ABCA7 and Pathogenic Pathways of Alzheimer’s Disease


Department of Neuroscience, Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL 32224, USA
*
Author to whom correspondence should be addressed.
Brain Sci. 2018, 8(2), 27; https://doi.org/10.3390/brainsci8020027
Submission received: 28 December 2017
/
Revised: 1 February 2018
/
Accepted: 3 February 2018
/
Published: 5 February 2018
{kind=link}
{kind=link}
Abstract
:The ATP-binding cassette (ABC) reporter family functions to regulate the homeostasis of phospholipids and cholesterol in the central nervous system, as well as peripheral tissues. ABCA7 belongs to the A subfamily of ABC transporters, which shares 54% sequence identity with ABCA1. While ABCA7 is expressed in a variety of tissues/organs, including the brain, recent genome-wide association studies (GWAS) have identified ABCA7 gene variants as susceptibility loci for late-onset Alzheimer’s disease (AD). More important, subsequent genome sequencing analyses have revealed that premature termination codon mutations in ABCA7 are associated with the increased risk for AD. Alzheimer’s disease is a progressive neurodegenerative disease and the most common cause of dementia, where the accumulation and deposition of amyloid-β (Aβ) peptides cleaved from amyloid precursor protein (APP) in the brain trigger the pathogenic cascade of the disease. In consistence with human genetic studies, increasing evidence has demonstrated that ABCA7 deficiency exacerbates Aβ pathology using in vitro and in vivo models. While ABCA7 has been shown to mediate phagocytic activity in macrophages, ABCA7 is also involved in the microglial Aβ clearance pathway. Furthermore, ABCA7 deficiency results in accelerated Aβ production, likely by facilitating endocytosis and/or processing of APP. Taken together, current evidence suggests that ABCA7 loss-of-function contributes to AD-related phenotypes through multiple pathways. A better understanding of the function of ABCA7 beyond lipid metabolism in both physiological and pathological conditions becomes increasingly important to explore AD pathogenesis.
Keywords:
ABCA1; amyloid-β; amyloid precursor protein; cholesterol; genetics; macrophage; microglia; neurons; phagocytosis; phospholipids1. Introduction
Alzheimer’s disease (AD) is the leading cause of dementia in the elderly, accounting for 60–80% of cases. Approximately 5.5 million individuals are living with Alzheimer’s dementia in the United States. This number is estimated to increase continuously due to the expansion of the aged population [1]. Alzheimer’s disease is pathologically characterized by the presence of amyloid-bearing plaques and neurofibrillary tangles, which are often accompanied by neuronal loss and trigger innate immune responses in the brain [2,3,4]. While AD is a neurodegenerative disease with complex pathogenesis, several genetic factors have been associated with the development of the disease [5]. Although a small population size (<0.5%), dominantly inherent mutations in three genes encoding amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2), cause familial AD, usually at a young age (30–50 years of age) [6]. Each of these genes has been shown to accelerate the production of neurotoxic amyloid-β (Aβ), leading to the accumulation and deposition of Aβ in the brain. On the other hand, the majority of AD cases are sporadic and late-onset, occuring in individuals over the age of 65, sharing the same clinical and pathological features with familial type early-onset AD [7]. Importantly, several genomic variations are attributable to 60–80% of cases of late-onset AD [8]. While the ε4 allele of APOE is the strongest genetic risk factor for late-onset AD [9,10,11,12,13,14,15,16], several gene loci in ABCA7 on chromosome 19p13.3 have also been recognized as novel risk factors for the disease [17]. ABCA7 codes ATP-binding cassette (ABC) transporter A7, which is a member of the A subfamily of (ABC) transporters. Consistently, accumulating in vitro and in vivo studies support the potential contribution of ABCA7 to AD-related phenotypes. Therefore, to explore the pathogenesis of AD, a greater understanding of the role of ABCA7 in physiological and pathological conditions might be important. In this review, we summarize current evidence for the risk of ABCA7 gene variants of AD development and discuss how ABCA7 is involved in the pathogenic pathways of AD.
2. ABCA7 Gene Variants and Alzheimer’s Disease
Common variants of ABCA7 with a minor allele frequency (MAF) of more than 5% have been implicated to associate with the risk for AD [17]. In 2011, Hollingworth et al. identified the common SNP (single nucleotide polymorphism) variant rs3764650, which is located in an ABCA7 intron, as one of the susceptibility loci for late-onset AD (odds ratio [OR] = 1.23; 95% CI = 1.17–1.28) with replication among independent Caucasian cohorts through a genome-wide association study (GWAS) [18]. Naj et al. also reported that ABCA7 SNP rs3752246, a missense variant (p.Gly1527Ala), is associated with the risk for late-onset AD (OR = 1.15; 95% CI = 1.09–1.21) [19]. Furthermore, a large meta-analysis of GWAS in individuals of European ancestry identified a new susceptibility variant rs4147929 in an ABCA7 intron (OR = 1.15; 95% CI = 1.11–1.19) [20]. Interestingly, ABCA7 rs3764650 has been associated with cortical and hippocampal atrophy in cognitively normal and mild cognitive impairment (MCI) subjects [21], as well as with memory decline in MCI and late-onset AD patients [22]. Therefore, ABCA7 is possibly responsible for both the development and progression of AD.
In an African American cohort, a coding variant of ABCA7 rs3764647 (p.His395Arg), located near rs3752246, has been associated with AD risk (OR = 1.32; 95% CI = 1.07–1.63), while no or a minimal significant association was detected in rs3752246 and rs3764650, respectively [23]. Another study in African Americans revealed that ABCA7 rs115550680 is linked to the development of late-onset AD, in which the effect size (OR = 1.79; 95% CI = 1.47–2.12) is comparable with that of APOE ε4 (OR = 2.31; 95% CI = 2.19–2.42) [24]. In addition, although ABCA7 rs142076058 (p.Arg578Alafs) is likely rare in Caucasians, it is relatively common in African Americans and has been identified as an AD risk allele; MAF 15.2% in AD vs. 9.74% in controls (OR = 2.13; 95% CI = 1.42–3.20) [25]. Thus, while increasing evidence clearly indicates that ABCA7 gene variants are involved in AD risk in both Caucasians and African Americans, there may be ethnic-dependent effects.
In addition to the common variants, whole genome sequencing, exome sequencing, and targeted resequencing have also demonstrated that some of the low frequency variants (MAF 1–5%) and rare variants (MAF < 1%) in ABCA7 have significant associations with the risk for AD. In a Belgian cohort, a low frequency variant, rs78117248, in an ABCA7 intron showed a strong association with AD even after adjustment for the common SNPs, rs3764650, rs4147929, and rs3752246 (OR = 2.00, 95% CI 1.22–3.26) [26]. A rare ABCA7 missense variant (rs3752239; p.Asn718Thr) was also shown to contribute to AD risk in African Americans [27]. On the other hand, another study showed that a low-frequency coding variant, rs72973581 (p.G215S), is a protective allele against AD (OR = 0.57; 95% CI = 0.41–0.80) in British and North-American ancestry, although the association is modest [28]. Of note, in 2015, Steinberg et al. comprehensively analyzed rare premature termination codon (PTC) mutations in ABCA7 using whole genome sequencing and demonstrated that they are associated with AD risk in an Icelandic population; when analyzed by combining those rare “loss-of-function” variants, the OR is calculated to be 2.12 [29]. Several independent studies have also confirmed the association of ABCA7 loss-of-function variants with increased AD risk [26,30,31,32,33,34,35]. Interestingly, long-read MinION cDNA sequencing has revealed that some of the ABCA7 loss-of-function variants receive exon skipping or alternative splicing, which likely allows the production of functional proteins and rescues the deleterious effects [34]. Taken together, accumulating evidence through genetic studies suggests that the contribution of ABCA7 to AD risk is mediated by the dysfunction or reduction of ABCA7. Indeed, a common ABCA7 variant, rs3764650, likely influences ABCA7 expression levels in the brain. Whereas ABCA7 mRNA expression is increased in AD brains compared to control individuals, carrying the protective rs3764650 (T) allele is associated with higher ABCA7 expression levels [36].
3. Biochemical and Functional Features of ABCA7
3.1. ABCA7 Structure
ATP-binding cassette (ABC) transporters constitute a superfamily of highly conserved proteins involved in the membrane transport of various substrates, such as ions, amino acids, lipids, and sterols across cell membranes [37]. Absolute ABC transporters are characterized by two nucleotide binding domains (NBD), which contain conserved Walker A and B motifs and conserved sequences, as well as two transmembrane domain bundles each composed of six membrane-spanning helices. The specificity of the transported molecules appears to be determined by the transmembrane domains, while ATP is required for the transport activity at the NBD [38]. ABCA7 possesses a typical ABC transporter structure, composed of 2146 amino acids with a molecular weight of approximately 220 kDa [39], which is mainly localized in the plasma membrane and the Golgi apparatus [40]. Of the 12 A class members, ABCA7 and ABCA1 are the closest homologues, sharing 54% sequence identity [39] (Figure 1). In vitro studies in HEK293 cells transfected with ABCA7 and ABCA1 have shown that ABCA7 also shares functional attributes with ABCA1; Apolipoprotein A (apoA)-I induces the release of cellular lipids including cholesterol and phospholipids through the two extracellular domains commonly preserved in ABCA7 and ABCA1 [41,42,43,44].
3.2. ABCA7 Expression and Organ/Tissue Distribution
In spite of their structural homology, the transcription of ABCA7 and ABCA1 is differently regulated. Although ABCA1 gene expression is upregulated through heterodimerization of the liver-X-receptor (LXR) and the retinoid-X-receptor (RXR) under conditions increasing cellular cholesterol accumulation [45,46], cellular cholesterol depletion has been shown to increase ABCA7 mRNA levels [47]. The similar regulation pattern is also observed in β-hydroxy β-methylglutaryl-CoA (HMG-CoA) reductase, low density lipoprotein receptor (LDLR), and sterol-responsive/regulatory element binding proteins (SREBPs), indicating the contribution of sterol regulatory element to transcriptional regulation [48]. Indeed, reporter cell assays have revealed that ABCA7 expression is positively regulated by sterol through the SREBP2 pathway, which is oppositely involved in ABCA1 transcription [47].
ABCA7 expression is detected in a variety of tissues/organs, which include brain, lung, adrenal gland, kidney, spleen, thymus, lymph node, testis, keratinocytes, and pancreatic islets, as well as blood cells (i.e., macrophages, erythrocytes, and platelets) [39,44,49,50]. In human brain cell cultures, ABCA7 mRNA is the most abundant in microglia compared to other cell types [51]. While in situ hybridization analysis has demonstrated that ABCA7 expression is higher in hippocampal neurons than other areas or cells in the mouse brain [51,52], single cell-type RNA-sequencing has detected Abca7, not only in neurons and microglia, but also in various cell types, including oligodendrocytes, endothelial cells, and astrocytes in the mouse cortex [53].
Interestingly, the ABCA7 gene is expressed as two variants, full-length cDNA (type I) and the shorter splicing variant cDNA (type II), in a tissue-specific manner. In peripheral tissues, type I ABCA7 expression is predominantly detected in bone marrow, whilst the type II ABCA7 is mainly expressed in the spleen and trachea [54]. In vitro experiments have shown that cells expressing type II ABCA7 less efficiently mediate lipid efflux compared to type I [54]. Thus, although further studies are needed, it has been hypothesized that the two ABCA7 splice variants might serve different functions depending on cell types, as well as organs/tissues.
3.3. ABCA7 and Lipid Metabolism
As predicted from the structure, the major function of ABCA7 is likely to regulate lipid metabolism. Serum levels of total cholesterol and high density lipoprotein (HDL) were lower in female ABCA7-knockout (KO) mice compared to wild-type mice under fasting conditions, while there was no significant difference in serum fatty acid levels [52]. Shotgun lipidomic analysis has also demonstrated that Abca7 deficiency alters the phospholipid profile in the mouse brain [55]. In human embryonic kidney (HEK)-293 cells, apolipoprotein-mediated efflux of cellular phospholipids was facilitated by expressing human ABCA7 [41,44,56,57]. While cholesterol and phosphatidylcholine (PC) are the major lipids exported by ABCA1, liquid chromatography-tandem mass spectrometry analyses revealed that lysophosphatidyl choline (LPC) and PC are predominantly released from BHK cells by ABCA7 overexpression [58]. However, in conflict with those results regarding the overexpression of ABCA7, a report shows that ABCA7 deficiency does not influence cholesterol and phospholipid efflux in mouse primary macrophages [52]. Thus, future studies should refine the physiological function of ABCA7 as a lipid transporter by assessing the effects depending on ABCA7 expression levels, cell types, and acceptors.
In addition to lipid efflux, the forced expression of ABCA7 has been shown to increase the amounts of intracellular/cell surface ceramide and intracellular phosphatidylserine (PS) in HeLa cells, resulting in cell cycle arrest [59]. Moreover, ABCA7 deficiency causes the disruption of lipid rafts on the plasma membrane of thymocytes and antigen presenting cells in mice, which is likely to be associated with the compromised development and function of natural killer T cells [60]. Together, these findings suggest that ABCA7 plays a role in maintaining intracellular lipid metabolism, thereby regulating cellular homeostasis.
3.4. ABCA7 and Phagocytosis
In Caenorhabditis elegans, CED-7 is one of the major adhesion molecules mediating the engulfment of apoptotic cells during embryogenesis [61]. As the orthologue of ced-7 in mammalians has been predicted to encode ABC transporters from sequence similarity [62], ABCA family members are likely involved in the regulation of phagocytosis. The ABCA7 protein also shares 24% sequence identity and 54% sequence similarity with the CED-7 protein [63]. Indeed, when ABCA7 was deleted in mouse embryonic fibroblast BALB/3T3 cells, their phagocytic activity to fluorescently-labeled latex beads was significantly decreased [47]. While the phagocytosis of fluorescent polystyrene microspheres was enhanced by apoA-I or apoA-II in mouse macrophage J774 cells, the effect was ablated by the knockdown of ABCA7, but not by ABCA1 [64,65]. Consistent with these results, an in vivo ink-engulfment assay has shown that Abca7 deficient peritoneal macrophages possess an impaired phagocytosis ability compared to those from wild-type mice [64]. Additionally, apoA-I-mediated phagocytosis of Staphylococcus aureus was suppressed by ABCA7 knockdown in J774 cells, reproducing the results using artificial polystyrene beads [64]. Another study also revealed that the phagocytosis of apoptotic neutrophils is reduced in macrophages from ABCA7 heteroinsufficient mice compared to control mice, while FcR-mediated phagocytosis for viable neutrophils coated with anti-CD18 antibody is not affected [66]. Although further studies are required to determine the molecular mechanism underlying the link between ABCA7 and phagocytosis, ERK signaling is likely involved in the pathway. Whereas the phosphorylation of extracellular signal-regulated kinase (ERK) is an important process for the phagocytosis of dying cells in response to apoptotic cells or the complement protein C1q, the event is diminished in ABCA7-deficient macrophages [66]. Thus, these results indicate that ABCA7 critically regulate phagocytic function in macrophages, contributing to immune responses along with the host defense system [65]. Because microglia are the resident macrophages of the central nervous system [67], it has been hypothesized that ABCA7 also mediates phagocytic activity in microglia, which may be involved in AD pathogenesis.
4. ABCA7 and Alzheimer Disease-Related Phenotypes
4.1. ABCA7, Neurobehaviors, and Aβ Pathology in Mouse Models
While the roles of ABCA7 in lipid metabolism and macrophage-mediated phagocytosis have been actively studied, its function in the central nervous system has received relatively less attention. Nonetheless, a study has shown that ABCA7 deficiency causes slight, but significant, effects on neurobehaviors in young mice [68]. Male ABCA7-KO mice failed to develop significant short-term novel object recognition at the age of 20 weeks, whereas anxiety, short-term spatial memory, and fear-associated learning were not affected [68]. The cheeseboard task test found that female ABCA7-KO mice had impaired spatial reference memory compared to control mice [68]. Although the sex-dependent phenotypes should be further elucidated, ABCA7 likely plays a role in maintaining neuronal homeostasis rather than neurogenesis [69]. In an aged mouse cohort composed of male and female mice (20–22 months old), spatial memory was significantly impaired in ABCA7-KO mice compared to control mice when analyzed with the Morris Water Maze test [55]. Thus, aging may be a critical factor exacerbating the deleterious effect on cognition caused by ABCA7 loss-of-function.
Several groups, including us, have demonstrated the contribution of ABCA7 to Aβ pathology by crossing ABCA7-KO mice with the amyloid AD model J20 [70], TgCRND8 [71], or APP/PS1 mice [55]. In J20 mice, ABCA7 deficiency aggravates amyloid plaque burden at around 17 months of age accompanied with increased insoluble Aβ levels but not soluble Aβ [70]. TgCRND8 mice lacking ABCA7 showed a substantial increase in the density of both diffuse and dense plaques at an early stage as young as 10 weeks old, where insoluble Aβ levels significantly increased but soluble Aβ was reduced [71]. Consistent with these results, our findings have also demonstrated that ABCA7 deficiency exacerbates amyloid plaque burden and increases soluble/insoluble Aβ42 in APP/PS1 mice at seven months of age [55]. Thus, accumulating evidence indicates that ABCA7 deficiency facilitates brain Aβ deposition in mouse models. In the following sections, we will discuss how ABCA7 is involved in the mechanisms of Aβ clearance and production.
4.2. ABCA7 and Microglial Aβ Clearance
As gene network analyses have demonstrated that microglial expressing genes including CR1, SPI1, the MS4As, TREM2, CD33, and INPP5D, as well as ABCA7, are involved in AD [17], contributions of microglia to the disease pathogenesis have become increasingly focused. While activated microglia produces pro-inflammatory cytokines and reactive oxygen species (ROS) in AD brains, microglia plays a critical role in the cellular uptake and proteostasis of Aβ [72]. Microglia can phagocytize Aβ aggregates, whereas soluble Aβ is taken up through fluid phase micropinocytosis [73]. As discussed above, ABCA7 has been shown to mediate phagocytosis in macrophages. Indeed, the capacity for macrophages [70,74] and microglia [74] from ABCA7-KO mice to take up oligomeric Aβ was significantly reduced compared to wild-type mice. Consistent with those results from in vitro experiments, the elimination of Aβ oligomers in the hippocampus is likely diminished in ABCA7-KO mice [74]. Since the number of plaque-associated Iba1-positive microglia is not affected by ABCA7 deficiency in APP mouse models [55,70], ABCA7 may directly regulate the phagocytic pathways, rather than migration ability, in microglia. In addition, in vivo microdialysis did not detect any significant difference in Aβ clearance from the interstitial fluid between control and ABCA7-KO mice with an APP/PS1 background [55]. Thus, it is possible that ABCA7 predominantly mediates the phagocytosis of Aβ aggregates, but not soluble Aβ species. Since other brain cell types, including astrocytes, neurons, and cerebrovascular cells, also play a critical role in cellular Aβ uptake and subsequent degradation [75], future studies should address how ABCA7 in those cells participates in brain Aβ elimination.
4.3. ABCA7 and APP Processing
Aβ is proteolytically cleaved from APP by processing through β- and γ-secretases [76]. Of note, ABCA7, ABCA1, and ABCG1 are likely involved in the APP processing pathway [77,78]. When ABCG1 or ABCA1 were overexpressed in a CHO cell line expressing human APP, Aβ generation was significantly reduced, although the transient expression of ABCA2 did not affect the Aβ level [78]. ABCA7 overexpression has also been demonstrated to reduce levels of the secreted sAPPα, sAPPβ, and Aβ in CHO-APP cells without affecting the activities of α-, β-, and γ-secretases [77]. On the other hand, suppressing endogenous ABCA7 by siRNA has been shown to facilitate β-secretase cleavage, resulting in increased secretions of sAPPβ, Aβ40, and Aβ42 in HeLa cells [71]. Consistent with these results, ABCA7 knockdown accelerates the production of murine Aβ40 and Aβ42 accompanied with increased β-site amyloid precursor protein cleaving enzyme 1 (BACE1) expression in mouse primary neurons [55]. These findings have been confirmed in ABCA7-KO mice, where ABCA7 deficiency facilitates APP processing and increases Aβ levels in mouse brains with or without the human APP transgene [55,71].
While the endocytosis of APP into endosomes is likely an important step for APP processing and Aβ generation [79], ABCA7 deficit results in enhanced APP endocytosis in microglia, which is predicted to account for increased Aβ production [71]. However, whether ABCA7 can directly interact with APP or indirectly regulate APP trafficking through alternate APP-interacting proteins, and if similar phenotypes are detected in other brain cell types, remains unclear.
In addition, ABCA7 deficiency induces endoplasmic reticulum (ER) stress, represented by the activation of the PERK-eIF2α pathway [55]. Since the phosphorylation of eIF2α has been shown to increase BACE1 levels [80], accelerated APP processing and Aβ production caused by ABCA7 deficiency may be partially explained through the ER stress-related pathway. While SREBP2 is a transcription factor for ABCA7 [47], SREBP2 levels are increased in brains from ABCA7-KO mice [55]. Interestingly, the activation of the SREBP2 pathway likely upregulates BACE1 expression [81], which also might be involved in the mechanisms of ABCA7 deficiency, resulting in enhanced Aβ generation. Therefore, although further studies are needed, increasing evidence suggests that ABCA7 plays a role in regulating APP processing likely through diverse mechanisms. Since the main function of ABCA7 may be linked to lipid metabolism, it is critical to determine if the accelerated APP cleavage induced through ABCA7 deficiency is mediated by an altered lipid profile. Indeed, increased levels of cellular cholesterol and phospholipids have been shown to regulate APP processing ranging/starting from the non-amyloidogenic α-secretase pathway to stimulation of the β- and γ-secretase pathways [82,83,84].
5. Summary and Perspective
Since the discovery of ABCA7 gene variants as susceptibility loci of AD from human genetics studies, a better understanding of the roles of ABCA7 in the central nervous system has been of high significance to explore the pathogenic pathways in AD. As PTC variants in ABCA7 are associated with an increased risk for AD, subsequent studies have proven that ABCA7 deficiency exacerbates brain Aβ accumulation and AD-related phenotype using in vitro and in vivo models. Studies to date have mainly implicated two possible mechanisms whereby ABCA7 loss-of-function contributes to AD pathology; disturbing microglial Aβ clearance and accelerating APP processing. Furthermore, ABCA7 deficiency in microglia may compromise the elimination of diverse brain debris including apoptotic cells during AD progression. It is also possible that ABCA7 deficiency makes brain cells more vulnerable to Aβ toxicity and neuroinflammation in AD (Figure 2). Future studies should clarify the molecular mechanisms underlying the link between ABCA7 and the pathogenic pathways with a specific focus on the potential contribution from lipid metabolism. ABCA7 is likely expressed not only in neurons and microglia, but also in several other brain cell types. Thus, comprehensive single-cell type transcriptome analyses in human and mouse brains, and studies using conditional ABCA7 knockout mice may be necessary to determine cell-type specific contributions of ABCA7 to AD pathogenesis. It is also desired to determine if there is a common pathway between ABCA7-mediated pathways and those of other AD risk genes including APOE4 and TREM2. These studies could provide us with novel insights to develop effective therapeutic strategies for AD. In addition, since lentivirus-mediated ABCA7 overexpression likely relieves the neurotoxicity of Aβ by promoting cell viability and reducing ER stress [85], the upregulation of ABCA7 through pharmacological approaches, including histone deacetylase inhibitors [86], may be beneficial in preventing and treating AD.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant R21AG054890 (to T.K.), a grant from Cure Alzheimer’s Foundation (to T.K.), and a Mayo Clinic Alzheimer’s Disease Research Center pilot grant (to T.A.).
Author Contributions
T.A., M.-L.H. and T.K. reviewed the literature, wrote parts of the text, and provided insights on the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| ABC | ATP binding cassette |
| ABCA7 | ATP-binding cassette transporter A7 |
| AD | Alzheimer’s disease |
| apoA | apolipoprotein A |
| apoE | apolipoprotein E |
| APP | amyloid precursor protein |
| Aβ | amyloid-β |
| BACE1 | β-site amyloid precursor protein cleaving enzyme 1 |
| ER | endoplasmic reticulum |
| ERK | extracellular signal-regulated kinase |
| GWAS | genome-wide association study |
| HMG-CoA | β-hydroxy β-methylglutaryl-CoA |
| HEK | human embryonic kidney |
| KO | knockout |
| LDLR | low density lipoprotein receptor |
| LPC | lysophosphatidyl choline |
| LXR | liver-X-receptor |
| MAF | minor allele frequency |
| MCI | mild cognitive impairment |
| NBD | nucleotide binding domain |
| OR | odds ratio |
| PC | phosphatidylcholine |
| PS | phosphatidylserine |
| PTC | premature termination codon |
| ROS | reactive oxygen species |
| RXR | retinoid-X-receptor |
| SNP | single nucleotide polymorphism |
| SREBP | sterol-responsive/regulatory element binding protein |
References
- Alzheimer’s Association. 2017 alzheimer’s disease facts and figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2016 alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [Google Scholar]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant alzheimer’s disease: A review and proposal for the prevention of alzheimer’s disease. Alzheimer’s Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of genes and environments for explaining alzheimer disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef] [PubMed]
- Bu, G. Apolipoprotein E and its receptors in alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Xu, H.; Bu, G. APOE and Aβ in alzheimer’s disease: Accidental encounters or partners? Neuron 2014, 81, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, R.L. Genome-wide association studies, alzheimer disease, and understudied populations. JAMA 2013, 309, 1527–1528. [Google Scholar] [CrossRef] [PubMed]
- Loy, C.T.; Schofield, P.R.; Turner, A.M.; Kwok, J.B. Genetics of dementia. Lancet 2014, 383, 828–840. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Herz, J.; Bu, G. Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006312. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and alzheimer disease. A meta-analysis. APOE and alzheimer disease meta analysis consortium. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, A.G.; Goate, A.M. Late onset alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, L.M.; Goukasian, N.; Porat, S.; Hwang, K.S.; Eastman, J.A.; Hurtz, S.; Wang, B.; Vang, N.; Sears, R.; Klein, E.; et al. Common variants in ABCA7 and MS4A6A are associated with cortical and hippocampal atrophy. Neurobiol. Aging 2016, 39, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, M.M.; Crook, J.E.; Pedraza, O.; Thomas, C.S.; Pankratz, V.S.; Allen, M.; Nguyen, T.; Malphrus, K.G.; Ma, L.; Bisceglio, G.D.; et al. Late-onset alzheimer’s risk variants in memory decline, incident mild cognitive impairment, and alzheimer’s disease. Neurobiol. Aging 2015, 36, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Logue, M.W.; Schu, M.; Vardarajan, B.N.; Buros, J.; Green, R.C.; Go, R.C.; Griffith, P.; Obisesan, T.O.; Shatz, R.; Borenstein, A.; et al. A comprehensive genetic association study of alzheimer disease in African Americans. Arch. Neurol. 2011, 68, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Jun, G.; Naj, A.; Rajbhandary, R.; Vardarajan, B.N.; Wang, L.S.; Valladares, O.; Lin, C.F.; Larson, E.B.; Graff-Radford, N.R.; et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset alzheimer disease in African Americans. JAMA 2013, 309, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Cukier, H.N.; Kunkle, B.W.; Vardarajan, B.N.; Rolati, S.; Hamilton-Nelson, K.L.; Kohli, M.A.; Whitehead, P.L.; Dombroski, B.A.; Van Booven, D.; Lang, R.; et al. ABCA7 frameshift deletion associated with alzheimer disease in African Americans. Neurol. Genet 2016, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; De Roeck, A.; Van den Bossche, T.; Van Cauwenberghe, C.; Bettens, K.; Vermeulen, S.; Mattheijssens, M.; Peeters, K.; Engelborghs, S.; Vandenbulcke, M.; et al. Mutations in ABCA7 in a Belgian cohort of alzheimer’s disease patients: A targeted resequencing study. Lancet Neurol. 2015, 14, 814–822. [Google Scholar] [CrossRef]
- N’Songo, A.; Carrasquillo, M.M.; Wang, X.; Burgess, J.D.; Nguyen, T.; Asmann, Y.W.; Serie, D.J.; Younkin, S.G.; Allen, M.; Pedraza, O.; et al. African American exome sequencing identifies potential risk variants at alzheimer disease loci. Neurol. Genet. 2017, 3, e141. [Google Scholar] [CrossRef] [PubMed]
- Sassi, C.; Nalls, M.A.; Ridge, P.G.; Gibbs, J.R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Brown, K.S.; et al. ABCA7 p.G215s as potential protective factor for alzheimer’s disease. Neurobiol. Aging 2016, 46, 235.e1–235.e9. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.; Stefansson, H.; Jonsson, T.; Johannsdottir, H.; Ingason, A.; Helgason, H.; Sulem, P.; Magnusson, O.T.; Gudjonsson, S.A.; Unnsteinsdottir, U.; et al. Loss-of-function variants in ABCA7 confer risk of alzheimer’s disease. Nat. Genet. 2015, 47, 445–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellenguez, C.; Charbonnier, C.; Grenier-Boley, B.; Quenez, O.; Le Guennec, K.; Nicolas, G.; Chauhan, G.; Wallon, D.; Rousseau, S.; Richard, A.C.; et al. Contribution to alzheimer’s disease risk of rare variants in TREM2, SORL1, and ABCA7 in 1779 cases and 1273 controls. Neurobiol. Aging 2017, 59, 220.e1–220.e9. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Lincoln, S.J.; Corda, M.; Watzlawik, J.O.; Carrasquillo, M.M.; Reddy, J.S.; Burgess, J.D.; Nguyen, T.; Malphrus, K.; Petersen, R.C.; et al. ABCA7 loss-of-function variants, expression, and neurologic disease risk. Neurol. Genet. 2017, 3, e126. [Google Scholar] [CrossRef] [PubMed]
- Le Guennec, K.; Nicolas, G.; Quenez, O.; Charbonnier, C.; Wallon, D.; Bellenguez, C.; Grenier-Boley, B.; Rousseau, S.; Richard, A.C.; Rovelet-Lecrux, A.; et al. ABCA7 rare variants and alzheimer disease risk. Neurology 2016, 86, 2134–2137. [Google Scholar] [CrossRef] [PubMed]
- Del-Aguila, J.L.; Fernandez, M.V.; Jimenez, J.; Black, K.; Ma, S.; Deming, Y.; Carrell, D.; Saef, B.; Alzheimer’s Disease Neuroimaging Initiative; Howells, B.; et al. Role of ABCA7 loss-of-function variant in alzheimer’s disease: A replication study in European-Americans. Alzheimer’s Res. Ther. 2015, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- De Roeck, A.; Van den Bossche, T.; van der Zee, J.; Verheijen, J.; De Coster, W.; Van Dongen, J.; Dillen, L.; Baradaran-Heravi, Y.; Heeman, B.; Sanchez-Valle, R.; et al. Deleterious ABCA7 mutations and transcript rescue mechanisms in early onset alzheimer’s disease. Acta Neuropathol. 2017, 134, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Vardarajan, B.N.; Ghani, M.; Kahn, A.; Sheikh, S.; Sato, C.; Barral, S.; Lee, J.H.; Cheng, R.; Reitz, C.; Lantigua, R.; et al. Rare coding mutations identified by sequencing of alzheimer disease genome-wide association studies loci. Ann. Neurol. 2015, 78, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Vasquez, J.B.; Fardo, D.W.; Estus, S. ABCA7 expression is associated with alzheimer’s disease polymorphism and disease status. Neurosci. Lett. 2013, 556, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Allikmets, R. Evolution of ATP-binding cassette transporter genes. Curr. Opin. Genet. Dev. 1995, 5, 779–785. [Google Scholar] [CrossRef]
- Kaminski, W.E.; Orso, E.; Diederich, W.; Klucken, J.; Drobnik, W.; Schmitz, G. Identification of a novel human sterol-sensitive ATP-binding cassette transporter (ABCA7). Biochem. Biophys. Res. Commun. 2000, 273, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef] [PubMed]
- Abe-Dohmae, S.; Ikeda, Y.; Matsuo, M.; Hayashi, M.; Okuhira, K.; Ueda, K.; Yokoyama, S. Human ABCA7 supports apolipoprotein-mediated release of cellular cholesterol and phospholipid to generate high density lipoprotein. J. Biol. Chem. 2004, 279, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M.L.; Morris, A.L.; Rhee, J.S.; Andersson, L.P.; Mendez, A.J.; Freeman, M.W. Naturally occurring mutations in the largest extracellular loops of abca1 can disrupt its direct interaction with apolipoprotein AI. J. Biol. Chem. 2002, 277, 33178–33187. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Silver, D.L.; Costet, P.; Tall, A.R. Specific binding of ApoA-I, enhanced cholesterol efflux, and altered plasma membrane morphology in cells expressing ABC1. J. Biol. Chem. 2000, 275, 33053–33058. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Gerbod-Giannone, M.; Linsel-Nitschke, P.; Jehle, A.W.; Chen, W.; Martinez, L.O.; Tall, A.R. ATP-binding cassette transporter A7 (ABCA7) binds apolipoprotein A-I and mediates cellular phospholipid but not cholesterol efflux. J. Biol. Chem. 2003, 278, 42906–42912. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F. Molecular basis of cholesterol homeostasis: Lessons from tangier disease and ABCA1. Trends Mol. Med. 2002, 8, 168–173. [Google Scholar] [CrossRef]
- Denis, M.; Bissonnette, R.; Haidar, B.; Krimbou, L.; Bouvier, M.; Genest, J. Expression, regulation, and activity of ABCA1 in human cell lines. Mol. Genet. Metab. 2003, 78, 265–274. [Google Scholar] [CrossRef]
- Iwamoto, N.; Abe-Dohmae, S.; Sato, R.; Yokoyama, S. ABCA7 expression is regulated by cellular cholesterol through the SREBP2 pathway and associated with phagocytosis. J. Lipid Res. 2006, 47, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Inoue, J.; Kawabe, Y.; Kodama, T.; Takano, T.; Maeda, M. Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. J. Biol. Chem. 1996, 271, 26461–26464. [Google Scholar] [CrossRef] [PubMed]
- Broccardo, C.; Osorio, J.; Luciani, M.F.; Schriml, L.M.; Prades, C.; Shulenin, S.; Arnould, I.; Naudin, L.; Lafargue, C.; Rosier, M.; et al. Comparative analysis of the promoter structure and genomic organization of the human and mouse ABCA7 gene encoding a novel ABCA transporter. Cytogenet. Cell Genet. 2001, 92, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Shoji, A.; Kubo, Y.; Nada, S.; Yamaguchi, A. Cloning of rat ABCA7 and its preferential expression in platelets. Biochem. Biophys. Res. Commun. 2003, 304, 777–782. [Google Scholar] [CrossRef]
- Kim, W.S.; Guillemin, G.J.; Glaros, E.N.; Lim, C.K.; Garner, B. Quantitation of ATP-binding cassette subfamily-A transporter gene expression in primary human brain cells. Neuroreport 2006, 17, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Fitzgerald, M.L.; Kang, K.W.; Okuhira, K.; Bell, S.A.; Manning, J.J.; Koehn, S.L.; Lu, N.F.; Moore, K.J.; Freeman, M.W. ABCA7 null mice retain normal macrophage phosphatidyleholine and cholesterol efflux activity despite alterations in adipose mass and serum cholesterol levels. J. Biol. Chem. 2005, 280, 3989–3995. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Abe-Dohmae, S.; Munehira, Y.; Aoki, R.; Kawamoto, S.; Furuya, A.; Shitara, K.; Amachi, T.; Kioka, N.; Matsuo, M.; et al. Posttranscriptional regulation of human abca7 and its function for the ApoA-I-dependent lipid release. Biochem. Biophys. Res. Commun. 2003, 311, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Sakae, N.; Liu, C.C.; Shinohara, M.; Frisch-Daiello, J.; Ma, L.; Yamazaki, Y.; Tachibana, M.; Younkin, L.; Kurti, A.; Carrasquillo, M.M.; et al. ABCA7 deficiency accelerates amyloid-beta generation and alzheimer’s neuronal pathology. J. Neurosci. 2016, 36, 3848–3859. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Abe-Dohmae, S.; Okazaki, M.; Ueda, K.; Yokoyama, S. Heterogeneity of high density lipoprotein generated by ABCA1 and ABCA7. J. Lipid Res. 2005, 46, 1703–1711. [Google Scholar] [CrossRef] [PubMed]
- Linsel-Nitschke, P.; Jehle, A.W.; Shan, J.; Cao, G.Q.; Bacic, D.; Lan, D.B.; Wang, N.; Tall, A.R. Potential role of ABCA7 in cellular lipid efflux to ApoA-I. J. Lipid Res. 2005, 46, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, M.; Toda, Y.; Manucat, N.B.; Akatsu, H.; Fukumoto, M.; Kono, N.; Arai, H.; Kioka, N.; Ueda, K. Lysophosphatidylcholine export by human ABCA7. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids. 2017, 1862, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Kielar, D.; Kaminski, W.E.; Liebisch, G.; Piehler, A.; Wenzel, J.J.; Mohle, C.; Heimerl, S.; Langmann, T.; Friedrich, S.O.; Bottcher, A.; et al. Adenosine triphosphate binding cassette (ABC) transporters are expressed and regulated during terminal keratinocyte differentiation: A potential role for ABCA7 in epidermal lipid reorganization. J. Investig. Dermatol. 2003, 121, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Nowyhed, H.N.; Chandra, S.; Kiosses, W.; Marcovecchio, P.; Andary, F.; Zhao, M.; Fitzgerald, M.L.; Kronenberg, M.; Hedrick, C.C. ATP binding cassette transporter ABCA7 regulates NKT cell development and function by controlling cd1d expression and lipid raft content. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Bratton, D.L.; Fadok, V.A. Apoptotic cell removal. Curr. Biol. 2001, 11, R795–R805. [Google Scholar] [CrossRef]
- Wu, Y.C.; Horvitz, H.R. The C. Elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 1998, 93, 951–960. [Google Scholar] [CrossRef]
- Li, H.Y.; Karl, T.; Garner, B. Understanding the function of ABCA7 in alzheimer’s disease. Biochem. Soc. Trans. 2015, 43, 920–923. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Abe-Dohmae, S.; Iwamoto, N.; Fitzgerald, M.L.; Yokoyama, S. Helical apolipoproteins of high-density lipoprotein enhance phagocytosis by stabilizing ATP-binding cassette transporter A7. J. Lipid Res. 2010, 51, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Abe-Dohmae, S.; Iwamoto, N.; Yokoyama, S. Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J. Atheroscler. Thromb. 2011, 18, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Jehle, A.W.; Gardai, S.J.; Li, S.; Linsel-Nitschke, P.; Morimoto, K.; Janssen, W.J.; Vandivier, R.W.; Wang, N.; Greenberg, S.; Dale, B.M.; et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J. Cell Biol. 2006, 174, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Logge, W.; Cheng, D.; Chesworth, R.; Bhatia, S.; Garner, B.; Kim, W.S.; Karl, T. Role of ABCA7 in mouse behaviours relevant to neurodegenerative diseases. PLoS ONE 2012, 7, e45959. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Karl, T.; Garner, B. ABCA7 deletion does not affect adult neurogenesis in the mouse. Biosci. Rep. 2016, 36, e00308. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Li, H.; Ruberu, K.; Chan, S.; Elliott, D.A.; Low, J.K.; Cheng, D.; Karl, T.; Garner, B. Deletion of ABCA7 increases cerebral amyloid-beta accumulation in the j20 mouse model of alzheimer’s disease. J. Neurosci. 2013, 33, 4387–4394. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Abe-Dohmae, S.; Yokoyama, S.; St George-Hyslop, P.; Fraser, P.E. ATP-binding cassette transporter A7 (ABCA7) loss of function alters alzheimer amyloid processing. J. Biol. Chem. 2015, 290, 24152–24165. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, S.; Jiang, Q.; Lee, C.Y.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Landreth, G.E. Microglia mediate the clearance of soluble abeta through fluid phase macropinocytosis. J. Neurosci. 2009, 29, 4252–4262. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Hsiao, J.H.; Paxinos, G.; Halliday, G.M.; Kim, W.S. ABCA7 mediates phagocytic clearance of amyloid-beta in the brain. J. Alzheimer’s Dis. 2016, 54, 569–584. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-beta clearance in alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of app. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Kim, W.S.; Kwok, J.B.; Hill, A.F.; Cappai, R.; Rye, K.A.; Garner, B. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J. Neurochem. 2008, 106, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Rahmanto, A.S.; Kamili, A.; Rye, K.A.; Guillemin, G.J.; Gelissen, I.C.; Jessup, W.; Hill, A.F.; Garner, B. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J. Biol. Chem. 2007, 282, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-beta in alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Guglielmotto, M.; Medana, C.; Catalano, M.G.; Cutrupi, S.; Borghi, R.; Tamagno, E.; Boccuzzi, G.; Aragno, M. Dysregulation of SREBP2 induces BACE1 expression. Neurobiol. Dis. 2011, 44, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Bodovitz, S.; Klein, W.L. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 1996, 271, 4436–4440. [Google Scholar] [CrossRef] [PubMed]
- Tun, H.; Marlow, L.; Pinnix, I.; Kinsey, R.; Sambamurti, K. Lipid rafts play an important role in a beta biogenesis by regulating the beta-secretase pathway. J. Mol. Neurosci. 2002, 19, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Vetrivel, K.S.; Cheng, H.; Kim, S.H.; Chen, Y.; Barnes, N.Y.; Parent, A.T.; Sisodia, S.S.; Thinakaran, G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J. Biol. Chem. 2005, 280, 25892–25900. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yuan, Y.; Hu, B.; Wu, L. Study on lentivirus-mediated ABCA7 improves neurocognitive function and related mechanisms in the C57BL/6 mouse model of alzheimer’s disease. J. Mol. Neurosci. 2017, 61, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Volmar, C.H.; Salah-Uddin, H.; Janczura, K.J.; Halley, P.; Lambert, G.; Wodrich, A.; Manoah, S.; Patel, N.H.; Sartor, G.C.; Mehta, N.; et al. M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing alzheimer’s disease genes and improving memory. Proc. Natl. Acad. Sci. USA 2017, 114, E9135–E9144. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Topological model of ABCA7. The full-length ABCA7 is predicted to possess two hydrophobic transmembrane domains and two large loops serving as substrate-binding domains by OCTOPUS [39]. In addition, ABCA7 has two nucleotide binding domains (NBDs) composed of three motifs: Walker A, Walker B, and the signature motifs [40]. Lipid species are transported across the membrane bilayer through binding of ATP to the NBDs.
Figure 1.
Topological model of ABCA7. The full-length ABCA7 is predicted to possess two hydrophobic transmembrane domains and two large loops serving as substrate-binding domains by OCTOPUS [39]. In addition, ABCA7 has two nucleotide binding domains (NBDs) composed of three motifs: Walker A, Walker B, and the signature motifs [40]. Lipid species are transported across the membrane bilayer through binding of ATP to the NBDs.
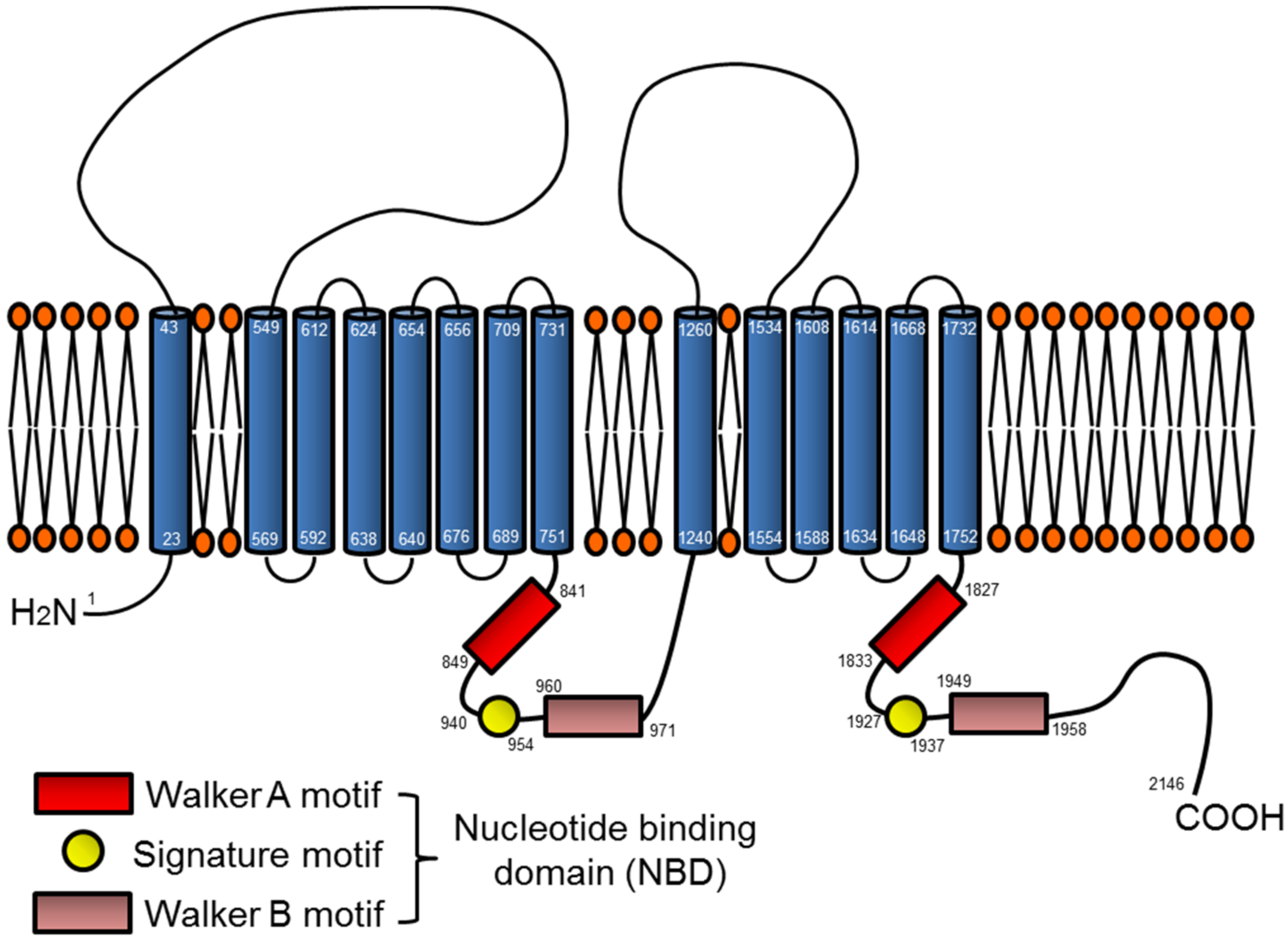
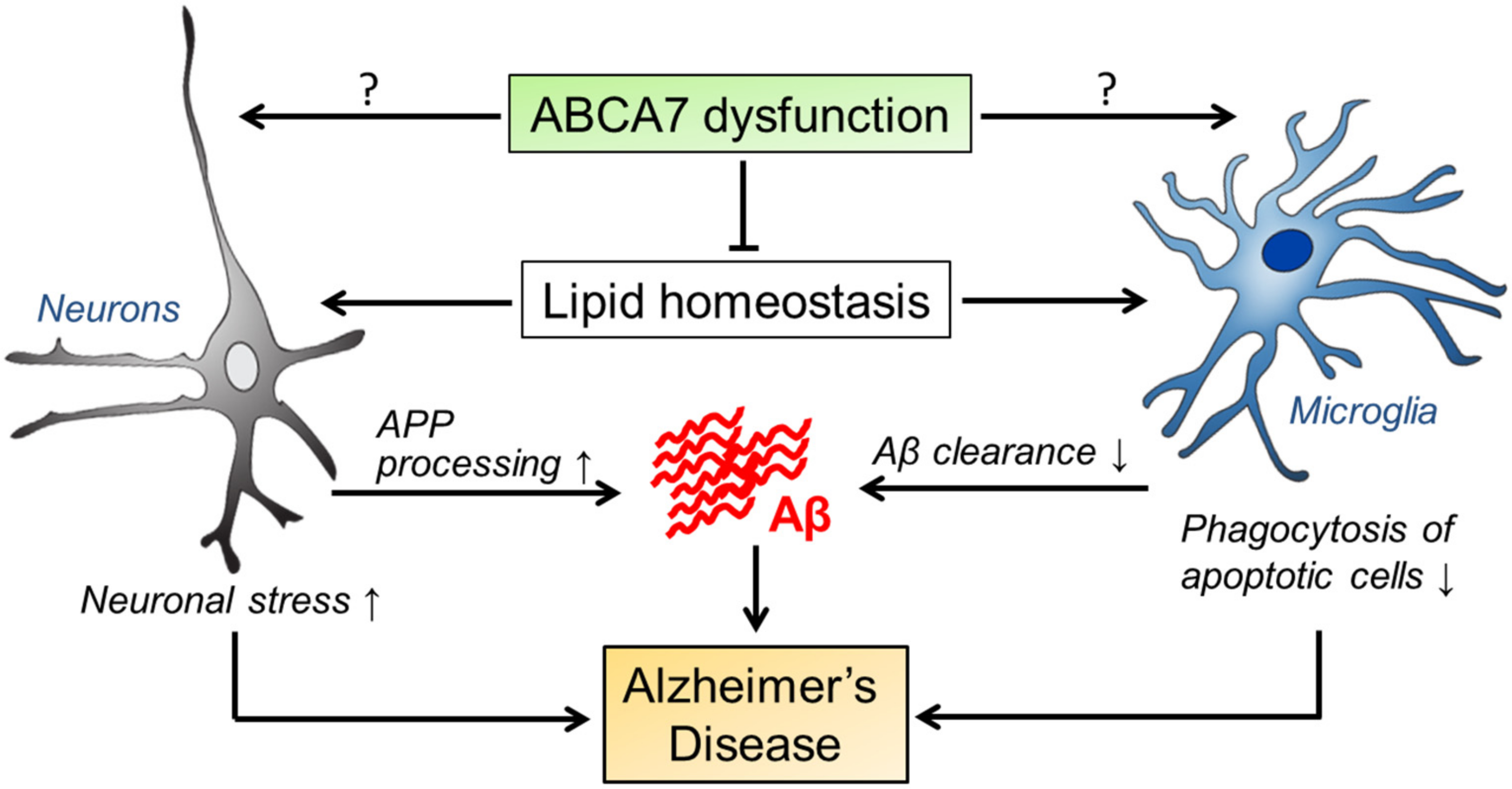
Figure 2.
Possible pathogenic pathways mediated by ABCA7 in AD. ABCA7 dysregulation may influence the properties of brain cell types, in particular neurons and microglia, by disturbing brain lipid homeostasis and/or through unknown direct mechanisms. Those alterations likely facilitate APP processing and suppress cellular Aβ clearance, contributing to AD development. During the disease progression, ABCA7 deficiency may also exacerbate neuronal damages and diminish microglial phagocytic ability.
Figure 2.
Possible pathogenic pathways mediated by ABCA7 in AD. ABCA7 dysregulation may influence the properties of brain cell types, in particular neurons and microglia, by disturbing brain lipid homeostasis and/or through unknown direct mechanisms. Those alterations likely facilitate APP processing and suppress cellular Aβ clearance, contributing to AD development. During the disease progression, ABCA7 deficiency may also exacerbate neuronal damages and diminish microglial phagocytic ability.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Aikawa, T.; Holm, M.-L.; Kanekiyo, T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sci. 2018, 8, 27. https://doi.org/10.3390/brainsci8020027
AMA Style
Aikawa T, Holm M-L, Kanekiyo T. ABCA7 and Pathogenic Pathways of Alzheimer’s Disease. Brain Sciences. 2018; 8(2):27. https://doi.org/10.3390/brainsci8020027
Chicago/Turabian StyleAikawa, Tomonori, Marie-Louise Holm, and Takahisa Kanekiyo. 2018. "ABCA7 and Pathogenic Pathways of Alzheimer’s Disease" Brain Sciences 8, no. 2: 27. https://doi.org/10.3390/brainsci8020027
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.