White Matter Loss in a Mouse Model of Periventricular Leukomalacia Is Rescued by Trophic Factors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Animals and Intraparenchymal Injection Procedures
2.2. Induction of Glutamate Excitotoxicity (GME)
2.3. Preparation of Trophic Factors
2.4. Collection and Examination of the Samples
2.5. Measurements of the Lateral Ventricles
2.6. In Situ Hybridization
2.7. Myelin Staining for Frozen Sections: Spielmeyer’s Method
2.8. Stereology
2.9. Examination of the Neuroprotective Effects of TSC1 against NMDA Excitotoxicity
2.10. Statistical Analysis
3. Results
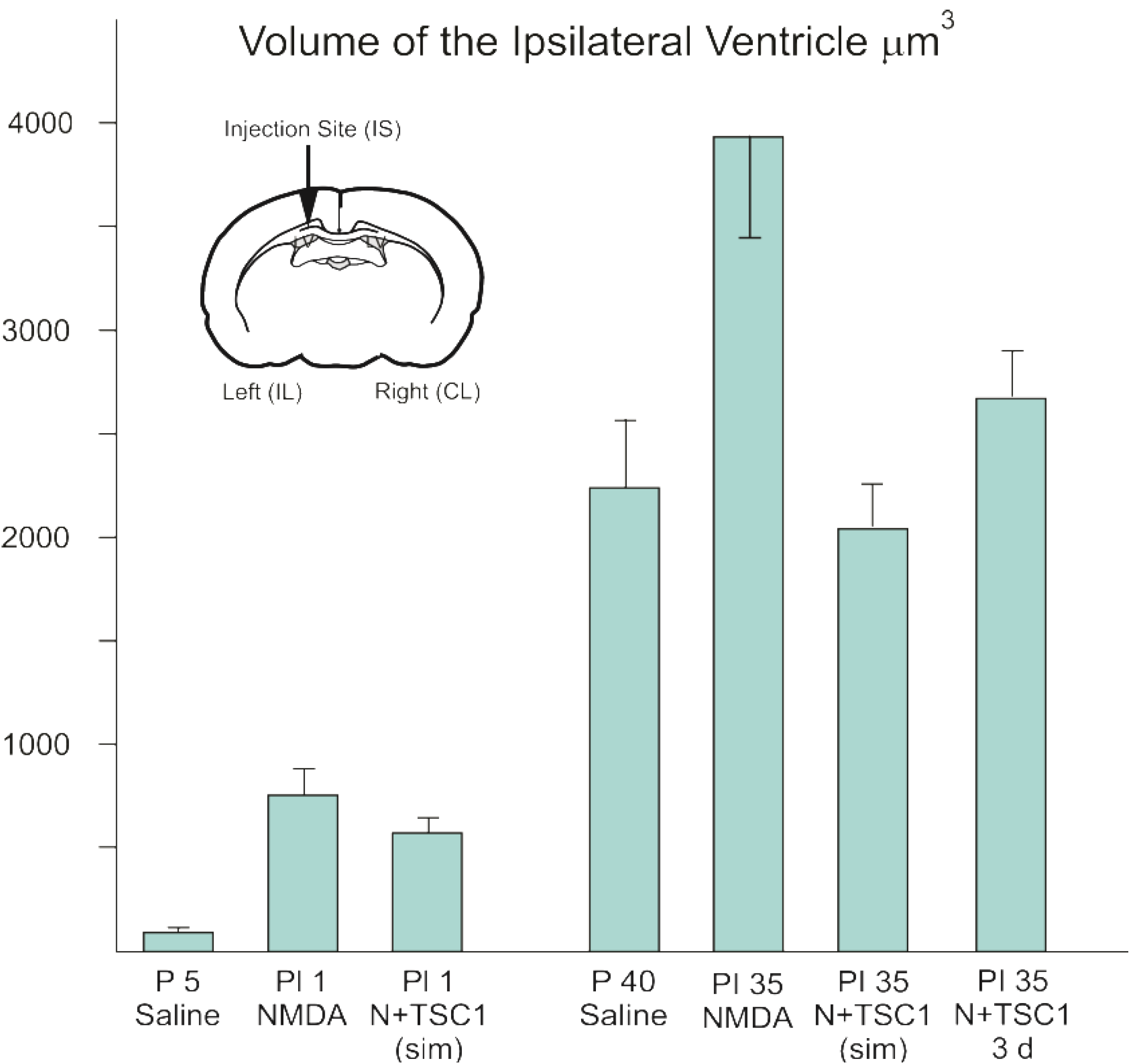
3.1. Effect of TSC1 on Ventriculomegaly

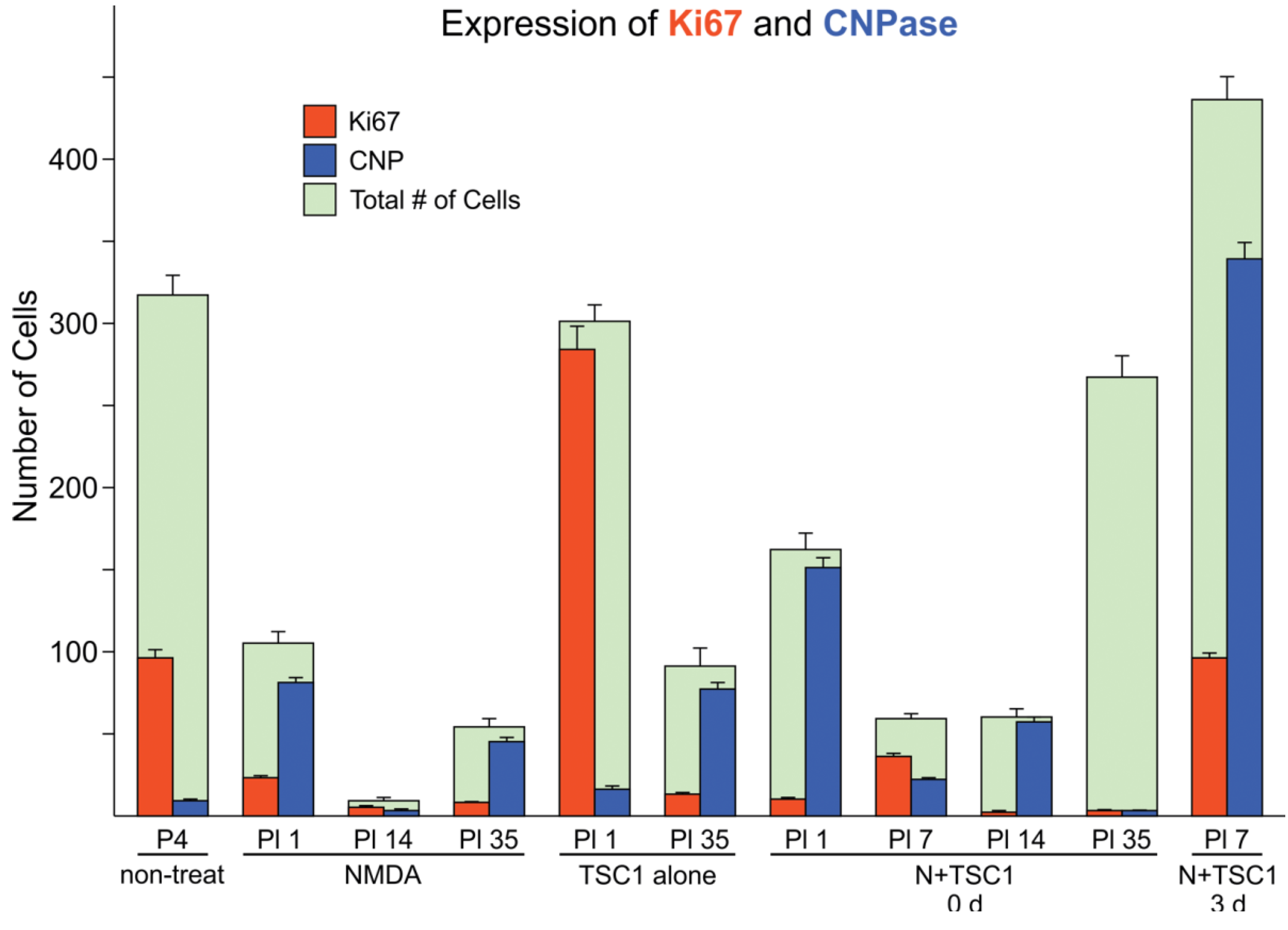
3.2. Effects of TSC1 on OL (Oligodendrocyte) Proliferation

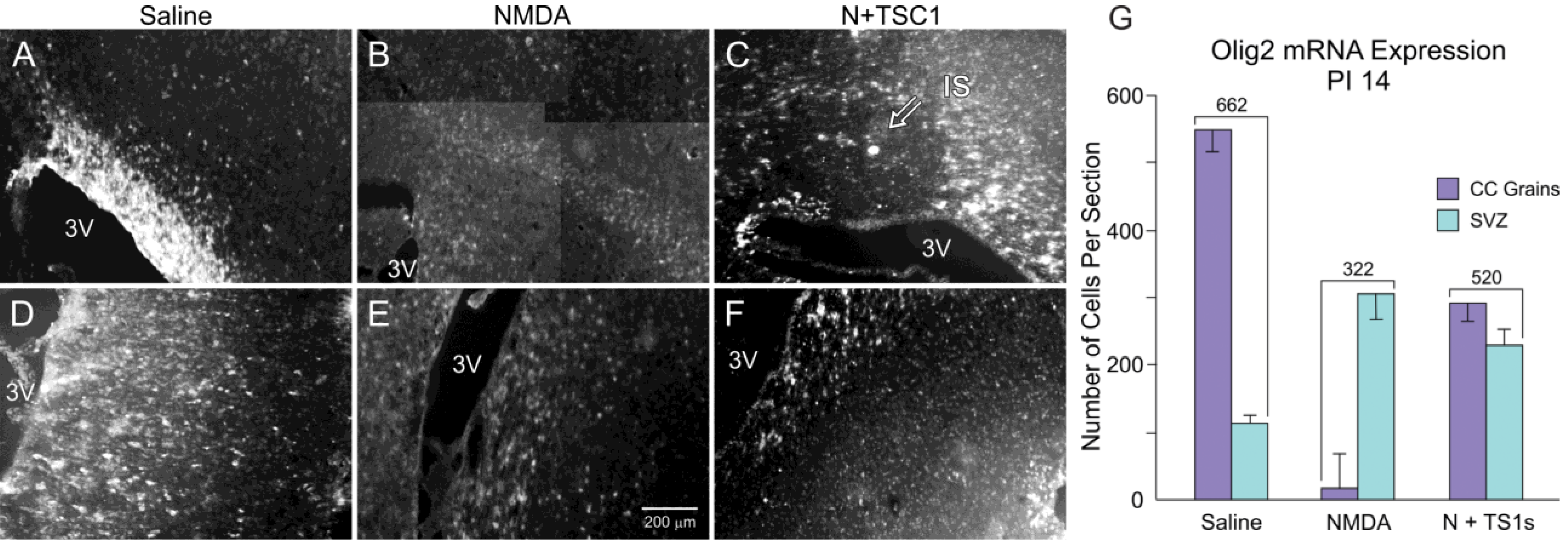
3.3. Olig2 Expression

3.4. Myelination after Treatment

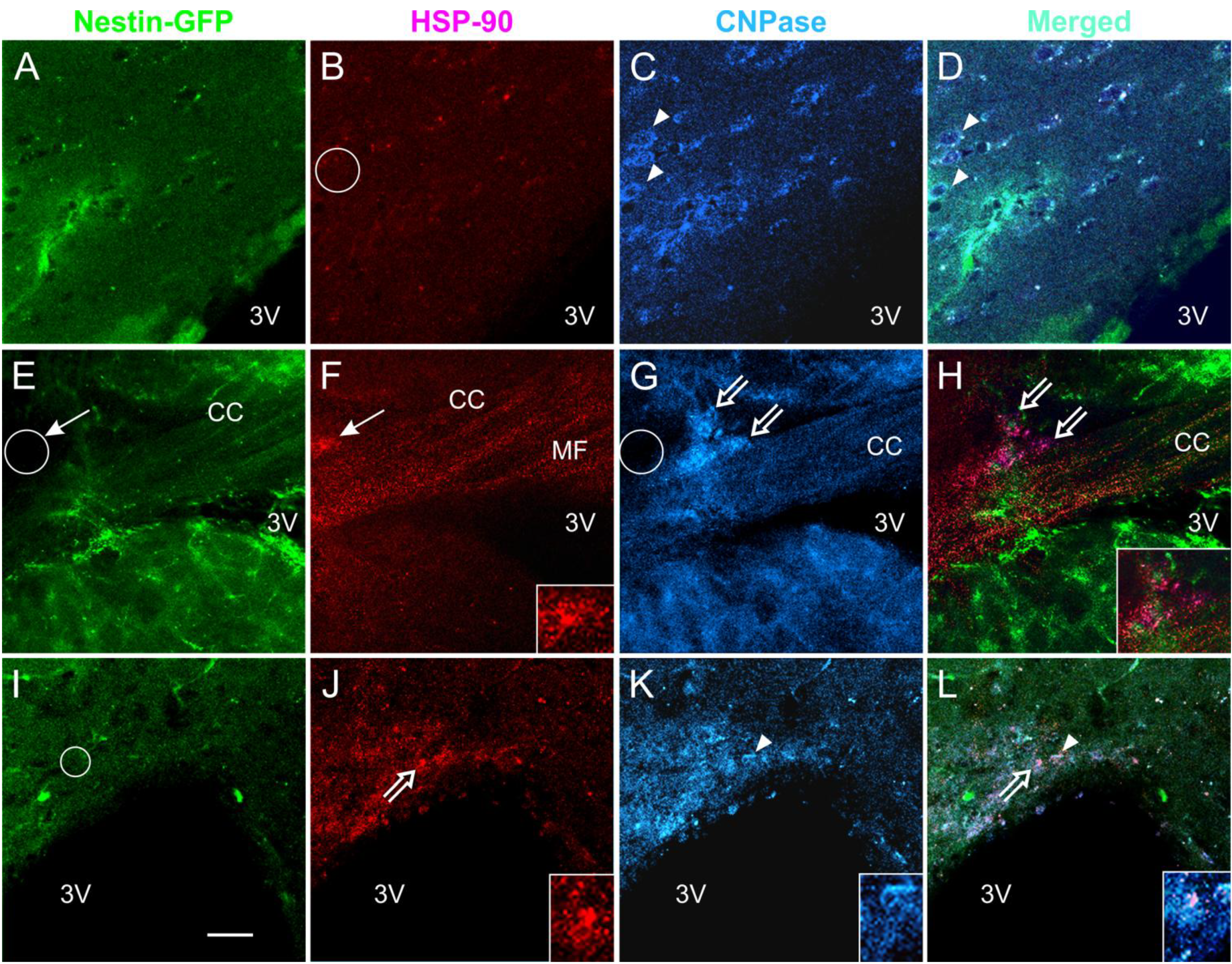
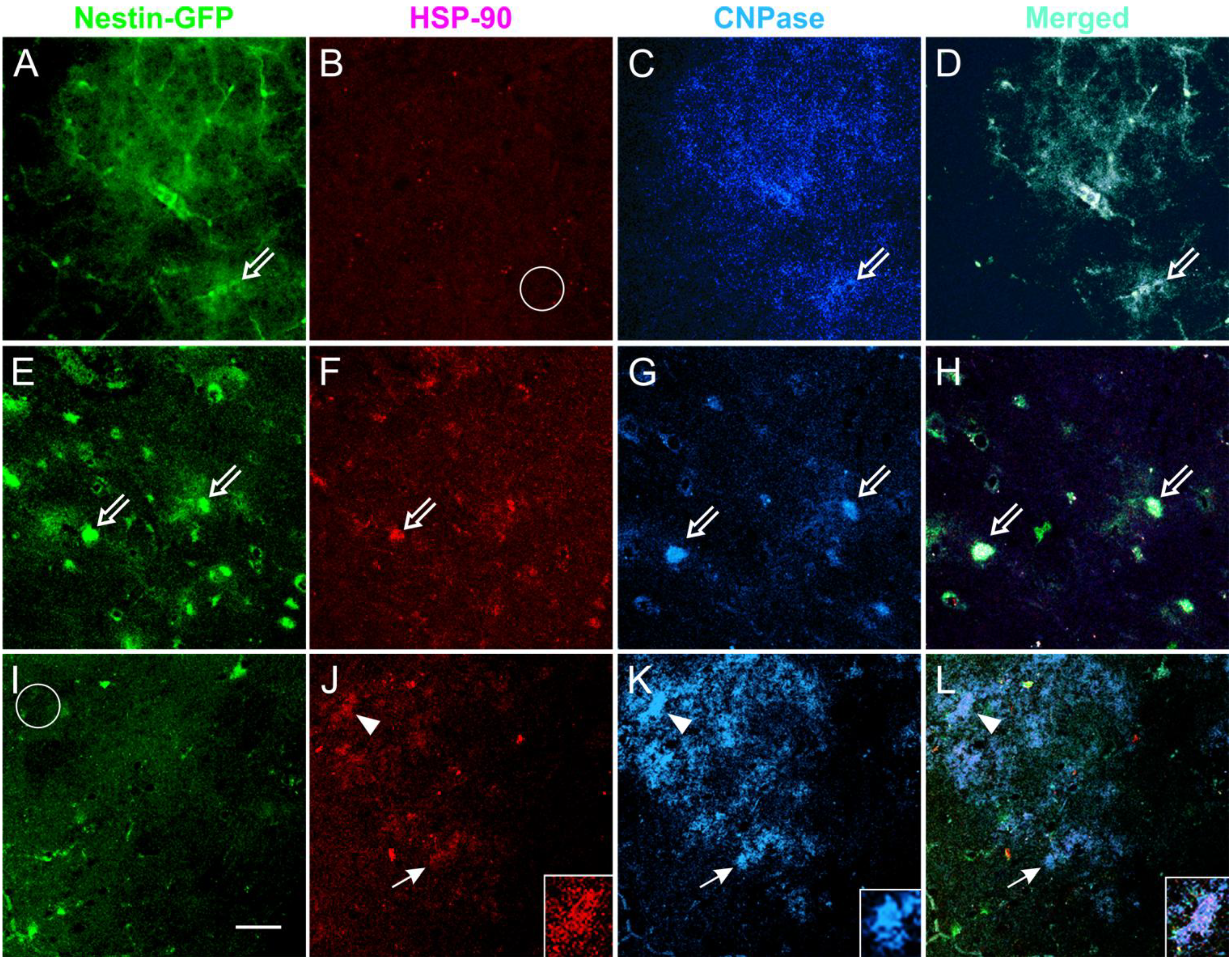
3.5. Expression of the Stress Protein HSP-90 in Acute Brain Slices

3.6. Coronal Views of the Caudate Putamen in Nestin-GFP Mouse Slices: Acute Treatment

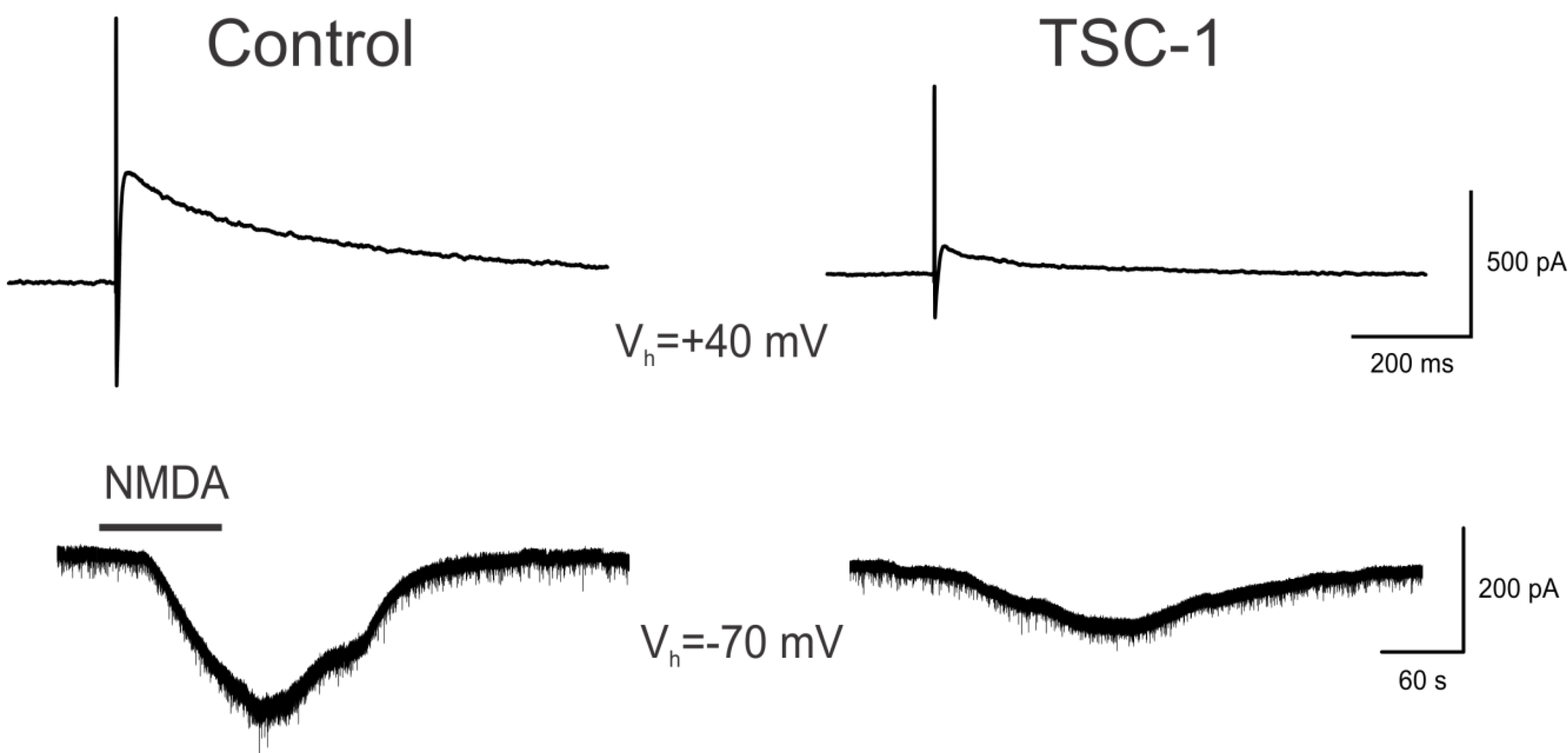
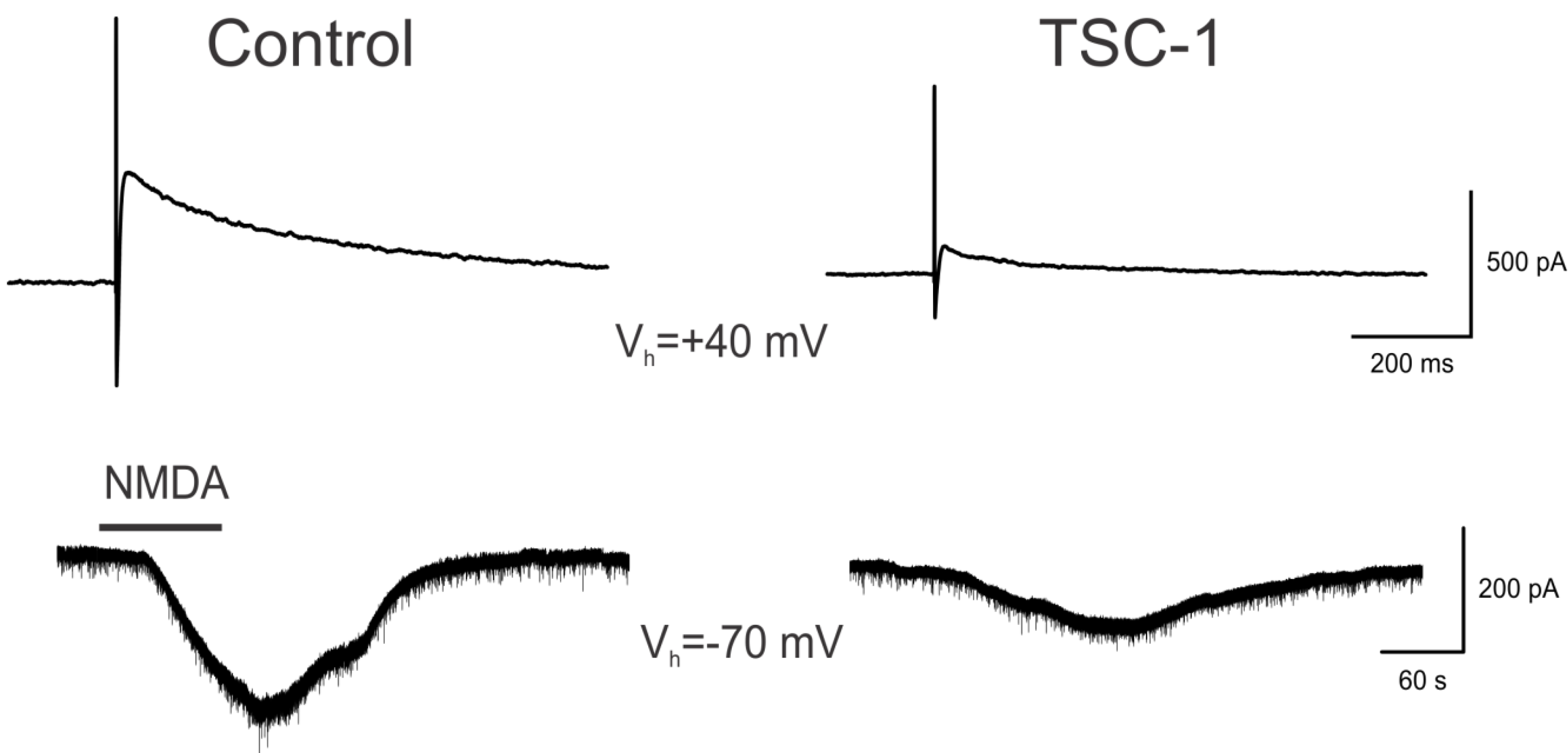
3.7. TSC1 Decreases NMDA-Induced Currents
4. Discussion
4.1. Proliferation of OLs after Treatment
4.2. TSC1 as Regulator of Myelination
4.3. Modeling and Rescue of Perinatal White Matter Damage
5. Conclusions
Treatment Challenges and Future Directions
Acknowledgments
Conflicts of Interest
References
- Nave, K.A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef]
- Blumenthal, I. Periventricular leucomalacia: A review. Eur. J. Pediatr. 2004, 163, 435–442. [Google Scholar] [CrossRef]
- Du Plessis, A.J.; Volpe, J.J. Perinatal brain injury in the preterm and term newborn. Curr. Opin. Neurol. 2002, 15, 151–157. [Google Scholar] [CrossRef]
- Goldberg, M.P.; Ransom, B.R. New light on white matter. Stroke 2003, 34, 330–332. [Google Scholar] [CrossRef]
- Deng, W. Neurobiology of injury to the developing brain. Nat. Rev. Neurol. 2010, 6, 328–336. [Google Scholar] [CrossRef]
- Dammann, O.; Leviton, A. Does prepregnancy bacterial vaginosis increase a mother’s risk of having a preterm infant with cerebral palsy? Dev. Med. Child Neurol. 1997, 39, 836–840. [Google Scholar] [CrossRef]
- Dammann, O.; Leviton, A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr. Res. 1997, 42, 1–8. [Google Scholar] [CrossRef]
- Volpe, J.J. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr. Res. 2001, 50, 553–562. [Google Scholar] [CrossRef]
- Brunssen, S.H.; Harry, G.J. Diffuse white matter injury and neurologic outcomes of infants born very preterm in the 1990s. J. Obstet. Gynecol. Neonatal Nur. 2007, 36, 386–395. [Google Scholar] [CrossRef]
- Back, S.A.; Rivkees, S.A. Emerging concepts in periventricular white matter injury. Semin. Perinatol. 2004, 28, 405–414. [Google Scholar] [CrossRef]
- Hagberg, H.; Peebles, D.; Mallard, C. Models of white matter injury: Comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 30–38. [Google Scholar] [CrossRef]
- Mesples, B.; Plaisant, F.; Fontaine, R.H.; Gressens, P. Pathophysiology of neonatal brain lesions: Lessons from animal models of excitotoxicity. Acta Paediatr. 2005, 94, 185–190. [Google Scholar] [CrossRef]
- Rees, S.; Inder, T. Fetal and neonatal origins of altered brain development. Early Hum. Dev. 2005, 81, 753–761. [Google Scholar] [CrossRef]
- Karadottir, R.; Cavelier, P.; Bergersen, J.H.; Attwell, D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 2005, 438, 1162–1166. [Google Scholar] [CrossRef]
- Salter, M.G.; Fern, R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 2005, 438, 1167–1171. [Google Scholar] [CrossRef]
- Kumar, S.; Macklin, W.B.; Gordon, M.N.; Espinosa de los Monteros, A.; Cole, R.; Scully, S.A.; de Vellis, J. Transcriptional regulation studies of myelin associated genes in md mutant rats. Dev. Neurosci. 1990, 12, 316–325. [Google Scholar] [CrossRef]
- Espinosa de los Monteros, A.; Kumar, S.; Scully, R.; Cole, R.; de Vellis, J. Transferrin gene expression and secretion by rat brain cells in vitro. J. Neurosci. Res. 1990, 25, 576–580. [Google Scholar] [CrossRef]
- Espinosa de los Monteros, A.; de Vellis, J. Myelin basic protein and transferrin characterize different subpopulations of oligodendrocytes in rat primary glial cultures. J. Neurosci. Res. 1988, 21, 181–187. [Google Scholar] [CrossRef]
- Espinosa de los Monteros, A.; Zhang, M.; Gordon, M.N.; Kumar, S.; Scully, S.; de Vellis, J. The myelin-deficient rat mutant: Partial recovery of oligodendrocyte maturation in vitro. Dev. Neurosci. 1990, 12, 326–339. [Google Scholar] [CrossRef]
- Espinosa de los Monteros, A.; Kumar, S.; Zhao, P.; Huang, J.C.; Nazarian, R.; Pan, T.; Scully, S.; Chang, R.; de Vellis, J. Transferrin is an essential factor for myelination. Neurochem. Res. 1999, 24, 235–248. [Google Scholar]
- Espinosa-Jeffrey, A.; Zhao, P.M.; Awosika, O.; Huang, A.; Chang, R.; de Vellis, J. Transferrin regulates transcription of the MBP gene and its action synergizes with IGF-1 to enhance myelinogenesis in the md rat. Dev. Neurosci. 2002, 24, 227–241. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Saito, H.; Suzuki, M.; Mori, K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport 2000, 11, 1991–1996. [Google Scholar] [CrossRef]
- Espinosa-Jeffrey, A.; Zhao, P.; Awosika, W.; Wu, N.; Macias, F.; Cepeda, C.; Levine, M.; de Vellis, J. Activation, proliferation and commitment of endogenous, stem/progenitor cells to the oligodendrocyte lineage by a combination of neurotrophic factors in a rat model of dysmyelination. Dev. Neurosci. 2006, 28, 488–498. [Google Scholar] [CrossRef]
- Espinosa de los Monteros, A.; Zhan, M.; Gordon, G.M.; Aymie, M.; Vellis, J. Transplantation of cultured premyelinating oligodendrocytes into normal and myelin-deficient rat brain. Dev. Neurosci. 1992, 14, 98–104. [Google Scholar] [CrossRef]
- Ma, J.; Matsumoto, M.; Tanaka, K.; Takebayashi, H.; Ikenaka, K. An animal model for late onset chronic demyelination disease caused by failed terminal differentiation of oligodendrocytes. Neuron Glia Biol. 2005, 2, 81–91. [Google Scholar] [CrossRef]
- Espinosa-Jeffrey, A.; Hitoshi, S.; Zhao, P.; Awosika, O.; Agbo, C.; Olaniyan, E.; Garcia, J.; Valera, R.; Thomassian, A.; Chang, W.R.; et al. Functional central nervous system myelin repair in an adult mouse model of demyelination caused by proteolipid protein overexpression. J. Neurosci. Res. 2010, 88, 1682–1694. [Google Scholar]
- Mallory, F.B. Pathological Technique: A Practical Manual for Workers in Pathological Histology Including Directions for the Performance of Autopsies and for Microphotography; W.B. Saunders: Philadelphia, PA, USA, 1942; p. 237. [Google Scholar]
- Fleiss, B.; Gressens, P. Tertiary mechanisms of brain damage: A new hope for treatment of cerebral palsy? Lancet Neurol. 2012, 11, 556–566. [Google Scholar] [CrossRef]
- Espinosa de los Monteros, A.; Baba, H.; Zhao, P.M.; Pan, T.; Chang, R.; de Vellis, J.; Ikenaka, K. Remyelination of the adult demyelinated mouse brain by grafted oligodendrocyte progenitors and the effect of B-104 cografts. Neurochem. Res. 2001, 26, 673–682. [Google Scholar] [CrossRef]
- Miller, S.P.; Ferriero, D.M. From selective vulnerability to connectivity: Insights from newborn brain imaging. Trends Neurosci. 2009, 32, 496–505. [Google Scholar] [CrossRef]
- Sprinkle, T.J. 2′,3′-Cyclic nucleotide 3′-phosphodiesterase, an oligodendrocyte-Schwann cell and myelin-associated enzyme of the nervous system. Crit. Rev. Neurobiol. 1989, 4, 235–301. [Google Scholar]
- Vogel, U.S.; Thompson, R.J. Molecular structure, localization, and possible functions of the myelin-associated enzyme 2′,3′-cyclic nucleotide 3′-phosphodiesterase. J. Neurochem. 1988, 50, 1667–1677. [Google Scholar] [CrossRef]
- Giulian, D.; Moore, S. Identification of 2′,3′-cyclic nucleotide 3′-phosphodiesterase in the vertebrate retina. J. Biol. Chem. 1980, 255, 5993–5995. [Google Scholar]
- Dreiling, C.E.; Schilling, R.J.; Reitz, R.C. 2′,3′-Cyclic nucleotide 3′-phosphohydrolase in rat liver mitochondrial membranes. Biochim. Biophys. Acta 1981, 640, 114–120. [Google Scholar] [CrossRef]
- Weissbarth, S.; Maker, H.S.; Raes, I.; Brannan, T.S.; Lapin, E.P.; Lehrer, G.M. The activity of 2′,3′-cyclic nucleotide 3′-phosphodiesterase in rat tissues. J. Neurochem. 1981, 37, 677–680. [Google Scholar]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar]
- Bartlett, W.P.; Li, X.S.; Connor, J.R. Expresion of transferring mRNA in the CNS of normal and jimpy mice. J. Neurochem. 1991, 57, 318–322. [Google Scholar] [CrossRef]
- Barlett, W.P.; Li, X.S.; Williams, M.; Benkovic, S. Localization of insulin-like growth factor-1 mRNA in murine central nervous system during postnatal development. Dev. Biol. 1991, 147, 239–250. [Google Scholar] [CrossRef]
- Escobar, O.P.; Bongarzone, E.R.; Soto, E.F.; Pasquini, J.M. Single intracerebral injection of apo-transferrin in young rats induces increased myelination. Dev. Neurosci. 1994, 16, 248–254. [Google Scholar] [CrossRef]
- Adamo, A.M.; Paez, P.M.; Escobar-Cabrera, O.R.; Wolfson, M; Franco, P.G.; Pasquini, J.M.; Soto, E.F. Remyelination after cuprizone-induced demyelination in the rat is stimulated by apotransferrin. Exp. Neuol. 2006, 198, 519–529. [Google Scholar]
- Saleh, M.C.; Espinosa de los Monteros, A.; de Arriba Zerpa, G.A.; Fontaine, I.; Piaud, O.; Djordjijevic, D.; Baroukh, N.; Garcia Otin, A.L.; Ortiz, E.; Lewis, S.; et al. Myelination and motor coordination are increased in transferrin transgenic mice. J. Neurosci. Res. 2003, 72, 587–594. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Alemán, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- McMorris, F.A.; Duboi-Dalcq, M. Insulin-like growth factor I promotes cell proliferation and oligodendroglial commitment in rat glial progenitor cells developing in vitro. J. Neurosci. Res. 1988, 21, 199–209. [Google Scholar] [CrossRef]
- McMorris, F.A.; McKinnon, R.D. Regulation of oligodendrocyte development and CNS myelinationby growth factors: Prospects for therapy of demyelinating disease. Brain Pathol. 1996, 6, 313–329. [Google Scholar] [CrossRef]
- Werner, H.; Woloschak, M.; Adamo, M.; Shen-Orr, Z.; Roberts, C.T., Jr.; LeRoith, D. Developmental regulation of the rat insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1989, 86, 7451–7455. [Google Scholar]
- Madathil, S.K.; Evans, H.N.; Saatman, K.E. Temporal and regional changes in IGF-1/IGF-1R signaling in the mouse brain after traumatic brain injury. J. Neurotrauma 2010, 27, 95–107. [Google Scholar] [CrossRef]
- Zapf, J.; Froesch, E.R. Insulin-like growth factor I actions on somatic growth. Compr. Physiol. 2011, 24, 663–699. [Google Scholar]
- Rosenberg, P.A.; Dai, W.; Gan, X.D.; Ali, S.; Fu, J.; Back, S.A; Sanchez, R.M.; Segal, M.M.; Follett, P.L.; Jensen, F.E.; et al. Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J. Neurosci. Res. 2003, 71, 237–245. [Google Scholar] [CrossRef]
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 2000, 92, 242–248. [Google Scholar] [CrossRef]
- Xiao, X.; Zuo, X.; Davis, A.A.; McMillan, D.R.; Curry, B.B.; Richardson, J.A.; Benjamin, I.J. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J. 1999, 18, 5943–5952. [Google Scholar] [CrossRef]
- Prodromou, C.; Panaretou, B.; Chohan, S.; Siligardi, G.; O’Brien, R.; Ladbury, J.E.; Roe, S.M.; Piper, P.W.; Pearl, L.H. The ATPase cycle of Hsp90 drives a molecular ‘clamp’ via transient dimerization of the N-terminal domains. EMBO J. 2000, 19, 4383–4392. [Google Scholar] [CrossRef]
- Panaretou, B.; Siligardi, G.; Meyer, P.; Maloney, A.; Sullivan, J.K.; Singh, S.; Millson, S.H.; Clarke, P.A.; Naaby-Hansen, S.; Stein, R.; et al. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1. Mol. Cell 2002, 10, 1307–1318. [Google Scholar] [CrossRef]
- Spence, R.D.; Hamby, M.E.; Umeda, E.; Itoh, N.; Du, J.; Wisdom, S.; Cao, Y.; Bondar, G.; Lam, J.; Ao, Y.; et al. Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 8867–8872. [Google Scholar] [CrossRef]
- Passinen, S.; Valkila, J.; Manninen, T.; Syvala, H.; Ylikomi, T. The C-terminal half of Hsp90 is responsible for its cytoplasmic localization. Eur. J. Biochem. 2001, 268, 5337–5342. [Google Scholar] [CrossRef]
- Picard, D.; Yamamoto, K.R. Two signals mediate hormone-dependent nuclear localization of the glucocorticoid receptor. EMBO J. 1987, 6, 3333–3340. [Google Scholar]
- Dickey, C.A.; Eriksen, J.; Kamal, A.; Burrows, F.; Kasibhatla, S.; Eckman, C.B.; Hutton, M.; Petrucelli, L. Development of a high throughput drug screening assay for the detection of changes in tau levels—Proof of concept with HSP90 inhibitors. Curr. Alzheimer Res. 2005, 2, 231–238. [Google Scholar] [CrossRef]
- Fujikake, N.; Nagai, H.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef]
- Auluck, P.K.; Meulener, M.C.; Bonini, N.M. Mechanisms of suppression of α-synuclein neurotoxicity by geldanamycin in drosophilia. J. Biol. Chem. 2005, 280, 2873–2878. [Google Scholar]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Tanaka, F; Sobue, G. Alleviating neurodegeneration by an anticancer agent: An Hsp90 inhibitor (17-AAG). Ann. N. Y. Acad. Sci. 2006, 1086, 21–34. [Google Scholar] [CrossRef]
- Chiosis, G.; Tao, H. Purine-scaffold Hsp90 inhibitors. IDrugs 2006, 9, 778–782. [Google Scholar]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting Hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef]
- Heath, E.L.; Gaskins, M.; Pitot, H.C.; Pili, R.; Tan, W.; Marschke, R.; Liu, G.; Hillman, D.; Sarkar, F.; Sheng, S.; et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Prostate Cancer 2005, 4, 138–141. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Trump, D.L.; Eiseman, J.L.; Belani, C.P.; Agarwala, S.S.; Zuhowski, E.G.; Lan, J.; Potter, D.M.; Ivy, S.P.; Ramalingam, S.; et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(allylamino)-17-demethoxygeldanamycin (17AAG, NSC 330507), a novel inhibitor of heat shock protein 90, in patients with refractory advanced cancers. Clin. Cancer Res. 2005, 11, 3385–3391. [Google Scholar] [CrossRef]
- DiFiglia, M. Excitotoxic injury of the neostriatum: A model for Huntington’s disease. Trends Neurosci. 1990, 7, 286–289. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Espinosa-Jeffrey, A.; Barajas, S.A.R.; Arrazola, A.R.; Taniguchi, A.; Zhao, P.M.; Bokhoor, P.; Holley, S.M.; Dejarme, D.P.; Chu, B.; Cepeda, C.; et al. White Matter Loss in a Mouse Model of Periventricular Leukomalacia Is Rescued by Trophic Factors. Brain Sci. 2013, 3, 1461-1482. https://doi.org/10.3390/brainsci3041461
Espinosa-Jeffrey A, Barajas SAR, Arrazola AR, Taniguchi A, Zhao PM, Bokhoor P, Holley SM, Dejarme DP, Chu B, Cepeda C, et al. White Matter Loss in a Mouse Model of Periventricular Leukomalacia Is Rescued by Trophic Factors. Brain Sciences. 2013; 3(4):1461-1482. https://doi.org/10.3390/brainsci3041461
Chicago/Turabian StyleEspinosa-Jeffrey, Araceli, Socorro A. R. Barajas, Alfonso R. Arrazola, Alana Taniguchi, Paul M. Zhao, Payam Bokhoor, Sandra M. Holley, Don P. Dejarme, Brian Chu, Carlos Cepeda, and et al. 2013. "White Matter Loss in a Mouse Model of Periventricular Leukomalacia Is Rescued by Trophic Factors" Brain Sciences 3, no. 4: 1461-1482. https://doi.org/10.3390/brainsci3041461