Cannabinoids: Well-Suited Candidates for the Treatment of Perinatal Brain Injury

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neonatal Hypoxic-Ischemic Encephalopathy (NHIE)
3. Neonatal Arterial Ischemic Stroke
4. The Endocannabinoid System
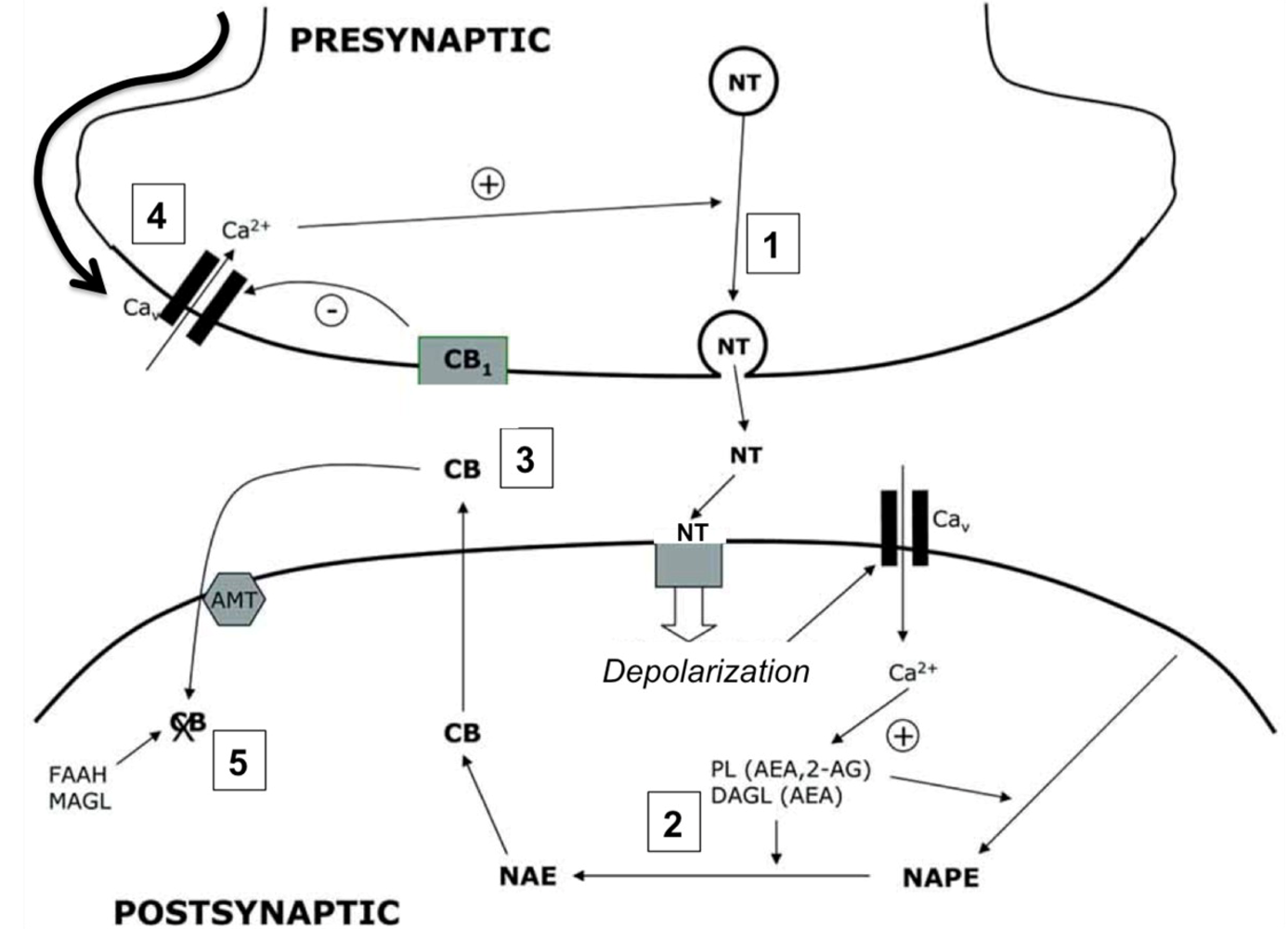
4.1. Cannabinergic Ligands
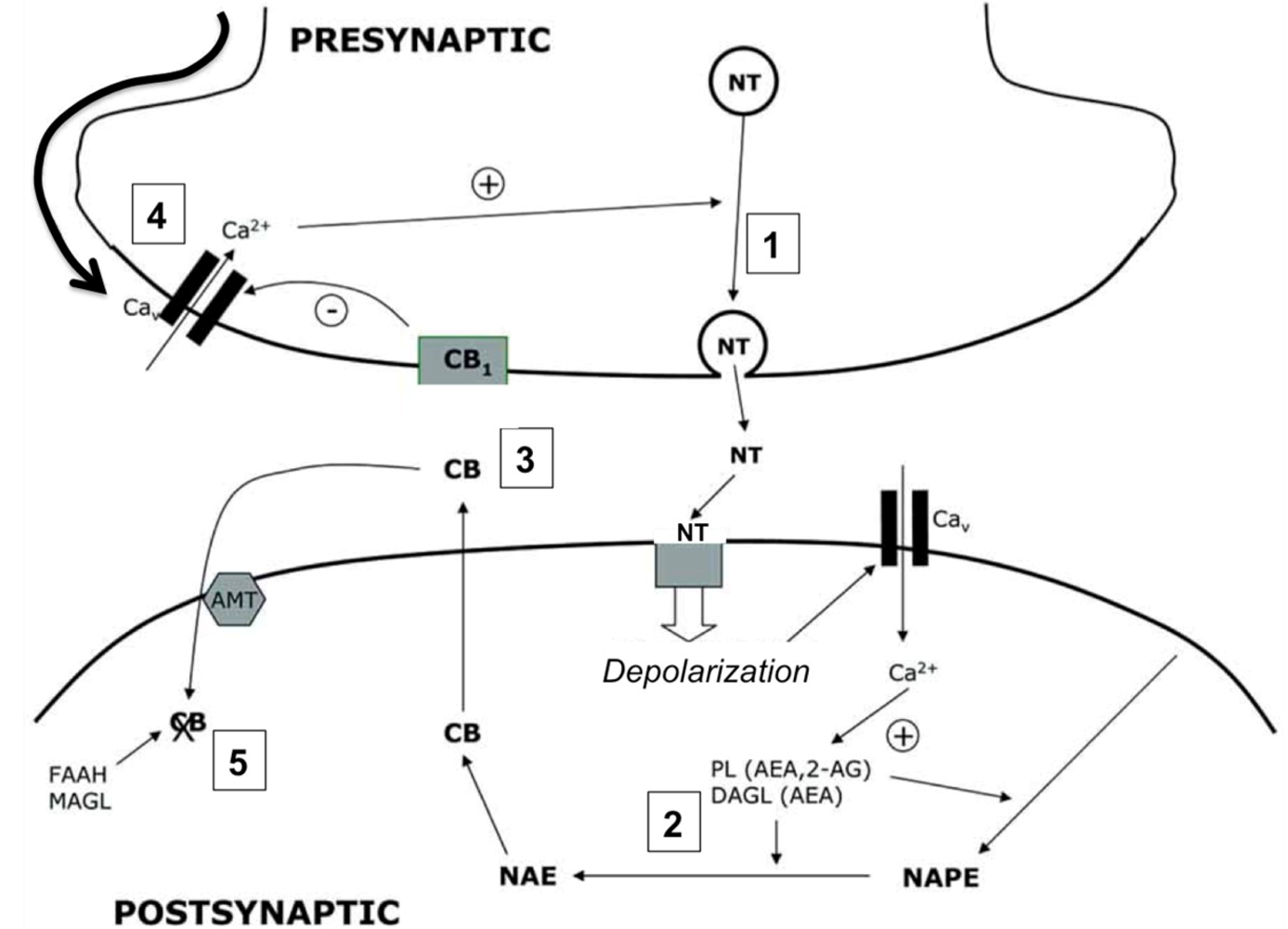
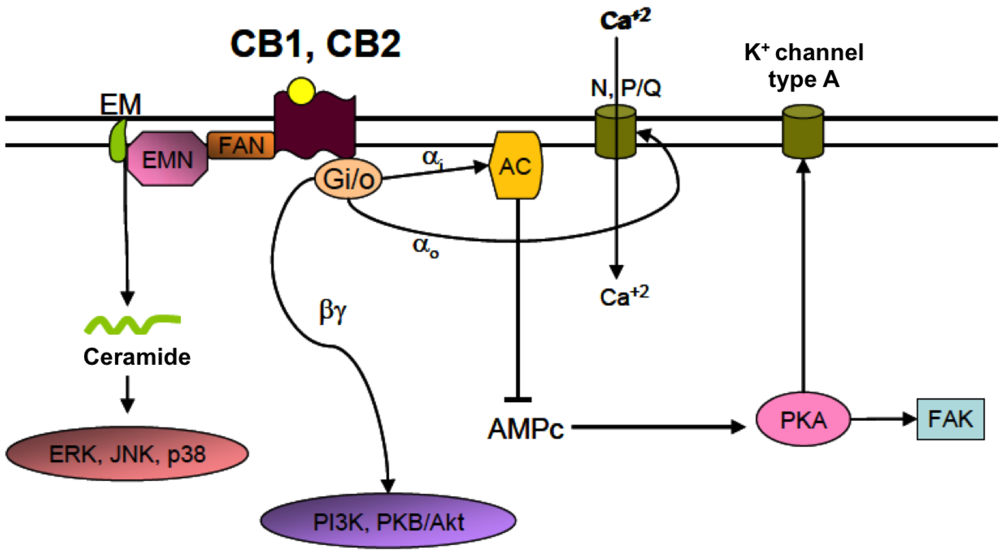
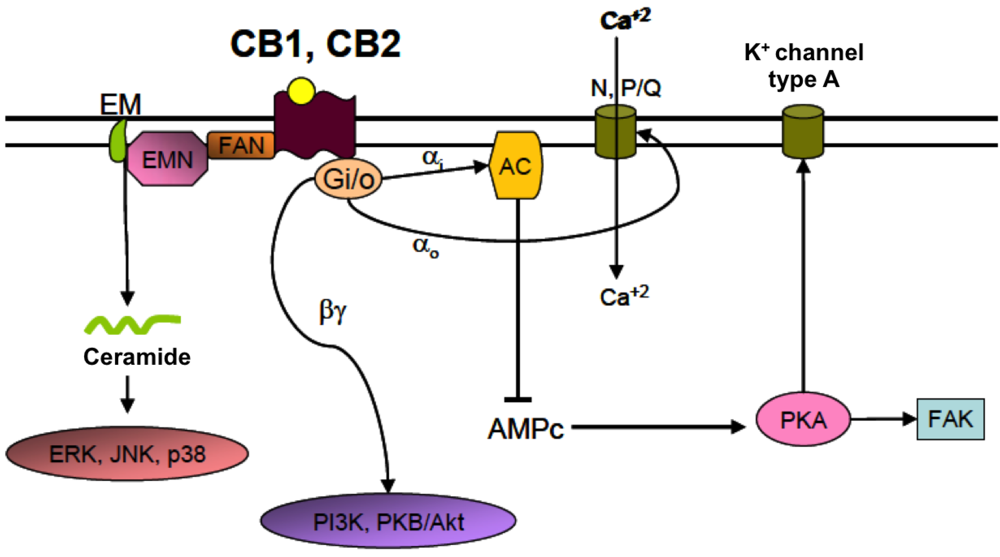
4.2. Cannabinoid Receptors (CBRs)
5. Mechanisms of Action of Cannabinoids in Experimental Models of Perinatal Brain Injury
5.1. Rodent Models of Neonatal Hypoxic-Ischemic Encephalopathy
5.2. Non-Rodent Models of Neonatal Hypoxic-Ischemic Encephalopathy
5.3. Rodent Model of Neonatal Stroke
6. Final Remarks
References
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef]
- Vannucci, R.C. Hypoxic-ischemic encephalopathy. Am. J. Perinatol. 2000, 17, 113–120. [Google Scholar] [CrossRef]
- Volpe, J.J. Perinatal brain injury: From pathogenesis to neuroprotection. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 56–64. [Google Scholar] [CrossRef]
- Oka, A.; Belliveau, M.J.; Rosenberg, P.A.; Volpe, J.J. Vulnerability of oligodendroglia to glutamate: Pharmacology, mechanisms, and prevention. J. Neurosci. 1993, 13, 1441–1453. [Google Scholar]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar]
- Kaur, C.; Ling, E.A. Periventricular white matter damage in the hypoxic neonatal brain: Role of microglial cells. Prog. Neurobiol. 2009, 87, 264–280. [Google Scholar] [CrossRef]
- Kirton, A.; Armstrong-Wells, J.; Chang, T.; Deveber, G.; Rivkin, M.J.; Hernandez, M.; Carpenter, J.; Yager, J.Y.; Lynch, J.K.; Ferriero, D.M. Symptomatic neonatal arterial ischemic stroke: The international pediatric stroke study. Pediatrics 2011, 128, e1402–e1410. [Google Scholar] [CrossRef]
- McQuillen, P.S.; Ferriero, D.M. Selective vulnerability in the developing central nervous system. Pediatr. Neurol. 2004, 30, 227–235. [Google Scholar] [CrossRef]
- Thornton, C.; Rousset, C.I.; Kichev, A.; Miyakuni, Y.; Vontell, R.; Baburamani, A.A.; Fleiss, B.; Gressens, P.; Hagberg, H. Molecular mechanisms of neonatal brain injury. Neurol. Res. Int. 2012, 2012, 506320. [Google Scholar]
- Manabat, C.; Han, B.H.; Wendland, M.; Derugin, N.; Fox, C.K.; Choi, J.; Holtzman, D.M.; Ferriero, D.M.; Vexler, Z.S. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke 2003, 34, 207–213. [Google Scholar] [CrossRef]
- Northington, F.J.; Graham, E.M.; Martin, L.J. Apoptosis in perinatal hypoxic-ischemic brain injury: How important is it and should it be inhibited? Brain Res. Brain Res. Rev. 2005, 50, 244–257. [Google Scholar] [CrossRef]
- Renolleau, S.; Fau, S.; Goyenvalle, C.; Joly, L.M.; Chauvier, D.; Jacotot, E.; Mariani, J.; Charriaut-Marlangue, C. Specific caspase inhibitor Q-VD-OPh prevents neonatal stroke in P7 rat: A role for gender. J. Neurochem. 2007, 100, 1062–1071. [Google Scholar] [CrossRef]
- Villapol, S.; Gelot, A.; Renolleau, S.; Charriaut-Marlangue, C. Astrocyte responses after neonatal ischemia: The yin and the yang. Neuroscientist 2008, 14, 339–344. [Google Scholar] [CrossRef]
- Plane, J.M.; Liu, R.; Wang, T.W.; Silverstein, F.S.; Parent, J.M. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol. Dis. 2004, 16, 585–595. [Google Scholar] [CrossRef]
- Ong, J.; Plane, J.M.; Parent, J.M.; Silverstein, F.S. Hypoxic-ischemic injury stimulates subventricular zone proliferation and neurogenesis in the neonatal rat. Pediatr. Res. 2005, 58, 600–606. [Google Scholar] [CrossRef]
- Yang, Z.; Covey, M.V.; Bitel, C.L.; Ni, L.; Jonakait, G.M.; Levison, S.W. Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann. Neurol. 2007, 61, 199–208. [Google Scholar] [CrossRef]
- Zaidi, A.U.; Bessert, D.A.; Ong, J.E.; Xu, H.; Barks, J.D.; Silverstein, F.S.; Skoff, R.P. New oligodendrocytes are generated after neonatal hypoxic-ischemic brain injury in rodents. Glia 2004, 46, 380–390. [Google Scholar] [CrossRef]
- Segovia, K.N.; McClure, M.; Moravec, M.; Luo, N.L.; Wan, Y.; Gong, X.; Riddle, A.; Craig, A.; Struve, J.; Sherman, L.S.; et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann. Neurol. 2008, 63, 520–530. [Google Scholar] [CrossRef]
- Gonzalez, F.F.; Ferriero, D.M. Neuroprotection in the newborn infant. Clin. Perinatol. 2009, 36, 859–880. [Google Scholar] [CrossRef]
- Marks, K.; Shany, E.; Shelef, I.; Golan, A.; Zmora, E. Hypothermia: A neuroprotective therapy for neonatal hypoxic ischemic encephalopathy. Isr. Med. Assoc. J. 2010, 12, 494–500. [Google Scholar]
- Shah, P.S. Hypothermia: A systematic review and meta-analysis of clinical trials. Semin. Fetal Neonatal Med. 2010, 15, 238–246. [Google Scholar] [CrossRef]
- Higgins, R.D.; Raju, T.; Edwards, A.D.; Azzopardi, D.V.; Bose, C.L.; Clark, R.H.; Ferriero, D.M.; Guillet, R.; Gunn, A.J.; Hagberg, H.; et al. Hypothermia and other treatment options for neonatal encephalopathy: An executive summary of the Eunice Kennedy Shriver NICHD workshop. J. Pediatr. 2011, 159, 851–858.e1. [Google Scholar] [CrossRef]
- Marret, S.; Gressens, P.; Gadisseux, J.F.; Evrard, P. Prevention by magnesium of excitotoxic neuronal death in the developing brain: An animal model for clinical intervention studies. Dev. Med. Child Neurol. 1995, 37, 473–484. [Google Scholar]
- Mayor, S. Xenon shows promise to prevent brain injury from lack of oxygen in newborns. BMJ 2010, 340, c2005. [Google Scholar] [CrossRef]
- Sfaello, I.; Baud, O.; Arzimanoglou, A.; Gressens, P. Topiramate prevents excitotoxic damage in the newborn rodent brain. Neurobiol. Dis. 2005, 20, 837–848. [Google Scholar] [CrossRef]
- Spandou, E.; Soubasi, V.; Papoutsopoulou, S.; Augoustides-Savvopoulou, P.; Loizidis, T.; Pazaiti, A.; Karkavelas, G.; Guiba-Tziampiri, O. Neuroprotective effect of long-term MgSO4 administration after cerebral hypoxia-ischemia in newborn rats is related to the severity of brain damage. Reprod. Sci. 2007, 14, 667–677. [Google Scholar] [CrossRef]
- Palmer, C.; Towfighi, J.; Roberts, R.L.; Heitjan, D.F. Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr. Res. 1993, 33, 405–411. [Google Scholar]
- Benders, M.J.; Bos, A.F.; Rademaker, C.M.; Rijken, M.; Torrance, H.L.; Groenendaal, F.; van Bel, F. Early postnatal allopurinol does not improve short term outcome after severe birth asphyxia. Arch. Dis. Child Fetal Neonatal Ed. 2006, 91, F163–F165. [Google Scholar]
- Paintlia, M.K.; Paintlia, A.S.; Barbosa, E.; Singh, I.; Singh, A.K. N-acetylcysteine prevents endotoxin-induced degeneration of oligodendrocyte progenitors and hypomyelination in developing rat brain. J. Neurosci. Res. 2004, 78, 347–361. [Google Scholar] [CrossRef]
- Buller, K.M.; Carty, M.L.; Reinebrant, H.E.; Wixey, J.A. Minocycline: A neuroprotective agent for hypoxic-ischemic brain injury in the neonate? J. Neurosci. Res. 2009, 87, 599–608. [Google Scholar] [CrossRef]
- Kumral, A.; Uysal, N.; Tugyan, K.; Sonmez, A.; Yilmaz, O.; Gokmen, N.; Kiray, M.; Genc, S.; Duman, N.; Koroglu, T.F.; et al. Erythropoietin improves long-term spatial memory deficits and brain injury following neonatal hypoxia-ischemia in rats. Behav. Brain Res. 2004, 153, 77–86. [Google Scholar] [CrossRef]
- Sun, Y.; Calvert, J.W.; Zhang, J.H. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke 2005, 36, 1672–1678. [Google Scholar] [CrossRef]
- Gonzalez, F.F.; McQuillen, P.; Mu, D.; Chang, Y.; Wendland, M.; Vexler, Z.; Ferriero, D.M. Erythropoietin enhances long-term neuroprotection and neurogenesis in neonatal stroke. Dev. Neurosci. 2007, 29, 321–330. [Google Scholar] [CrossRef]
- Alonso-Alconada, D.; Alvarez, A.; Hilario, E. Cannabinoid as a neuroprotective strategy in perinatal hypoxic-ischemic injury. Neurosci. Bull. 2011, 27, 275–285. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Aguado, T.; Palazuelos, J.; Guzman, M. The endocannabinoid system and neurogenesis in health and disease. Neuroscientist 2007, 13, 109–114. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Aguado, T.; Palazuelos, J.; Guzman, M. Mechanisms of control of neuron survival by the endocannabinoid system. Curr. Pharm. Des. 2008, 14, 2279–2288. [Google Scholar] [CrossRef]
- Lee, J.; Croen, L.A.; Lindan, C.; Nash, K.B.; Yoshida, C.K.; Ferriero, D.M.; Barkovich, A.J.; Wu, Y.W. Predictors of outcome in perinatal arterial stroke: A population-based study. Ann. Neurol. 2005, 58, 303–308. [Google Scholar] [CrossRef]
- Nelson, K.B.; Lynch, J.K. Stroke in newborn infants. Lancet Neurol. 2004, 3, 150–158. [Google Scholar] [CrossRef]
- Kirton, A.; deVeber, G. Advances in perinatal ischemic stroke. Pediatr. Neurol. 2009, 40, 205–214. [Google Scholar] [CrossRef]
- Benders, M.J.; Groenendaal, F.; Uiterwaal, C.S.; Nikkels, P.G.; Bruinse, H.W.; Nievelstein, R.A.; de Vries, L.S. Maternal and infant characteristics associated with perinatal arterial stroke in the preterm infant. Stroke 2007, 38, 1759–1765. [Google Scholar] [CrossRef]
- deVeber, G.; Roach, E.S.; Riela, A.R.; Wiznitzer, M. Stroke in children: Recognition, treatment, and future directions. Semin. Pediatr. Neurol. 2000, 7, 309–317. [Google Scholar] [CrossRef]
- Westmacott, R.; MacGregor, D.; Askalan, R.; deVeber, G. Late emergence of cognitive deficits after unilateral neonatal stroke. Stroke 2009, 40, 2012–2019. [Google Scholar] [CrossRef]
- Kamath, B.D.; Todd, J.K.; Glazner, J.E.; Lezotte, D.; Lynch, A.M. Neonatal outcomes after elective cesarean delivery. Obstet. Gynecol. 2009, 113, 1231–1238. [Google Scholar]
- Wegener, N.; Koch, M. Neurobiology and systems physiology of the endocannabinoid system. Pharmacopsychiatry 2009, 42 (Suppl. 1), S79–S86. [Google Scholar] [CrossRef]
- Fowler, C.J. Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J. 2013, 280, 1895–1904. [Google Scholar] [CrossRef]
- Breivogel, C.S.; Sim-Selley, L.J. Basic neuroanatomy and neuropharmacology of cannabinoids. Int. Rev. Psychiatry 2009, 21, 113–121. [Google Scholar] [CrossRef]
- Rodriguez de Fonseca, F.; Del Arco, I.; Bermudez-Silva, F.J.; Bilbao, A.; Cippitelli, A.; Navarro, M. The endocannabinoid system: Physiology and pharmacology. Alcohol Alcohol. 2005, 40, 2–14. [Google Scholar]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Endocannabinoids and synaptic function in the CNS. Neuroscientist 2007, 13, 127–137. [Google Scholar] [CrossRef]
- Pertwee, R.G. Cannabis and cannabinoids: Pharmacology and rationale for clinical use. Forsch Komplementarmed 1999, 6 (Suppl. 3), 12–15. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Ross, R.A. Cannabinoid receptors and their ligands. Prostaglandins Leukot. Essent. Fatty Acids 2002, 66, 101–121. [Google Scholar] [CrossRef]
- Benito, C.; Tolon, R.M.; Pazos, M.R.; Nunez, E.; Castillo, A.I.; Romero, J. Cannabinoid CB2 receptors in human brain inflammation. Br. J. Pharmacol. 2008, 153, 277–285. [Google Scholar] [CrossRef]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar]
- Castillo, P.E.; Younts, T.J.; Chavez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef]
- Varga, E.V.; Georgieva, T.; Tumati, S.; Alves, I.; Salamon, Z.; Tollin, G.; Yamamura, H.I.; Roeske, W.R. Functional selectivity in cannabinoid signaling. Curr. Mol. Pharmacol. 2008, 1, 273–284. [Google Scholar]
- Basu, S.; Dittel, B.N. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol. Res. 2011, 51, 26–38. [Google Scholar] [CrossRef]
- Tanasescu, R.; Constantinescu, C.S. Cannabinoids and the immune system: An overview. Immunobiology 2010, 215, 588–597. [Google Scholar] [CrossRef]
- Cabral, G.A.; Griffin-Thomas, L. Emerging role of the cannabinoid receptor CB2 in immune regulation: Therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11, e3. [Google Scholar] [CrossRef]
- Kim, S.H.; Won, S.J.; Mao, X.O.; Jin, K.; Greenberg, D.A. Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol. Pharmacol. 2006, 69, 691–696. [Google Scholar]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutierrez, S.O.; van der Stelt, M.; et al. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Van der Stelt, M.; Veldhuis, W.B.; Maccarrone, M.; Bar, P.R.; Nicolay, K.; Veldink, G.A.; Di Marzo, V.; Vliegenthart, J.F. Acute neuronal injury, excitotoxicity, and the endocannabinoid system. Mol. Neurobiol. 2002, 26, 317–346. [Google Scholar] [CrossRef]
- Stella, N. Endocannabinoid signaling in microglial cells. Neuropharmacology 2009, 56 (Suppl. 1), 244–253. [Google Scholar] [CrossRef]
- Guzman, M.; Sanchez, C.; Galve-Roperh, I. Cannabinoids and cell fate. Pharmacol. Ther. 2002, 95, 175–184. [Google Scholar]
- Guzman, M.; Sanchez, C.; Galve-Roperh, I. Control of the cell survival/death decision by cannabinoids. J. Mol. Med. (Berl.) 2001, 78, 613–625. [Google Scholar] [CrossRef]
- Palazuelos, J.; Aguado, T.; Egia, A.; Mechoulam, R.; Guzman, M.; Galve-Roperh, I. Non-psychoactive CB2 cannabinoid agonists stimulate neural progenitor proliferation. FASEB J. 2006, 20, 2405–2407. [Google Scholar] [CrossRef]
- Aguado, T.; Monory, K.; Palazuelos, J.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzman, M.; Galve-Roperh, I. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 2005, 19, 1704–1706. [Google Scholar]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzman, M.; Galve-Roperh, I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef]
- Aguado, T.; Romero, E.; Monory, K.; Palazuelos, J.; Sendtner, M.; Marsicano, G.; Lutz, B.; Guzman, M.; Galve-Roperh, I. The CB1 cannabinoid receptor mediates excitotoxicity-induced neural progenitor proliferation and neurogenesis. J. Biol. Chem. 2007, 282, 23892–23898. [Google Scholar] [CrossRef]
- Van der Stelt, M.; Di Marzo, V. Cannabinoid receptors and their role in neuroprotection. Neuromolecular Med. 2005, 7, 37–50. [Google Scholar] [CrossRef]
- Ramirez, S.H.; Hasko, J.; Skuba, A.; Fan, S.; Dykstra, H.; McCormick, R.; Reichenbach, N.; Krizbai, I.; Mahadevan, A.; Zhang, M.; et al. Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood-brain barrier dysfunction under inflammatory conditions. J. Neurosci. 2012, 32, 4004–4016. [Google Scholar] [CrossRef]
- Murikinati, S.; Juttler, E.; Keinert, T.; Ridder, D.A.; Muhammad, S.; Waibler, Z.; Ledent, C.; Zimmer, A.; Kalinke, U.; Schwaninger, M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2010, 24, 788–798. [Google Scholar] [CrossRef]
- Zhuang, S.Y.; Bridges, D.; Grigorenko, E.; McCloud, S.; Boon, A.; Hampson, R.E.; Deadwyler, S.A. Cannabinoids produce neuroprotection by reducing intracellular calcium release from ryanodine-sensitive stores. Neuropharmacology 2005, 48, 1086–1096. [Google Scholar] [CrossRef]
- Bacci, A.; Huguenard, J.R.; Prince, D.A. Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature 2004, 431, 312–316. [Google Scholar]
- Esposito, G.; Izzo, A.A.; Di Rosa, M.; Iuvone, T. Selective cannabinoid CB1 receptor-mediated inhibition of inducible nitric oxide synthase protein expression in C6 rat glioma cells. J. Neurochem. 2001, 78, 835–841. [Google Scholar] [CrossRef]
- Sheng, W.S.; Hu, S.; Min, X.; Cabral, G.A.; Lokensgard, J.R.; Peterson, P.K. Synthetic cannabinoid WIN55,212-2 inhibits generation of inflammatory mediators by IL-1beta-stimulated human astrocytes. Glia 2005, 49, 211–219. [Google Scholar] [CrossRef]
- Fernandez-Lopez, D.; Martinez-Orgado, J.; Nunez, E.; Romero, J.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Characterization of the neuroprotective effect of the cannabinoid agonist WIN-55212 in an in vitro model of hypoxic-ischemic brain damage in newborn rats. Pediatr. Res. 2006, 60, 169–173. [Google Scholar] [CrossRef]
- Fernandez-Lopez, D.; Martinez-Orgado, J.; Casanova, I.; Bonet, B.; Leza, J.C.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Immature rat brain slices exposed to oxygen-glucose deprivation as an in vitro model of neonatal hypoxic-ischemic encephalopathy. J. Neurosci. Methods 2005, 145, 205–212. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Connor, J.R.; Mauger, D.T.; Palmer, C.; Smith, M.B.; Towfighi, J.; Vannucci, S.J. Rat model of perinatal hypoxic-ischemic brain damage. J. Neurosci. Res. 1999, 55, 158–163. [Google Scholar] [CrossRef]
- Fernandez-Lopez, D.; Pazos, M.R.; Tolon, R.M.; Moro, M.A.; Romero, J.; Lizasoain, I.; Martinez-Orgado, J. The cannabinoid agonist WIN55212 reduces brain damage in an in vivo model of hypoxic-ischemic encephalopathy in newborn rats. Pediatr. Res. 2007, 62, 255–260. [Google Scholar] [CrossRef]
- Palazuelos, J.; Ortega, Z.; Diaz-Alonso, J.; Guzman, M.; Galve-Roperh, I. CB2 cannabinoid receptors promote neural progenitor cell proliferation via mTORC1 signaling. J. Biol. Chem. 2012, 287, 1198–1209. [Google Scholar]
- Butti, E.; Bacigaluppi, M.; Rossi, S.; Cambiaghi, M.; Bari, M.; Cebrian Silla, A.; Brambilla, E.; Musella, A.; de Ceglia, R.; Teneud, L.; et al. Subventricular zone neural progenitors protect striatal neurons from glutamatergic excitotoxicity. Brain 2012, 135, 3320–3335. [Google Scholar] [CrossRef]
- Fernandez-Lopez, D.; Pradillo, J.M.; Garcia-Yebenes, I.; Martinez-Orgado, J.A.; Moro, M.A.; Lizasoain, I. The cannabinoid WIN55212-2 promotes neural repair after neonatal hypoxia-ischemia. Stroke 2010, 41, 2956–2964. [Google Scholar] [CrossRef]
- Arevalo-Martin, A.; Garcia-Ovejero, D.; Rubio-Araiz, A.; Gomez, O.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoids modulate olig2 and polysialylated neural cell adhesion molecule expression in the subventricular zone of post-natal rats through cannabinoid receptor 1 and cannabinoid receptor 2. Eur. J. Neurosci. 2007, 26, 1548–1559. [Google Scholar] [CrossRef]
- Gomez, O.; Sanchez-Rodriguez, A.; Le, M.; Sanchez-Caro, C.; Molina-Holgado, F.; Molina-Holgado, E. Cannabinoid receptor agonists modulate oligodendrocyte differentiation by activating PI3K/Akt and the mammalian target of rapamycin (mTOR) pathways. Br. J. Pharmacol. 2011, 163, 1520–1532. [Google Scholar] [CrossRef]
- Gomez, O.; Arevalo-Martin, A.; Garcia-Ovejero, D.; Ortega-Gutierrez, S.; Cisneros, J.A.; Almazan, G.; Sanchez-Rodriguez, M.A.; Molina-Holgado, F.; Molina-Holgado, E. The constitutive production of the endocannabinoid 2-arachidonoylglycerol participates in oligodendrocyte differentiation. Glia 2010, 58, 1913–1927. [Google Scholar] [CrossRef]
- Castillo, A.; Tolon, M.R.; Fernandez-Ruiz, J.; Romero, J.; Martinez-Orgado, J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol. Dis. 2010, 37, 434–440. [Google Scholar] [CrossRef]
- Pazos, M.R.; Cinquina, V.; Gomez, A.; Layunta, R.; Santos, M.; Fernandez-Ruiz, J.; Martinez-Orgado, J. Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 2012, 63, 776–783. [Google Scholar] [CrossRef]
- Alvarez, F.J.; Lafuente, H.; Rey-Santano, M.C.; Mielgo, V.E.; Gastiasoro, E.; Rueda, M.; Pertwee, R.G.; Castillo, A.I.; Romero, J.; Martinez-Orgado, J. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic-ischemic newborn piglets. Pediatr. Res. 2008, 64, 653–658. [Google Scholar] [CrossRef]
- Lafuente, H.; Alvarez, F.J.; Pazos, M.R.; Alvarez, A.; Rey-Santano, M.C.; Mielgo, V.; Murgia-Esteve, X.; Hilario, E.; Martinez-Orgado, J. Cannabidiol reduces brain damage and improves functional recovery after acute hypoxia-ischemia in newborn pigs. Pediatr. Res. 2011, 70, 272–277. [Google Scholar]
- Alonso-Alconada, D.; Alvarez, A.; Alvarez, F.J.; Martinez-Orgado, J.A.; Hilario, E. The cannabinoid WIN 55212-2 mitigates apoptosis and mitochondrial dysfunction after hypoxia ischemia. Neurochem. Res. 2012, 37, 161–170. [Google Scholar] [CrossRef]
- Derugin, N.; Ferriero, D.M.; Vexler, Z.S. Neonatal reversible focal cerebral ischemia: A new model. Neurosci. Res. 1998, 32, 349–353. [Google Scholar] [CrossRef]
- Fernandez-Lopez, D.; Faustino, J.; Derugin, N.; Wendland, M.; Lizasoain, I.; Moro, M.A.; Vexler, Z.S. Reduced infarct size and accumulation of microglia in rats treated with WIN 55,212-2 after neonatal stroke. Neuroscience 2012, 207, 307–315. [Google Scholar] [CrossRef]
- Denker, S.P.; Ji, S.; Dingman, A.; Lee, S.Y.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Macrophages are comprised of resident brain microglia not infiltrating peripheral monocytes acutely after neonatal stroke. J. Neurochem. 2007, 100, 893–904. [Google Scholar] [CrossRef]
- Zarruk, J.G.; Fernandez-Lopez, D.; Garcia-Yebenes, I.; Garcia-Gutierrez, M.S.; Vivancos, J.; Nombela, F.; Torres, M.; Burguete, M.C.; Manzanares, J.; Lizasoain, I.; et al. Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 2012, 43, 211–219. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fernández-López, D.; Lizasoain, I.; Moro, M.Á.; Martínez-Orgado, J. Cannabinoids: Well-Suited Candidates for the Treatment of Perinatal Brain Injury. Brain Sci. 2013, 3, 1043-1059. https://doi.org/10.3390/brainsci3031043
Fernández-López D, Lizasoain I, Moro MÁ, Martínez-Orgado J. Cannabinoids: Well-Suited Candidates for the Treatment of Perinatal Brain Injury. Brain Sciences. 2013; 3(3):1043-1059. https://doi.org/10.3390/brainsci3031043
Chicago/Turabian StyleFernández-López, David, Ignacio Lizasoain, Maria Ángeles Moro, and José Martínez-Orgado. 2013. "Cannabinoids: Well-Suited Candidates for the Treatment of Perinatal Brain Injury" Brain Sciences 3, no. 3: 1043-1059. https://doi.org/10.3390/brainsci3031043