Molecular Dissection of Cyclosporin A’s Neuroprotective Effect Reveals Potential Therapeutics for Ischemic Brain Injury

Abstract
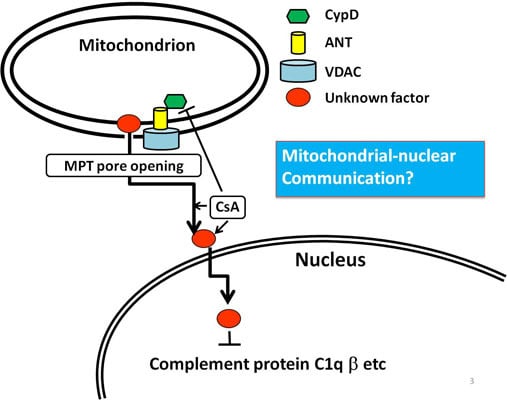
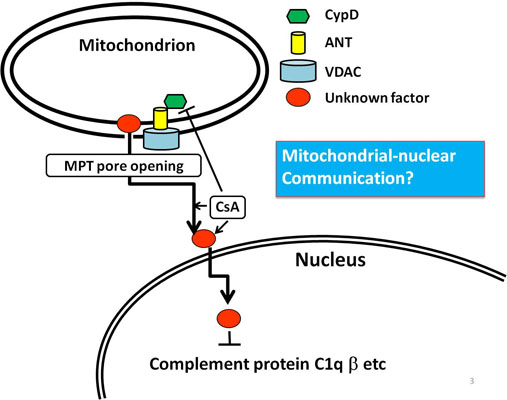
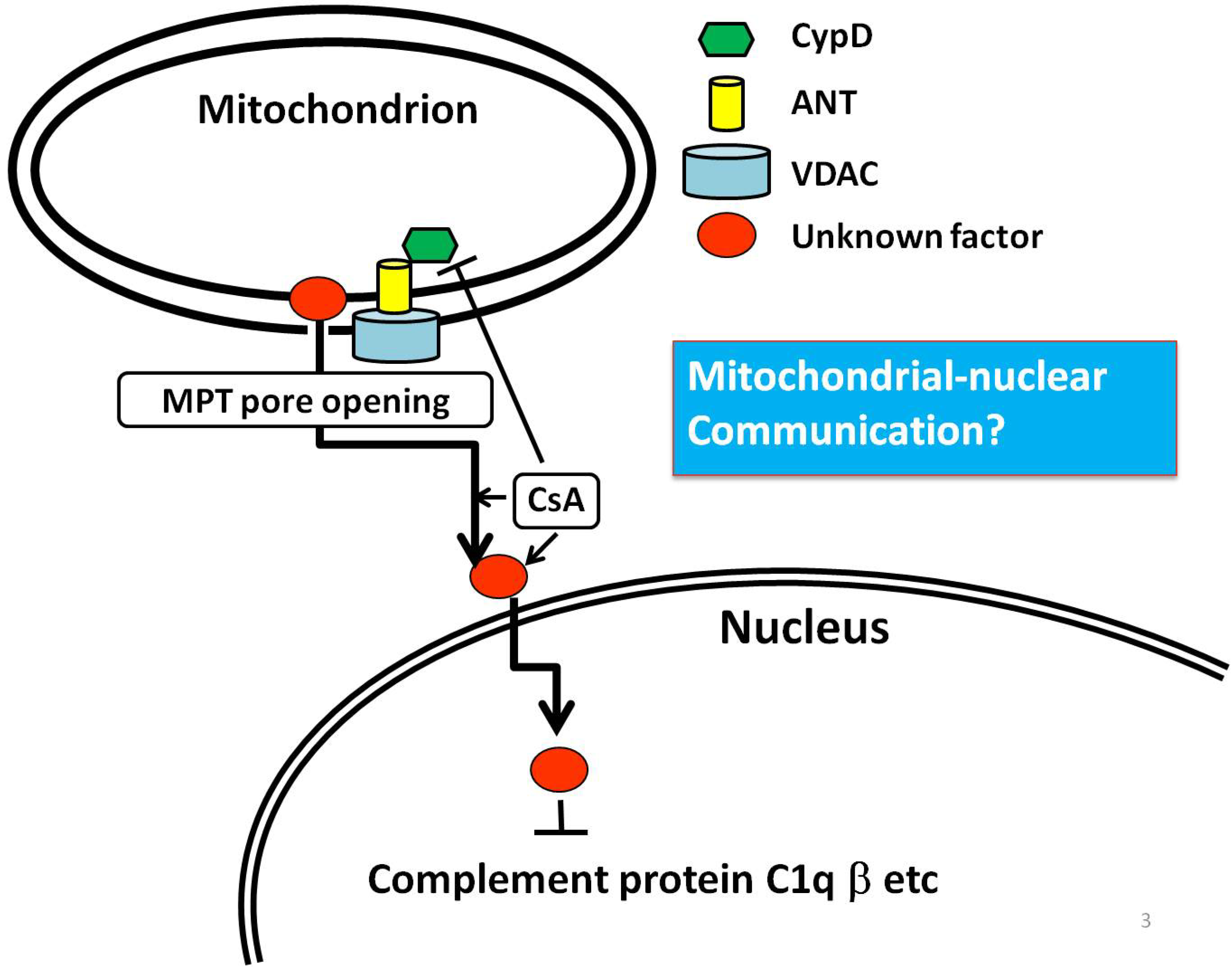
:
1. Introduction
2. Neuroprotective Drugs
2.1. Antioxidants
2.1.1. NXY-059 (Cerovive)

{kind=link}
{kind=link}
| Categories | Drugs | Current status/Result of clinical trial |
|---|---|---|
| Antioxidants | NXY-059 (Cerovive) | Trial in a larger number of cases indicated no significant activity. The program was terminated. |
| Tirilazad | The drug increased the combined endpoint of “death or disability” by about one-fifth. | |
| Nicaraven | The drug did not demonstrate enough therapeutic efficacy in the treatment of acute ischemic stroke. | |
| Ebselen | A phase III trial exploring the efficacy in patients with cortical infarct is under way. | |
| SUN-N8075 | A phase I trial is under way in the United States. | |
| Calcium Antagonists | Nimodipine | A clinical trial once suggested a beneficial effect, but none of the subsequent trials confirmed this result. |
| Monosialoganglioside GM1 | There is not enough evidence to conclude that gangliosides are beneficial in acute stroke. | |
| Nootropil (Piracetam) | There is not enough evidence to assess the effect of piracetam in acute ischemic stroke. | |
| E-2051 | A phase I clinical trial is under way in Europe. | |
| N-methyl-D-aspartate (NMDA) Receptor Antagonists | Selfotel | The drug was not effective and might have a neurotoxic effect in brain ischemia. |
| Gavestinel | There is no evidence of benefit. Further development has been discontinued. | |
| Magnesium | No beneficial effects on the functional outcome of stroke in patients were accrued except in cases of lacunar syndromes. | |
| GABA Agonists | Clomethiazole | The drug did not improve outcome in patients with major ischemic stroke. |
| Sodium Channel Blockers | lubeluzole | The drug failed to show the efficacy in the treatment of acute stroke. |
| Opioid Antagonists | nalmefene | A phase III study was completed, but the results were not published. |
| Membrane Stabilizers | Citicoline | The drug was not efficacious in the treatment of moderate-to-severe acute ischemic stroke. |
| DP-b99 | The Phase III trial is under way. | |
| Poly ADP-ribose Polymerase (PARP) Inhibitors | ONO-2231 | A Phase I clinical trial is under way in the UK. |
| AX200 (Granulocyte Colony-Stimulating Factor (G-CSF)) | The drug failed to show the efficacy in the treatment of acute stroke. | |
| Immunosuppressants | FK506 (Tacrolimus) | A human stroke trial was stopped in phase II. |
| Cyclosporin A | A Phase II clinical trial, Neuroprotection Impact of Cyclosporin A in Cerebral Infarction (CsAStroke), is under way. | |
| PTP inhibitors | Pramipexole(Mirapex) | No information about clinical trial for an inhibitor of PTP. |
| S-15176 | No information about clinical trial for an inhibitor of PTP. | |
| Others | Albumin | A Phase III randomized multicenter clinical trial of high-dose human albumin therapy for neuroprotection in acute ischemic stroke. |
| ONO-2506 (Proglia) | A Phase III crinical trial in acute stroke patients was completed and failed to show the efficacy of the drug in the treatment of acute stroke. | |
| SUN-N4057 (Piclozotan) | Phase II clinical trials are under way in the USA and Europe. | |
| TS-011 | Phase I clinical trials of TS-011 are under way. |
2.1.2. Tirilazad
2.1.3. Nicaraven
2.1.4. Ebselen
2.1.5. SUN-N8075
2.2. Calcium Antagonists
2.2.1. Nimodipine
2.2.2. Monosialoganglioside GM1
2.2.3. Nootropil (Piracetam)
2.2.4. E-2051
2.3. N-Methyl-D-Aspartate (NMDA) Receptor Antagonists
2.3.1. Selfotel
2.3.2. Gavestinel
2.3.3. Magnesium
2.4.GABA Agonists: Clomethiazole
2.5.Sodium Channel Blockers: Lubeluzole
2.6.Opioid Antagonists: Nalmefene
2.7. Membrane Stabilizers
2.7.1. Citicoline
2.7.2. DP-b99
2.8. Poly ADP-Ribose Polymerase (PARP) Inhibitors
2.8.1. ONO-2231
2.8.2. AX200 (Granulocyte Colony-Stimulating Factor (G-CSF))
2.9. Albumin
2.10. ONO-2506 (Proglia)
2.11. SUN-N4057 (Piclozotan)
2.12. TS-011
3. Immunosuppressants
3.1. FK506 (Tacrolimus)
3.2. Cyclosporin A
- Acute: TBI, SCI, stroke, and cerebral vasospasm following subarachnoid hemorrhage (SAH).
- Chronic: neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), Parkinson’s disease (PD), and Alzheimer’s disease (AD).
- Selective neuronal brain protection from radiotherapy for brain cancer.
4. Agents That Modulates/Inhibits PTP
4.1. CsA as a Neuroprotective Agent
- (1)
- CypD deficiency resulted in a significant reduction of hypoxic-ischemic brain injury in adult mice but worsened injury in neonates.
- (2)
- CypD-deficient cells also responded to various apoptotic stimuli in a manner similar to that of the WT, suggesting that CypD is not a central component of the apoptotic death pathway.
4.2. Other PTP Inhibitors
4.2.1. Pramipexole (Mirapex)
4.2.2. S-15176
5. CsA’s Effect on Transcriptional Regulation
5.1. Analysis of the Effects of CsA Administration on Gene Expression
- easy and cost-effective
- applicable when levels of starting material are minimal
- does not require prior knowledge of the transcriptomes
- can detect not only mRNA but also a variety of other RNA species (e.g., miRNA, siRNA)
- can obtain candidate cDNA clones during the analysis.
- Gene-chip is fast, simple, and shows more coverage
- Gene-chip does not need cloning or sequence steps.
5.2. Materials and Methods
5.2.1. CsA Administration
5.2.2. Tissue Preparation and RNA Preparation
5.2.3. cDNA Subtraction and Isolation of the Candidate Clones
5.3. Results of cDNA Subtraction Experiment
5.3.1. Significantly Upregulated Genes
5.3.2. Significantly Downregulated Genes (Group 1)
5.3.3. Significantly Downregulated Genes (Group 2)
5.4. Discussion
- CsA appears to affect the expression of many genes related to neuronal cell survival and regeneration.
- Many of the significantly upregulated genes identified in the present study are reported to be neurotrophic or have roles in the regeneration of damaged neurons.
- CsA reduces the expression of some genes that were reported to be detrimental to neuronal cells.
- CsA reduces the expression of certain genes that appear to be important for oxidative metabolism.
6. An Example of Mitochondria-Nuclear Communication; Prohibitin 1 (PHB1)
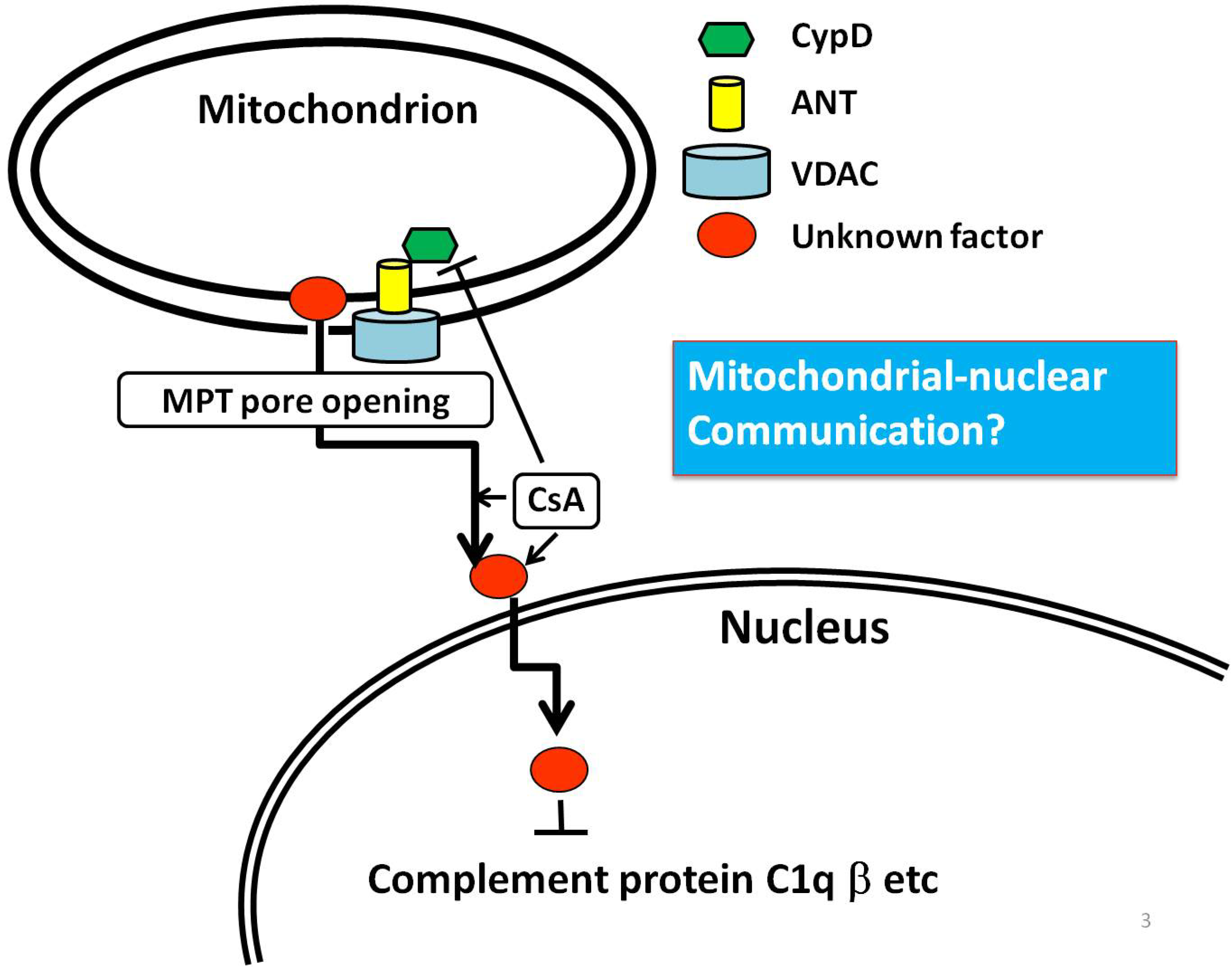
7. Conclusion
Abbreviations
| AD: | Alzheimer’s disease |
| ALS: | amyotrophic lateral sclerosis |
| ANT: | adenosine nucleotide translocase |
| BDNF: | brain-derived neurotrophic factor |
| CaM: | calmodulin |
| CCP: | classical complement pathway |
| CsA: | Cyclosporin A |
| CypD: | cyclophilin-D |
| G-CSF: | granulocyte-colony stimulating factor |
| HD: | Huntington’s disease |
| HETE: | hydroxyeicosatetraenoic acid |
| HIF-1: | hypoxia Inducible factor-1 |
| IL-1: | interleukin-1 |
| IL-2: | interleukin-2 |
| MCAO: | middle cerebral artery occlusion |
| MDH: | malate dehydrogenase |
| MPT: | mitochondrial membrane permeability transition |
| MS: | multiple sclerosis |
| NF-AT: | nuclear factor of activated T-cells |
| NMDA: | N-methyl-D-aspartate |
| PARP: | poly ADP-ribose polymerase |
| PD: | Parkinson’s disease |
| PGK-1: | phosphoglycerate kinase 1 |
| SAH: | subarachnoid hemorrhage |
| SCI: | spinal cord injury |
| STAT3: | signal transducers and activator of transcription 3 |
| TBI: | traumatic brain injury |
| VDAC: | voltage dependent anion channel |
| VDC: | voltage-dependent calcium channel |
Acknowledgments
Conflicts of Interest
References
- Katsura, K.; Suda, S.; Abe, A.; Kanamaru, T.; Toda, Y.; Katayama, Y. Brain protection therapy in acute cerebral infarction. J. Nippon Med. Sch. 2012, 79, 104–110. [Google Scholar] [CrossRef]
- Sutherland, B.A.; Minnerup, J.; Balami, J.S.; Arba, F.; Buchan, A.M.; Kleinschnitz, C. Neuroprotection for ischaemic stroke: Translation from the bench to the bedside. Int. J. Stroke 2012, 7, 407–418. [Google Scholar] [CrossRef]
- Widgerow, A.D. Ischemia-reperfusion injury: Influencing the microcirculatory and cellular environment. Ann. Plast. Surg. 2012. [Google Scholar] [CrossRef]
- Durukan, A.; Tatlisumak, T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol. Biochem. Behav. 2007, 87, 179–197. [Google Scholar] [CrossRef]
- Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc. Dis. 2003, 15, 222–229. [CrossRef]
- Shinohara, Y.; Saito, I.; Kobayashi, S.; Uchiyama, S. Edaravone (radical scavenger) versus sodium ozagrel (antiplatelet agent) in acute noncardioembolic ischemic stroke (edo trial). Cerebrovasc. Dis. 2009, 27, 485–492. [Google Scholar] [CrossRef]
- Watanabe, T.; Tanaka, M.; Watanabe, K.; Takamatsu, Y.; Tobe, A. Research and development of the free radical scavenger edaravone as a neuroprotectant. Yakugaku Zasshi 2004, 124, 99–111. [Google Scholar] [CrossRef]
- Higashi, Y.; Jitsuiki, D.; Chayama, K.; Yoshizumi, M. Edaravone (3-methyl-1-phenyl-2-pyrazolin-5-one), a novel free radical scavenger, for treatment of cardiovascular diseases. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 85–93. [Google Scholar]
- Yoshida, H.; Yanai, H.; Namiki, Y.; Fukatsu-Sasaki, K.; Furutani, N.; Tada, N. Neuroprotective effects of edaravone: A novel free radical scavenger in cerebrovascular injury. CNS Drug Rev. 2006, 12, 9–20. [Google Scholar] [CrossRef]
- Lees, K.R.; Zivin, J.A.; Ashwood, T.; Davalos, A.; Davis, S.M.; Diener, H.C.; Grotta, J.; Lyden, P.; Shuaib, A.; Hardemark, H.G.; et al. NXY-059 for acute ischemic stroke. N. Engl. J. Med. 2006, 354, 588–600. [Google Scholar] [CrossRef]
- Lees, K.R.; Davalos, A.; Davis, S.M.; Diener, H.C.; Grotta, J.; Lyden, P.; Shuaib, A.; Ashwood, T.; Hardemark, H.G.; Wasiewski, W.; et al. Additional outcomes and subgroup analyses of NXY-059 for acute ischemic stroke in the saint i trial. Stroke 2006, 37, 2970–2978. [Google Scholar] [CrossRef]
- Tirilazad international steering committee. Tirilazad mesylate in acute ischemic stroke: A systematic review. Stroke 2000, 31, 2257–2265. [CrossRef]
- Goto, F.; Takakura, K.; Shinohara, Y. Usefulness of avs (nicaraven) for acute stage of cerebrovascular disorder. Studies by multi-institutional double blind test. Rinsyo Kenkyu 1989, 66, 3577–3596. [Google Scholar]
- Sakai, F.; Goto, F.; Takakura, K. Kyuu-seiki nou syukketsu nou kousoku kanjya ni taisuru nicaraven no shiteki youryou no kentou. Kouki dai II sou nijyuu mouken shiken. (in Japanese). Igaku No Ayumi 1995, 175, 767–798. [Google Scholar]
- Minnerup, J.; Sutherland, B.A.; Buchan, A.M.; Kleinschnitz, C. Neuroprotection for stroke: Current status and future perspectives. Int. J. Mol. Sci. 2012, 13, 11753–11772. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sano, K.; Takakura, K.; Saito, I.; Shinohara, Y.; Asano, T.; Yasuhara, H. Ebselen in acute ischemic stroke: A placebo-controlled, double-blind clinical trial. Ebselen study group. Stroke 1998, 29, 12–17. [Google Scholar] [CrossRef]
- Yamaguchi, T. Stroke Trials Registry Home Page. Available online: http://www.strokecenter.org/trials/clinicalstudies/list?utf8=%E2%9C%93&filters[name_or_brief_summary_or_detailed_description_or_acronym_contains]=Ebselen&filters[overall_status_name_equals]=&filters[phase_equals]=&filters[clinicalstudies_registries_registry_id_equals]=&filters[conditions_id_equals_any]=&filters[interventions_id_equals_any]=&filters[studydesigns_allocation_equals]=&filters[studydesigns_primary_purpose_equals]=&filters[studydesigns_time_perspective_equals]=&filters[studydesigns_intervention_model_equals]=&filters[studydesigns_masking_equals]=&filters[studydesigns_observational_model_equals]=&filters[studydesigns_endpoint_classification_equals]=&filters[gender_equals]=&filters[results_available_equals]=0 (accessed on 28 January 2013).
- Akane, M.; Shimazawa, M.; Inokuchi, Y.; Tsuruma, K.; Hara, H. SUN N8075, a novel radical scavenger, protects against retinal cell death in mice. Neurosci. Lett. 2011, 488, 87–91. [Google Scholar] [CrossRef]
- Gelmers, H.J.; Gorter, K.; de Weerdt, C.J.; Wiezer, H.J. A controlled trial of nimodipine in acute ischemic stroke. N. Engl. J. Med. 1988, 318, 203–207. [Google Scholar] [CrossRef]
- Gelmers, H.J.; Hennerici, M. Effect of nimodipine on acute ischemic stroke. Pooled results from five randomized trials. Stroke 1990, 21, IV81–IV84. [Google Scholar]
- Horn, J.; Limburg, M. Calcium antagonists for ischemic stroke: A systematic review. Stroke 2001, 32, 570–576. [Google Scholar] [CrossRef]
- Candelise, L.; Ciccone, A. Gangliosides for acute ischemic stroke. Stroke 2002, 33, 2336. [Google Scholar] [CrossRef]
- Ricci, S.; Celani, M.G.; Cantisani, T.A.; Righetti, E. Piracetam for acute ischaemic stroke. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef]
- Katsura, K.; Katayama, Y. Kyoketsusei shinkei saiboushi no bunshi kikou to nou hogo ryouhou. Shinki nou hogoyaku he no kitai. (in Japanese). Nou Jyunkan Taisya 2006, 18, 73–77. [Google Scholar]
- Davis, S.M.; Lees, K.R.; Albers, G.W.; Diener, H.C.; Markabi, S.; Karlsson, G.; Norris, J. Selfotel in acute ischemic stroke: Possible neurotoxic effects of an NMDA antagonist. Stroke 2000, 31, 347–354. [Google Scholar] [CrossRef]
- Muir, K.W.; Lees, K.R. Excitatory amino acid antagonists for acute stroke. Cochrane Database Syst. Rev. 2003. [Google Scholar] [CrossRef]
- Lees, K.R.; Asplund, K.; Carolei, A.; Davis, S.M.; Diener, H.C.; Kaste, M.; Orgogozo, J.M.; Whitehead, J. Glycine antagonist (gavestinel) in neuroprotection (GAIN international) in patients with acute stroke: A randomised controlled trial. GAIN international investigators. Lancet 2000, 355, 1949–1954. [Google Scholar] [CrossRef]
- Sacco, R.L.; DeRosa, J.T.; Haley, E.C., Jr.; Levin, B.; Ordronneau, P.; Phillips, S.J.; Rundek, T.; Snipes, R.G.; Thompson, J.L. Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: A randomized controlled trial. JAMA 2001, 285, 1719–1728. [Google Scholar] [CrossRef]
- Singh, H.; Jalodia, S.; Gupta, M.S.; Talapatra, P.; Gupta, V.; Singh, I. Role of magnesium sulfate in neuroprotection in acute ischemic stroke. Ann. Indian Acad. Neurol. 2012, 15, 177–180. [Google Scholar] [CrossRef]
- Muir, K.W.; Lees, K.R.; Ford, I.; Davis, S. Magnesium for acute stroke (intravenous magnesium efficacy in stroke trial): Randomised controlled trial. Lancet 2004, 363, 439–445. [Google Scholar] [CrossRef]
- Lyden, P.; Shuaib, A.; Ng, K.; Levin, K.; Atkinson, R.P.; Rajput, A.; Wechsler, L.; Ashwood, T.; Claesson, L.; Odergren, T.; et al. Clomethiazole acute stroke study in ischemic stroke (CLASS-I): Final results. Stroke 2002, 33, 122–128. [Google Scholar] [CrossRef]
- Wahlgren, N.G.; Ranasinha, K.W.; Rosolacci, T.; Franke, C.L.; van Erven, P.M.; Ashwood, T.; Claesson, L. Clomethiazole acute stroke study (CLASS): Results of a randomized, controlled trial of clomethiazole versus placebo in 1360 acute stroke patients. Stroke 1999, 30, 21–28. [Google Scholar] [CrossRef]
- Diener, H.C. Multinational randomised controlled trial of lubeluzole in acute ischaemic stroke. European and australian lubeluzole ischaemic stroke study group. Cerebrovasc. Dis. 1998, 8, 172–181. [Google Scholar] [CrossRef]
- Grotta, J. Lubeluzole treatment of acute ischemic stroke. The US and Canadian Lubeluzole Ischemic Stroke Study Group. Stroke 1997, 28, 2338–2346. [Google Scholar] [CrossRef]
- Diener, H.C.; Cortens, M.; Ford, G.; Grotta, J.; Hacke, W.; Kaste, M.; Koudstaal, P.J.; Wessel, T. Lubeluzole in acute ischemic stroke treatment: A double-blind study with an 8-hour inclusion window comparing a 10-mg daily dose of lubeluzole with placebo. Stroke 2000, 31, 2543–2551. [Google Scholar] [CrossRef]
- Gandolfo, C.; Sandercock, P.; Conti, M. Lubeluzole for acute ischaemic stroke. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef]
- Clark, W.M.; Raps, E.C.; Tong, D.C.; Kelly, R.E. Cervene (Nalmefene) in acute ischemic stroke: Final results of a phase III efficacy study. The Cervene Stroke Study Investigators. Stroke 2000, 31, 1234–1239. [Google Scholar] [CrossRef]
- Zweifler, R.M. Membrane stabilizer: Citicoline. Curr. Med. Res. Opin. 2002, 18, s14–s17. [Google Scholar] [CrossRef]
- Davalos, A.; Alvarez-Sabin, J.; Castillo, J.; Diez-Tejedor, E.; Ferro, J.; Martinez-Vila, E.; Serena, J.; Segura, T.; Cruz, V.T.; Masjuan, J.; et al. Citicoline in the treatment of acute ischaemic stroke: An international, randomised, multicentre, placebo-controlled study (ICTUS trial). Lancet 2012, 380, 349–357. [Google Scholar] [CrossRef]
- Clark, W.M.; Williams, B.J.; Selzer, K.A.; Zweifler, R.M.; Sabounjian, L.A.; Gammans, R.E. A randomized efficacy trial of citicoline in patients with acute ischemic stroke. Stroke 1999, 30, 2592–2597. [Google Scholar] [CrossRef]
- Tazaki, Y.; Sakai, F.; Otomo, E.; Kutsuzawa, T.; Kameyama, M.; Omae, T.; Fujishima, M.; Sakuma, A. Treatment of acute cerebral infarction with a choline precursor in a multicenter double-blind placebo-controlled study. Stroke 1988, 19, 211–216. [Google Scholar] [CrossRef]
- Clark, W.M.; Wechsler, L.R.; Sabounjian, L.A.; Schwiderski, U.E. A phase III randomized efficacy trial of 2000 mg citicoline in acute ischemic stroke patients. Neurology 2001, 57, 1595–1602. [Google Scholar] [CrossRef]
- Diener, H.C.; Schneider, D.; Lampl, Y.; Bornstein, N.M.; Kozak, A.; Rosenberg, G. DP-b99, a membrane-activated metal ion chelator, as neuroprotective therapy in ischemic stroke. Stroke 2008, 39, 1774–1778. [Google Scholar] [CrossRef]
- Rosenberg, G.; Bornstein, N.; Diener, H.C.; Gorelick, P.B.; Shuaib, A.; Lees, K. The membrane-activated chelator stroke intervention (macsi) trial of DP-b99 in acute ischemic stroke: A randomized, double-blind, placebo-controlled, multinational pivotal phase III study. Int. J. Stroke 2011, 6, 362–367. [Google Scholar] [CrossRef]
- Jain, K. The Handbook of Neuroprotection; Humana Press: New York, NY, USA, 2011. [Google Scholar]
- Ringelstein, B.; Thjis, V.; Chamorro, A.; Grond, M.; Saver, J.; Norrving, B.; Aichner, F. New Drug Doesn’t Improve Disability among Stroke Patients. Available online: http://newsroom.heart.org/news/new-drug-doesn-t-improve-disability-228205 (accessed on 28 January 2013).
- Prajapati, K.D.; Sharma, S.S.; Roy, N. Current perspectives on potential role of albumin in neuroprotection. Rev. Neurosci. 2011, 22, 355–363. [Google Scholar]
- Ginsberg, M.D.; Hill, M.D.; Palesch, Y.Y.; Ryckborst, K.J.; Tamariz, D. The alias pilot trial: A dose-escalation and safety study of albumin therapy for acute ischemic stroke—I: Physiological responses and safety results. Stroke 2006, 37, 2100–2106. [Google Scholar] [CrossRef]
- Palesch, Y.Y.; Hill, M.D.; Ryckborst, K.J.; Tamariz, D.; Ginsberg, M.D. The alias pilot trial: A dose-escalation and safety study of albumin therapy for acute ischemic stroke—II: Neurologic outcome and efficacy analysis. Stroke 2006, 37, 2107–2114. [Google Scholar] [CrossRef]
- Ginsberg, M.D.; Palesch, Y.Y.; Martin, R.H.; Hill, M.D.; Moy, C.S.; Waldman, B.D.; Yeatts, S.D.; Tamariz, D.; Ryckborst, K.; Investigators, A. The albumin in acute stroke (ALIAS) multicenter clinical trial: Safety analysis of part 1 and rationale and design of part 2. Stroke 2011, 42, 119–127. [Google Scholar] [CrossRef]
- Hill, M.D.; Martin, R.H.; Palesch, Y.Y.; Tamariz, D.; Waldman, B.D.; Ryckborst, K.J.; Moy, C.S.; Barsan, W.G.; Ginsberg, M.D.; Investigators, A.; et al. The albumin in acute stroke part 1 trial: An exploratory efficacy analysis. Stroke 2011, 42, 1621–1625. [Google Scholar] [CrossRef]
- Tateishi, N.; Mori, T.; Kagamiishi, Y.; Satoh, S.; Katsube, N.; Morikawa, E.; Morimoto, T.; Matsui, T.; Asano, T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: Suppression of astrocytic activation by a novel agent (R)-(−)-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J. Cereb. Blood Flow Metab. 2002, 22, 723–724. [Google Scholar]
- Ono Pharmaceutical Co. Ltd. Announcement on Results of Phase II/III Clinical Study in Japan with Arocyte for Injection for Treatment of Acute Ischemic Stroke. Available online: http://www.ono.co.jp/eng/news/pdf/sm_cn081001.pdf (accessed on 28 January 2013).
- Kamei, K.; Maeda, N.; Ogino, R.; Koyama, M.; Nakajima, M.; Tatsuoka, T.; Ohno, T.; Inoue, T. New 5-HT1A receptor agonists possessing 1,4-benzoxazepine scaffold exhibit highly potent anti-ischemic effects. Bioorg. Med. Chem. Lett. 2001, 11, 595–598. [Google Scholar] [CrossRef]
- Miyata, N.; Roman, R.J. Role of 20-hydroxyeicosatetraenoic acid (20-HETE) in vascular system. J. Smooth Muscle Res. 2005, 41, 175–193. [Google Scholar] [CrossRef]
- Ganong, W.F. Review of Medical Physiology, 22nd ed.; McGraw-Hill: New York, NY, USA, 2003. [Google Scholar]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Sharkey, J.; Butcher, S.P. Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature 1994, 371, 336–339. [Google Scholar] [CrossRef]
- Arii, T.; Kamiya, T.; Arii, K.; Ueda, M.; Nito, C.; Katsura, K.I.; Katayama, Y. Neuroprotective effect of immunosuppressant FK506 in transient focal ischemia in rat: Therapeutic time window for FK506 in transient focal ischemia. Neurol. Res. 2001, 23, 755–760. [Google Scholar] [CrossRef]
- Katsura, K.; Kurihara, J.; Hiraide, T.; Takahashi, K.; Kato, H.; Katayama, Y. Effects of FK506 on the translocation of protein kinase C and CaM kinase Ii in the gerbil hippocampal CA1 neurons. Neurol. Res. 2003, 25, 522–527. [Google Scholar] [CrossRef]
- Katsura, K.; Takahashi, K.; Asoh, S.; Watanabe, M.; Sakurazawa, M.; Ohsawa, I.; Mori, T.; Igarashi, H.; Ohkubo, S.; Katayama, Y.; et al. Combination therapy with transductive anti-death FNK protein and FK506 ameliorates brain damage with focal transient ischemia in rat. J. Neurochem. 2008, 106, 258–270. [Google Scholar] [CrossRef]
- Gold, B.G. Fk506 and the role of immunophilins in nerve regeneration. Mol. Neurobiol. 1997, 15, 285–306. [Google Scholar] [CrossRef]
- Steiner, J.P.; Connolly, M.A.; Valentine, H.L.; Hamilton, G.S.; Dawson, T.M.; Hester, L.; Snyder, S.H. Neurotrophic actions of nonimmunosuppressive analogues of immunosuppressive drugs FK506, rapamycin and cyclosporin A. Nat. Med. 1997, 3, 421–428. [Google Scholar] [CrossRef]
- Furuichi, Y.; Noto, T.; Li, J.Y.; Oku, T.; Ishiye, M.; Moriguchi, A.; Aramori, I.; Matsuoka, N.; Mutoh, S.; Yanagihara, T. Multiple modes of action of tacrolimus (FK506) for neuroprotective action on ischemic damage after transient focal cerebral ischemia in rats. Brain Res. 2004, 1014, 120–130. [Google Scholar] [CrossRef]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of bad. Science 1999, 284, 339–343. [Google Scholar] [CrossRef]
- Yousuf, S.; Atif, F.; Kesherwani, V.; Agrawal, S.K. Neuroprotective effects of tacrolimus (FK-506) and cyclosporin (CSA) in oxidative injury. Brain Behav. 2011, 1, 87–94. [Google Scholar] [CrossRef]
- Zawadzka, M.; Kaminska, B. A novel mechanism of FK506-mediated neuroprotection: Downregulation of cytokine expression in glial cells. Glia 2005, 49, 36–51. [Google Scholar] [CrossRef]
- Gold, B.G.; Voda, J.; Yu, X.; McKeon, G.; Bourdette, D.N. FK506 and a nonimmunosuppressant derivative reduce axonal and myelin damage in experimental autoimmune encephalomyelitis: Neuroimmunophilin ligand-mediated neuroprotection in a model of multiple sclerosis. J. Neurosci. Res. 2004, 77, 367–377. [Google Scholar] [CrossRef]
- Laupacis, A.; Keown, P.A.; Ulan, R.A.; McKenzie, N.; Stiller, C.R. Cyclosporin A: A powerful immunosuppressant. Can. Med. Assoc. J. 1982, 126, 1041–1046. [Google Scholar]
- Li, P.A.; Uchino, H.; Elmer, E.; Siesjo, B.K. Amelioration by cyclosporin A of brain damage following 5 or 10 min of ischemia in rats subjected to preischemic hyperglycemia. Brain Res. 1997, 753, 133–140. [Google Scholar] [CrossRef]
- Uchino, H.; Elmer, E.; Uchino, K.; Lindvall, O.; Siesjo, B.K. Cyclosporin A dramatically ameliorates CA1 hippocampal damage following transient forebrain ischaemia in the rat. Acta Physiol. Scand. 1995, 155, 469–471. [Google Scholar] [CrossRef]
- Uchino, H.; Elmer, E.; Uchino, K.; Li, P.A.; He, Q.P.; Smith, M.L.; Siesjo, B.K. Amelioration by cyclosporin A of brain damage in transient forebrain ischemia in the rat. Brain Res. 1998, 812, 216–226. [Google Scholar] [CrossRef]
- Mirzayan, M.J.; Klinge, P.M.; Ude, S.; Hotop, A.; Samii, M.; Brinker, T.; Korkmaz, Z.; Meyer, G.J.; Knapp, W.H.; Samii, A. Modified calcium accumulation after controlled cortical impact under cyclosporin A treatment: A 45Ca autoradiographic study. Neurol. Res. 2008, 30, 476–479. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef]
- Mbye, L.H.; Singh, I.N.; Carrico, K.M.; Saatman, K.E.; Hall, E.D. Comparative neuroprotective effects of cyclosporin A and NIM811, a nonimmunosuppressive cyclosporin A analog, following traumatic brain injury. J. Cereb. Blood Flow Metab. 2009, 29, 87–97. [Google Scholar] [CrossRef]
- Signoretti, S.; Marmarou, A.; Tavazzi, B.; Dunbar, J.; Amorini, A.M.; Lazzarino, G.; Vagnozzi, R. The protective effect of cyclosporin A upon N-acetylaspartate and mitochondrial dysfunction following experimental diffuse traumatic brain injury. J. Neurotrauma 2004, 21, 1154–1167. [Google Scholar]
- Van Den Heuvel, C.; Donkin, J.J.; Finnie, J.W.; Blumbergs, P.C.; Kuchel, T.; Koszyca, B.; Manavis, J.; Jones, N.R.; Reilly, P.L.; Vink, R. Downregulation of amyloid precursor protein (APP) expression following post-traumatic cyclosporin-A administration. J. Neurotrauma 2004, 21, 1562–1572. [Google Scholar]
- Okonkwo, D.O.; Melon, D.E.; Pellicane, A.J.; Mutlu, L.K.; Rubin, D.G.; Stone, J.R.; Helm, G.A. Dose-response of cyclosporin A in attenuating traumatic axonal injury in rat. Neuroreport 2003, 14, 463–466. [Google Scholar]
- Mazzeo, A.T.; Beat, A.; Singh, A.; Bullock, M.R. The role of mitochondrial transition pore, and its modulation, in traumatic brain injury and delayed neurodegeneration after TBI. Exp. Neurol. 2009, 218, 363–370. [Google Scholar] [CrossRef]
- Hatton, J.; Rosbolt, B.; Empey, P.; Kryscio, R.; Young, B. Dosing and safety of cyclosporine in patients with severe brain injury. J. Neurosurg. 2008, 109, 699–707. [Google Scholar] [CrossRef]
- Mazzeo, A.T.; Alves, O.L.; Gilman, C.B.; Hayes, R.L.; Tolias, C.; Niki Kunene, K.; Ross Bullock, M. Brain metabolic and hemodynamic effects of cyclosporin A after human severe traumatic brain injury: A microdialysis study. Acta Neurochir. 2008, 150, 1019–1031, discussion 1031. [Google Scholar] [CrossRef]
- Nighoghossian, N. Neuroprotection Impact of Cyclosporin A in Cerebral Infarction (Csastroke). Available online: http://clinicaltrials.gov/show/NCT01527240 (accessed on 28 January 2013).
- Cho, B.B.; Toledo-Pereyra, L.H. Caspase-independent programmed cell death following ischemic stroke. J. Invest. Surg. 2008, 21, 141–147. [Google Scholar] [CrossRef]
- Yakel, J.L. Calcineurin regulation of synaptic function: From ion channels to transmitter release and gene transcription. Trends Pharmacol. Sci. 1997, 18, 124–134. [Google Scholar] [CrossRef]
- Morioka, M.; Hamada, J.; Ushio, Y.; Miyamoto, E. Potential role of calcineurin for brain ischemia and traumatic injury. Prog. Neurobiol. 1999, 58, 1–30. [Google Scholar] [CrossRef]
- Clemens, J.A. Cerebral ischemia: Gene activation, neuronal injury, and the protective role of antioxidants. Free Radic. Biol. Med. 2000, 28, 1526–1531. [Google Scholar] [CrossRef]
- Shibasaki, F.; Kondo, E.; Akagi, T.; McKeon, F. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature 1997, 386, 728–731. [Google Scholar] [CrossRef]
- Uchino, H.; Kuroda, Y.; Morota, S.; Hirabayashi, G.; Ishii, N.; Shibasaki, F.; Ikeda, Y.; Hansson, M.J.; Elmer, E. Probing the molecular mechanisms of neuronal degeneration: Importance of mitochondrial dysfunction and calcineurin activation. J. Anesth. 2008, 22, 253–262. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Uchino, H.; He, Q.P.; Li, P.A.; Siesjo, B.K. Cyclosporin A, but not FK506, prevents the downregulation of phosphorylated Akt after transient focal ischemia in the rat. Brain Res. 2001, 899, 148–158. [Google Scholar] [CrossRef]
- Wang, X.; Carlsson, Y.; Basso, E.; Zhu, C.; Rousset, C.I.; Rasola, A.; Johansson, B.R.; Blomgren, K.; Mallard, C.; Bernardi, P.; et al. Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci. 2009, 29, 2588–2596. [Google Scholar] [CrossRef]
- Hubble, J.P. Pre-clinical studies of pramipexole: Clinical relevance. Eur. J. Neurol. 2000, 7, 15–20. [Google Scholar] [CrossRef]
- Abramova, N.A.; Cassarino, D.S.; Khan, S.M.; Painter, T.W.; Bennett, J.P., Jr. Inhibition by R(+) or S(−) pramipexole of caspase activation and cell death induced by methylpyridinium ion or beta amyloid peptide in SH-SY5Y neuroblastoma. J. Neurosci. Res. 2002, 67, 494–500. [Google Scholar] [CrossRef]
- Danzeisen, R.; Schwalenstoecker, B.; Gillardon, F.; Buerger, E.; Krzykalla, V.; Klinder, K.; Schild, L.; Hengerer, B.; Ludolph, A.C.; Dorner-Ciossek, C.; Kussmaul, L. Targeted antioxidative and neuroprotective properties of the dopamine agonist pramipexole and its nondopaminergic enantiomer SND919CL2x [(+)2-amino-4,5,6,7-tetrahydro-6-Lpropylamino-benzathiazole dihydrochloride]. J. Pharmacol. Exp. Ther. 2006, 316, 189–199. [Google Scholar]
- Sayeed, I.; Parvez, S.; Winkler-Stuck, K.; Seitz, G.; Trieu, I.; Wallesch, C.W.; Schönfeld, P.; Siemen, D. Patch clamp reveals powerful blockade of the mitochondrial permeability transition pore by the D2-receptor agonist pramipexole. FASEB J. 2006, 20, 556–558. [Google Scholar]
- Ferrari-Toninelli, G.; Maccarinelli, G.; Uberti, D.; Buerger, E.; Memo, M. Mitochondria-targeted antioxidant effects of S(−) and R(+) pramipexole. BMC Pharmacol. 2010, 10. [Google Scholar] [CrossRef]
- Settaf, A.; Zahidy, M.; Elimadi, A.; Sapena, R.; Alsamad, I.A.; Tillement, J.; Morin, D. S-15176 reduces the hepatic injury in rats subjected to experimental ischemia and reperfusion. Eur. J. Pharmacol. 2000, 406, 281–292. [Google Scholar] [CrossRef]
- Rupp, H.; Zarain-Herzberg, A.; Maisch, B. The use of partial fatty acid oxidation inhibitors for metabolic therapy of angina pectoris and heart failure. Herz 2002, 27, 621–636. [Google Scholar] [CrossRef]
- Elimadi, A.; Jullien, V.; Tillement, J.P.; Morin, D. S-15176 inhibits mitochondrial permeability transition via a mechanism independent of its antioxidant properties. Eur. J. Pharmacol. 2003, 468, 93–101. [Google Scholar] [CrossRef]
- Miyata, K.; Omori, N.; Uchino, H.; Yamaguchi, T.; Isshiki, A.; Shibasaki, F. Involvement of the brain-derived neurotrophic factor/TrkB pathway in neuroprotecive effect of cyclosporin A in forebrain ischemia. Neuroscience 2001, 105, 571–578. [Google Scholar] [CrossRef]
- Kawakami, M.; Yoshimoto, T.; Nakagata, N.; Yamamura, K.; Siesjo, B.K. Effects of cyclosporin A administration on gene expression in rat brain. Brain Inj. 2011, 25, 614–623. [Google Scholar] [CrossRef]
- Diatchenko, L.; Lau, Y.F.; Campbell, A.P.; Chenchik, A.; Moqadam, F.; Huang, B.; Lukyanov, S.; Lukyanov, K.; Gurskaya, N.; Sverdlov, E.D.; et al. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cdna probes and libraries. Proc. Natl. Acad. Sci. USA 1996, 93, 6025–6030. [Google Scholar] [CrossRef]
- Mathur, S.; Cleary, K.R.; Inamdar, N.; Kim, Y.H.; Steck, P.; Frazier, M.L. Overexpression of elongation factor-1gamma protein in colorectal carcinoma. Cancer 1998, 82, 816–821. [Google Scholar] [CrossRef]
- Cavallius, J.; Rattan, S.I.; Clark, B.F. Changes in activity and amount of active elongation factor 1 alpha in aging and immortal human fibroblast cultures. Exp. Gerontol. 1986, 21, 149–157. [Google Scholar] [CrossRef]
- Shepherd, J.C.; Walldorf, U.; Hug, P.; Gehring, W.J. Fruit flies with additional expression of the elongation factor EF-1 alpha live longer. Proc. Natl. Acad. Sci. USA 1989, 86, 7520–7521. [Google Scholar] [CrossRef]
- Wang, J.F.; Engelsberg, B.N.; Johnson, S.W.; Witmer, C.; Merrick, W.C.; Rozmiarek, H.; Billings, P.C. DNA binding activity of the mammalian translation elongation complex: Recognition of chromium- and transplatin-damaged DNA. Arch. Toxicol. 1997, 71, 450–454. [Google Scholar] [CrossRef]
- Jung, M.; Kondratyev, A.D.; Dritschilo, A. Elongation factor 1 delta is enhanced following exposure to ionizing radiation. Cancer Res. 1994, 54, 2541–2543. [Google Scholar]
- DeWille, J.W.; Farmer, S.J. Quaking phenotype influences brain lipid-related mRNA levels. Neurosci. Lett. 1992, 141, 195–198. [Google Scholar] [CrossRef]
- DeWille, J.W.; Farmer, S.J. Postnatal dietary fat influences mRNAs involved in myelination. Dev. Neurosci. 1992, 14, 61–68. [Google Scholar] [CrossRef]
- Zumkeller, W. The effect of insulin-like growth factors on brain myelination and their potential therapeutic application in myelination disorders. Eur. J. Paediatr. Neurol. 1997, 1, 91–101. [Google Scholar] [CrossRef]
- Kubota, H.; Yokota, S.; Yanagi, H.; Yura, T. Structures and co-regulated expression of the genes encoding mouse cytosolic chaperonin CCT subunits. Eur. J. Biochem. 1999, 262, 492–500. [Google Scholar] [CrossRef]
- Stoldt, V.; Rademacher, F.; Kehren, V.; Ernst, J.F.; Pearce, D.A.; Sherman, F. Review: The CCT eukaryotic chaperonin subunits of saccharomyces cerevisiae and other yeasts. Yeast 1996, 12, 523–529. [Google Scholar] [CrossRef]
- Creutz, C.E.; Liou, A.; Snyder, S.L.; Brownawell, A.; Willison, K. Identification of the major chromaffin granule-binding protein, chromobindin A, as the cytosolic chaperonin CCT (chaperonin containing TCP-1). J. Biol. Chem. 1994, 269, 32035–32038. [Google Scholar]
- Roobol, A.; Holmes, F.E.; Hayes, N.V.; Baines, A.J.; Carden, M.J. Cytoplasmic chaperonin complexes enter neurites developing in vitro and differ in subunit composition within single cells. J. Cell Sci. 1995, 108, 1477–1488. [Google Scholar]
- Bloom, L.; Horvitz, H.R. The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc. Natl. Acad. Sci. USA 1997, 94, 3414–3419. [Google Scholar] [CrossRef]
- Kuroda, S.; Nakagawa, N.; Tokunaga, C.; Tatematsu, K.; Tanizawa, K. Mammalian homologue of the Caenorhabditis elegans unc-76 protein involved in axonal outgrowth is a protein kinase C zeta-interacting protein. J. Cell Biol. 1999, 144, 403–411. [Google Scholar] [CrossRef]
- Aitken, A. 14-3-3 proteins on the map. Trends Biochem. Sci. 1995, 20, 95–97. [Google Scholar] [CrossRef]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996, 87, 619–628. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, J.; Fu, H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 8511–8515. [Google Scholar] [CrossRef]
- Hara, H.; Friedlander, R.M.; Gagliardini, V.; Ayata, C.; Fink, K.; Huang, Z.; Shimizu-Sasamata, M.; Yuan, J.; Moskowitz, M.A. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 2007–2012. [Google Scholar] [CrossRef]
- Yaoita, H.; Ogawa, K.; Maehara, K.; Maruyama, Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 1998, 97, 276–281. [Google Scholar] [CrossRef]
- Cheng, Y.; Deshmukh, M.; D’Costa, A.; Demaro, J.A.; Gidday, J.M.; Shah, A.; Sun, Y.; Jacquin, M.F.; Johnson, E.M.; Holtzman, D.M. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J. Clin. Invest. 1998, 101, 1992–1999. [Google Scholar] [CrossRef]
- Chen, J.; Nagayama, T.; Jin, K.; Stetler, R.A.; Zhu, R.L.; Graham, S.H.; Simon, R.P. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J. Neurosci. 1998, 18, 4914–4928. [Google Scholar]
- Rabuffetti, M.; Sciorati, C.; Tarozzo, G.; Clementi, E.; Manfredi, A.A.; Beltramo, M. Inhibition of caspase-1-like activity by Ac-Tyr-Val-Ala-Asp-chloromethyl ketone induces long-lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J. Neurosci. 2000, 20, 4398–4404. [Google Scholar]
- Martinou, J.C.; Dubois-Dauphin, M.; Staple, J.K.; Rodriguez, I.; Frankowski, H.; Missotten, M.; Albertini, P.; Talabot, D.; Catsicas, S.; Pietra, C.; et al. Overexpression of Bcl-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 1994, 13, 1017–1030. [Google Scholar] [CrossRef]
- Wiessner, C.; Allegrini, P.R.; Rupalla, K.; Sauer, D.; Oltersdorf, T.; McGregor, A.L.; Bischoff, S.; Bottiger, B.W.; van der Putten, H. Neuron-specific transgene expression of Bcl-XL but not Bcl-2 genes reduced lesion size after permanent middle cerebral artery occlusion in mice. Neurosci. Lett. 1999, 268, 119–122. [Google Scholar] [CrossRef]
- Xing, H.; Zhang, S.; Weinheimer, C.; Kovacs, A.; Muslin, A.J. 14-3-3 proteins block apoptosis and differentially regulate MAPK cascades. EMBO J. 2000, 19, 349–358. [Google Scholar] [CrossRef]
- Landgraf, P.; Wahle, P.; Pape, H.C.; Gundelfinger, E.D.; Kreutz, M.R. The survival-promoting peptide Y-P30 enhances binding of pleiotrophin to syndecan-2 and -3 and supports its neuritogenic activity. J. Biol. Chem. 2008, 283, 25036–25045. [Google Scholar] [CrossRef]
- Landgraf, P.; Sieg, F.; Wahle, P.; Meyer, G.; Kreutz, M.R.; Pape, H.C. A maternal blood-borne factor promotes survival of the developing thalamus. FASEB J. 2005, 19, 225–227. [Google Scholar]
- Zhao, Y.; Lang, G.; Ito, S.; Bonnet, J.; Metzger, E.; Sawatsubashi, S.; Suzuki, E.; Le Guezennec, X.; Stunnenberg, H.G.; Krasnov, A.; et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Mol. Cell 2008, 29, 92–101. [Google Scholar] [CrossRef]
- Nagy, Z.; Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007, 26, 5341–5357. [Google Scholar] [CrossRef]
- Leng, N.; Gu, G.; Simerly, R.B.; Spindel, E.R. Molecular cloning and characterization of two putative g protein-coupled receptors which are highly expressed in the central nervous system. Brain Res. Mol. Brain Res. 1999, 69, 73–83. [Google Scholar] [CrossRef]
- Marazziti, D.; Gallo, A.; Golini, E.; Matteoni, R.; Tocchini-Valentini, G.P. Molecular cloning and chromosomal localization of the mouse Gpr37 gene encoding an orphan G-protein-coupled peptide receptor expressed in brain and testis. Genomics 1998, 53, 315–324. [Google Scholar] [CrossRef]
- Rezgaoui, M.; Susens, U.; Ignatov, A.; Gelderblom, M.; Glassmeier, G.; Franke, I.; Urny, J.; Imai, Y.; Takahashi, R.; Schaller, H.C. The neuropeptide head activator is a high-affinity ligand for the orphan G-protein-coupled receptor Gpr37. J. Cell Sci. 2006, 119, 542–549. [Google Scholar] [CrossRef]
- Bodenmuller, H.; Schaller, H.C. Conserved amino acid sequence of a neuropeptide, the head activator, from coelenterates to humans. Nature 1981, 293, 579–580. [Google Scholar] [CrossRef]
- Berman, S.B.; Hastings, T.G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: Implications for parkinson’s disease. J. Neurochem. 1999, 73, 1127–1137. [Google Scholar] [CrossRef]
- Famulski, J.K.; Vos, L.; Sun, X.; Chan, G. Stable hZW10 kinetochore residency, mediated by hZwint-1 interaction, is essential for the mitotic checkpoint. J. Cell Biol. 2008, 180, 507–520. [Google Scholar] [CrossRef]
- Van Vlijmen, T.; Vleugel, M.; Evers, M.; Mohammed, S.; Wulf, P.S.; Heck, A.J.; Hoogenraad, C.C.; van der Sluijs, P. A unique residue in rab3c determines the interaction with novel binding protein Zwint-1. FEBS Lett. 2008, 582, 2838–2842. [Google Scholar] [CrossRef]
- Rogers, J.; Cooper, N.R.; Webster, S.; Schultz, J.; McGeer, P.L.; Styren, S.D.; Civin, W.H.; Brachova, L.; Bradt, B.; Ward, P.; et al. Complement activation by beta-amyloid in alzheimer disease. Proc. Natl. Acad. Sci. USA 1992, 89, 10016–10020. [Google Scholar] [CrossRef]
- Jiang, H.; Burdick, D.; Glabe, C.G.; Cotman, C.W.; Tenner, A.J. Beta-amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q a chain. J. Immunol. 1994, 152, 5050–5059. [Google Scholar]
- Afagh, A.; Cummings, B.J.; Cribbs, D.H.; Cotman, C.W.; Tenner, A.J. Localization and cell association of C1q in Alzheimer’s disease brain. Exp. Neurol. 1996, 138, 22–32. [Google Scholar] [CrossRef]
- Yao, J.; Harvath, L.; Gilbert, D.L.; Colton, C.A. Chemotaxis by a CNS macrophage, the microglia. J. Neurosci. Res. 1990, 27, 36–42. [Google Scholar] [CrossRef]
- O’Barr, S.; Cooper, N.R. The C5a complement activation peptide increases IL-1beta and IL-6 release from amyloid-beta primed human monocytes: Implications for Alzheimer’s disease. J. Neuroimmunol. 2000, 109, 87–94. [Google Scholar] [CrossRef]
- Benveniste, E.N.; Nguyen, V.T.; O’Keefe, G.M. Immunological aspects of microglia: Relevance to Alzheimer’s disease. Neurochem. Int. 2001, 39, 381–391. [Google Scholar] [CrossRef]
- Tenner, A.J. Complement in Alzheimer’s disease: Opportunities for modulating protective and pathogenic events. Neurobiol. Aging 2001, 22, 849–861. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Zhou, J.; Botto, M.; Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004, 24, 6457–6465. [Google Scholar] [CrossRef]
- Pedersen, E.D.; Waje-Andreassen, U.; Vedeler, C.A.; Aamodt, G.; Mollnes, T.E. Systemic complement activation following human acute ischaemic stroke. Clin. Exp. Immunol. 2004, 137, 117–122. [Google Scholar] [CrossRef]
- Arumugam, T.V.; Magnus, T.; Woodruff, T.M.; Proctor, L.M.; Shiels, I.A.; Taylor, S.M. Complement mediators in ischemia-reperfusion injury. Clin. Chim. Acta 2006, 374, 33–45. [Google Scholar] [CrossRef]
- Ten, V.S.; Sosunov, S.A.; Mazer, S.P.; Stark, R.I.; Caspersen, C.; Sughrue, M.E.; Botto, M.; Connolly, E.S., Jr.; Pinsky, D.J. C1q-deficiency is neuroprotective against hypoxic-ischemic brain injury in neonatal mice. Stroke 2005, 36, 2244–2250. [Google Scholar] [CrossRef]
- Ten, V.S.; Yao, J.; Ratner, V.; Sosunov, S.; Fraser, D.A.; Botto, M.; Sivasankar, B.; Morgan, B.P.; Silverstein, S.; Stark, R.; et al. Complement component C1q mediates mitochondria-driven oxidative stress in neonatal hypoxic-ischemic brain injury. J. Neurosci. 2010, 30, 2077–2087. [Google Scholar] [CrossRef]
- Girod, A.; Storrie, B.; Simpson, J.C.; Johannes, L.; Goud, B.; Roberts, L.M.; Lord, J.M.; Nilsson, T.; Pepperkok, R. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat. Cell Biol. 1999, 1, 423–430. [Google Scholar] [CrossRef]
- White, J.; Johannes, L.; Mallard, F.; Girod, A.; Grill, S.; Reinsch, S.; Keller, P.; Tzschaschel, B.; Echard, A.; Goud, B.; et al. Rab6 coordinates a novel Golgi to er retrograde transport pathway in live cells. J. Cell Biol. 1999, 147, 743–760. [Google Scholar] [CrossRef]
- McConlogue, L.; Castellano, F.; deWit, C.; Schenk, D.; Maltese, W.A. Differential effects of a Rab6 mutant on secretory versus amyloidogenic processing of Alzheimer’s beta-amyloid precursor protein. J. Biol. Chem. 1996, 271, 1343–1348. [Google Scholar] [CrossRef]
- Scheper, W.; Hoozemans, J.J.; Hoogenraad, C.C.; Rozemuller, A.J.; Eikelenboom, P.; Baas, F. Rab6 is increased in Alzheimer’s disease brain and correlates with endoplasmic reticulum stress. Neuropathol. Appl. Neurobiol. 2007, 33, 523–532. [Google Scholar]
- Lin, L.; Jeanclos, E.M.; Treuil, M.; Braunewell, K.H.; Gundelfinger, E.D.; Anand, R. The calcium sensor protein visinin-like protein-1 modulates the surface expression and agonist sensitivity of the alpha 4beta 2 nicotinic acetylcholine receptor. J. Biol. Chem. 2002, 277, 41872–41878. [Google Scholar]
- Schnurra, I.; Bernstein, H.G.; Riederer, P.; Braunewell, K.H. The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: A link between calcium sensors and Alzheimer’s disease? Neurobiol. Dis. 2001, 8, 900–909. [Google Scholar] [CrossRef]
- Mali, Y.; Zisapels, N. Gain of interaction of ALS-linked G93A superoxide dismutase with cytosolic malate dehydrogenase. Neurobiol. Dis. 2008, 32, 133–141. [Google Scholar] [CrossRef]
- Volkert, M.R.; Elliott, N.A.; Housman, D.E. Functional genomics reveals a family of eukaryotic oxidation protection genes. Proc. Natl. Acad. Sci. USA 2000, 97, 14530–14535. [Google Scholar] [CrossRef]
- Natoli, R.; Provis, J.; Valter, K.; Stone, J. Expression and role of the early-response gene Oxr1 in the hyperoxia-challenged mouse retina. Invest. Ophthalmol. Vis. Sci. 2008, 49, 4561–4567. [Google Scholar] [CrossRef]
- Inouye, S.; Seo, M.; Yamada, Y.; Nakazawa, A. Increase of adenylate kinase isozyme 1 protein during neuronal differentiation in mouse embryonal carcinoma p19 cells and in rat brain primary cultured cells. J. Neurochem. 1998, 71, 125–133. [Google Scholar]
- O’Rourke, J.F.; Pugh, C.W.; Bartlett, S.M.; Ratcliffe, P.J. Identification of hypoxically inducible mRNAs in HeLa cells using differential-display PCR. Role of hypoxia-inducible factor-1. Eur. J. Biochem. 1996, 241, 403–410. [Google Scholar] [CrossRef]
- Shahjahan, M.; Yamada, M.; Tanaka, H.; Nakazawa, A. Cloning and characterization of the gene encoding bovine mitochondrial adenylate kinase isozyme 3. Gene 1991, 107, 313–317. [Google Scholar] [CrossRef]
- Popanda, O.; Fox, G.; Thielmann, H.W. Modulation of DNA polymerases alpha, delta and epsilon by lactate dehydrogenase and 3-phosphoglycerate kinase. Biochim. Biophys. Acta 1998, 1397, 102–117. [Google Scholar]
- Ogino, T.; Iwama, M.; Kinouchi, J.; Shibagaki, Y.; Tsukamoto, T.; Mizumoto, K. Involvement of a cellular glycolytic enzyme, phosphoglycerate kinase, in sendai virus transcription. J. Biol. Chem. 1999, 274, 35999–36008. [Google Scholar]
- Shetty, S.; Muniyappa, H.; Halady, P.K.; Idell, S. Regulation of urokinase receptor expression by phosphoglycerate kinase. Am. J. Respir. Cell Mol. Biol. 2004, 31, 100–106. [Google Scholar] [CrossRef]
- Lay, A.J.; Jiang, X.M.; Kisker, O.; Flynn, E.; Underwood, A.; Condron, R.; Hogg, P.J. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature 2000, 408, 869–873. [Google Scholar] [CrossRef]
- Gold, B.G.; Gordon, H.S.; Wang, M.S. Efficacy of delayed or discontinuous FK506 administrations on nerve regeneration in the rat sciatic nerve crush model: Lack of evidence for a conditioning lesion-like effect. Neurosci. Lett. 1999, 267, 33–36. [Google Scholar] [CrossRef]
- Friberg, H.; Ferrand-Drake, M.; Bengtsson, F.; Halestrap, A.P.; Wieloch, T. Cyclosporin A, but not FK506, protects mitochondria and neurons against hypoglycemic damage and implicates the mitochondrial permeability transition in cell death. J. Neurosci. 1998, 18, 5151–5159. [Google Scholar]
- Nicolli, A.; Basso, E.; Petronilli, V.; Wenger, R.M.; Bernardi, P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, and cyclosporin A-sensitive channel. J. Biol. Chem. 1996, 271, 2185–2192. [Google Scholar] [CrossRef]
- Baumann, G.; Zenke, G.; Wenger, R.; Hiestand, P.; Quesniaux, V.; Andersen, E.; Schreier, M.H. Molecular mechanisms of immunosuppression. J. Autoimmun. 1992, 5, 67–72. [Google Scholar] [CrossRef]
- Hansson, M.J.; Mattiasson, G.; Mansson, R.; Karlsson, J.; Keep, M.F.; Waldmeier, P.; Ruegg, U.T.; Dumont, J.M.; Besseghir, K.; Elmer, E. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 2004, 36, 407–413. [Google Scholar] [CrossRef]
- Tiepolo, T.; Angelin, A.; Palma, E.; Sabatelli, P.; Merlini, L.; Nicolosi, L.; Finetti, F.; Braghetta, P.; Vuagniaux, G.; Dumont, J.M.; et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1−/− myopathic mice. Br. J. Pharmacol. 2009, 157, 1045–1052. [Google Scholar] [CrossRef]
- Giorgio, V.; Soriano, M.E.; Basso, E.; Bisetto, E.; Lippe, G.; Forte, M.A.; Bernardi, P. Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 2010, 1797, 1113–1118. [Google Scholar]
- Theiss, A.L.; Sitaraman, S.V. The role and therapeutic potential of prohibitin in disease. Biochim. Biophys. Acta 2011, 1813, 1137–1143. [Google Scholar] [CrossRef]
- Altus, M.S.; Wood, C.M.; Stewart, D.A.; Roskams, A.J.; Friedman, V.; Henderson, T.; Owens, G.A.; Danner, D.B.; Jupe, E.R.; Dell’Orco, R.T.; et al. Regions of evolutionary conservation between the rat and human prohibitin-encoding genes. Gene 1995, 158, 291–294. [Google Scholar] [CrossRef]
- Nijtmans, L.G.; de Jong, L.; Artal Sanz, M.; Coates, P.J.; Berden, J.A.; Back, J.W.; Muijsers, A.O.; van der Spek, H.; Grivell, L.A. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000, 19, 2444–2451. [Google Scholar] [CrossRef]
- Sharma, A.; Qadri, A. VI polysaccharide of Salmonella typhi targets the prohibitin family of molecules in intestinal epithelial cells and suppresses early inflammatory responses. Proc. Natl. Acad. Sci. USA 2004, 101, 17492–17497. [Google Scholar] [CrossRef]
- Rastogi, S.; Joshi, B.; Fusaro, G.; Chellappan, S. Camptothecin induces nuclear export of prohibitin preferentially in transformed cells through a CRM-1-dependent mechanism. J. Biol. Chem. 2006, 281, 2951–2959. [Google Scholar]
- Wang, S.; Fusaro, G.; Padmanabhan, J.; Chellappan, S.P. Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene 2002, 21, 8388–8396. [Google Scholar] [CrossRef]
- Sripathi, S.R.; He, W.; Atkinson, C.L.; Smith, J.J.; Liu, Z.; Elledge, B.M.; Jahng, W.J. Mitochondrial-nuclear communication by prohibitin shuttling under oxidative stress. Biochemistry 2011, 50, 8342–8351. [Google Scholar] [CrossRef]
- Sanchez-Quiles, V.; Santamaria, E.; Segura, V.; Sesma, L.; Prieto, J.; Corrales, F.J. Prohibitin deficiency blocks proliferation and induces apoptosis in human hepatoma cells: Molecular mechanisms and functional implications. Proteomics 2010, 10, 1609–1620. [Google Scholar] [CrossRef]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1,026 experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [CrossRef]
- Seaton, T.A.; Cooper, J.M.; Schapira, A.H. Cyclosporin inhibition of apoptosis induced by mitochondrial complex I toxins. Brain Res. 1998, 809, 12–17. [Google Scholar] [CrossRef]
- Tatton, W.G.; Chalmers-Redman, R.M. Mitochondria in neurodegenerative apoptosis: An opportunity for therapy? Ann. Neurol. 1998, 44, S134–S141. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Swerdlow, R.H.; Parks, J.K.; Parker, W.D., Jr.; Bennett, J.P., Jr. . Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer’s disease cybrids. Biochem. Biophys. Res. Commun. 1998, 248, 168–173. [Google Scholar] [CrossRef]
- Wong, A.; Cortopassi, G. Mtdna mutations confer cellular sensitivity to oxidant stress that is partially rescued by calcium depletion and cyclosporin A. Biochem. Biophys. Res. Commun. 1997, 239, 139–145. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Lai, M.M.; Snyder, S.H. Neural roles of immunophilins and their ligands. Mol. Neurobiol. 1997, 15, 223–239. [Google Scholar] [CrossRef]
- Miller, R.G.; Sharma, K.R.; Pavlath, G.K.; Gussoni, E.; Mynhier, M.; Lanctot, A.M.; Greco, C.M.; Steinman, L.; Blau, H.M. Myoblast implantation in duchenne muscular dystrophy: The San Francisco study. Muscle Nerve 1997, 20, 469–478. [Google Scholar] [CrossRef]
- Sharma, K.R.; Mynhier, M.A.; Miller, R.G. Cyclosporine increases muscular force generation in duchenne muscular dystrophy. Neurology 1993, 43, 527–532. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kawakami, M. Molecular Dissection of Cyclosporin A’s Neuroprotective Effect Reveals Potential Therapeutics for Ischemic Brain Injury. Brain Sci. 2013, 3, 1325-1356. https://doi.org/10.3390/brainsci3031325
Kawakami M. Molecular Dissection of Cyclosporin A’s Neuroprotective Effect Reveals Potential Therapeutics for Ischemic Brain Injury. Brain Sciences. 2013; 3(3):1325-1356. https://doi.org/10.3390/brainsci3031325
Chicago/Turabian StyleKawakami, Minoru. 2013. "Molecular Dissection of Cyclosporin A’s Neuroprotective Effect Reveals Potential Therapeutics for Ischemic Brain Injury" Brain Sciences 3, no. 3: 1325-1356. https://doi.org/10.3390/brainsci3031325