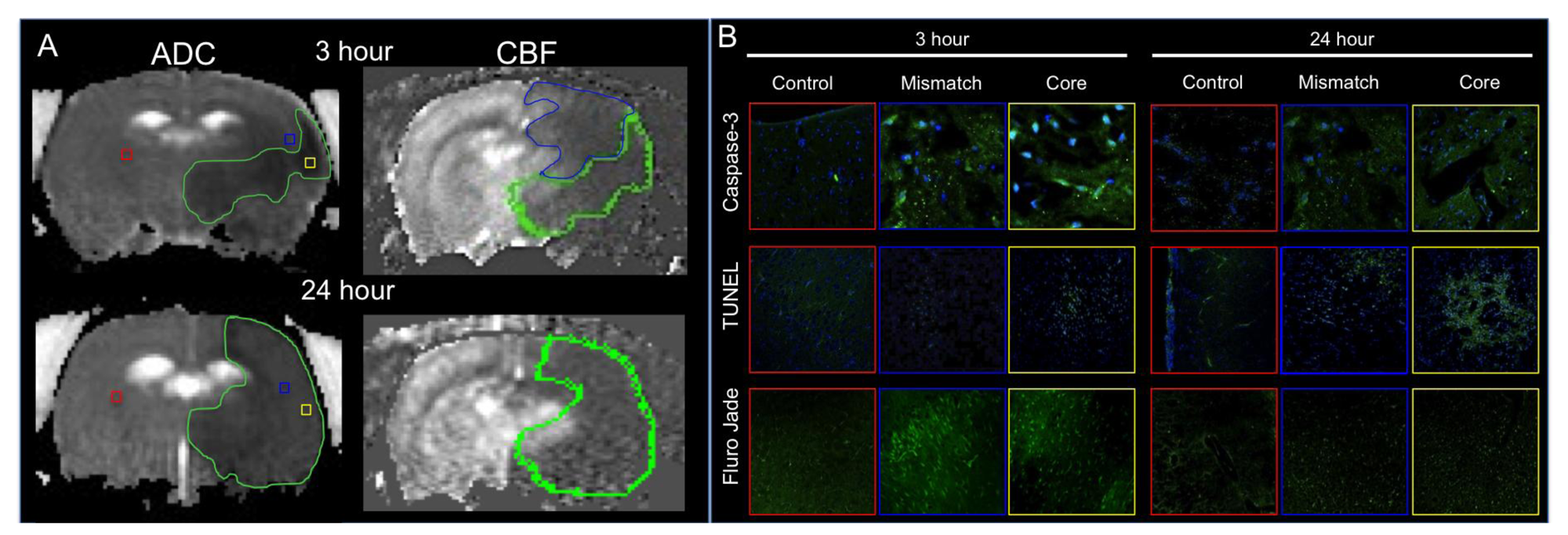
3. Current Therapeutic Approaches for Treatment of Stroke
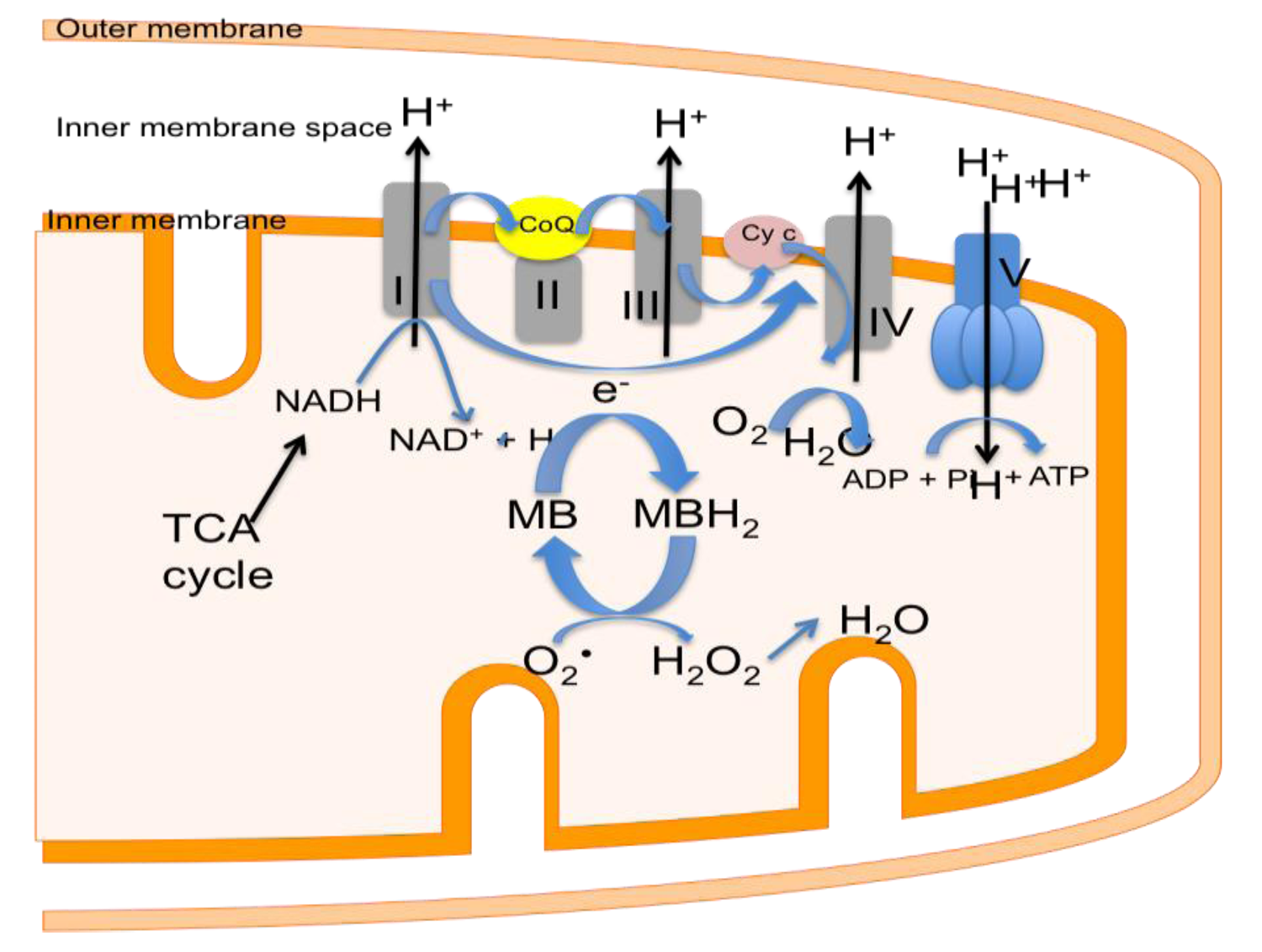
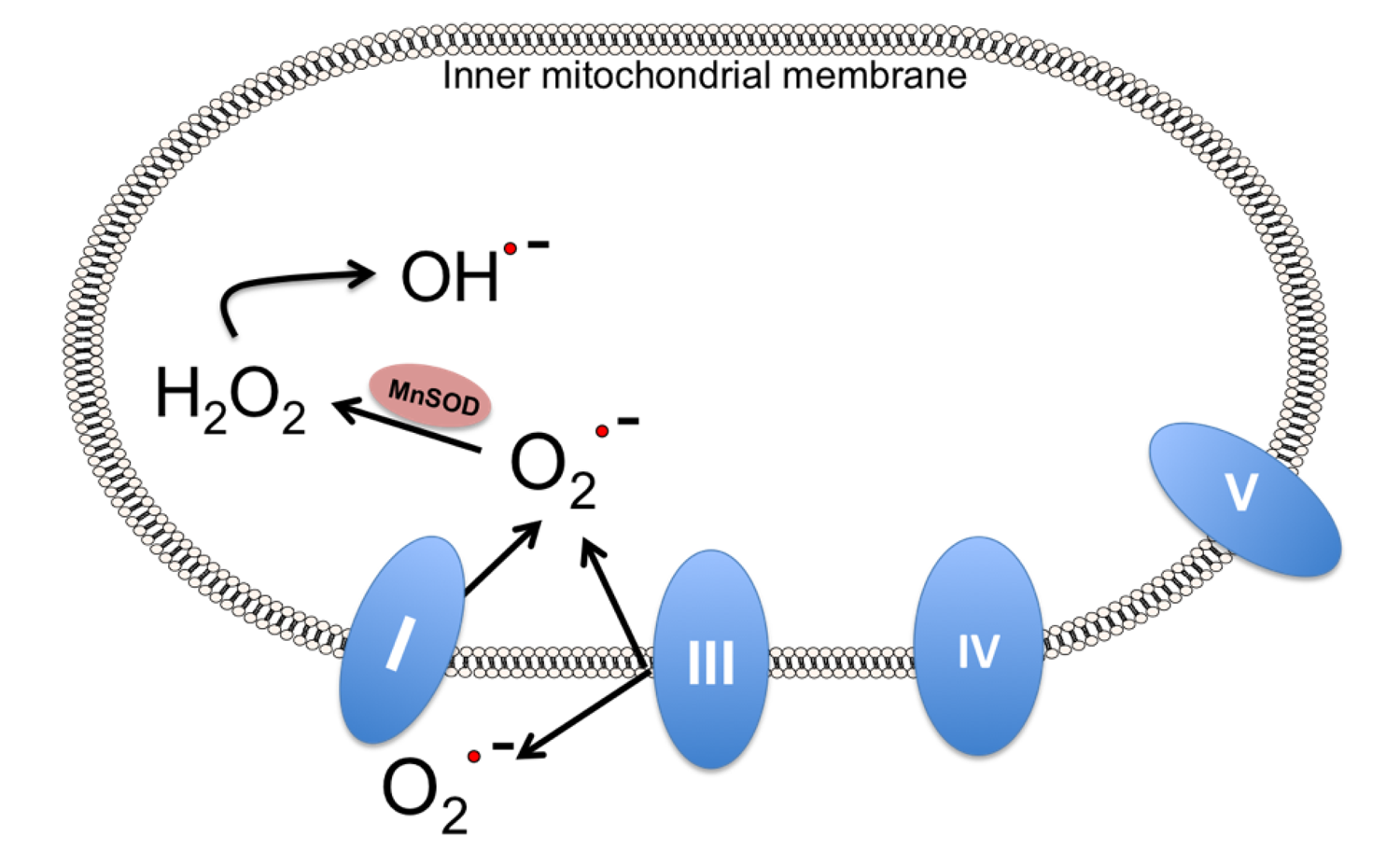
4. Mitochondrial Dysfunction in Stroke
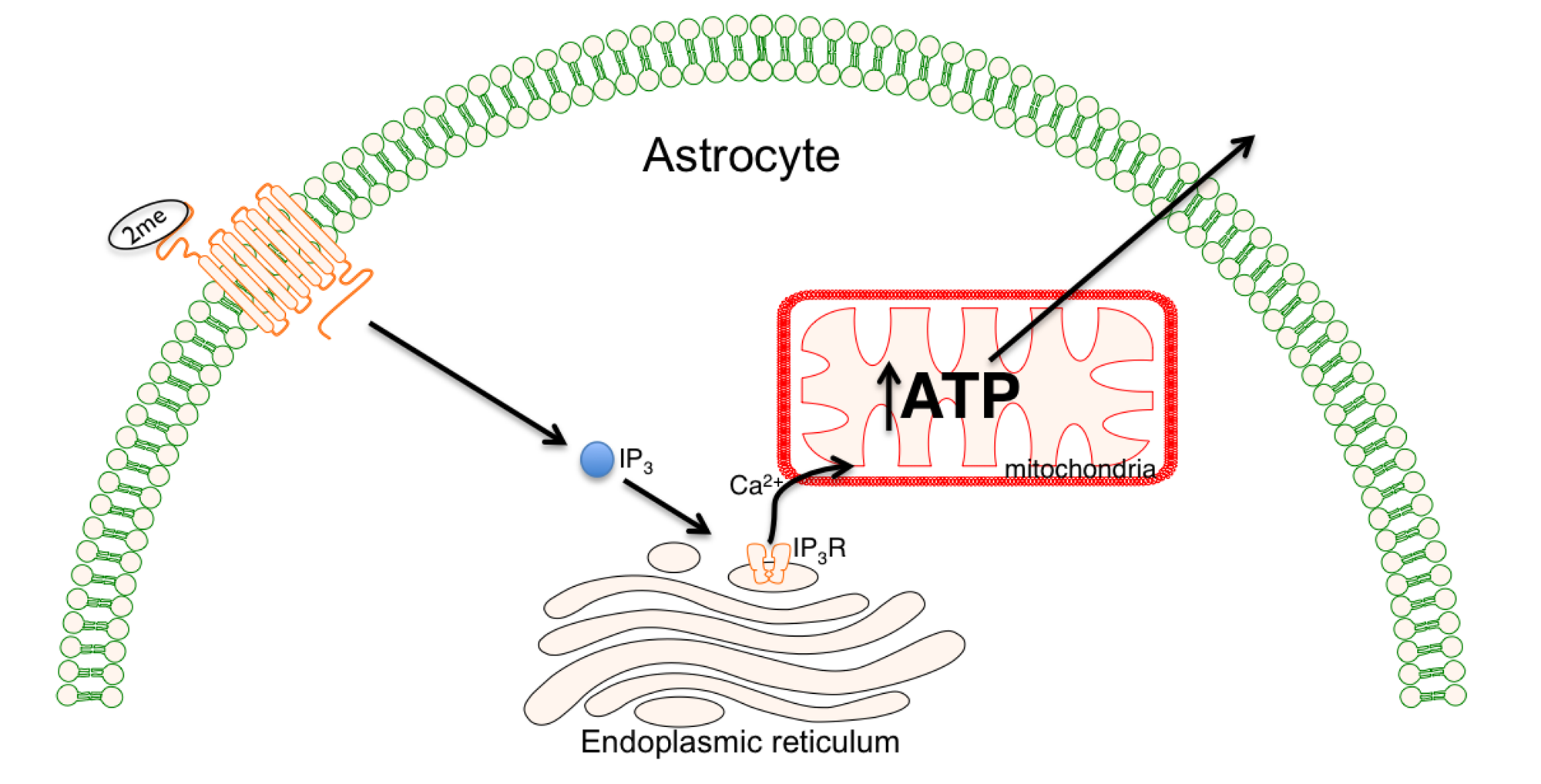
5. Increasing ATP Production with Purinergic Receptor Stimulation
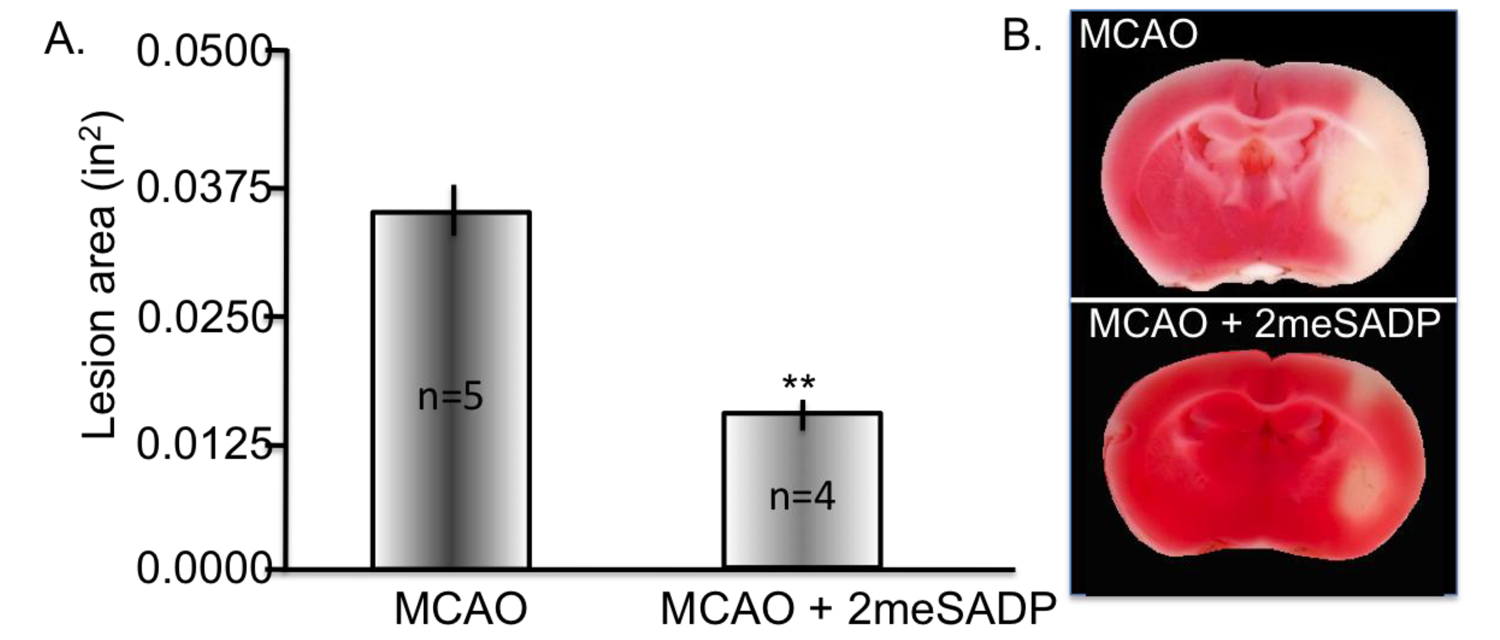
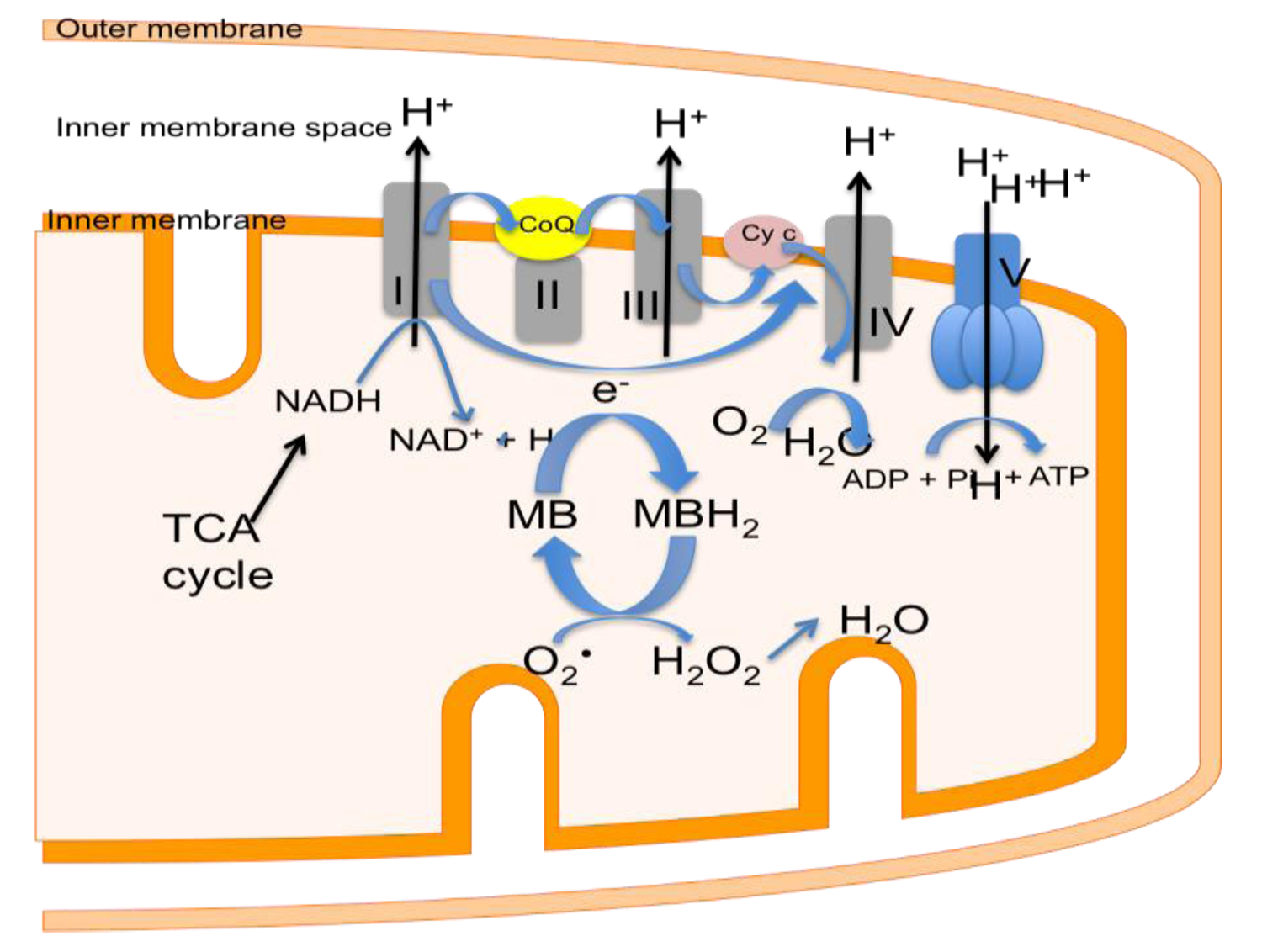
6. Decreasing Superoxide Production with Methylene Blue
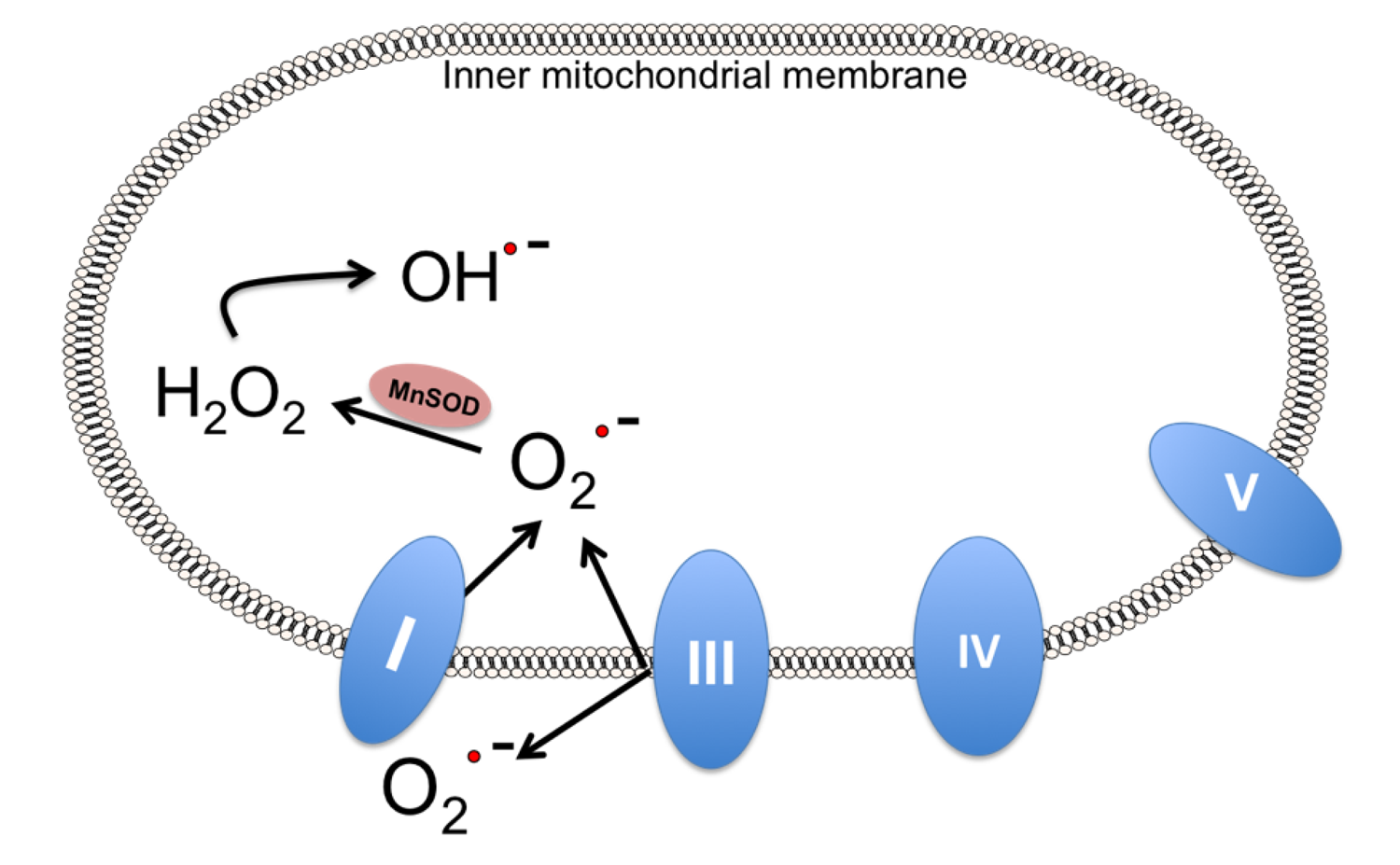
7. Alternatives to Endogenous MnSOD: MnSOD Mimetics
8. Conclusion
Acknowledgments
Conflict of Interest
References
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Adams, R.J.; Berry, J.D.; Brown, T.M.; Carnethon, M.R.; Dai, S.; de Simone, G.; Ford, E.S.; et al. Heart disease and stroke statistics—2011 update: A report from the American Heart Association. Circulation 2011, 123, e18–e209. [Google Scholar] [CrossRef]
- Shang, W.; Liu, J. Stroke subtype classification: A comparative study of ASCO and modified TOAST. J. Neurol. Sci. 2012, 314, 66–70. [Google Scholar]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Globus, M.Y.; Busto, R.; Lin, B.; Schnippering, H.; Ginsberg, M.D. Detection of free radical activity during transient global ischemia and recirculation: Effects of intraischemic brain temperature modulation. J. Neurochem. 1995, 65, 1250–1256. [Google Scholar]
- Sherman, D.G.; Easton, J.D. Cerebral edema in stroke: A common, often fatal complication. Postgrad. Med. 1980, 68, 107–113, 116, 119–120. [Google Scholar]
- Brouns, R.; Wauters, A.; de Surgeloose, D.; Marien, P.; de Deyn, P.P. Biochemical markers for blood-brain barrier dysfunction in acute ischemic stroke correlate with evolution and outcome. Eur. Neurol. 2011, 65, 23–31. [Google Scholar] [CrossRef]
- Hakim, A. The cerebral ischemic penumbra. Can. J. Neurol. Sci. 1987, 14, 557–559. [Google Scholar]
- Schlaug, G.; Benfield, A.; Baird, A.; Siewert, B.; Lövblad, K.; Parker, R.; Edelman, R.; Warach, S. The ischemic penumbra. Neurology 1999, 53, 1528–1528. [Google Scholar]
- Guadagno, J.V.; Donnan, G.A.; Markus, R.; Gillard, J.H.; Baron, J.C. Imaging the ischaemic penumbra. Curr. Opin. Neurol. 2004, 17, 61–67. [Google Scholar] [CrossRef]
- Soares, B.P.; Chien, J.D.; Wintermark, M. MR and CT Monitoring of Recanalization, Reperfusion, and Penumbra Salvage. Stroke 2009, 40, S24–S27. [Google Scholar]
- Ferrer, I.; Planas, A.M. Signaling of cell death and cell survival following focal cerebral ischemia: Life and death struggle in the penumbra. J. Neuropathol. Exp. Neurol. 2003, 62, 329–339. [Google Scholar]
- Wass, C.T.; Lanier, W.L. Glucose modulation of ischemic brain injury: Review and clinical recommendations. Mayo Clin. Proc. 1996, 71, 801–812. [Google Scholar] [CrossRef]
- Bruno, A.; Biller, J.; Adams, H.P., Jr.; Clarke, W.R.; Woolson, R.F.; Williams, L.S.; Hansen, M.D. Acute blood glucose level and outcome from ischemic stroke. Trial of ORG 10172 in Acute Stroke Treatment (TOAST) Investigators. Neurology 1999, 52, 280–284. [Google Scholar] [CrossRef]
- Reith, J.; Jorgensen, H.S.; Pedersen, P.M.; Nakayama, H.; Raaschou, H.O.; Jeppesen, L.L.; Olsen, T.S. Body temperature in acute stroke: Relation to stroke severity, infarct size, mortality, and outcome. Lancet 1996, 347, 422–425. [Google Scholar]
- Garcia, J.H.; Lassen, N.A.; Weiller, C.; Sperling, B.; Nakagawara, J. Ischemic stroke and incomplete infarction. Stroke 1996, 27, 761–765. [Google Scholar] [CrossRef]
- Del Zoppo, G.J.; Pessin, M.S.; Mori, E.; Hacke, W. Thrombolytic intervention in acute thrombotic and embolic stroke. Semin. Neurol. 1991, 11, 368–384. [Google Scholar] [CrossRef]
- Siesjo, B.K. Cell damage in the brain: A speculative synthesis. J. Cereb. Blood Flow Metab. 1981, 1, 155–185. [Google Scholar] [CrossRef]
- Siesjo, B.K.; Agardh, C.D.; Bengtsson, F. Free radicals and brain damage. Cerebrovasc. Brain Metab. Rev. 1989, 1, 165–211. [Google Scholar]
- Rothman, S.M.; Olney, J.W. Glutamate and the pathophysiology of hypoxic—ischemic brain damage. Ann. Neurol. 1986, 19, 105–111. [Google Scholar] [CrossRef]
- Becker, K.J. Inflammation and acute stroke. Curr. Opin. Neurol. 1998, 11, 45–49. [Google Scholar] [CrossRef]
- Hademenos, G.J.; Massoud, T.F. Biophysical mechanisms of stroke. Stroke 1997, 28, 2067–2077. [Google Scholar] [CrossRef]
- DeGraba, T.J. The role of inflammation after acute stroke: Utility of pursuing anti-adhesion molecule therapy. Neurology 1998, 51, S62–S68. [Google Scholar] [CrossRef]
- Kroemer, G.; Petit, P.; Zamzami, N.; Vayssiere, J.L.; Mignotte, B. The biochemistry of programmed cell death. FASEB J. 1995, 9, 1277–1287. [Google Scholar]
- Garcia, J.H.; Liu, K.-F.; Ho, K.-L. Neuronal necrosis after middle cerebral artery occlusion in Wistar rats progresses at different time intervals in the audoputamen and the cortex. Stroke 1995, 26, 636–643. [Google Scholar] [CrossRef]
- Davis, S.M.; Donnan, G.A. 4.5 hours: The new time window for tissue plasminogen activator in stroke. Stroke 2009, 40, 2266–2267. [Google Scholar] [CrossRef]
- Kleindorfer, D.; Kissela, B.; Schneider, A.; Woo, D.; Khoury, J.; Miller, R.; Alwell, K.; Gebel, J.; Szaflarski, J.; Pancioli, A.; et al. Eligibility for recombinant tissue plasminogen activator in acute ischemic stroke: A population-based study. Stroke 2004, 35, e27–e29. [Google Scholar] [CrossRef]
- Crumrine, R.C.; Marder, V.J.; Taylor, G.M.; Lamanna, J.C.; Tsipis, C.P.; Scuderi, P.; Petteway, S.R., Jr.; Arora, V. Intra-arterial administration of recombinant tissue-type plasminogen activator (rt-PA) causes more intracranial bleeding than does intravenous rt-PA in a transient rat middle cerebral artery occlusion model. Exp. Transl. Stroke Med. 2011, 3, 10. [Google Scholar] [CrossRef]
- Hacke, W.; Donnan, G.; Fieschi, C.; Kaste, M.; von Kummer, R.; Broderick, J.P.; Brott, T.; Frankel, M.; Grotta, J.C.; Haley, E.C., Jr.; et al. Association of outcome with early stroke treatment: Pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004, 363, 768–774. [Google Scholar] [CrossRef]
- Wahlgren, N.; Ahmed, N.; Davalos, A.; Ford, G.A.; Grond, M.; Hacke, W.; Hennerici, M.G.; Kaste, M.; Kuelkens, S.; Larrue, V.; et al. Thrombolysis with alteplase for acute ischaemic stroke in the Safe Implementation of Thrombolysis in Stroke-Monitoring Study (SITS-MOST): An observational study. Lancet 2007, 369, 275–282. [Google Scholar]
- Suzuki, S.; Saver, J.L.; Scott, P.; Jahan, R.; Duckwiler, G.; Starkman, S.; Su, Y.; Kidwell, C.S. Access to intra-arterial therapies for acute ischemic stroke: An analysis of the US population. AJNR Am. J. Neuroradiol. 2004, 25, 1802–1806. [Google Scholar]
- Chih, C.P.; Roberts, E.L. Energy substrates for neurons during neural activity: A critical review of the astrocyte-neuron lactate shuttle hypothesis. J. Cereb. Blood Flow Metab. 2003, 23, 1263–1281. [Google Scholar] [CrossRef]
- Swanson, R.A.; Benington, J. Astrocyte glucose metabolism under normal and pathological conditions in vitro. Dev. Neurosci. 1996, 18, 515–521. [Google Scholar] [CrossRef]
- Swanson, R.A.; Yu, A.C.H.; Sharp, F.R.; Chan, P.H. Regulation of glycogen content in primary astrocyte culture: Effects of glucose analogues, phenobarbital, and methionine sulfoximine. J. Neurochem. 1989, 52, 1359–1365. [Google Scholar] [CrossRef]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: From basic research to clinical application. Am. J. Med. 1991, 91, S31–S38. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Brookes, P.S. Mitochondrial H+ leak and ROS generation: An odd couple. Free Radic. Biol. Med. 2005, 38, 12–23. [Google Scholar] [CrossRef]
- Nohl, H.; Gille, L.; Staniek, K. Intracellular generation of reactive oxygen species by mitochondria. Biochem. Pharmacol. 2005, 69, 719–723. [Google Scholar]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, and apoptosis. Am. J. Med. Genet. 2001, 106, 62–70. [Google Scholar] [CrossRef]
- Pastorino, J.G.; Snyder, J.W.; Serroni, A.; Hoek, J.B.; Farber, J.L. Cyclosporin and carnitine prevent the anoxic death of cultured hepatocytes by inhibiting the mitochondrial permeability transition. J. Biol. Chem. 1993, 268, 13791–13798. [Google Scholar]
- Zahrebelski, G.; Nieminen, A.L.; Al-Ghoul, K.; Qian, T.; Herman, B.; Lemasters, J.J. Progression of subcellular changes during chemical hypoxia to cultured rat hepatocytes: A laser scanning confocal microscopic study. Hepatology 1995, 21, 1361–1372. [Google Scholar]
- Beal, M.F.; Brouillet, E.; Jenkins, B.G.; Ferrante, R.J.; Kowall, N.W.; Miller, J.M.; Storey, E.; Srivastava, R.; Rosen, B.R.; Hyman, B.T. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J. Neurosci. 1993, 13, 4181–4192. [Google Scholar]
- Dawson, T.L.; Gores, G.J.; Nieminen, A.L.; Herman, B.; Lemasters, J.J. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. Am. J. Physiol. 1993, 264, C961–C967. [Google Scholar]
- Newmeyer, D.D.; Farschon, D.M.; Reed, J.C. Cell-free apoptosis in Xenopus egg extracts: Inhibition by Bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell 1994, 79, 353–364. [Google Scholar] [CrossRef]
- Zamzami, N.; Susin, S.A.; Marchetti, P.; Hirsch, T.; Gomez-Monterrey, I.; Castedo, M.; Kroemer, G. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 1996, 183, 1533–1544. [Google Scholar]
- Petit, P.X.; Lecoeur, H.; Zorn, E.; Dauguet, C.; Mignotte, B.; Gougeon, M.L. Alterations in mitochondrial structure and function are early events of dexamethasone-induced thymocyte apoptosis. J. Cell Biol. 1995, 130, 157–167. [Google Scholar] [CrossRef]
- Kroemer, G. Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ. 1997, 4, 443–456. [Google Scholar]
- Chan, K.; Han, X.D.; Kan, Y.W. An important function of Nrf2 in combating oxidative stress: Detoxification of acetaminophen. Proc. Natl. Acad. Sci. USA 2001, 98, 4611–4616. [Google Scholar] [CrossRef]
- Sawada, M.; Sester, U.; Carlson, J.C. Superoxide radical formation and associated biochemical alterations in the plasma membrane of brain, heart, and liver during the lifetime of the rat. J. Cell. Biochem. 1992, 48, 296–304. [Google Scholar]
- Antier, D.; Carswell, H.V.; Brosnan, M.J.; Hamilton, C.A.; Macrae, I.M.; Groves, S.; Jardine, E.; Reid, J.L.; Dominiczak, A.E. Increased levels of superoxide in brains from old female rats. Free Radic. Res. 2004, 38, 177–183. [Google Scholar]
- Dienel, G.A.; Hertz, L. Astrocytic contributions to bioenergetics of cerebral ischemia. Glia 2005, 50, 362–388. [Google Scholar]
- Bernardi, M.L.; Flechon, J.E.; Delouis, C. Influence of culture system and oxygen tension on the development of ovine zygotes matured and fertilized in vitro. J. Reprod. Fertil. 1996, 106, 161–167. [Google Scholar] [CrossRef]
- Bains, J.S.; Shaw, C.A. Neurodegenerative disorders in humans: The role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Brain Res. Rev. 1997, 25, 335–358. [Google Scholar]
- Robb, S.J.; Connor, J.R. An in vitro model for analysis of oxidative death in primary mouse astrocytes. Brain Res. 1998, 788, 125–132. [Google Scholar] [CrossRef]
- Amin, N.; Pearce, B. Glutamate toxicity in neuron-enriched and neuron-astrocyte co-cultures: Effect of the glutamate uptake inhibitor l-trans-pyrrolidine-2,4-dicarboxylate. Neurochem. Int. 1997, 30, 271–276. [Google Scholar] [CrossRef]
- Winn, H.R.; Rubio, R.; Berne, R.M. Brain adenosine production in the rat during 60 seconds of ischemia. Circ. Res. 1979, 45, 486–492. [Google Scholar] [CrossRef]
- Schultz, S.G. Pump-leak parallelism in sodium-absorbing epithelia: The role of ATP-regulated potassium channels. J. Exp. Zool. 1997, 279, 476–483. [Google Scholar] [CrossRef]
- Liu, S.; Levine, S.R.; Winn, H.R. Targeting ischemic penumbra: Part I—from pathophysiology to therapeutic strategy. J. Exp. Stroke Transl. Med. 2010, 3, 47–55. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G. Purinergic signalling: Pathophysiological roles. Jpn. J. Pharmacol. 1998, 78, 113–145. [Google Scholar] [CrossRef]
- Fredholm, B.B. Purinoceptors in the nervous system. Pharmacol. Toxicol. 1995, 76, 228–239. [Google Scholar] [CrossRef]
- Kowaluk, E.A.; Bhagwat, S.S.; Jarvis, M.F. Adenosine kinase inhibitors. Curr. Pharm. Des. 1998, 4, 403–416. [Google Scholar]
- Rudolphi, K.A.; Schubert, P.; Parkinson, F.E.; Fredholm, B.B. Neuroprotective role of adenosine in cerebral ischaemia. Trends Pharmacol. Sci. 1992, 13, 439–445. [Google Scholar]
- Williams, M.; Burnstock, G. Purinergic Neurotransmission and Neuromodulation: A Historical Perspective; Wiley-Liss: New York, NY, USA, 1997. [Google Scholar]
- Winn, H.R.; Rubio, G.R.; Berne, R.M. The role of adenosine in the regulation of cerebral blood flow. J. Cereb. Blood Flow Metab. 1981, 1, 239–244. [Google Scholar] [CrossRef]
- Zhou, G.; Smith, J.L.; Zalkin, H. Binding of purine nucleotides to two regulatory sites results in synergistic feedback inhibition of glutamine 5-phosphoribosylpyrophosphate amidotransferase. J. Biol. Chem. 1994, 269, 6784–6789. [Google Scholar]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar]
- Verkhratsky, A.; Kettenmann, H. Calcium signalling in glial cells. Trends Neurosci. 1996, 19, 346–352. [Google Scholar] [CrossRef]
- James, G.; Butt, A.M. P2Y and P2X purinoceptor mediated Ca2+ signalling in glial cell pathology in the central nervous system. Eur. J. Pharmacol. 2002, 447, 247–260. [Google Scholar] [CrossRef]
- Denton, R.M.; McCormack, J.G. Physiological role of Ca2+ transport by mitochondria. Nature 1985, 315, 635. [Google Scholar] [CrossRef]
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 1990, 70, 391–425. [Google Scholar]
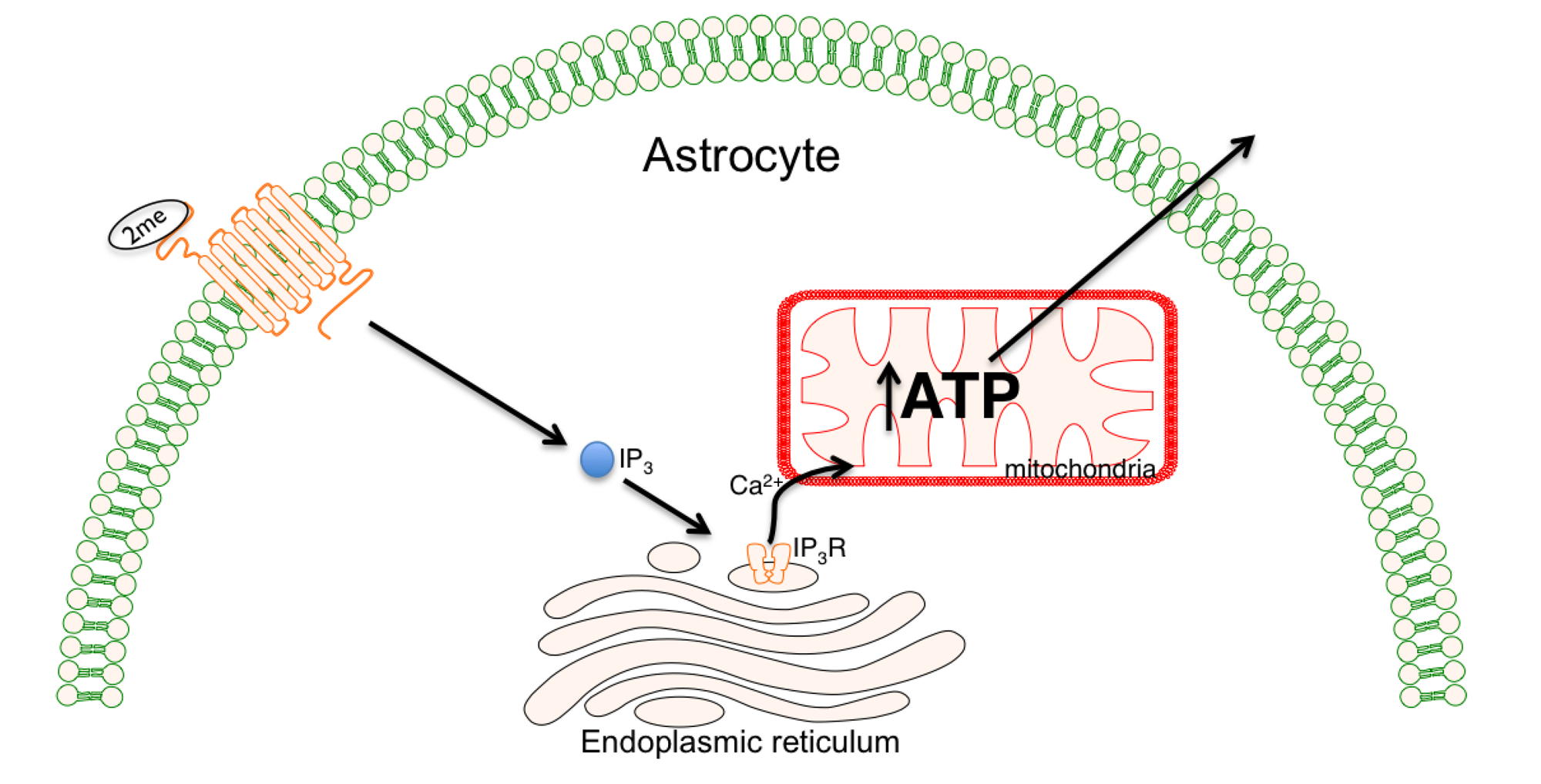
- Hajnoczky, G.; Csordas, G.; Krishnamurthy, R.; Szalai, G. Mitochondrial calcium signaling driven by the IP3 receptor. J. Bioenerg. Biomembr. 2000, 32, 15–25. [Google Scholar] [CrossRef]
- Hajnoczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef]
- Sak, K.; Webb, T.E. A retrospective of recombinant P2Y receptor subtypes and their pharmacology. Arch. Biochem. Biophys. 2002, 397, 131–136. [Google Scholar] [CrossRef]
- Burnstock, G. P2X receptors in sensory neurones. Br. J. Anaesth. 2000, 84, 476–488. [Google Scholar]
- Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar]
- Fabre, J.E.; Nguyen, M.; Latour, A.; Keifer, J.A.; Audoly, L.P.; Coffman, T.M.; Koller, B.H. Decreased platelet aggregation, increased bleeding time and resistance to thromboembolism in P2Y1-deficient mice. Nat. Med. 1999, 5, 1199–1202. [Google Scholar]
- Leon, C.; Hechler, B.; Freund, M.; Eckly, A.; Vial, C.; Ohlmann, P.; Dierich, A.; LeMeur, M.; Cazenave, J.P.; Gachet, C. Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y1 receptor-null mice. J. Clin. Invest. 1999, 104, 1731–1737. [Google Scholar]
- Wu, O.; Sumii, T.; Asahi, M.; Sasamata, M.; Ostergaard, L.; Rosen, B.R.; Lo, E.H.; Dijkhuizen, R.M. Infarct prediction and treatment assessment with MRI-based algorithms in experimental stroke models. J. Cereb. Blood Flow Metab. 2007, 27, 196–204. [Google Scholar]
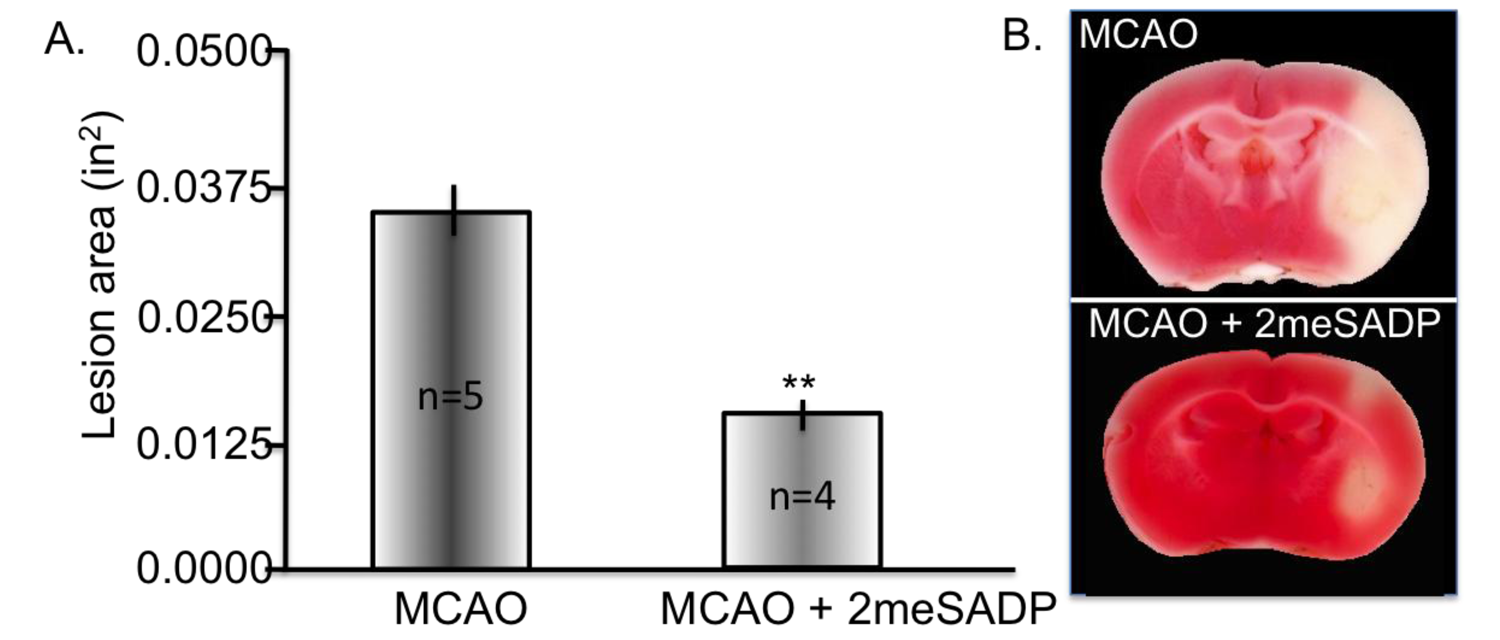
- Zheng, W.; Watts, L.T.; Holstein, D.M.; Prajapati, S.I.; Keller, C.; Grass, E.H.; Walter, C.A.; Lechleiter, J.D. Purinergic receptor stimulation reduces cytotoxic edema and brain infarcts in mouse induced by photothrombosis by energizing glial mitochondria. PLos One 2010, 5, e14401. [Google Scholar]
- Zheng, W.; Talley Watts, L.; Holstein, D.M.; Wewer, J.; Lechleiter, J.D. P2Y1R-initiated, IP3R-dependent stimulation of astrocyte mitochondrial metabolism reduces and partially reverses ischemic neuronal damage in mouse. J. Cereb. Blood Flow Metab. 2012, 33, 600–611. [Google Scholar]
- Owens, A.P., III; Mackman, N. Tissue factor and thrombosis: The clot starts here. Thromb. Haemost. 2010, 104, 432–439. [Google Scholar] [CrossRef]
- Wu, J.; Holstein, J.D.; Upadhyay, G.; Lin, D.T.; Conway, S.; Muller, E.; Lechleiter, J.D. Purinergic receptor-stimulated IP3-mediated Ca2+ release enhances neuroprotection by increasing astrocyte mitochondrial metabolism during aging. J. Neurosci. 2007, 27, 6510–6520. [Google Scholar]
- Savi, P.; Beauverger, P.; Labouret, C.; Delfaud, M.; Salel, V.; Kaghad, M.; Herbert, J.M. Role of P2Y1 purinoceptor in ADP-induced platelet activation. FEBS Lett. 1998, 422, 291–295. [Google Scholar] [CrossRef]
- Jin, J.; Daniel, J.L.; Kunapuli, S.P. Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J. Biol. Chem. 1998, 273, 2030–2034. [Google Scholar] [CrossRef]
- Jin, J.; Kunapuli, S.P. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc. Natl. Acad. Sci. USA 1998, 95, 8070–8074. [Google Scholar] [CrossRef]
- Hechler, B.; Leon, C.; Vial, C.; Vigne, P.; Frelin, C.; Cazenave, J.P.; Gachet, C. The P2Y1 receptor is necessary for adenosine 5′-diphosphate-induced platelet aggregation. Blood 1998, 92, 152–159. [Google Scholar]
- Jarvis, G.E.; Humphries, R.G.; Robertson, M.J.; Leff, P. ADP can induce aggregation of human platelets via both P2Y1 and P2T receptors. Br. J. Pharmacol. 2000, 129, 275–282. [Google Scholar] [CrossRef]
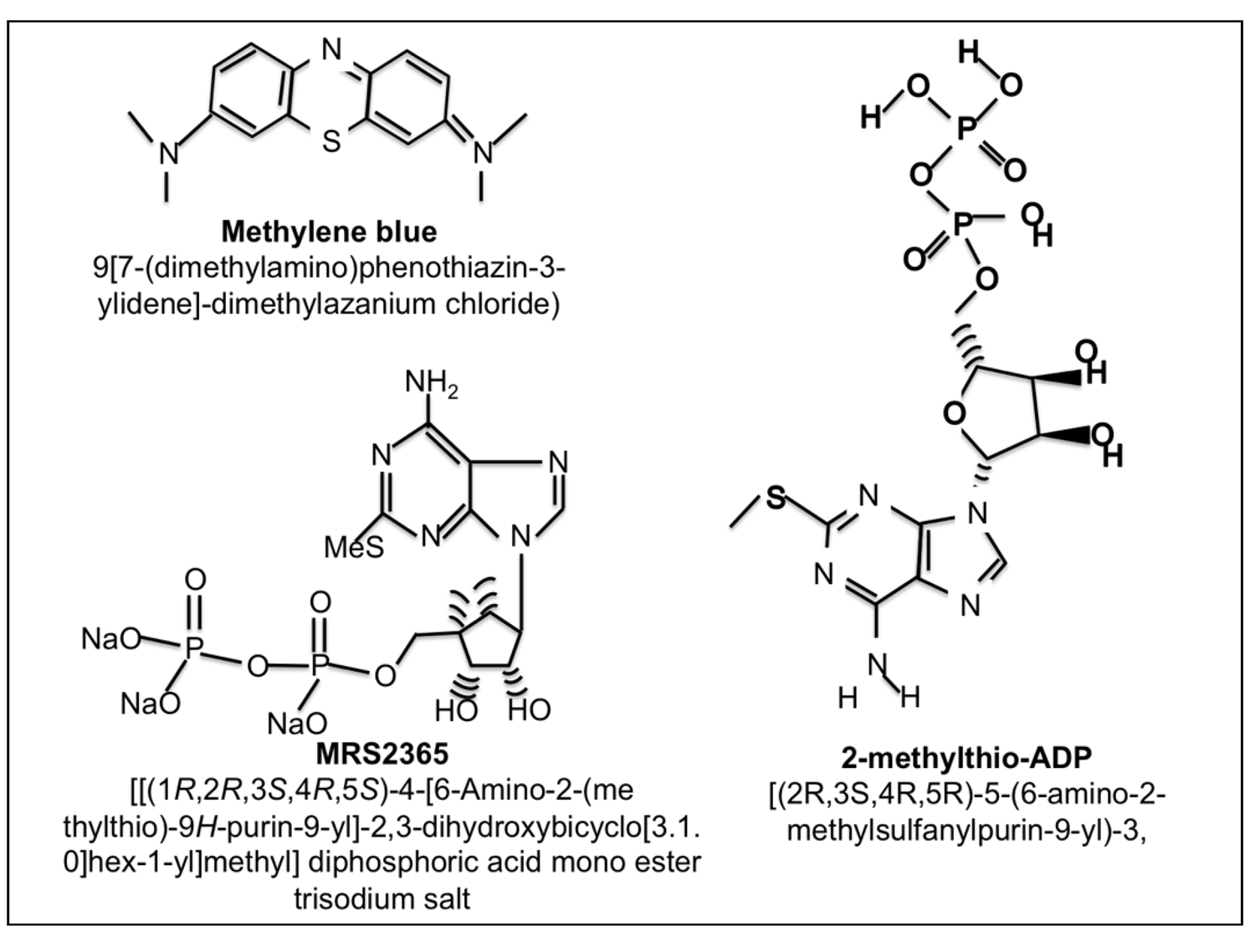
- Chhatriwala, M.; Ravi, R.G.; Patel, R.I.; Boyer, J.L.; Jacobson, K.A.; Harden, T.K. Induction of novel agonist selectivity for the ADP-activated P2Y1 receptor versus the ADP-activated P2Y12 and P2Y13 receptors by conformational constraint of an ADP analog. J. Pharmacol. Exp. Ther. 2004, 311, 1038–1043. [Google Scholar] [CrossRef]
- Ravi, R.G.; Kim, H.S.; Servos, J.; Zimmermann, H.; Lee, K.; Maddileti, S.; Boyer, J.L.; Harden, T.K.; Jacobson, K.A. Adenine nucleotide analogues locked in a Northern methanocarba conformation: Enhanced stability and potency as P2Y(1) receptor agonists. J. Med. Chem. 2002, 45, 2090–2100. [Google Scholar]
- Lu, M.; Zhang, R.L.; Zhang, Z.G.; Yang, J.J.; Chopp, M. Linkage of cell cycle kinetics between embryonic and adult stroke models: An analytical approach. J. Neurosci. Methods 2007, 161, 323–330. [Google Scholar] [CrossRef]
- Cavaliere, F.; Florenzano, F.; Amadio, S.; Fusco, F.R.; Viscomi, M.T.; D’Ambrosi, N.; Vacca, F.; Sancesario, G.; Bernardi, G.; Molinari, M.; Volonte, C. Up-regulation of P2X2, P2X4 receptor and ischemic cell death: prevention by P2 antagonists. Neuroscience 2003, 120, 85–98. [Google Scholar] [CrossRef]
- Rojas, J.C.; Bruchey, A.K.; Gonzalez-Lima, F. Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog. Neurobiol. 2012, 96, 32–45. [Google Scholar] [CrossRef]
- Guttmann, P.; Ehrlich, P. ber die Wirkung des Methylenblau bei Malaria. Berl. Klin. Wochenschr. 1891, 28, 953–956. [Google Scholar]
- Kupfer, A.; Aeschlimann, C.; Cerny, T. Methylene blue and the neurotoxic mechanisms of ifosfamide encephalopathy. Eur. J. Clin. Pharmacol. 1996, 50, 249–252. [Google Scholar] [CrossRef]
- Wainwright, M.; Crossley, K.B. Methylene Blue—A therapeutic dye for all seasons? J. Chemother. 2002, 14, 431–443. [Google Scholar]
- Naylor, G.J.; Martin, B.; Hopwood, S.E.; Watson, Y. A two-year double-blind crossover trial of the prophylactic effect of methylene blue in manic-depressive psychosis. Biol. Psychiatry 1986, 21, 915–920. [Google Scholar] [CrossRef]
- Zhang, X.; Rojas, J.C.; Gonzalez-Lima, F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox. Res. 2006, 9, 47–57. [Google Scholar] [CrossRef]
- Peter, C.; Hongwan, D.; Kupfer, A.; Lauterburg, B.H. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur. J. Clin. Pharmacol. 2000, 56, 247–250. [Google Scholar] [CrossRef]
- Oz, M.; Lorke, D.E.; Petroianu, G.A. Methylene blue and Alzheimer’s disease. Biochem. Pharmacol. 2009, 78, 927–932. [Google Scholar]
- Wiklund, L.; Basu, S.; Miclescu, A.; Wiklund, P.; Ronquist, G.; Sharma, H.S. Neuro- and cardioprotective effects of blockade of nitric oxide action by administration of methylene blue. Ann. N. Y. Acad. Sci. 2007, 1122, 231–244. [Google Scholar]
- Miclescu, A.; Basu, S.; Wiklund, L. Cardio-cerebral and metabolic effects of methylene blue in hypertonic sodium lactate during experimental cardiopulmonary resuscitation. Resuscitation 2007, 75, 88–97. [Google Scholar] [CrossRef]
- Kelner, M.J.; Bagnell, R.; Hale, B.; Alexander, N.M. Potential of methylene blue to block oxygen radical generation in reperfusion injury. Basic Life Sci. 1988, 49, 895–898. [Google Scholar]
- Miclescu, A.; Basu, S.; Wiklund, L. Methylene blue added to a hypertonic-hyperoncotic solution increases short-term survival in experimental cardiac arrest. Crit. Care Med. 2006, 34, 2806–2813. [Google Scholar] [CrossRef]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar] [CrossRef]
- Lin, A.L.; Poteet, E.; Du, F.; Gourav, R.C.; Liu, R.; Wen, Y.; Bresnen, A.; Huang, S.; Fox, P.T.; Yang, S.H.; Duong, T.Q. Methylene blue as a cerebral metabolic and hemodynamic enhancer. Plos One 2012, 7, e46585. [Google Scholar]
- Huang, S.; Du, F.; Shih, Y.I.; Shen, Q.; Gonzalez-Lima, F.; Duong, T.Q. Methylene blue potentiates stimulus-evoked fMRI responses and cerebral oxygen consumption during normoxia and hypoxia. NeuroImage 2013, 72, 237–242. [Google Scholar] [CrossRef]
- Devasagayam, T.P.; Tilak, J.C.; Boloor, K.K.; Sane, K.S.; Ghaskadbi, S.S.; Lele, R.D. Free radicals and antioxidants in human health: Current status and future prospects. J. Assoc. Physicians India 2004, 52, 794–804. [Google Scholar]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef]
- Niizuma, K.; Endo, H.; Chan, P.H. Oxidative stress and mitochondrial dysfunction as determinants of ischemic neuronal death and survival. J. Neurochem. 2009, 109 (Suppl. 1), 133–138. [Google Scholar] [CrossRef]
- Holley, A.K.; Dhar, S.K.; St Clair, D.K. Manganese superoxide dismutase vs. p53: Regulation of mitochondrial ROS. Mitochondrion 2010, 10, 649–661. [Google Scholar] [CrossRef]
- Shmonin, A.; Melnikova, E.; Galagudza, M.; Vlasov, T. Characteristics of cerebral ischemia in major rat stroke models of middle cerebral artery ligation through craniectomy. Int. J. Stroke 2012. [Google Scholar] [CrossRef]
- Maier, C.M.; Hsieh, L.; Crandall, T.; Narasimhan, P.; Chan, P.H. A new approach for the investigation of reperfusion-related brain injury. Biochem. Soc. Trans. 2006, 34, 1366–1369. [Google Scholar] [CrossRef]
- Ivanovi-BurmazoviĆ, I.; FilipoviĆ, M. Reactivity of manganese superoxide dismutase mimics toward superoxide and nitric oxide: Selectivity versus cross-reactivity. Adv.Inorg. Chem. 2012, 64, 53–95. [Google Scholar] [CrossRef]
- Friedel, F.C.; Lieb, D.; Ivanović-Burmazović, I. Comparative studies on manganese-based SOD mimetics, including the phosphate effect, by using global spectral analysis. J. Inorg. Biochem. 2012, 109, 26–32. [Google Scholar] [CrossRef]
- Park, W.; Lim, D. Synthesis and SOD activity of manganese complexes of pentaaza macrocycles containing amino- and guanidino-auxiliary. Bull. Korean Chem. Soc. 2001, 32, 3787. [Google Scholar]
- Huang, H.F.; Guo, F.; Cao, Y.Z.; Shi, W.; Xia, Q. Neuroprotection by manganese superoxide dismutase (MnSOD) mimics: Antioxidant effect and oxidative stress regulation in acute experimental stroke. CNS Neurosci. Ther. 2012, 18, 811–818. [Google Scholar]
- Kelso, G.F.; Maroz, A.; Cocheme, H.M.; Logan, A.; Prime, T.A.; Peskin, A.V.; Winterbourn, C.C.; James, A.M.; Ross, M.F.; Brooker, S.; et al. Mitochondria-Targeted Macrocyclic Mn(II) Superoxide Dismutase Mimetic. Chem. Biol. 2012, 19, 1237–1246. [Google Scholar] [CrossRef]
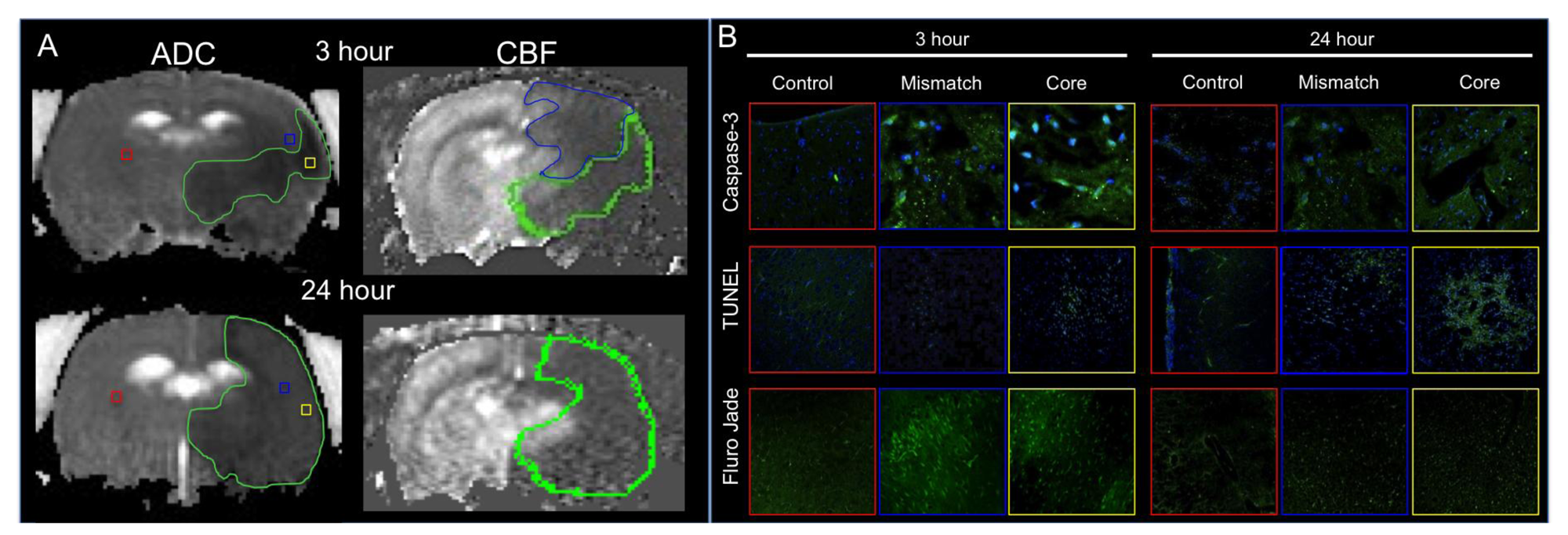
- Shen, Q.; Fisher, M.; Sotak, C.H.; Duong, T.Q. Effects of reperfusion on ADC and CBF pixel-by-pixel dynamics in stroke: Characterizing tissue fates using quantitative diffusion and perfusion imaging. J. Cereb. Blood Flow Metab. 2004, 24, 280–290. [Google Scholar]
- Shen, Q.; Huang, S.; Du, F.; Duong, T.Q. Probing ischemic tissue fate with bold fMRI of brief oxygen challenge. Brain Res. 2011, 1425, 132–141. [Google Scholar]
- Shen, Q.; Meng, X.; Fisher, M.; Sotak, C.H.; Duong, T.Q. Pixel-by-pixel spatiotemporal progression of focal ischemia derived using quantitative perfusion and diffusion imaging. J.Cereb. BloodFlow Metab. 2003, 23, 1479–1488. [Google Scholar]
- Shen, Q.; Ren, H.; Cheng, H.; Fisher, M.; Duong, T.Q. Functional, perfusion and diffusion MRI of acute focal ischemic brain injury. J. Cereb. Blood Flow Metab. 2005, 25, 1265–1279. [Google Scholar]
- Meng, X.; Fisher, M.; Shen, Q.; Sotak, C.H.; Duong, T.Q. Characterizing the diffusion/perfusion mismatch in experimental focal cerebral ischemia. Ann. Neurol. 2004, 55, 207–212. [Google Scholar] [CrossRef]
- Hui, E.S.; Du, F.; Huang, S.; Shen, Q.; Duong, T.Q. Spatiotemporal dynamics of diffusional kurtosis, mean diffusivity and perfusion changes in experimental stroke. Brain Res. 2012, 1451, 100–109. [Google Scholar]
- Sicard, K.M.; Henninger, N.; Fisher, M.; Duong, T.Q.; Ferris, C.F. Long-term changes of functional MRI based brain function, behavioral status, and histopathology after transient focal cerebral ischemia in rats. Stroke 2006, 37, 2593–2600. [Google Scholar] [CrossRef]
- Sicard, K.M.; Henninger, N.; Fisher, M.; Duong, T.Q.; Ferris, C.F. Differential recovery of multimodal mri and behavior after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2006, 26, 1451–1462. [Google Scholar] [CrossRef]
- Tanaka, Y.; Ishii, H.; Hiraoka, M.; Miyasaka, N.; Kuroiwa, T.; Hajjar, K.A.; Nagaoka, T.; Duong, T.Q.; Ohno, K.; Yoshida, M. Efficacy of recombinant annexin 2 for fibrinolytic therapy in a rat embolic stroke model: A magnetic resonance imaging study. Brain Res. 2007, 1165, 135–143. [Google Scholar]
- Shen, Q.; Ren, H.; Fisher, M.; Bouley, J.; Duong, T.Q. Dynamic tracking of acute ischemic tissue fates using improved unsupervised isodata analysis of high-resolution quantitative perfusion and diffusion data. J. Cereb. Blood Flow Metab. 2004, 24, 887–897. [Google Scholar]
- Shen, Q.; Ren, H.; Fisher, M.; Duong, T.Q. Statistical prediction of tissue fate in acute ischemic brain injury. J. Cereb. Blood Flow Metab. 2005, 25, 1336–1345. [Google Scholar] [CrossRef]
- Huang, S.; Shen, Q.; Duong, T.Q. Artificial neural network prediction of ischemic tissue fate in acute stroke imaging. J. Cereb. Blood Flow Metab. 2010, 30, 1661–1670. [Google Scholar] [CrossRef]
- Huang, S.; Shen, Q.; Duong, T.Q. Quantitative prediction of acute ischemic tissue fate using support vector machine. Brain Res. 2011, 1045, 77–84. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Watts, L.T.; Lloyd, R.; Garling, R.J.; Duong, T. Stroke Neuroprotection: Targeting Mitochondria. Brain Sci. 2013, 3, 540-560. https://doi.org/10.3390/brainsci3020540
Watts LT, Lloyd R, Garling RJ, Duong T. Stroke Neuroprotection: Targeting Mitochondria. Brain Sciences. 2013; 3(2):540-560. https://doi.org/10.3390/brainsci3020540
Chicago/Turabian StyleWatts, Lora Talley, Reginald Lloyd, Richard Justin Garling, and Timothy Duong. 2013. "Stroke Neuroprotection: Targeting Mitochondria" Brain Sciences 3, no. 2: 540-560. https://doi.org/10.3390/brainsci3020540