Drug-Induced Apoptosis: Mechanism by which Alcohol and Many Other Drugs Can Disrupt Brain Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fundamental Features of Developmental Drug-Induced Apoptosis
2.1. Neuroapoptosis
2.2. Oligoapoptosis
2.3. Windows of Vulnerability
3. Relationship between Drug-Induced and Natural Apoptosis
4. Similarities and Differences between Alcohol and Other Apoptogenic Drugs
4.1. Suppression of Neural Activity—A Common Denominator
4.2. Competing Hypotheses
4.3. Intracellular Signaling Pathways
4.4. Toxic Synergism of Drug Combinations
4.5. Relating Patterns of Cell Loss to Long-Term Neurobehavioral Disturbances
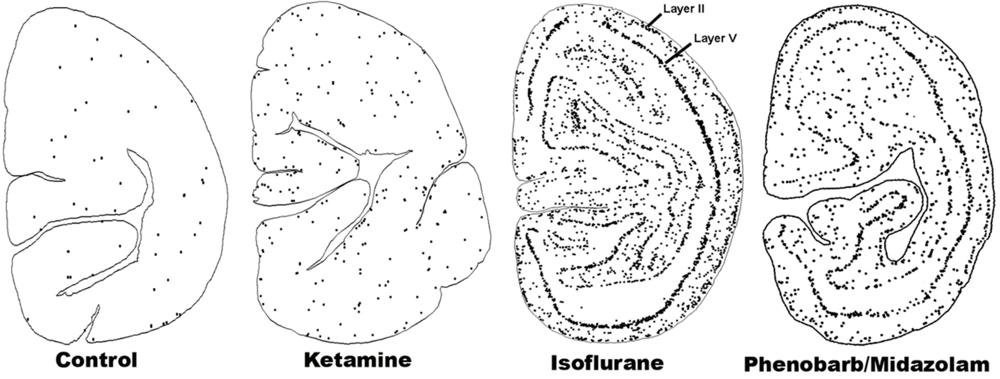

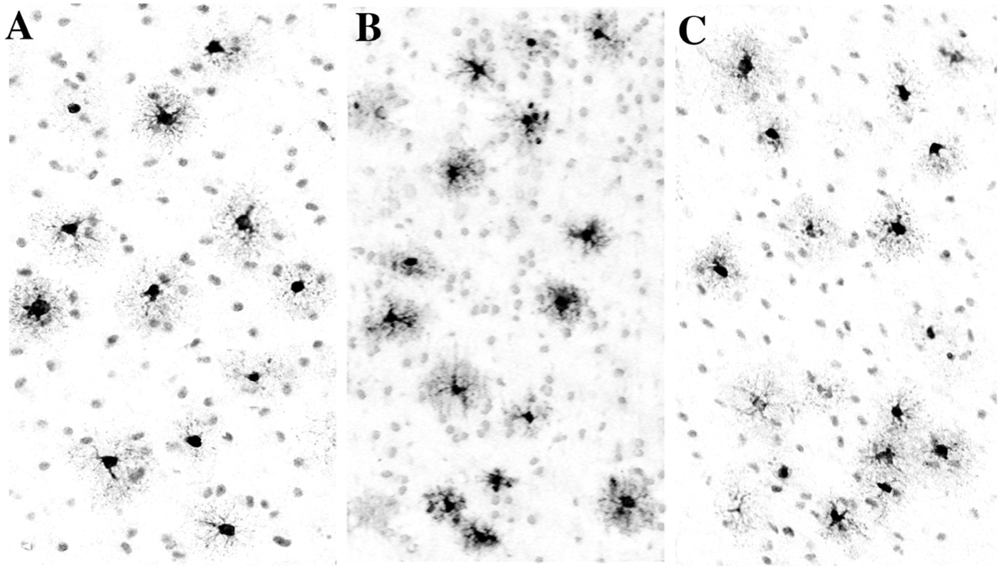
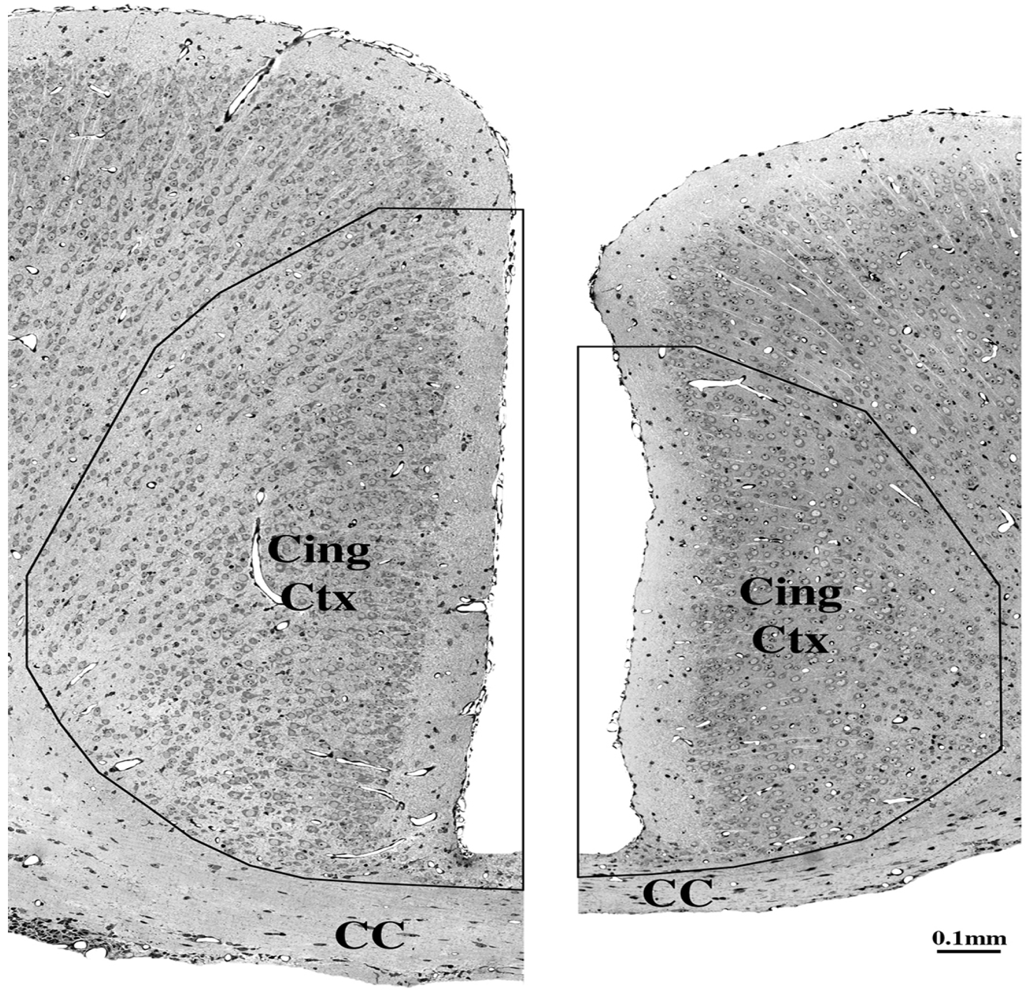
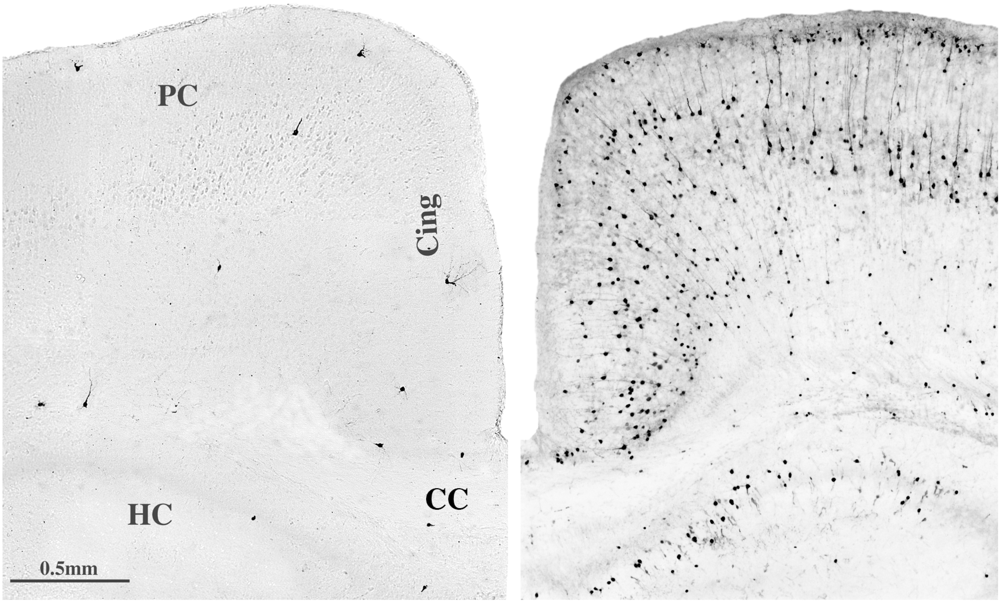
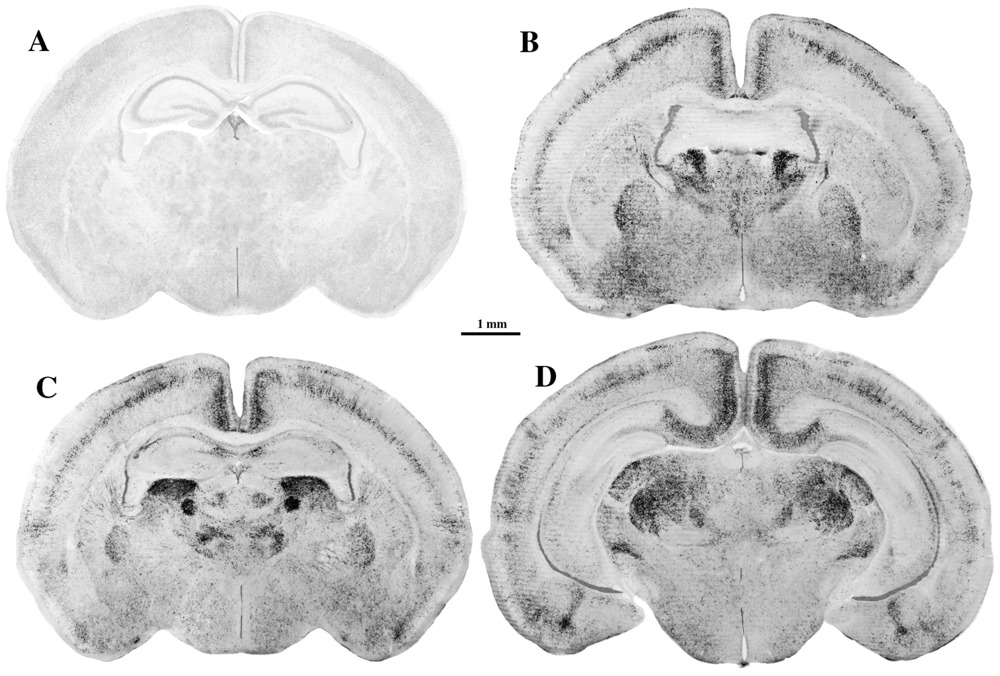
5. Can Alcohol Apoptogenicity Explain Signs and Symptoms of FASD?
5.1. Reduction in Brain Mass
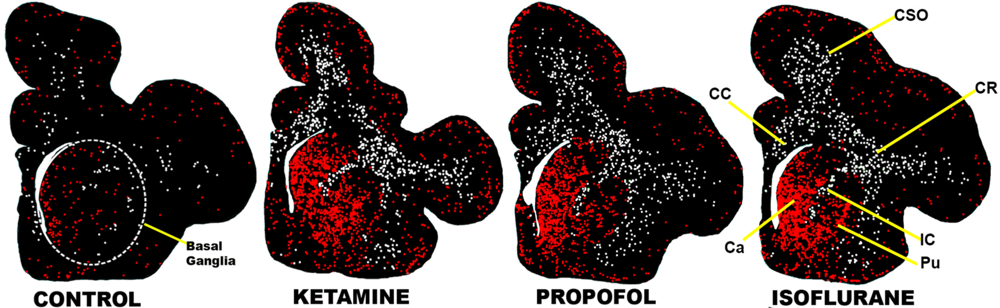
5.2. Focal Impact on Basal Ganglia

5.3. Impact on Corpus Callosum
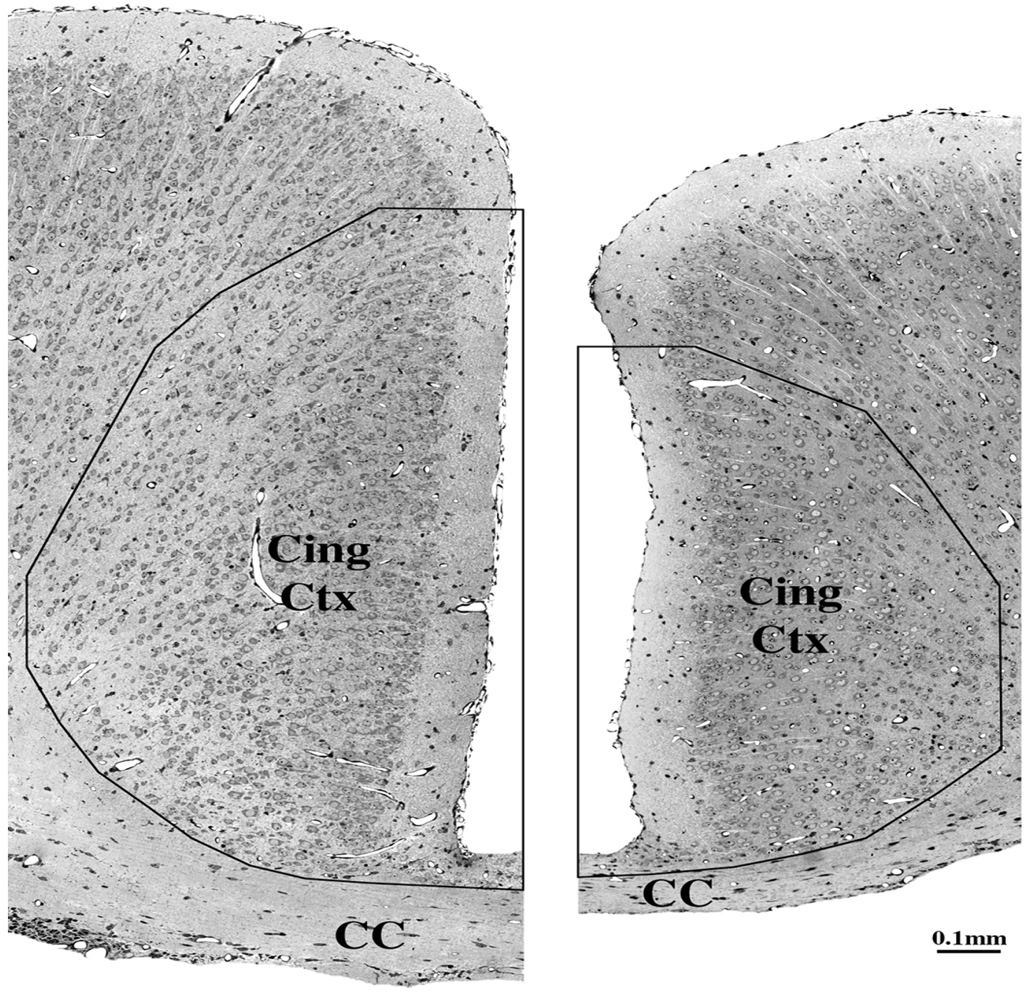
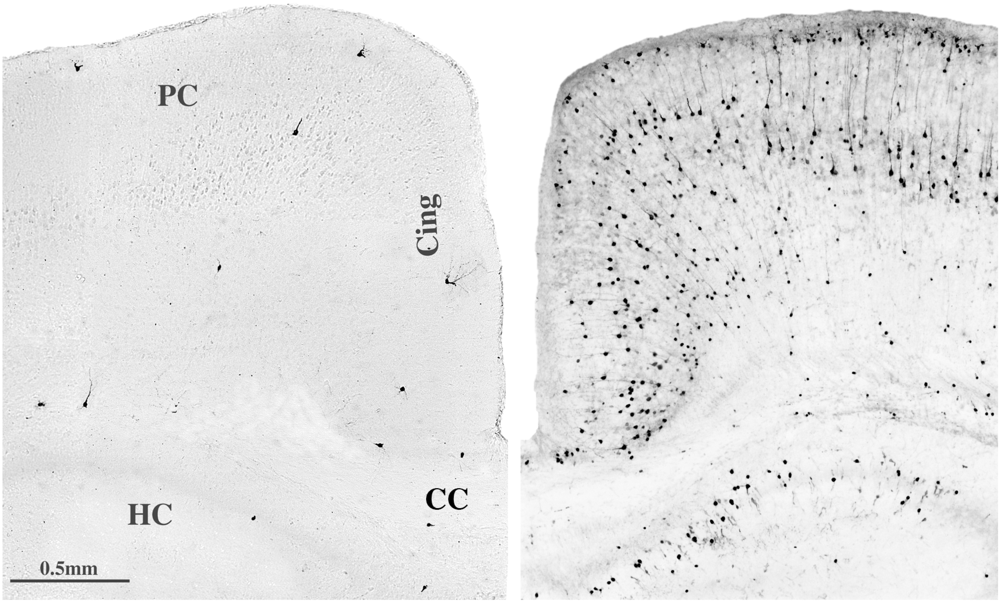
5.4. Long-Term Neurobehavioral Disturbances
5.5. Craniofacial Dysmorphism
6. Can Other Apoptogenic Drugs Cause FASD-Like Syndromes?
6.1. Anti-Epileptic Drugs (AEDs)
6.1.1. Late Gestation Structural Brain Changes, Including Focal Impact on Basal Ganglia
6.1.2. Early Gestation Teratogenic Effects
6.1.3. Neurobehavioral Disturbances
6.2. Gerneral Anesthetics (GAs)
6.2.1. Late Gestation Structural Brain Changes, Including Focal Impact on Basal Ganglia
6.2.2. Early Gestation Teratogenic Effects
6.2.3. Neurobehavioral Disturbances
6.2.4. Tentative Conclusion
7. Excessive Cell Death by Apoptosis
7.1. Excessive Cell Death during Early Embryogenesis
7.2. Excessive Cell Death during Later Stages of Gestation
7.3. Apoptosis—A Unifying Concept
8. Summary and Conclusions
Acknowledgments
Conflict of Interest
References
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vöckler, J.; Dikranian, K.; Tenkova, T.; Stevoska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar]
- Ikonomidou, C.; Bittigau, P.; Ishimaru, M.J.; Wozniak, D.F.; Koch, C.; Genz, K.; Price, M.T.; Stefovska, V.; Hörster, F.; Tenkova, T.; et al. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science 2000, 287, 1056–1060. [Google Scholar] [CrossRef]
- Bittigau, P.; Sifringer, M.; Genz, K.; Reith, E.; Pospischil, D.; Govindarajalu, S.; Dzietko, M.; Pesditschek, S.; Mai, I.; Dikranian, K.; et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15089–15094. [Google Scholar] [CrossRef]
- Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Nemmers, B.; Olney, J.W. Ethanol-induced neuroapoptosis in the developing rodent cerebellum and related brain stem structures. Dev. Brain Res. 2005, 155, 1–13. [Google Scholar]
- Istaphanous, G.K.; Howard, J.; Nan, X.; Hughes, E.A.; McCann, J.C.; McAuliffe, J.J.; Danzer, S.C.; Loepke, A.W. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology 2011, 114, 578–587. [Google Scholar] [CrossRef]
- Jevtovic-Todorovic, V.; Hartman, R.E.; Izumi, Y.; Benshoff, N.D.; Dikranian, K.; Zorumski, C.F.; Olney, J.W.; Wozniak, D.F. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J. Neurosci. 2003, 23, 876–882. [Google Scholar]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef]
- Rizzi, S.; Carter, L.B.; Ori, C.; Jevtovic-Todorovic, V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathol. 2008, 18, 198–210. [Google Scholar]
- Rizzi, S.; Ori, C.; Jevtovic-Todorovic, V. Timing versus duration: determinants of anesthesia-induceddevelopmental apoptosis in the young mammalian brain. Ann. N. Y. Acad. Sci. 2010, 1199, 43–51. [Google Scholar]
- Tenkova, T.; Young, C.; Dikranian, K.; Labruyere, J.; Olney, J.W. Ethanol-induced apoptosis in the visual system during synaptogenesis. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2809–2817. [Google Scholar]
- Young, C.; Jevtovic-Todorovic, V.; Qin, Y.Q.; Tenkova, T.; Wang, H.; Labruyere, J.; Olney, J.W. Potential of ketamine and midazolam, individually or in combination, to induce apoptotic neurodegeneration in the infant mouse brain. Br. J. Pharmacol. 2005, 146, 189–197. [Google Scholar]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Zhang, X.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology 2010, 112, 834–841. [Google Scholar] [CrossRef]
- Brambrink, A.M.; Evers, A.S.; Avidan, M.S.; Farber, N.B.; Smith, D.J.; Martin, L.D.; Dissen, G.A.; Creeley, C.E.; Olney, J.W. Ketamine-induced neuroapoptosis in the fetal and neonatal rhesus macaque brain. Anesthesiology 2012, 116, 372–384. [Google Scholar] [CrossRef]
- Brambrink, A.M.; Dissen, G.A.; Martin, L.D.; Creeley, C.E.; Olney, J.W. Propofol-Induced Apoptosis of Neurons and Oligodendrocytes in Neonatal Macaque Brain. In Proceedings of the American Society of Anesthesiologists, Washington, DC, USA, 13–17 October 2012. Abstract Number A103.
- Farber, N.B.; Creeley, C.E.; Olney, J.W. Alcohol-induced neuroapoptosis in the fetal macaque brain. Neurobiol. Dis. 2010, 40, 200–206. [Google Scholar]
- Paule, M.G.; Li, M.; Allen, R.R.; Liu, F.; Zou, X.; Hotchkiss, C.; Hanig, J.P.; Patterson, T.A.; Slikker, W., Jr.; Wang, C. Ketamine anesthesia during the first week of life can cause long-lasting cognitive deficits in rhesus monkeys. Neurotoxicol. Teratol. 2011, 33, 220–230. [Google Scholar]
- Slikker, W., Jr.; Zou, X.; Hotchkiss, C.E.; Divine, R.L.; Sadovova, N.; Twaddle, N.C.; Doerge, D.R.; Scallet, A.C.; Patterson, T.A.; Hanig, J.P.; et al. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol. Sci. 2007, 98, 145–158. [Google Scholar]
- Zou, X.; Liu, F.; Zhang, X.; Patterson, T.A.; Callicott, R.; Liu, S.; Hanig, J.P.; Paule, M.G.; Slikker, W.; Wang, C. Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicol. Teratol. 2011, 33, 592–597. [Google Scholar]
- Zou, X.; Patterson, T.A.; Divine, R.L.; Sadova, N.; Zhang, X.; Hanig, J.P.; Paule, M.G.; Slikker, W.; Wang, C. Prolonged exposure to ketamine increases neurodegeneration in the developing monkey brain. Int. J. Dev. Neurosci. 2009, 27, 727–731. [Google Scholar]
- Brambrink, A.M.; Back, S.A.; Riddle, A.; Gong, X.; Moravec, M.D.; Dissen, G.A.; Creeley, C.E.; Dikranian, K.; Olney, J.W. Isoflurane-induced apoptosis of oligodendrocytes in the neonatal primate brain. Ann. Neurol. 2012, 72, 525–535. [Google Scholar] [CrossRef]
- Creeley, C.E.; Dikranian, K.T.; Johnson, S.A.; Farber, N.B.; Olney, J.W. Alcohol-induced apoptosis of oligodendrocytes in the fetal macaque brain. Acta Neuropathol. Commun. 2013, 1, 23. [Google Scholar] [CrossRef]
- Fredriksson, A.; Archer, T. Neurobehavioural deficits associated with apoptotic neurodegeneration and vulnerability for ADHD. Neurotox. Res. 2004, 6, 435–456. [Google Scholar] [CrossRef]
- Fredriksson, A.; Ponten, E.; Gordh, T.; Eriksson, P. Neonatal exposure to a combination of N-methyl-d-aspartate and γ-aminobutyric acid type A receptor anesthetic agents potentiates apoptotic neurodegeneration and persistent behavioral deficits. Anesthesiology 2007, 107, 427–436. [Google Scholar] [CrossRef]
- Satomoto, M.; Satoh, Y.; Terui, K.; Miyao, H.; Takishima, K.; Ito, M.; Imaki, J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology 2009, 110, 628–637. [Google Scholar] [CrossRef]
- Zhu, C.; Gao, J.; Karlsson, N.; Li, Q.; Zhang, Y.; Huang, Z.; Li, H.; Kuhn, H.G.; Blomgren, K. Isoflurane anesthesia induced persistent, progressive memory impairment, caused a loss of neural stem cells, and reduced neurogenesis in young, but not adult, rodents. J. Cereb. Blood Flow Metab. 2010, 30, 1017–1030. [Google Scholar] [CrossRef]
- Wozniak, D.F.; Hartman, R.E.; Boyle, M.P.; Vogt, S.K.; Brooks, A.R.; Tenkova, T.; Young, C.; Olney, J.W.; Muglia, L.J. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol. Dis. 2004, 17, 403–414. [Google Scholar] [CrossRef]
- Dikranian, K.; Ishimaru, M.J.; Tenkova, T.; Labruyere, J.; Qin, Y.Q.; Ikonomidou, C.; Olney, J.W. Apoptosis in the in vivo mammalian forebrain. Neurobiol. Dis. 2001, 8, 359–379. [Google Scholar] [CrossRef]
- Nikizad, H.; Yon, J.H.; Carter, L.B.; Jevtovic-Todorovic, V. Early exposure to general anesthesia causes significant neuronal deletion in the developing rat brain. Ann. N. Y. Acad. Sci. 2007, 1122, 69–82. [Google Scholar]
- Lunardi, N.; Ori, C.; Erisir, A.; Jevtovic-Todorovic, V. General anesthesia causes long-lasting disturbances in the ultrastructural properties of developing synapses in young rats. Neurotox. Res. 2010, 17, 179–188. [Google Scholar] [CrossRef]
- Sanders, R.D.; Xu, J.; Shu, Y.; Fidalgo, A.; Ma, D.; Maze, M. General anesthetics induce apoptotic neurodegeneration in the neonatal rat spinal cord. Anesth. Analg. 2008, 106, 1708–1711. [Google Scholar] [CrossRef]
- Young, C.; Klocke, J.; Tenkova, T.; Choi, J.; Labruyere, J.; Qin, Y.Q.; Holtzman, D.M.; Roth, K.A.; Olney, J.W. Ethanol-induced neuronal apoptosis in the in vivo developing mouse brain is BAX dependent. Cell Death Differ. 2003, 10, 1148–1155. [Google Scholar] [CrossRef]
- Young, C.; Straiko, M.M.W.; Johnson, S.A.; Creeley, C.; Olney, J.W. Ethanol causes and lithium prevents neuroapoptosis and suppression of pERK in the infant mouse brain. Neurobiol. Dis. 2008, 31, 355–360. [Google Scholar] [CrossRef]
- Straiko, M.M.W.; Young, C.; Cattano, D.; Creeley, C.E.; Wang, H.; Smith, D.J.; Johnson, S.A.; Li, E.S.; Olney, J.W. Lithium protects against anesthesia-induced developmental neuroapoptosis. Anesthesiology 2009, 110, 662–668. [Google Scholar]
- Sanders, R.D.; Sun, P.; Patel, S.; Li, M.; Maze, M.; Ma, D. Dexmedetomidine provides cortical neuroprotection: Impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta Anaesthesiol. Scand. 2010, 54, 710–716. [Google Scholar]
- Yon, J.H.; Carter, L.B.; Jevtovic-Todorovic, V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol. Dis. 2006, 21, 522–530. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Muglia, L.J.; Jermakowicz, W.J.; D’Sa, C.; Roth, K.A. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol. Dis. 2002, 9, 205–219. [Google Scholar] [CrossRef]
- Young, C.; Roth, K.A.; Klocke, B.J.; West, T.; Holtzman, D.M.; Labruyere, J.; Qin, Y.Q.; Dikranian, K.; Olney, J.W. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol. Dis. 2005, 20, 608–614. [Google Scholar] [CrossRef]
- Yon, J.H.; Carter, L.B.; Reiter, R.J.; Jevtovic-Todorovic, V. Anesthesia induces suicide in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience 2005, 135, 815–827. [Google Scholar] [CrossRef]
- Cattano, D.; Young, C.; Olney, J.W. Sub-anesthetic doses of propofol induce neuroapoptosis in the infant mouse brain. Anesth. Analg. 2008, 106, 1712–1714. [Google Scholar] [CrossRef]
- Ma, D.; Williamson, P.; Januszewski, A.; Nogaro, M.C.; Hossain, M.; Ong, L.P.; Shu, Y.; Franks, N.P.; Maze, M. Xenon mitigates isoflurane-induced neuronal apoptosis in the developing rodent brain. Anesthesiology 2007, 106, 746–753. [Google Scholar] [CrossRef]
- Johnson, S.A.; Young, C.; Olney, J.W. Isoflurane-induced neuroapoptosis in the developing brain of non-hypoglycemic mice. J. Neurosurg. Anesth. 2008, 20, 21–28. [Google Scholar] [CrossRef]
- Sanders, R.D.; Xu, J.; Shu, Y.; Januszewski, A.; Halder, S.; Fidalgo, A.; Sun, P.; Hossain, M.; Ma, D.; Maze, M. Dexmedetomidine attenuates isoflurane-induced neurocognitive impairment in neonatal rats. Anesthesiology 2009, 110, 11077–11085. [Google Scholar]
- Zhang, X.; Xue, Z.; Sun, A. Subclinical concentration of sevoflurane potentiates neuronal apoptosis in the developing C57BL/6 mouse brain. Neurosci. Lett. 2008, 447, 109–114. [Google Scholar] [CrossRef]
- Cattano, D.; Williamson, P.; Fukui, K.; Avidan, M.; Evers, A.S.; Olney, J.W.; Young, C. Potential of xenon to induce or to protect against neuroapoptosis in the developing mouse brain. Can. J. Anesth. 2008, 55, 429–436. [Google Scholar] [CrossRef]
- Brambrink, A.M.; Back, S.A.; Avidan, M.S.; Creeley, C.E.; Olney, J.W. Ketamine and Isoflurane Anesthesia Triggers Neuronal and Glial Apoptosis in the Neonatal Macaque. In Proceedings of the American Society of Anesthesiologists, San Diego, CA, USA, 16–20 October 2010. Abstract Number A375.
- Brambrink, A.M.; Dissen, G.A.; Martin, L.D.; Creeley, C.E.; Olney, J.W. Neuronal and glial apoptosis observed after intravenous propofol anesthesia in neonatal macaques. J. Neurosurg. Anesthesiol. 2012, 24, 494. [Google Scholar]
- Brambrink, A.M.; Dikranian, K.; Evers, A.S.; Creeley, C.E.; Olney, J.W. Isoflurane-Induced Apoptosis of Neurons and Oligodendrocytes in the Fetal Rhesus Macaque Brain. In Proceedings of the American Society of Anesthesiologists, Washington, DC, USA, 13–17 October 2012. Abstract Number LBB10.
- Forcelli, P.A.; Janssen, M.J.; Vicini, S.; Gale, K. Neonatal exposure to antiepileptic drugs disrupts striatal synaptic development. Ann. Neurol. 2012, 72, 363–372. [Google Scholar] [CrossRef]
- Boscolo, A.; Starr, J.A.; Sanchez, V.; Lunardi, N.; DiGruccio, M.R.; Ori, C.; Erisir, A.; Trimmer, P.; Bennett, J.; Jevtovic-Todorovic, V. The abolishment of anesthesia-induced cognitive impairment by timely protection of mitochondria in the developing rat brain: The importance of free oxygen radicals and mitochondrial integrity. Neurobiol. Dis. 2012, 45, 1031–1041. [Google Scholar] [CrossRef]
- Leraci, A.; Herrera, D.G. Nicoinamide protects against ethanol-induced apoptotic neurodegeneration in the developing mouse brain. PLoS Med. 2006, 4, e101. [Google Scholar] [CrossRef]
- Manthey, D.; Asimiadou, S.; Stefovska, V.; Kaindl, A.M.; Fassbender, J.; Ikonomidou, C.; Bittigau, P. Sulthiame but not levetiracetam exerts neurotoxic effect in the developing rat brain. Exp. Neurol. 2005, 193, 497–503. [Google Scholar] [CrossRef]
- Ramantani, G.; Ikonomidou, C.; Walter, B.; Rating, D.; Dinger, J. Levetiracetam: Safety and efficacy in neonatal seizures. Eur. J. Paediatr. Neurol. 2011, 15, 1–7. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Volpe, J.J.; Kinney, H.C. Arrested oligodendrocyte lineage progression during human cerebral white matter development: Dissociation between the timing of progenitor differentiation and myelinogenesis. J. Neuropath. Exp. Neurol. 2002, 61, 197–211. [Google Scholar]
- Creeley, C.E.; Dikranian, K.T.; Back, S.A.; Olney, J.W.; Brambrink, A.M. Isoflurane-induced apoptosis of neurons and oligodendrocytes in the fetal rhesus macaque brain. Anesthesiology 2013, in press. [Google Scholar]
- Cowan, W.M.; Fawcett, J.W.; O’Leary, D.D.; Stanfield, B.B. Regressive events in neurogenesis. Science 1984, 225, 1258–1265. [Google Scholar]
- Oppenheim, R.W. Cell death during development of the nervous system. Ann. Rev. Neurosci. 1991, 14, 453–501. [Google Scholar] [CrossRef]
- Raff, M.C.; Barres, B.A.; Burne, J.F.; Coles, H.S.; Ishizaki, Y.; Jacobson, M.D. Programmed cell death and the control of survival: Lessons from the nervous system. Science 1993, 262, 695–700. [Google Scholar]
- Ferrer, I.; Soriano, E.; del Rio, J.A.; Alcantara, S.; Auladell, C. Cell death and removal in the cerebral cortex during development. Prog. Neurobiol. 1992, 39, 1–43. [Google Scholar] [CrossRef]
- Blaschke, A.J.; Staley, K.; Chun, J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996, 122, 1165–1174. [Google Scholar]
- Thomaidou, D.; Mione, M.C.; Cavanagh, J.F.; Parnavelas, J.G. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J. Neurosci. 1997, 17, 1075–1085. [Google Scholar]
- Sulik, K.K. Genesis of alcohol-induced craniofacial dysmorphism. Exp. Biol. Med. 2005, 230, 366–375. [Google Scholar]
- Dunty, W.C., Jr.; Chen, S.Y.; Zucker, R.M.; Dehart, D.B.; Sulik, K.K. Selective vulnerability of embryonic cell populations to ethanol-induced apoptosis: Implications for alcohol-related birth defects and neurodevelopmental disorder. Alcohol. Clin. Exp. Res. 2001, 25, 1523–1535. [Google Scholar] [CrossRef]
- Gallo, V.; Mangin, J.M.; Kukley, M.; Dietrich, D. Synapses on NG2-expressing progenitors in the brain: Multiple functions? J. Physiol. 2008, 16, 3767–3781. [Google Scholar]
- Cherubini, E.; Rovira, C.; Gaiarsa, J.L.; Corradetti, R.; Ben Ari, Y. GABA mediated excitation in immature rat CA3 hippocampal neurons. Int. J. Dev. Neurosci. 1990, 8, 481–490. [Google Scholar] [CrossRef]
- Liu, F.; Patterson, T.A.; Sadovova, N.; Zhang, X.; Liu, S.; Zou, X.; Hanig, J.P.; Paule, M.G.; Slikker, W.; Wang, C. Ketamine-induced neuronal damage and altered NMDA receptor function in rat primary forebrain culture. Toxicol. Sci. 2013, 131, 548–557. [Google Scholar] [CrossRef]
- Wang, C.; Sadovova, N.; Patteron, T.A.; Zou, X.; Fu, X.; Hanig, J.P.; Paule, M.G.; Ali, S.F.; Zhang, X.; Slikker, W. Protective effects of 7-nitroindazole on ketamine-induced neurotoxicity in rat forebrain culture. Neurtoxicology 2008, 29, 613–620. [Google Scholar] [CrossRef]
- Xu, W.; Cormier, R.; Fu, T.; Covey, D.F.; Isenberg, K.E.; Zorumski, C.F.; Mennerick, S. Slow death of postnatal hippocampal neurons by GABAA receptor overactivation. J. Neurosci. 2000, 20, 3147–3158. [Google Scholar]
- Hwang, J.Y.; Kim, Y.H.; Ahn, Y.H.; Wie, M.B.; Koh, J.Y. N-Methyl-daspartate receptor blockade induces neuronal apoptosis in cortical culture. Exp. Neurol. 1999, 159, 124–130. [Google Scholar] [CrossRef]
- Turner, C.P.; Debenedetto, D.; Liu, C. NMDAR blockade-induced neonatal brain injury: Reversal by the calcium channel agonist BayK 8644. Neurosci. Lett. 2009, 450, 292–295. [Google Scholar] [CrossRef]
- Ishimaru, M.J.; Ikonomidou, C.; Tenkova, T.I.; Der, T.C.; Dikranian, K.; Sesma, M.; Olney, J.W. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J. Comp. Neurol. 1999, 408, 461–476. [Google Scholar] [CrossRef]
- Klintsova, A.Y.; Helfer, J.L.; Calizo, L.H.; Dong, W.K.; Goodlett, C.R.; Greenough, W.T. Persistent impairment of hippocampal neurogenesis in young adult rats following early postnatal alcohol exposure. Alcohol. Clin. Exp. Res. 2007, 31, 2073–2082. [Google Scholar] [CrossRef]
- Stratmann, G.; Sall, J.W.; May, L.D.; Bell, J.S.; Magnusson, K.R.; Rau, V.; Visrodia, K.H.; Alvi, R.S.; Ku, B.; Lee, M.T.; et al. Isoflurane differentially affects neurogenesis and long-term neurocognitive function in 60-day-old and 7-day-old rats. Anesthesiology 2009, 110, 834–848. [Google Scholar] [CrossRef]
- Stefovska, V.G.; Uckermann, O.; Czuczwar, M.; Smitka, M.; Czuczwar, P.; Kis, J.; Kaindl, A.M.; Turski, L.; Turski, W.A.; Ikonomidou, C. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann. Neurol. 2008, 64, 434–445. [Google Scholar] [CrossRef]
- Jevtovic-Todorovic, V.; Boscolo, A.; Sanchez, V.; Lunardi, N. Anesthesia-induced developmental neurodegeneration: The role of neuronal organelles. Front. Neurol. 2012, 3, 1–7. [Google Scholar]
- Kang, S.H.; Lee, Y.A.; Won, S.J.; Rhee, K.-H.; Gwag, B.J. Caffeine-induced neuronal death in neonatal rat brain and cortical cell cultures. Neuroreport 2002, 13, 1945–1950. [Google Scholar] [CrossRef]
- Black, A.M.; Pandya, S.; Clark, D.; Armstrong, E.A.; Yager, J.Y. Effect of caffeine and morphine on the developing pre-mature brain. Brain Res. 2008, 1219, 136–142. [Google Scholar]
- Yuede, C.M.; Creeley, C.E.; Olney, J.W. Long-Term Behavioral Effects of the Interaction between NMDA Antagonists and Caffeine in the Developing Mouse Brain. In Proceedings of the Society for Neuroscience, Washington, DC, USA, 12–16 November 2011. Abstract Number 33.01.
- Creeley, C.E.; Yuede, C.M.; Olney, J.W. Long-Term Behavioral Effects of the Interaction between GABA Agonists and Caffeine in the Developing Mouse Brain. In Proceedings of the Society for Neuroscience, Washington, DC, USA, 12–16 November 2011. Abstract Number 33.02.
- Katz, L.; Kim, J.; Gale, K.; Kondratyev, A. Effects of lamotrigine alone and in combination with MK-801, phenobarbital, or phentoin on cell death on the neonatal rat brain. J. Pharmacol. Exp. Ther. 2002, 322, 494–500. [Google Scholar]
- Lohaugen, G.C.; Gramstad, A.; Evensen, K.A.; Martinussen, M.; Lindqvist, S.; Indredavik, M.; Vik, T.; Brubakk, A.M.; Skranes, J. Cognitive profile in young adults born preterm at very low birthweight. Dev. Med. Child Neurol. 2010, 52, 1078–1079. [Google Scholar] [CrossRef]
- Anand, K.J.S.; Soriano, S.G. Anesthetic agents and the immature brain: Are these toxic or therapeutic? Anesthesiology 2004, 101, 527–530. [Google Scholar] [CrossRef]
- Shu, Y.; Zhou, Z.; Wan, Y.; Sanders, R.D.; Li, M.; Pac-Soo, C.K.; Maze, M.; Ma, D. Nociceptive stimuli enhance anesthetic-induced neuroapoptosis in the rat developing brain. Neurobiol. Dis. 2012, 45, 743–750. [Google Scholar] [CrossRef]
- Streissguth, A.P.; O’Malley, K. Neuropsychiatric implications and long-term consequences of fetal alcohol spectrum disorders. Semin. Clin. Neuropsychiatry 2000, 5, 177–190. [Google Scholar]
- Riley, E.P.; McGee, C.L. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp. Biol. Med. 2005, 230, 357–365. [Google Scholar]
- Famy, C.; Streissguth, A.P.; Unis, A.S. Mental illness in adults with fetal alcohol syndrome or fetal alcohol effects. Am. J. Psychiatry 1998, 155, 552–554. [Google Scholar]
- Archibald, S.L.; Fennema-Notestine, C.; Gamst, A.; Riley, E.P.; Mattson, S.N.; Jernigan, T.L. Brain dysmorphology in individuals with severe prenatal alcohol exposure. Dev. Med. Child Neurol. 2001, 43, 148–154. [Google Scholar]
- Goodlett, C.R.; Marcussen, B.L.; West, J.R. A single day of alcohol exposure during the brain growth spurt induces brain weight restriction and cerebellar Purkinje cell loss. Alcohol 1990, 7, 107–114. [Google Scholar] [CrossRef]
- Livy, D.J.; Elberger, A.J. Effect of prenatal alcohol exposure on midsagittal commissure size in rats. Teratology 2001, 63, 15–22. [Google Scholar] [CrossRef]
- Creeley, C.E.; Olney, J.W.; Brambrink, A.M. Acute apoptosis of neurons and oligodendrocytes induced in the developing rhesus macaque brain by anti-epileptic drugs. Soc. Neurosci. Abstr. 2013, in press. [Google Scholar]
- Ikonomidou, C.; Scheer, I.; Wilhelm, T.; Juengling, F.D.; Titze, K.; Stöver, B.; Lehmkuhl, U.; Koch, S.; Kassubek, J. Brain morphology alterations in the basal ganglia and the hypothalamus following prenatal exposure to antiepileptic drugs. Eur. J. Paediatr. Neurol. 2007, 11, 297–301. [Google Scholar] [CrossRef]
- Farwell, J.R.; Lee, Y.J.; Hirtz, D.G.; Sulzbacher, S.I.; Ellenberg, J.H.; Nelson, K.B. Phenbarbital for febrile seizures—effects on intelligence and on seizure recurrence. N. Engl. J. Med. 1990, 322, 364–369. [Google Scholar] [CrossRef]
- Sulzbacher, S.; Farwell, J.R.; Temkin, N.; Lu, A.S.; Hirtz, D.G. Late cognitive effects of early treatment with phenobarbital. Clin. Pediatr. 1999, 38, 387–394. [Google Scholar] [CrossRef]
- Meador, K.J.; NEAD Study Group. Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs. N. Engl. J. Med. 2009, 360, 1597–1605. [Google Scholar] [CrossRef]
- Meador, K.J.; NEAD Study Group. Effects of fetal antiepileptic drug exposure: Outcomes at age 4.5 years. Neurology 2012, 78, 1207–1214. [Google Scholar] [CrossRef]
- Forcelli, P.A.; Kim, J.; Kondrayev, A.; Gale, K. Effects of neonatal antiepileptic drug exposure on cognitive, emotional and motor function in adult rats. J. Pharmacol. Exp. Ther. 2012, 340, 558–566. [Google Scholar] [CrossRef]
- DiMaggio, C.; Sun, L.S.; Kakavouli, A.; Burne, M.W.; Li, G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J. Neurosurg. Anesthesiol. 2009, 4, 286–291. [Google Scholar]
- DiMaggio, C.; Sun, L.; Li, G. Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesth. Analg. 2011, 113, 1143–1151. [Google Scholar] [CrossRef]
- Wilder, R.T.; Flick, R.P.; Sprung, J.; Katusic, S.K.; Barbaresi, W.J.; Mickelson, C.; Gleich, S.J.; Schroeder, D.R.; Weaver, A.L.; Warner, D.O. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology 2009, 110, 796–804. [Google Scholar] [CrossRef]
- Flick, R.P.; Katusic, S.K.; Colligan, R.C.; Wilder, R.T.; Voigt, R.G.; Olson, M.D.; Sprung, J.; Weaver, A.L.; Schroeder, D.R.; Warner, D.O. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics 2011, 128, 1053–1061. [Google Scholar] [CrossRef]
- Sprung, J.; Flick, R.P.; Katusic, S.K.; Colligan, R.C.; Barbaresi, W.J.; Bojanic, K.; Welch, T.L.; Olson, M.D.; Hanson, A.C.; Schroeder, D.R.; et al. Attention-deficit/hyperactivity disorder after early exposure to procedures requiring general anesthesia. Mayo Clin. Proc. 2012, 87, 120–129. [Google Scholar] [CrossRef]
- Ing, C.; DiMaggio, C.; Whitehouse, A.; Hegarty, M.K.; Brady, J.; von Ungern-Sternberg, B.S.; Davidson, A.; Wood, A.J.J.; Li, G.; Sun, L.S. Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics 2012, 130, 476–485. [Google Scholar] [CrossRef]
- Block, R.I.; Thomas, J.J.; Bayman, E.O.; Choi, J.W.; Kimble, K.K.; Todd, M.M. Are anesthesia and surgery during infancy associated with altered academic performance during childhood? Anesthesiology 2012, 117, 494–503. [Google Scholar] [CrossRef]
- Cheek, T.G.; Baird, E. Anesthesia for nonobstetric surgery: Maternal and fetal considerations. Clin. Obstet. Gynecol. 2009, 52, 535–545. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Creeley, C.E.; Olney, J.W. Drug-Induced Apoptosis: Mechanism by which Alcohol and Many Other Drugs Can Disrupt Brain Development. Brain Sci. 2013, 3, 1153-1181. https://doi.org/10.3390/brainsci3031153
Creeley CE, Olney JW. Drug-Induced Apoptosis: Mechanism by which Alcohol and Many Other Drugs Can Disrupt Brain Development. Brain Sciences. 2013; 3(3):1153-1181. https://doi.org/10.3390/brainsci3031153
Chicago/Turabian StyleCreeley, Catherine E., and John W. Olney. 2013. "Drug-Induced Apoptosis: Mechanism by which Alcohol and Many Other Drugs Can Disrupt Brain Development" Brain Sciences 3, no. 3: 1153-1181. https://doi.org/10.3390/brainsci3031153