
The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk

1
Center for Translational Medicine, The First Affiliated Hospital, Sun Yat-Sen University, Guangzhou 510080, China
2
Department of Pathology and the Vascular Biology and Therapeutics Program, Yale University School of Medicine, New Haven, CT 06520, USA
*
Author to whom correspondence should be addressed.
Antioxidants 2017, 6(2), 42; https://doi.org/10.3390/antiox6020042
Submission received: 18 April 2017
/
Revised: 31 May 2017
/
Accepted: 2 June 2017
/
Published: 8 June 2017
(This article belongs to the Special Issue ROS Derived from NADPH Oxidase (NOX) in Angiogenesis)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) family is the major source of reactive oxygen species (ROS) in the vascular system. In this family, NOX4, a constitutive active form of NOXs, plays an important role in angiogenesis. Thioredoxin 2 (TRX2) is a key mitochondrial redox protein that maintains normal protein function and also provides electrons to peroxiredoxin 3 (PRX3) to scavenge H2O2 in mitochondria. Angiogenesis, a process of new blood vessel formation, is involved in a variety of physiological processes and pathological conditions. It seems to be paradoxical for ROS-producing NOX4 and ROS-scavenging TRX2 to have a similar role in promoting angiogenesis. In this review, we will focus on data supporting the role of NOX4 and TRX2 in angiogenesis and their cross-talks and discuss how ROS can positively or negatively regulate angiogenesis, depending on their species, levels and locations. NOX4 and TRX2-mediated ROS signaling could be promising targets for the treatment of angiogenesis-related diseases.
1. Introduction
Angiogenesis, a process of new blood vessel formation, is involved in a variety of physiological processes and pathological conditions [1,2,3]. Excessive angiogenesis can cause cancer, diabetic retinopathy and atherosclerosis, while insufficient angiogenesis links peripheral arterial disease and myocardial infarction. It is reported that reactive oxygen species (ROS) can regulate angiogenesis in both positive and negative manners. In vascular cells, ROS are generated from a number of sources, including the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, xanthine oxidase, the uncoupling of NO synthase and mitochondria [2,3,4,5]. NADPH oxidases have been considered as the major sources of ROS in the vasculature [6]. Recent reports suggest that ROS generated from mitochondria gravely regulate endothelial cell (EC) function [2,3,7,8]. Numerous cardiovascular risk factors contribute to mitochondria malfunction, inducing overproduction of ROS. Under physiological conditions, ROS are known to serve as second messengers in signal transduction that regulate EC growth, proliferation, apoptosis, barrier function, vasodilatation and vascular remodeling [9,10,11]. This is well demonstrated from in vitro hypoxia and in vivo ischemia on angiogenesis. However, excessive ROS production resulting from mitochondrial dysfunction can inhibit reparative angiogenesis by inducing endothelial dysfunction and cell apoptosis under pathological conditions such as diabetes and myocardial infarction.
Angiogenesis is delicately co-regulated by ROS producing oxidases and ROS scavenging enzymes. NOX4 is the major isoform of NADPH oxidases expressed in vascular cells and predominantly produces ROS, which plays an important role in angiogenesis. Thioredoxin 2 (TRX2) is the main ROS-scavenging enzyme in mitochondria that balances the ROS levels and maintains mitochondrial function in various cells. TRX2 also positively regulates ischemia-induced angiogenesis. The aim of this review is to briefly summarize recent progress and information on the redox signaling in angiogenesis with a focus on NOX4 and TRX2.
2. NADPH Oxidase Family
The NADPH oxidase (NOX) family consists of seven members, including NOX1–5 and the dual oxidases (Duox) 1 and 2. NOX1, NOX2, NOX4 and NOX5 are expressed in the vascular system [12]. Except for uncoupled endothelial nitric oxide synthase (eNOS) and mitochondria, the major vascular sources of ROS are the NOX family [12,13]. All five NOX enzymes are transmembrane oxidoreductases containing dual heme, which span the membrane six times. The electrons from NADPH transfer to the two heme residues via flavin adenine dinucleotide (FAD) and ultimately, to O2 to generate ROS [14]. Superoxide anions (O2−) are generated in this process, which can further react to hydrogen peroxide (H2O2) or to peroxynitrite (ONOO−) in the presence of nitric oxide (NO). Recently, NOX4 gained substantial attention because it is readily distinguished from the other NOX isoforms by its activation, type of ROS released, subcellular localization, tissue-specific expression and influence over signaling pathways.
2.1. NOX4
The activation of NOX1–3 depends on phosphorylation and protein-protein interactions of cytosolic subunits, while NOX5 and Duox1 and 2 are Ca2+-activated. In contrast, NOX4 is constitutively activated and can produce ROS in the absence of cytosolic subunits, due to the unique intrinsically-activated NOX4 dehydrogenase (DH) domain, which promotes the constitutive electrons transfer from NADPH to FAD [15]. ROS generation by NOX enzymes occurs through electrons transfer from NADPH to O2 and thus yields O2−. While NOX1–3 and NOX5 appear to release O2−, NOX4 predominantly produces H2O2. Preferential production of H2O2 by NOX4 is attributed to a highly conserved histidine in the third extracytosolic loop (E-loop) of NOX4 that accelerates spontaneous dismutation of superoxide to form H2O2 before it leaves the enzyme [16]. NOX4 directly interacts with p22phox [17], which is a prerequisite for H2O2 generation [18]. The subcellular localization of NOX4 has been reported in nucleus [19,20], focal adhesions [21], endoplasmic reticulum (ER) [22], plasma membrane (PM) [23] and mitochondria [24], depending on specific cell types [14,25]. Recently, another report shows that NOX4 protein contains a 73 amino acids-long mitochondrial localization signal at the N-terminus [26]. The expression of NOX4 in ECs in vivo is higher than other NOX isoforms [27]. However, the cellular localization and function of NOX4 in EC need further investigations.
Compelling evidence demonstrates that NOX4 and its generated H2O2 play an important role in cell proliferation, migration, apoptosis and oxygen sensing, which has been reviewed in detail elsewhere [28]. NOX4 regulated specific signaling pathways and cellular function depends on the level of NOX4 expression, the intracellular location and the cell types. Low level H2O2 derived from NOX4 can activate mitogen-activated protein kinase (MAPK) family members, the transforming growth factor-β1 (TGF-β1)/SMAD2/3 pathway [29] or phosphatidylinositol 3-kinase (PI3K)/ Protein Kinase B (Akt) signaling [30] to promote cell proliferation. In contrast, NOX4 also plays a negative role in hepatocyte proliferation and inhibits liver cancer progression [31]. Increasing evidence suggests that NOX4 participates in promoting cell migration in different cells, which has been reviewed in detail elsewhere [28]. Both cell proliferation and cell migration are crucial during angiogenesis and vascular development.
2.2. The Role of NOX4 in Angiogenesis
Angiogenesis is a tightly regulated multistage process, including vessel sprouting, lumen formation and maturation [32]. Upon pro-angiogenic stimulation, ECs firstly sprout from the pre-existing vascellum after the degradation of extracellular matrix (ECM). Afterwards, these ECs undergo proliferation, migration and differentiation and recruit smooth muscle cells (SMCs) or pericytes to cover the newly-formed vessels to promote their maturation. The essential role of NOX4 in angiogenesis has been the subject of research for years. Hepatopulmonary syndrome (HPS) affects 10–30% of patients with cirrhosis and portal hypertension, and pulmonary angiogenesis contributes to the development of HPS [33]. Two NOX4 single nucleotide polymorphisms (SNPs) (rs585197 and rs2164521) have been found in patients with cirrhosis being evaluated for liver transplantation [33]. NOX4−/− mice exhibit attenuated angiogenesis, reduction of endothelial nitric oxide synthase expression, nitric oxide production and heme oxygenase-1 (HO-1) expression [34]. In contrast, endothelial-specific NOX4 transgenic mice exhibit enhanced angiogenesis and blood flow recovery under ischemia in an eNOS-dependent manner [35]. Expression studies suggest that NOX4 expression predominates in ECs [36] and is lower in the smooth muscle layer of the vessel [34]. It is noted that NOX4 expression in SMCs can be increased in response to injury and decreased under normal conditions in the rat carotid artery [37]. Moreover, NOX4 is an important source of ROS in human ECs [35]. Mechanistic studies indicate that NOX4 promotes angiogenic responses, at least partly, via enhanced activation of receptor tyrosine kinases and the downstream extracellular signal-regulated kinase (ERK) pathway. NOX4 expression also promotes proliferation and migration of endothelial cells and reduces serum deprivation-induced apoptosis [38].
2.3. NOX4 Signaling Pathways and Regulation in Angiogenesis
NOX4-mediated angiogenesis is regulated by many factors, including hypoxia, ischemia, vascular endothelial growth factor (VEGF), fibroblast growth factor-1 and -2 (FGF-1 and FGF-2), tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and TGF-β1.
2.3.1. Hypoxia
Accumulating lines of evidence suggest that hypoxia promotes angiogenesis by increasing NOX4 expression in different cells. In lung tissue from mice and isolated pulmonary artery smooth muscle cells (PASMCs), hypoxia rapidly enhances NOX4 mRNA and protein levels [39]. Overexpression of hypoxia inducible factor 1 alpha (HIF-1α) increases NOX4 expression, while HIF-1α depletion prevents this response [40]. Induction of NOX4 by HIF-1α contributes to maintain ROS levels after hypoxia and hypoxia-induced proliferation of human pulmonary artery SMCs (HPASMCs). Interestingly, exposure of human pulmonary artery endothelial cells (HPAECs) to hyperoxia also enhances mRNA and protein expression of NOX4, and NOX4 siRNA attenuates hyperoxia-induced ROS production, cell migration and capillary tube formation [20]. In cardiac microvascular endothelial cells (CMECs), NOX4 plays a vital role against hypoxia/reoxygenation (H/R) injury by inhibiting apoptosis and promoting migration and angiogenesis via inhibition of prolyl hydroxylase 2 (PHD2)-dependent upregulation of HIF-1α/VEGF proangiogenic signaling in vitro [41]. Hypoxia also leads to a 30–50% increase in angiogenesis and cell migration by signal transducer and activator of transcription 3 (STAT3) phosphorylation in glioblastoma cells [42]. Activated STAT3 leads to HIF-1α and VEGF expression and angiogenesis. In addition, the expression of NOX4 is increased at both mRNA and protein levels in hypoxic glioblastoma cells [43]. The elevated ROS production by NOX4 plays a vital role for STAT3 activation and angiogenesis in hypoxic glioblastoma cells.
Mounting evidence demonstrates that NOX4 plays an important role in the development of pulmonary vascular remodeling and hypertension caused by hypoxia. NOX4 is exclusively upregulated in the pulmonary arterial vessels of mice with chronic exposure to hypoxia and in the vascular lesions of patients with idiopathic pulmonary arterial hypertension (IPAH) [39]. In isolated PASMCs, the expression of NOX4 is increased after exposure to hypoxia in vitro. In another mouse model, chronic intermittent hypoxia (CIH)-induced pulmonary hypertension is associated with increased lung levels of the NOX4 and p22phox, reduced NO bioavailability, increased activity of platelet-derived growth factor receptor β (PDGFRβ) and downstream effector, Akt kinase [44]. On the contrary, activation of peroxisome proliferator–activated receptor γ (PPARγ) with the synthetic ligand rosiglitazone attenuates hypoxia-induced increases in mouse lung NOX4 expression, ROS generation and pulmonary vascular remodeling and hypertension [45]. Hypoxia increases the binding of the nuclear factor-κB (NF-κB) subunit, p65, to the NOX4 promoter in HPASMCs, while PPARγ activation reduces the binding of p65 to the NOX4 promoter [46]. Consistent with this notion, the NOX4 inhibitor GKT137831 attenuates hypoxia-induced H2O2 release, proliferation and TGF-β1 expression and blunts reductions in PPARγ in vitro and in vivo [47]. Collectively, these findings indicate that functional interference with NOX4 may provide a novel therapeutic approach for the treatment of attenuate hypoxia-induced pulmonary hypertension.
2.3.2. Ischemia
Ischemic diseases are characterized by an impaired supply of nutrients and oxygen resulting from narrowed or blocked arteries [48]. Despite considerable advances in the clinic, vascular occlusion and/or microcirculation impairment put patients at constant risk of ischemia and the consequent detrimental effects on quality-of-life and longevity [49]. Blood flow recovery is significantly attenuated in global NOX4−/− mice, as well as in inducible NOX4−/− mice after femoral artery ligation [34]. In contrast, the endothelial-specific NOX4 overexpression mice accelerate blood flow recovery from hind limb ischemia and increase angiogenesis [35]. Tube formation in cultured NOX4−/− lung endothelial cells (LECs) is attenuated and restored by low concentrations of H2O2, while polyethylene glycol (PEG)-catalase attenuates tube formation in control LECs [34]. Mechanistically, loss of NOX4 leads to a reduction of endothelial nitric oxide synthase (eNOS) expression, nitric oxide (NO) production and heme oxygenase-1 (HO-1) expression. The expression of HO-1 is controlled by the redox-sensitive transcription factor Nuclear factor (erythroid-derived 2)-like 2 (Nrf-2), which is stabilized in response to oxidative stress [50]. H2O2, derived from vascular NOX4, prevents Nrf2 degradation [34]. Nrf-2-driven HO-1 expression contributes to the protective effects of NOX4. In conclusion, NOX4 protects vascular function through H2O2 generation, maintenance of NO production and antioxidant HO-1 expression. Consistent with this notion, endogenous EC-derived H2O2 plays a crucial role in reparative neovascularization in response to ischemia by activating eNOS in ECs [51]. NOX4 is also upregulated in neurons under ischemic conditions and plays a role in ischemia-induced brain angiogenesis [52].
2.3.3. VEGF
VEGF, a well-known target of HIF1α, is one of the most potent angiogenesis growth factors and stimulates proliferation, migration and tube formation of ECs and angiogenesis in vivo [53]. VEGF binding VEGF receptor 2 (VEGFR2) initiates tyrosine phosphorylation of VEGFR2 and activates downstream signaling, including ERK1/2, Akt and eNOS, which contribute to angiogenic-related responses in EC [54]. NOX4 is shown to mediate VEGF–induced angiogenesis. In HUVECs, VEGF-activated VEGFR2 induces Rac1 to form a complex with NOX4, resulting in a burst of ROS that further promotes angiogenesis [55]. Conversely, NOX4 can induce HIF-1α and HIF-1α-dependent VEGF expression and angiogenesis [42,43]. In cardiac microvascular ECs, NOX4 plays a protective role against H/R injury by inhibiting apoptosis and promoting migration and angiogenesis via a PHD2-dependent upregulation of the HIF-1α/VEGF proangiogenic signaling [41]. Similarly, NOX4 promotes tumor angiogenesis through stabilization of HIF-1α and induction of VEGF expression [56].
2.3.4. TRAIL
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is a member of the TNF family of cytokines. In human umbilical vein endothelial cells (HUVECs), recombinant TRAIL induces a proangiogenic phenotype, which includes both early (increase in migration, invasion and proliferation) and late (differentiation into vascular cords) angiogenic events in vitro [57]. More importantly, TRAIL is angiogenic to a degree comparable to VEGF. Using Trail−/− mice, the authors demonstrate that TRAIL plays an important role in ischemia-induced angiogenesis in vivo. The angiogenic effect of TRAIL on human microvascular endothelial cell-1 cells lies downstream of FGF-2. Furthermore, TRAIL also promotes SMCs proliferation after arterial injury [58]. Recently, a report demonstrates that TRAIL promotes angiogenesis in vitro by modulating H2O2, eNOS phosphorylation at Ser-1177 and NO production via NOX4 [49].
2.3.5. TGF-β1
Transforming growth factor-β1 (TGF-β1) is a multifunctional growth factor, which plays a vital role in many biological processes including embryonic development, cell proliferation, migration, extracellular matrix production and differentiation of a variety of cell types [29]. TGF-β1 induces the expression of NOX4 and ROS-dependent proliferation in human pulmonary artery SMCs (HPASMCs) [59] and human airway SMCs (HAWSMCs) [60]. TGF-β1 promotes NOX4 expression through the activation of SMAD2/3 and ERK1/2 [59]. In another study, TGF-β1 enhances the phosphorylation of SMAD2 and ERK1/2 to promote proliferation of human microvascular endothelial cells (HMECs) [61]. The activator protein (AP)-1/SMAD binding box located between 3.97 kb and 4.76 kb upstream of the transcriptional start site (TSS) of the human NOX4 promoter is fundamental for the transcription of NOX4 gene induced by TGF-β1 in human lung fibroblasts [62]. TGF-β1 also stimulates NOX4 expression and ROS formation in ECs via the SMAD2 pathway to promote angiogenesis, which is dependent on TGF-β1 receptors I activin receptor-like kinase 5 (ALK5) activity [29]. In murine heart ECs (MHECs) from NOX4-deficient mice, TGF-β1-induced cell proliferation, migration and tube formation are abolished. In vivo, TGF-β1-induced angiogenesis is markedly reduced in NOX4 knockout mice.
On the other hand, NOX4 regulates ischemia-induced angiogenesis through H2O2− and TGF-β1-mediated cell signaling pathways in endothelial specific NOX4 transgenic mouse lines [63]. Application of TGF-β1 increases both VEGFR2 and eNOS expression levels, which are critical for angiogenesis. Targeting NOX4 with a novel inhibitor GKT137831 [2-(2-chlorophenyl)-4-[3-(dimethylamino)phenyl]-5-methyl-1H-pyrazolo[4,3-c]pyridine-3,6(2H,5H)-dione] attenuates hypoxia-induced H2O2 release, proliferation and TGF-β1 expression in human pulmonary artery endothelial cells (HPAECs) and pulmonary arterial smooth muscle cells (HPASMCs) in vitro [47]. Therefore, TGF-β1 can serve as both the upstream and downstream of NOX4 signaling (Figure 1). NOX4-derived angiogenesis is also regulated by many other factors: ubiquitination of p300-histone acetyltransferase (p300-HAT) [64], prostacyclin [65], notch [32], insulin [66], exercise [67], chronic stresses [68] and brain-derived neurotrophic factor (BDNF) [69].
2.4. Role of NOX4-Mediated Angiogenesis in Cancer
Angiogenesis is necessary for the invasive growth and metastasis of tumors and is an important target in the control of cancer progression [70]. Compelling evidence demonstrates that NOX4 and its generated ROS have a close relation to tumor angiogenesis in different cancers. In a carcinogen 3-methylcholanthrene (MCA)-induced fibrosarcoma mice model, there is a significant 38% reduction in tumor vascularization in fibrosarcomas of NOX4−/− mice [56]. The accumulation of HIF-1α and the expression of the HIF-1α-dependent pro-angiogenic genes such as VEGF-A, glucose transporter 1 (GLUT-1) and adrenomedullin are indeed significantly attenuated in tumors of NOX4−/− mice compared to wild type (WT) mice. HIF1α is a key transcription factor of angiogenesis in solid tumors [71,72] and tumors lacking HIF-1α show significantly reduced vascularization [73]. NOX4 promotes tumor angiogenesis by stabilization of HIF-1α and induction of VEGF expression [56]. In von Hippel Lindau (VHL)-deficient renal cell carcinoma (RCC), NOX4 also promotes renal tumorigenesis in a similar signal pathway via nuclear accumulation of HIF-2α, not HIF-1α [74]. HIF1α expression is biased toward HIF2α [75], whose transcriptional activity is dependent on NOX4 in VHL-deficient RCC [76]. Stable NOX4 knockdown by shRNA significantly reduces ROS production and suppresses glioblastoma cells proliferation and invasion and tumor-associated angiogenesis [77]. Taken together, NOX4 is implicated in tumor angiogenesis of different cancer types, including fibrosarcoma, VHL-deficient RCC, glioblastoma and human astroglioma [78]. Therefore, NOX4 might be a promising target for anti-angiogenic tumor therapy.
3. The Thioredoxin System
The thioredoxin (TRX) system belongs to thiol-disulfide oxide reductase that can decrease the level of ROS in cells. The antioxidant function of the TRX system is through reducing peroxiredoxins (Prx) to scavenge ROS [79]. The TRX system can be classified into different isoforms according to the subcellular localization. The TRX1 isoform is distributed in the cytoplasm and nucleus, and the TRX2 isoform is specifically identified in the mitochondria. Homozygous knockout of any isoform in mice can cause early embryonic lethality [80,81]. TRX molecules can be detected throughout the whole process of development, revealing that the TRX system is necessary for life [82,83,84]. TRX proteins contain a conserved active site Trp-Cys-Gly-Pro-Cys [85] that reduces ROS and other oxidized proteins. Using the electron donor NADPH, TRX reductase-1(TRXR1) and TRX reductase-2 (TRXR2) can regenerate TRX1 and TRX2. The catalytic motif of TRX isoforms can be modulated by thioredoxin-interacting protein (TXNIP), inhibiting the function of TRX isoforms to decrease disulfides of the substrates and withstand reversible oxidation. It has been confirmed in several cell lines in vitro [86,87,88], but the results have not been confirmed in vivo [89,90]. All of these traits indicate that TXNIP belong to the endogenous inhibitors of the TRX system.
TRX1 is ubiquitously expressed. TRX2 precursor protein has a mitochondrial localization sequence, and the mature TRX2 protein is localized in mitochondria [91]. In mitochondria, the TRX2 system includes TRX2, TRX2 reductase (TRXR2) and TRX2 dependent PRX3. TRX2 is widely expressed in human tissues, especially enhanced expression in metabolically exuberant organs, like heart, brain and liver [92]. TRX1/2 regulate their target proteins to affect cell growth, apoptosis, inflammatory response and other physiological processes [93]. The redox status of the cysteine groups in certain nucleus transcription factors directly affects their DNA binding capacity, and TRX1 in the nucleus participates in the maintenance of the reduction state of the cysteine residues to enhance the transcription factor activity. Similar to TRX1, TRX2 regulates the transcription factor NF-κB [94] and apoptosis signaling kinase-1 (ASK1). Both TRX1 and TRX2 also regulate ASK1, but in distinct mechanisms. TRX1 interacts with ASK1, promoting ASK1 ubiquitination and degradation to suppress ASK1-mediated apoptosis [95,96]. TRX2 can bind to ASK1 located in the mitochondria and block its activity. Auranofin is a metal phosphine complex initially developed for treatment of rheumatoid arthritis, which has been widely used as an inhibitor of TRX2 to promote ECs apoptosis [97]. TXNIP can also bind to the reduced TRX2, reducing its activity [98]. In the normal growth state of the cells, TXNIP is mainly located in the nucleus, while TRX2 binds ASK1 to block its protein kinase activity in mitochondria [99]. When the cells are under oxidative stress, TXNIP is localized from the nucleus to mitochondria and competes with ASK1 to bind TRX2, causing ASK1 to change from an inhibited state to an active state to induce cell apoptosis. That suggests TRX2 is an endogenous inhibitor of ASK1. Consistently, TRX2 knockdown in EC promotes ASK1 activation and cell apoptosis [99]. Conversely, ECs isolated from TRX2-TG show more resistance to ASK1 activation-induced oxidative stress and apoptosis [100].
3.1. TRX1/2 and Angiogenesis
TRX1/2 in ROS scavenging, anti-apoptosis and NF-κB activation implicate their potential function in angiogenesis. In this regard, TRX2 is better understood than TRX1. EC-specific transgenesis of TRX2 (TRX2-TG) has been constructed [101]. By reducing oxidative stress, EC-specific transgenesis of TRX2 (TRX2-TG) reduces atherosclerotic lesions at aortic roots and improves aortic EC function in an ApoE-deficient mouse model [101]. Using TRX2-TG mice, the critical role of TRX2 is to enhance ischemia-mediated arteriogenesis and angiogenesis by increasing NO bioavailability and decreasing apoptosis in EC [100]. The phenotype of TRX2-TG mice is similar to the EC-specific eNOS transgenic mice (eNOS-TG). More importantly, expression of TRX2 in eNOS-KO mice partly rescues the angiogenic defects of the eNOS-KO mice, indicating that that TRX2-augmented NO bioactivity contributes significantly to enhanced angiogenesis in TRX2-TG mice [100]. It is TRX2, but not TRX1, that is downregulated in ischemia. Ischemia appears to regulate TRX2 at the transcriptional level. Therefore, TRX2 plays a critical role in ischemia-mediated arteriogenesis and angiogenesis by decreasing oxidative stress and increasing NO bioactivity (Figure 2).
Apoptosis and cell survival are critical components in angiogenesis and vascular remodeling, which can be regulated by the TRX system. TRX1/2 inhibit apoptosis in both a redox-dependent (by scavenging ROS enhancing NO activity) and a redox activity independent manner (by inhibiting ASK1) [96]. This was elucidated by crossing TRX2-TG with eNOS-deficient or ASK1-deficient mice [100]. Conversely, overexpression of ASK1 in ECs inhibits cell migration and vascular network formation induced by VEGF [102]. Of note, ASK1-interating protein-1(AIP1)-deficient mice have enhanced ischemia and inflammatory angiogenesis in ischemic hind limb and sponge granuloma models [103].
The transcription factor NF-κB regulates various angiogenesis-related genes and proteins, including Akt activation of VEGF, which plays an important role in ECs proliferation, migration and survival [104,105]. NF-κB can directly promote vascular sprouting and neovascularization. TRX1 can be transferred from the cytoplasm to the nucleus, reducing the cysteine residues of NF-κB and activator protein-1(AP-1) nucleoprotein to enhance their transcriptional activity [106,107]. The TRX2 system also regulates NF-κB activity by a yet-to-be-defined mechanism. It is certain that NF-κB contributes to TRX1/2-mediated angiogenesis.
3.2. Role of TRX1/2-Mediated Angiogenesis in Cancer
Compelling evidence from both clinical and laboratory studies demonstrates that the TRX system presents as potential target for anticancer drug development [108,109,110]. Enhanced levels of the TRX system are observed in many aggressive tumors [111,112,113]. Moreover, transfection with dominant-negative mutant TRX or depletion of TRXR results in retardation in tumor progression, metastasis and tumor-derived angiogenesis [114,115,116,117].
Recently some cationic triphenylmethanes such as brilliant green (BG) and gentian violet have shown potent antitumor and antiangiogenic activity in mice and humans [118,119]. Mechanistic studies indicate that triphenylmethane dyes induce TRX2 oxidized and degraded by mitochondrial Lon protease [120]. BG can kill cells at nanomolar concentrations and target mitochondrial TRX2, which was oxidized and degraded. In HeLa cells, TRX2 down-regulation by siRNA results in increased sensitivity to BG, whereas for fibroblasts, the same treatments have no effect. BG accumulates in mitochondria and causes a rapid and dramatic decrease in mitochondrial TRX2 protein. Treatment with BG causes oxidation of both TRX1 and TRX2, followed by release of cytochrome c and apoptosis-inducing factor from the mitochondria into the cytosol. Furthermore, this treatment results in an elevation of the mRNA level of Lon protease, a protein quality control enzyme in the mitochondrial matrix, suggesting that the oxidized TRX2 may be degraded by Lon protease. Recently, we have shown that auranofin, a metal phosphine complex, decreases cellular survival protein TRXR2, TRX2 and transcription factor NF-κB whereas increases stress signaling p38MAPK, leading to EC apoptosis at high doses (≥1 μΜ) [97]. In another study, we show that 1,2-bis(methylsulfonyl)-1-[(methylamino)carbonyl] hydrazine (101MDCE), an analog of laromustine that generates only methyl isocyanate, activates ASK1-JNK/p38 signaling in EC [121]. Mechanistically, methyl isocyanate induces dissociation of ASK1 from Trx1 either directly by carbamoylating the critical Cys groups in the ASK1-Trx1 complex or indirectly by inhibiting TRXR1. Our study supports that 101MDCE induces EC death through a non-apoptotic (necroptotic) pathway leading to inhibition of angiogenesis in vitro. Furthermore, one preliminary study finds that TRX2-transgenic mice have a slightly higher incidence of cancer than wild-type mice at old age (24–26 months) [122]. Therefore, the anti-tumor effects of anti-TRX system agents may be largely due to their anti-angiogenic activities. However, the role of TRX1/2-mediated angiogenesis in cancer is not well studied at this moment. The potential effect of TRX1/2-mediated angiogenesis in cancer tumor development in humans needs to be very carefully addressed.
4. The Potential Cross-Talk between NOX4 and TRX2 in Angiogenesis
The similarities and differences between NOX4 and TRX2-mediated angiogenesis can be readily compared because both NOX4 [35] and TRX2 [100,101] endothelial-specific expression mice have been generated. Both NOX4 and TRX2 play import roles in hindlimb ischemia-induced angiogenesis. TRX2 maintains EC function by two parallel pathways: scavenging ROS to increase NO bioavailability and inhibiting ASK1 activity to enhance EC survival [100]. Similarly, NOX4 promotes angiogenesis and recovery from hypoxia through enhancing eNOS activation and blocking ASK1 activity [35]. However, TRX2 and NOX4 regulate these two common targets (NO and ASK1) by distinct mechanisms. ECs overexpressing TRX2 may regulate NO bioavailability without significant effects on eNOS expression, phosphorylation and/or enzymatic activity. In cultured ECs overexpressing NOX4, eNOS protein expression and activity significantly increase. Endothelial eNOS is a key regulator of angiogenesis [123]. For ASK1, TRX2 directly binds to mitochondria-located ASK1 and blocks mitochondrial apoptotic signaling. NOX4-generated ROS also stimulate phosphorylation of Akt, which in turn inactivates ASK1 by phosphorylating ASK1 on Ser-83, leading to an antiapoptotic effect [124]. Therefore, it is plausible that the TRX2 and NOX4 pathways synergistically promote angiogenesis. However, TRX2 and NOX4 genes are distinctly regulated under pathogenesis. Expression of TRX2 is drastically reduced in response to hypoxia in vitro and in vivo [125]. In contrast, NOX4 increases in response to hypoxia in vitro and in vivo [35]. Similarly, hypoxia inhibits TRX2 expression, but promotes NOX4 expression of pulmonary hypertension [39,125]. The reciprocal expression of TRX2 and NOX4 under pathological conditions suggests that TRX2 and NOX4 exhibit synergistic effects in physiological angiogenesis when both TRX2 and NOX4 are expressed. However, NOX4 may play a more predominant role for pathological angiogenesis with the reduced or absent of TRX2. Moreover, NOX4 may compensate TRX2 function under pathological settings. In the heart tissues from cardiac-specific TRX2 knockout mice, gene analyses by NanoString technology show that NOX4 expression increases [126]. Consistent with our findings, expression of a dominant negative C93S TRX2 mutant that mimics TRX2 oxidation exacerbated hypoxia-induced increases in HPASMC H2O2 levels and cell proliferation [125], which may be a result of damaged TRX2-derived increase of NOX4 expression. It is unclear how TRX2 deficiency induces NOX4 expression, and it is likely that mitochondria-derived ROS could directly activate NOX4 or indirectly increase NOX4 expression. It has also been reported that NOX4 can be located in the mitochondria where NOX4 may regulate mitochondria ROS generation. NOX4 and TRX1/2 are also regulated by similar angiogenic factors such as hypoxia, ischemia and VEGF. NOX4-mediated angiogenesis is also regulated by many other factors, including FGF-1, FGF-2, TRAIL and TGF-β1. An important question is to address if these angiogenic factors contribute to TRX2-mediated angiogenesis. The mechanism responsible for the cross-talk between NOX4 and TRX2 requires further investigations (Figure 3).
5. Conclusions
The effects of ROS on angiogenesis are dependent on the amount, species and site of production, as well as the balance of pro-oxidant and antioxidant enzyme activity. The mechanisms of NOX4 in all forms of angiogenesis have been the subject of research for years. However, NOX4-specific inhibitors or activators are urgently required. Although the role of TRX2 in ischemia-induced angiogenesis has been studied, further research is required to define the mechanism of TRX2 regulation in various pathological settings. In spite of increasing lines of evidence suggesting that NOX4 and TRX2 may have a cross-talk in angiogenesis, more studies are needed to illustrate the underlying mechanisms, which may provide that NOX4 and TRX2 are potential therapeutic targets for the treatment of angiogenesis-dependent diseases.
Acknowledgments
This work was partly supported by National Key Research and Development Program of China (2016YFC1300600), the National Natural Science Foundation of China (No. 91539110) and the Scientific Grants of Guangdong (Nos. 2015B020225002 and 2015A050502018) to Wang Min, R01 HL109420 and R01 HL115148 and Connecticut Stem Cell Innovation Award (Established Investigator Grant) 14-SCB-YALE-17 to Wang Min.
Author Contributions
Chaofei Chen, Li Li, Huanjiao Jenny Zhou and Wang Min contributed to writing the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Ballinger, S.W.; Patterson, C.; Knight-Lozano, C.A.; Burow, D.L.; Conklin, C.A.; Hu, Z.; Reuf, J.; Horaist, C.; Lebovitz, R.; Hunter, G.C.; et al. Mitochondrial integrity and function in atherogenesis. Circulation 2002, 106, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Wolin, M.S. Interactions of oxidants with vascular signaling systems. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ. Res. 2005, 96, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Levonen, A.L.; Brookes, P.S.; Ceaser, E.; Shiva, S.; Barone, M.C.; Darley-Usmar, V. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Radic. Biol. Med. 2002, 33, 1465–1474. [Google Scholar] [CrossRef]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; Subbaram, S.; Regan, K.J.; Nelson, K.K.; Mazurkiewicz, J.E.; Bartholomew, P.J.; Aplin, A.E.; Tai, Y.T.; Aguirre-Ghiso, J.; Flores, S.C.; et al. Mitochondrial H2O2 regulates the angiogenic phenotype via pten oxidation. J. Biol. Chem. 2005, 280, 16916–16924. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. NADPH oxidases in cardiovascular disease. Free Radic. Biol. Med. 2010, 49, 687–706. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J. From form to function: The role of NOX4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef] [PubMed]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Gorshkova, I.A.; Usatyuk, P.V.; He, D.; Pennathur, A.; Lambeth, J.D.; Thannickal, V.J.; Natarajan, V. Role of NOX4 and NOX2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid. Redox. Signal. 2009, 11, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vas. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Van Buul, J.D.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and localization of Nox2 and Nox4 in primary human endothelial cells. Antioxid. Redox Signal. 2005, 7, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Von Lohneysen, K.; Noack, D.; Jesaitis, A.J.; Dinauer, M.C.; Knaus, U.G. Mutational analysis reveals distinct features of the Nox4-p22phox complex. J. Biol. Chem. 2008, 283, 35273–35282. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Block, K.; Gorin, Y.; Abboud, H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.A.; Kulawiec, M.; Owens, K.M.; Li, X.; Desouki, M.M.; Chandra, D.; Singh, K.K. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol. Ther. 2010, 10, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of Nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Chen, X. The human Nox4: Gene, structure, physiological function and pathological significance. J. Drug Target. 2015, 23, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Peshavariya, H.M.; Chan, E.C.; Liu, G.S.; Jiang, F.; Dusting, G.J. Transforming growth factor-beta1 requires NADPH oxidase 4 for angiogenesis in vitro and in vivo. J. Cell. Mol. Med. 2014, 18, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lan, T.; Hou, J.; Li, J.; Fang, R.; Yang, Z.; Zhang, M.; Liu, J.; Liu, B. Nox4 promotes non-small cell lung cancer cell proliferation and metastasis through positive feedback regulation of PI3K/Akt signaling. Oncotarget 2014, 5, 4392–4405. [Google Scholar] [CrossRef] [PubMed]
- Crosas-Molist, E.; Bertran, E.; Sancho, P.; Lopez-Luque, J.; Fernando, J.; Sanchez, A.; Fernandez, M.; Navarro, E.; Fabregat, I. The NADPH oxidase Nox4 inhibits hepatocyte proliferation and liver cancer progression. Free Radic. Biol. Med. 2014, 69, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.X.; Liang, L.; Wang, L.; Han, J.T.; Zhu, X.X.; Han, H.; Hu, D.H.; Zhang, P. Inhibition of notch signaling leads to increased intracellular ROS by up-regulating Nox4 expression in primary HUVECs. Cell. Immunol. 2014, 287, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.E.; Kawut, S.M.; Krowka, M.J.; Brown, R.S.; Trotter, J.F.; Shah, V.; Peter, I.; Tighiouart, H.; Mitra, N.; Handorf, E.; et al. Genetic risk factors for hepatopulmonary syndrome in patients with advanced liver disease. Gastroenterology 2010, 139, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F. NADPH Oxidase 4 Promotes Endothelial Angiogenesis Through Endothelial Nitric Oxide Synthase Activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K.; Lassegue, B.; Sorescu, D.; Hilenski, L.L.; Valppu, L.; Couse, T.L.; Wilcox, J.N.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Roth, M.; Konig, P.; Hofmann, S.; Dony, E.; Goyal, P.; Selbitz, A.C.; Schermuly, R.T.; Ghofrani, H.A.; Kwapiszewska, G.; et al. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit Nox4 in the pulmonary vasculature. Circ. Res. 2007, 101, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The NADPH oxidase subunit Nox4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell. 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hong, Z.; Zeng, C.; Yu, Q.; Wang, H. NADPH oxidase 4 promotes cardiac microvascular angiogenesis after hypoxia/reoxygenation in vitro. Free Radic. Biol. Med. 2014, 69, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Yu, M.O.; Park, K.J.; Chi, S.G.; Park, D.H.; Chung, Y.G. Activated STAT3 regulates hypoxia-induced angiogenesis and cell migration in human glioblastoma. Neurosurgery 2010, 67, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.O.; Park, K.J.; Park, D.H.; Chung, Y.G.; Chi, S.G.; Kang, S.H. Reactive oxygen species production has a critical role in hypoxia-induced Stat3 activation and angiogenesis in human glioblastoma. J. Neurooncol. 2015, 125, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Graves, A.S.; Kleinhenz, D.J.; Rupnow, H.L.; Reed, A.L.; Fan, T.H.; Mitchell, P.O.; Sutliff, R.L.; Hart, C.M. The role of NADPH oxidase in chronic intermittent hypoxia-induced pulmonary hypertension in mice. Am. J. Respir. Cell. Mol. Biol. 2009, 40, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.E.; Bland, J.M.; Kleinhenz, D.J.; Mitchell, P.O.; Walp, E.R.; Sutliff, R.L.; Hart, C.M. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am. J. Respir. Cell. Mol. Biol. 2010, 42, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Murphy, T.C.; Nanes, M.S.; Hart, C.M. PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2010, 299, L559–L566. [Google Scholar] [CrossRef] [PubMed]
- Green, D.E.; Murphy, T.C.; Kang, B.Y.; Kleinhenz, J.M.; Szyndralewiez, C.; Page, P.; Sutliff, R.L.; Hart, C.M. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell. Mol. Biol. 2012, 47, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; Madeddu, P. Angiogenesis gene therapy to rescue ischaemic tissues: Achievements and future directions. Br. J. Pharmacol. 2001, 133, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolo, B.A.; Cartland, S.P.; Prado-Lourenco, L.; Griffith, T.S.; Gentile, C.; Ravindran, J.; Azahri, N.S.; Thai, T.; Yeung, A.W.; Thomas, S.R.; et al. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Promotes Angiogenesis and Ischemia-Induced Neovascularization Via NADPH Oxidase 4 (Nox4) and Nitric Oxide-Dependent Mechanisms. J. Am. Heart. Assoc. 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Srisook, K.; Kim, C.; Cha, Y.N. Molecular mechanisms involved in enhancing HO-1 expression: De-repression by heme and activation by Nrf2, the “one-two” punch. Antioxid. Redox. Signal. 2005, 7, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Sudhahar, V.; Kim, S.J.; Chen, G.F.; McKinney, R.D.; Kojda, G.; Fukai, T.; Ushio-Fukai, M. Critical role of endothelial hydrogen peroxide in post-ischemic neovascularization. PLoS ONE 2013, 8, e57618. [Google Scholar] [CrossRef] [PubMed]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.P.; Szanto, I. Neuronal expression of the NADPH oxidase Nox4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Zachary, I.; Gliki, G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc. Res. 2001, 49, 568–581. [Google Scholar] [CrossRef]
- Ushio-Fukai, M.; Alexander, R.W. Reactive oxygen species as mediators of angiogenesis signaling: Role of NAD(P)H oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Jiang, T.; Lu, D.; Luo, Y.; Zheng, C.; Feng, J.; Yang, D.; Chen, C.; Yan, X. NADPH oxidase 4 mediates reactive oxygen species induction of CD146 dimerization in VEGF signal transduction. Free Radic. Biol. Med. 2010, 49, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Helfinger, V.; Henke, N.; Harenkamp, S.; Walter, M.; Epah, J.; Penski, C.; Mittelbronn, M.; Schroder, K. The NADPH Oxidase Nox4 mediates tumour angiogenesis. Acta. Physiol. 2016, 216, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Gonelli, A.; Carnevale, E.; Corallini, F.; Rizzardi, C.; Zacchigna, S.; Melato, M.; Zauli, G. Evidence for a proangiogenic activity of TNF-related apoptosis-inducing ligand. Neoplasia 2004, 6, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Prado-Lourenco, L.; Khachigian, L.M.; Bennett, M.R.; Di Bartolo, B.A.; Kavurma, M.M. Trail promotes VSMC proliferation and neointima formation in a FGF-2-, Sp1 phosphorylation-, and NFKappaB-dependent manner. Circ. Res. 2010, 106, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Cahill, B.; Norman, K.; Huecksteadt, T.P.; Hill, K.; Sanders, K.; Karwande, S.V.; Stringham, J.C.; Bull, D.A.; Gleich, M.; et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L661–L673. [Google Scholar] [CrossRef] [PubMed]
- Sturrock, A.; Huecksteadt, T.P.; Norman, K.; Sanders, K.; Murphy, T.M.; Chitano, P.; Wilson, K.; Hoidal, J.R.; Kennedy, T.P. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. AJP Lung Cell. Mol. Physiol. 2007, 292, L1543–L1555. [Google Scholar] [CrossRef] [PubMed]
- Hakami, N.Y.; Wong, H.; Shah, M.H.; Dusting, G.J.; Jiang, F.; Peshavariya, H.M. Smad-independent pathway involved in transforming growth factor beta1-induced Nox4 expression and proliferation of endothelial cells. Naunyn-Schmiedeberg Arch. Pharmacol. 2015, 388, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Bai, G.; Hock, T.D.; Logsdon, N.; Zhou, Y.; Thannickal, V.J. A far-upstream AP-1/Smad binding box regulates human NOX4 promoter activation by transforming growth factor-beta. Gene 2014, 540, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiao, J.; Kuroda, J.; Ago, T.; Sadoshima, J.; Cohen, R.A.; Tong, X. Both hydrogen peroxide and transforming growth factor beta 1 contribute to endothelial Nox4 mediated angiogenesis in endothelial Nox4 transgenic mouse lines. Biochim. Biophys. Acta 2014, 1842, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Hakami, N.Y.; Dusting, G.J.; Peshavariya, H.M. Trichostatin A, a histone deacetylase inhibitor suppresses NADPH Oxidase 4-Derived Redox Signalling and Angiogenesis. J. Cell. Mol. Med. 2016, 20, 1932–1944. [Google Scholar] [CrossRef] [PubMed]
- Peshavariya, H.M.; Liu, G.S.; Chang, C.W.; Jiang, F.; Chan, E.C.; Dusting, G.J. Prostacyclin signaling boosts NADPH oxidase 4 in the endothelium promoting cytoprotection and angiogenesis. Antioxid. Redox Signal. 2014, 20, 2710–2725. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Mei, A.; Liu, J.; Kang, X.; Shi, X.; Qian, R.; Chen, S. NADPH oxidase 4 mediates insulin-stimulated HIF-1alpha and VEGF expression, and angiogenesis in vitro. PLoS ONE 2012, 7, e48393. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Kruse, C.; Zhang, M.; Schroder, K. Nox4 supports proper capillary growth in exercise and retina neo-vascularization. J. Physiol. 2015, 593, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Brewer, A.C.; Schroder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Naruo, A.; Okada, M.; Hayabe, Y.; Yamawaki, H. Brain-derived neurotrophic factor promotes angiogenic tube formation through generation of oxidative stress in human vascular endothelial cells. Acta Physiol. 2014, 211, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Role of angiogenesis in tumor growth and metastasis. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.L.; Wang, S.; Klco, J.M.; Kaelin, W.G.; Livingston, D.M. Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat. Med. 2000, 6, 1335–1340. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.; Giaccia, A.J.; Denko, N.C. The hypoxic tumor microenvironment and gene expression. Semin. Radiat. Oncol. 2004, 14, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 394, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Gregg, J.L.; Turner, R.M.; Chang, G.; Joshi, D.; Zhan, Y.; Chen, L.; Maranchie, J.K. NADPH Oxidase NOX4 Supports Renal Tumorigenesis by Promoting the Expression and Nuclear Accumulation of HIF2. Cancer Res. 2014, 74, 3501–3511. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kim, W.Y.; Lechpammer, M.; Kaelin, W.G. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003, 1, E83. [Google Scholar] [CrossRef] [PubMed]
- Maranchie, J.K.; Zhan, Y. Nox4 is critical for hypoxia-inducible factor 2-alpha transcriptional activity in von Hippel-Lindau-deficient renal cell carcinoma. Cancer Res. 2005, 65, 9190–9193. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, N.; Yin, T.; Huang, L.; Liu, S.; Liu, D.; Xie, C.; Zhang, M. Lentivirus-Mediated Nox4 shRNA Invasion and Angiogenesis and Enhances Radiosensitivity in Human Glioblastoma. Oxid. Med. Cell. Longev. 2014, 2014, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.S.; Ahn, Y.H.; Moon, B.I.; Kim, H.S. Exogenous C2 Ceramide Suppresses Matrix Metalloproteinase Gene Expression by Inhibiting ROS Production and MAPK Signaling Pathways in PMA-Stimulated Human Astroglioma Cells. Int. J. Mol. Sci. 2016, 17, 477. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Chae, H.Z.; Seo, M.S.; Kim, K.; Baines, I.C.; Rhee, S.G. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J. Biol. Chem. 1998, 273, 6297–6302. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Oshima, M.; Oshima, H.; Takaku, K.; Maruyama, T.; Yodoi, J.; Taketo, M.M. Early embryonic lethality caused by targeted disruption of the mouse thioredoxin gene. Dev. Biol 1996, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Nonn, L.; Williams, R.R.; Erickson, R.P.; Powis, G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 2003, 23, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.C.; Moore, E.C.; Reichard, P. Enzymatic Synthesis of Deoxyribonucleotides. Iv. Isolation and Characterization of Thioredoxin, the Hydrogen Donor from Escherichia Coli B. J. Biol. Chem. 1964, 239, 3436–3444. [Google Scholar] [PubMed]
- Moore, E.C. A thioredoxin—Thioredoxin reductase system from rat tumor. Biochem. Biophys. Res. Commun. 1967, 29, 264–268. [Google Scholar] [CrossRef]
- Clarke, F.M.; Orozco, C.; Perkins, A.V.; Cock, I.; Tonissen, K.F.; Robins, A.J.; Wells, J.R. Identification of molecules involved in the “early pregnancy factor” phenomenon. J. Reprod. Fertil. 1991, 93, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Spyrou, G.; Enmark, E.; Miranda-Vizuete, A.; Gustafsson, J. Cloning and expression of a novel mammalian thioredoxin. J. Biol. Chem. 1997, 272, 2936–2941. [Google Scholar] [CrossRef] [PubMed]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular shuttling and mitochondrial function of thioredoxin-interacting protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Liu, H.; Choe, E.; Yoshioka, J.; Shalev, A.; Bloch, K.D.; Lee, R.T. Nitric oxide-dependent suppression of thioredoxin-interacting protein expression enhances thioredoxin activity. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2666–2672. [Google Scholar] [CrossRef] [PubMed]
- Turturro, F.; Friday, E.; Welbourne, T. Hyperglycemia regulates thioredoxin-ROS activity through induction of thioredoxin-interacting protein (TXNIP) in metastatic breast cancer-derived cells MDA-MB-231. BMC Cancer 2007, 7, 96. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, J.; Imahashi, K.; Gabel, S.A.; Chutkow, W.A.; Burds, A.A.; Gannon, J.; Schulze, P.C.; MacGillivray, C.; London, R.E.; Murphy, E.; et al. Targeted deletion of thioredoxin-interacting protein regulates cardiac dysfunction in response to pressure overload. Circ. Res. 2007, 101, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Chutkow, W.A.; Patwari, P.; Yoshioka, J.; Lee, R.T. Thioredoxin-interacting protein (Txnip) is a critical regulator of hepatic glucose production. J. Biol. Chem. 2008, 283, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Damdimopoulos, A.E.; Miranda-Vizuete, A.; Pelto-Huikko, M.; Gustafsson, J.A.; Spyrou, G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J. Biol. Chem. 2002, 277, 33249–33257. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules—From biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.M.; Zhang, H.; Jones, D.P. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol. Sci. 2006, 91, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Min, W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ. Res. 2002, 90, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, H.J.; Huang, Q.; Lu, L.; Min, W. Novel action and mechanism of auranofin in inhibition of vascular endothelial growth factor receptor-3-dependent lymphangiogenesis. Anti-Cancer Agent Med. Chem. 2014, 14, 946–954. [Google Scholar] [CrossRef]
- Chen, J.; Hui, S.T.; Couto, F.M.; Mungrue, I.N.; Davis, D.B.; Attie, A.D.; Lusis, A.J.; Davis, R.A.; Shalev, A. Thioredoxin-interacting protein deficiency induces Akt/Bcl-xL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008, 22, 3581–3594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Al-Lamki, R.; Bai, L.; Streb, J.W.; Miano, J.M.; Bradley, J.; Min, W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ. Res. 2004, 94, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; He, Y.; Zhang, H.; Yu, L.; Wan, T.; Xu, Z.; Jones, D.; Chen, H.; Min, W. Endothelial-specific expression of mitochondrial thioredoxin promotes ischemia-mediated arteriogenesis and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, Y.; Zhang, W.; He, Y.; Dai, S.; Zhang, R.; Huang, Y.; Bernatchez, P.; Giordano, F.J.; Shadel, G.; et al. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am. J. Pathol. 2007, 170, 1108–1120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; He, X.; Liu, W.; Lu, M.; Hsieh, J.T.; Min, W. AIP1 mediates TNF-alpha-induced ASK1 activation by facilitating dissociation of ASK1 from its inhibitor 14-3-3. J. Clin. Investig. 2003, 111, 1933–1943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; He, Y.; Dai, S.; Xu, Z.; Luo, Y.; Wan, T.; Luo, D.; Jones, D.; Tang, S.; Chen, H.; et al. AIP1 functions as an endogenous inhibitor of VEGFR2-mediated signaling and inflammatory angiogenesis in mice. J. Clin. Investig. 2008, 118, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Romashkova, J.A.; Makarov, S.S. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.R.; Wakasugi, N.; Virelizier, J.L.; Yodoi, J.; Hay, R.T. Thioredoxin regulates the DNA binding activity of NF-kappa B by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 1992, 20, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Matsui, M.; Iwata, S.; Nishiyama, A.; Mori, K.; Yodoi, J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA 1997, 94, 3633–3638. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Tonissen, K.F.; Di Trapani, G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 2009, 53, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Shytaj, I.L.; Stamler, J.S.; Savarino, A. Dual targeting of the thioredoxin and glutathione systems in cancer and HIV. J. Clin. Investig. 2016, 126, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Diccianni, M.B.; Tanaka, T.; Gribi, R.; Yu, A.L.; Pullen, J.D.; Camitta, B.M.; Yu, J. Thioredoxin expression in primary T-cell acute lymphoblastic leukemia and its therapeutic implication. Cancer Res. 2001, 61, 7333–7338. [Google Scholar] [PubMed]
- Berggren, M.; Gallegos, A.; Gasdaska, J.R.; Gasdaska, P.Y.; Warneke, J.; Powis, G. Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res. 1996, 16, 3459–3466. [Google Scholar] [PubMed]
- Lincoln, D.T.; Ali Emadi, E.M.; Tonissen, K.F.; Clarke, F.M. The thioredoxin-thioredoxin reductase system: Over-expression in human cancer. Anticancer Res. 2003, 23, 2425–2433. [Google Scholar] [PubMed]
- Gallegos, A.; Gasdaska, J.R.; Taylor, C.W.; Paine-Murrieta, G.D.; Goodman, D.; Gasdaska, P.Y.; Berggren, M.; Briehl, M.M.; Powis, G. Transfection with human thioredoxin increases cell proliferation and a dominant-negative mutant thioredoxin reverses the transformed phenotype of human breast cancer cells. Cancer Res. 1996, 56, 5765–5770. [Google Scholar] [PubMed]
- Yoo, M.H.; Xu, X.M.; Carlson, B.A.; Gladyshev, V.N.; Hatfield, D.L. Thioredoxin reductase 1 deficiency reverses tumor phenotype and tumorigenicity of lung carcinoma cells. J. Biol. Chem. 2006, 281, 13005–13008. [Google Scholar] [CrossRef] [PubMed]
- Hellfritsch, J.; Kirsch, J.; Schneider, M.; Fluege, T.; Wortmann, M.; Frijhoff, J.; Dagnell, M.; Fey, T.; Esposito, I.; Kolle, P.; et al. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid. Redox. Signal. 2015, 22, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Fink, E.E.; Mannava, S.; Bagati, A.; Bianchi-Smiraglia, A.; Nair, J.R.; Moparthy, K.; Lipchick, B.C.; Drokov, M.; Utley, A.; Ross, J.; et al. Mitochondrial thioredoxin reductase regulates major cytotoxicity pathways of proteasome inhibitors in multiple myeloma cells. Leukemia 2015. [Google Scholar] [CrossRef] [PubMed]
- Perry, B.N.; Govindarajan, B.; Bhandarkar, S.S.; Knaus, U.G.; Valo, M.; Sturk, C.; Carrillo, C.O.; Sohn, A.; Cerimele, F.; Dumont, D.; et al. Pharmacologic blockade of angiopoietin-2 is efficacious against model hemangiomas in mice. J. Investig. Dermatol. 2006, 126, 2316–2322. [Google Scholar] [CrossRef] [PubMed]
- Lapidoth, M.; Ben-Amitai, D.; Bhandarkar, S.; Fried, L.; Arbiser, J.L. Efficacy of topical application of eosin for ulcerated hemangiomas. J. Am. Acad. Dermatol. 2009, 60, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, Y.; Fried, L.E.; Du, Y.; Montano, S.J.; Sohn, A.; Lefkove, B.; Holmgren, L.; Arbiser, J.L.; Holmgren, A.; et al. Disruption of the mitochondrial thioredoxin system as a cell death mechanism of cationic triphenylmethanes. Free Radic. Biol. Med. 2011, 50, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Yang, M.; Praggastis, A.; Li, Y.; Zhou, H.J.; He, Y.; Ghazvinian, R.; Cincotta, D.J.; Rice, K.P.; Min, W. Carbamoylating activity associated with the activation of the antitumor agent laromustine inhibits angiogenesis by inducing ASK1-dependent endothelial cell death. PLoS ONE 2014, 9, e103224. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, G.M.; Roman, M.G.; Flores, L.C.; Hubbard, G.B.; Salmon, A.B.; Zhang, Y.; Gelfond, J.; Ikeno, Y. The paradoxical role of thioredoxin on oxidative stress and aging. Arch. Biochem. Biophys. 2015, 576, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.C.; Salyapongse, A.N.; Bragdon, G.A.; Shears, L.L.; Watkins, S.C.; Edington, H.D.; Billiar, T.R. Impaired wound healing and angiogenesis in eNOS-deficient mice. Am. J. Physiol. 1999, 277, H1600–H1608. [Google Scholar] [PubMed]
- Mochizuki, T.; Furuta, S.; Mitsushita, J.; Shang, W.H.; Ito, M.; Yokoo, Y.; Yamaura, M.; Ishizone, S.; Nakayama, J.; Konagai, A.; et al. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene 2006, 25, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Adesina, S.E.; Wade, B.E.; Bijli, K.M.; Kang, B.Y.; Williams, C.R.; Ma, J.; Go, Y.M.; Hart, C.M.; Sutliff, R.L. Hypoxia inhibits expression and function of mitochondrial thioredoxin 2 to promote pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L599–L608. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 Inhibits Mitochondrial Reactive Oxygen Species Generation and Apoptosis Stress Kinase-1 Activity to Maintain Cardiac Function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [PubMed]
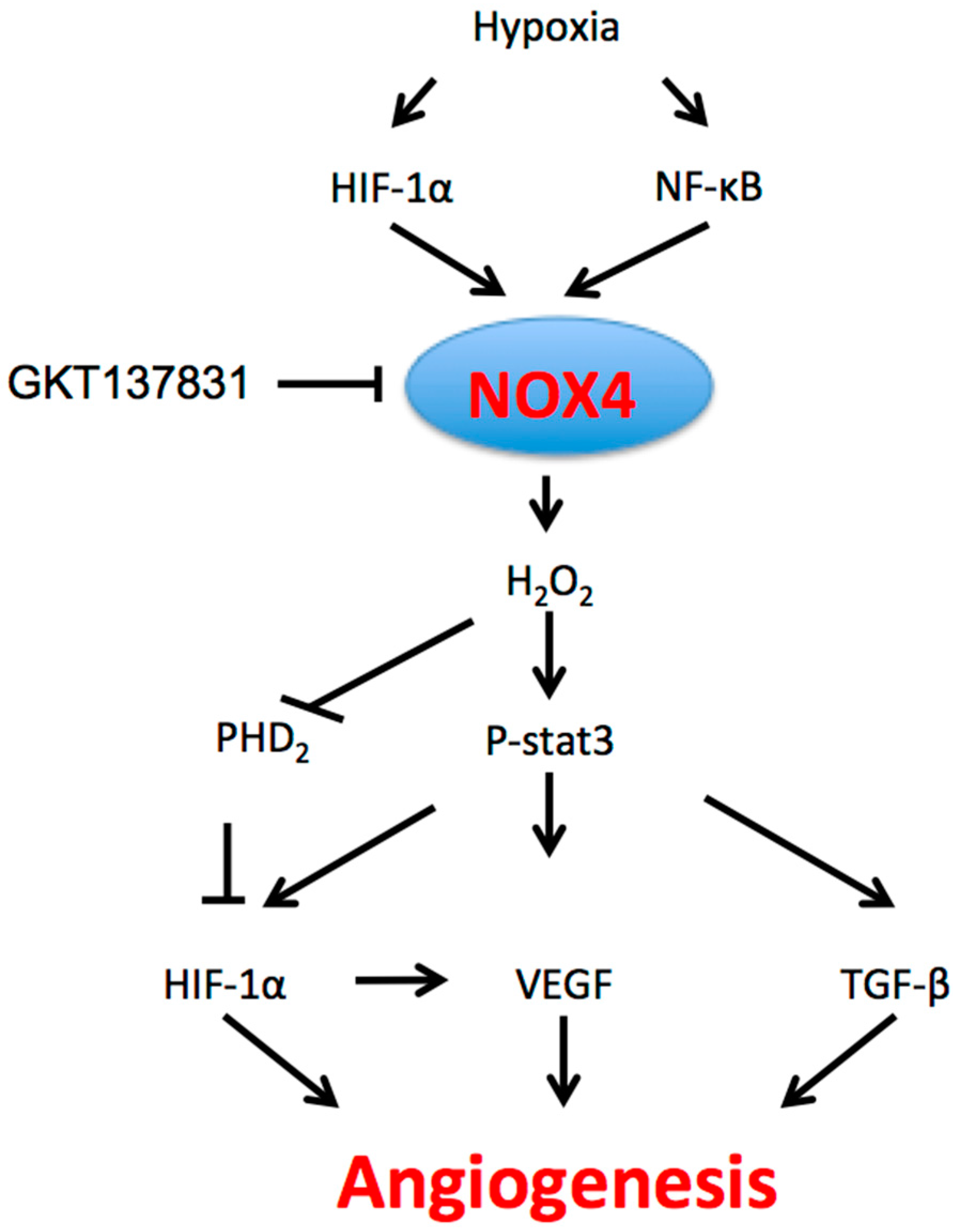
Figure 1.
The role of NOX4 in angiogenesis. Hypoxia through HIF-1α and NF-κB induces NOX4 expression; NOX4 generates cytosolic ROS, which in turn activate multiple intracellular angiogenic pathways, including STAT3, VEGF and TGF-β signaling. ROS: reactive oxygen species; STAT3: signal transducer and activator of transcription 3; VEGF: vascular endothelial growth factor; TGF-β: transforming growth factor-β.
Figure 1.
The role of NOX4 in angiogenesis. Hypoxia through HIF-1α and NF-κB induces NOX4 expression; NOX4 generates cytosolic ROS, which in turn activate multiple intracellular angiogenic pathways, including STAT3, VEGF and TGF-β signaling. ROS: reactive oxygen species; STAT3: signal transducer and activator of transcription 3; VEGF: vascular endothelial growth factor; TGF-β: transforming growth factor-β.
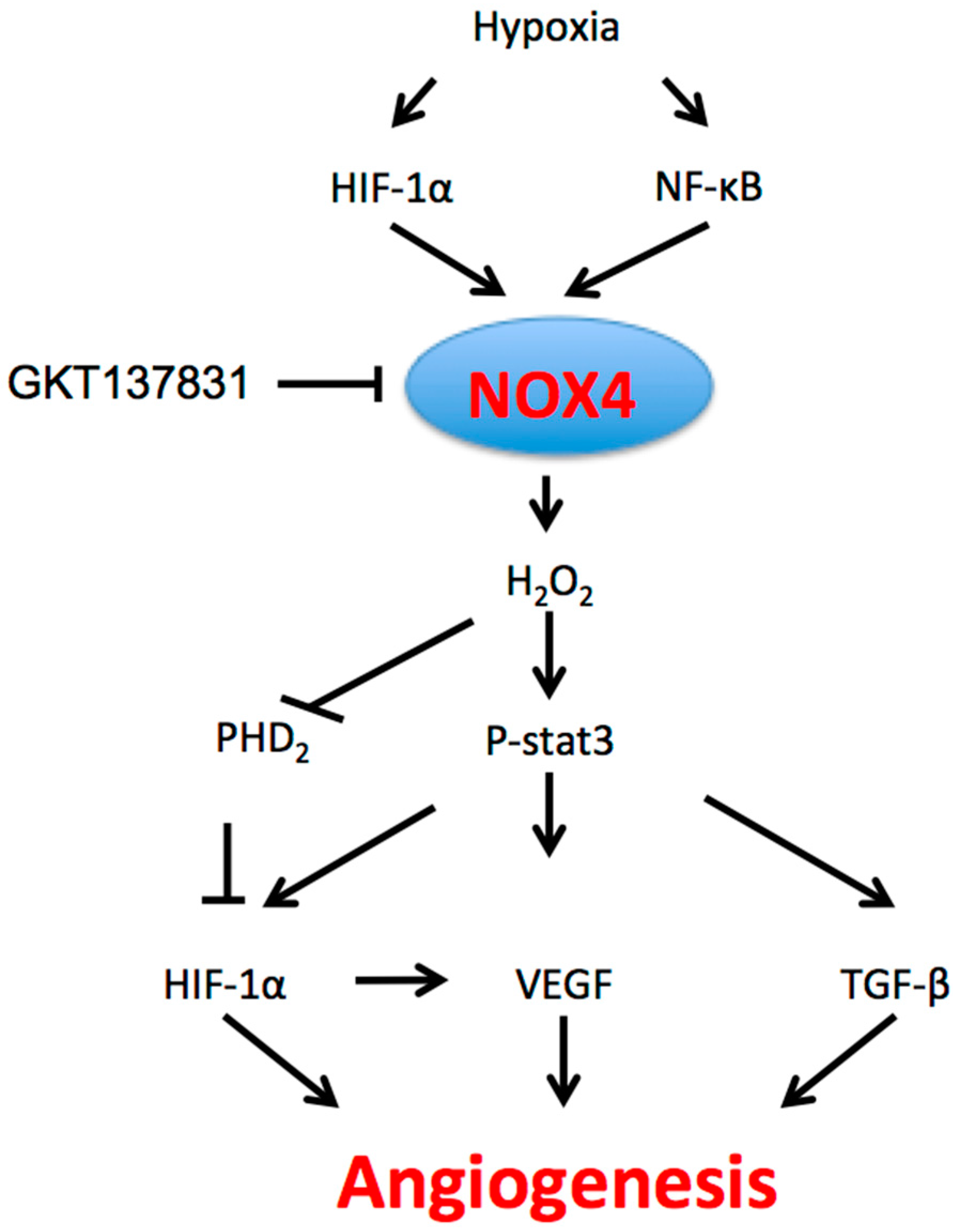
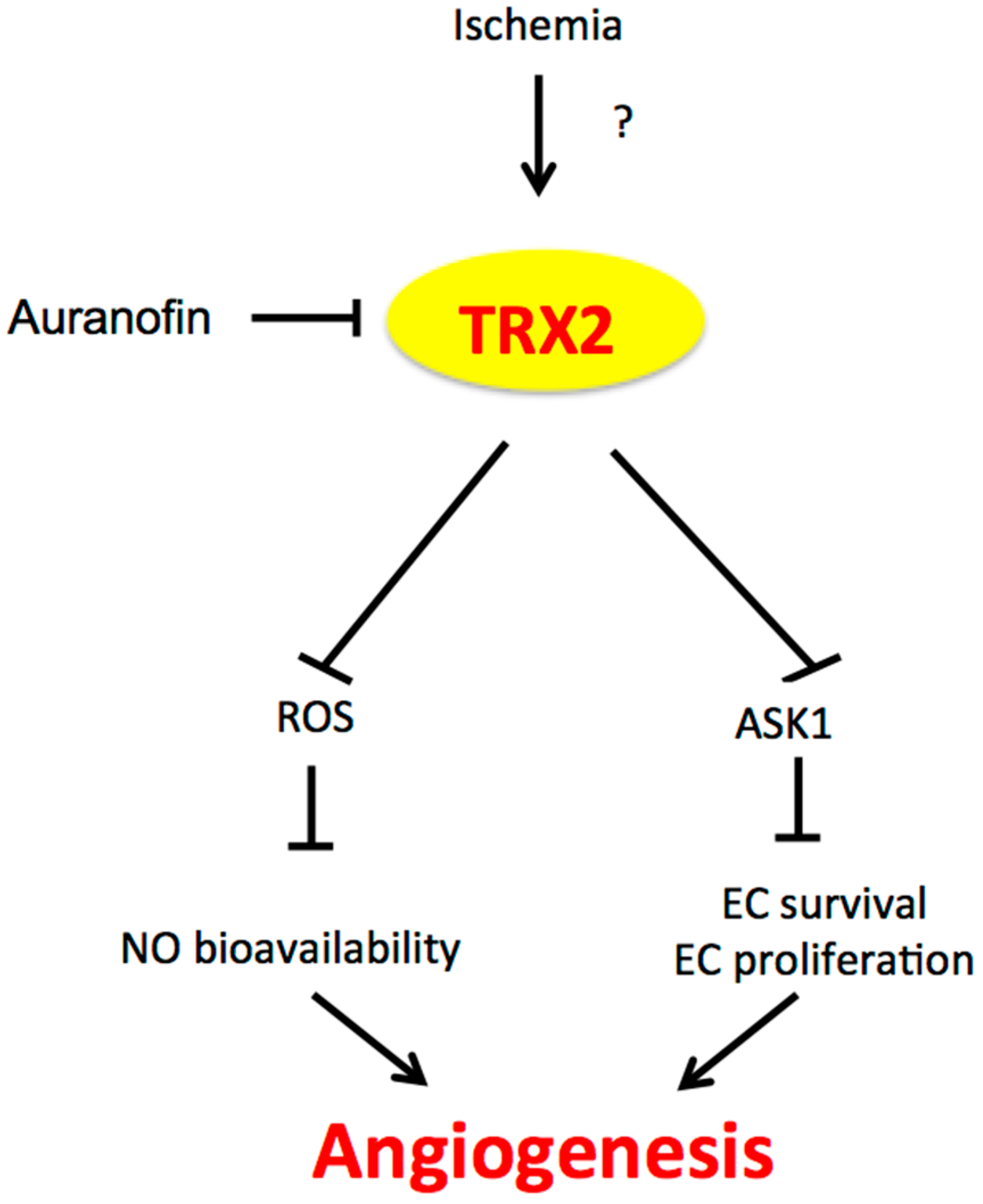
Figure 2.
The role of TRX2 in angiogenesis. Ischemia induces TRX2 expression by an unknown mechanism. TRX2 in the mitochondria inhibits mitochondrial ROS production and mitochondrial ASK1-mediated apoptosis, leading to increased NO bioavailability and EC survival/proliferation and augmented angiogenesis. TRX2: Thioredoxin 2; ASK1: apoptosis signaling kinase-1; EC: endothelial cell.
Figure 2.
The role of TRX2 in angiogenesis. Ischemia induces TRX2 expression by an unknown mechanism. TRX2 in the mitochondria inhibits mitochondrial ROS production and mitochondrial ASK1-mediated apoptosis, leading to increased NO bioavailability and EC survival/proliferation and augmented angiogenesis. TRX2: Thioredoxin 2; ASK1: apoptosis signaling kinase-1; EC: endothelial cell.
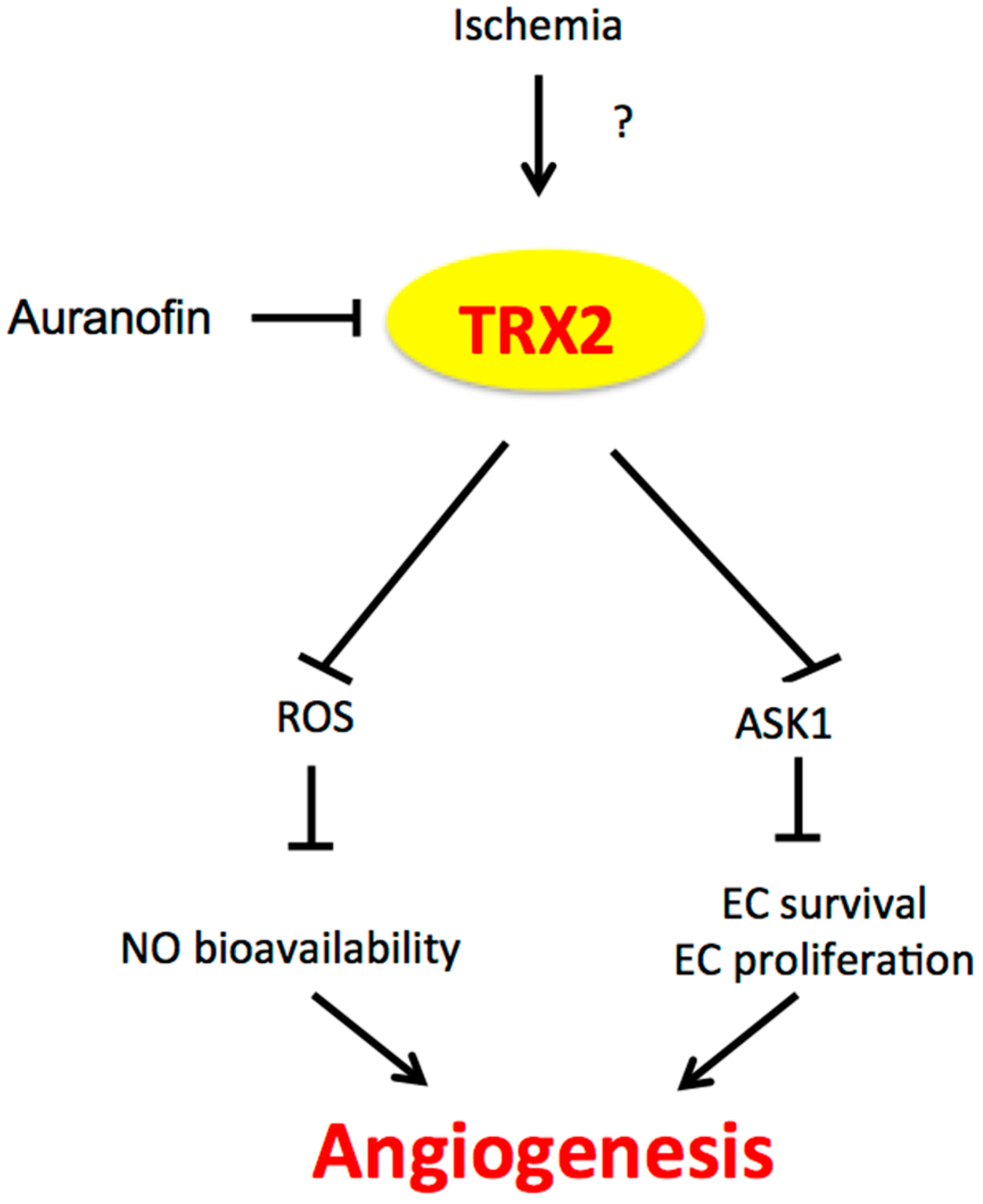
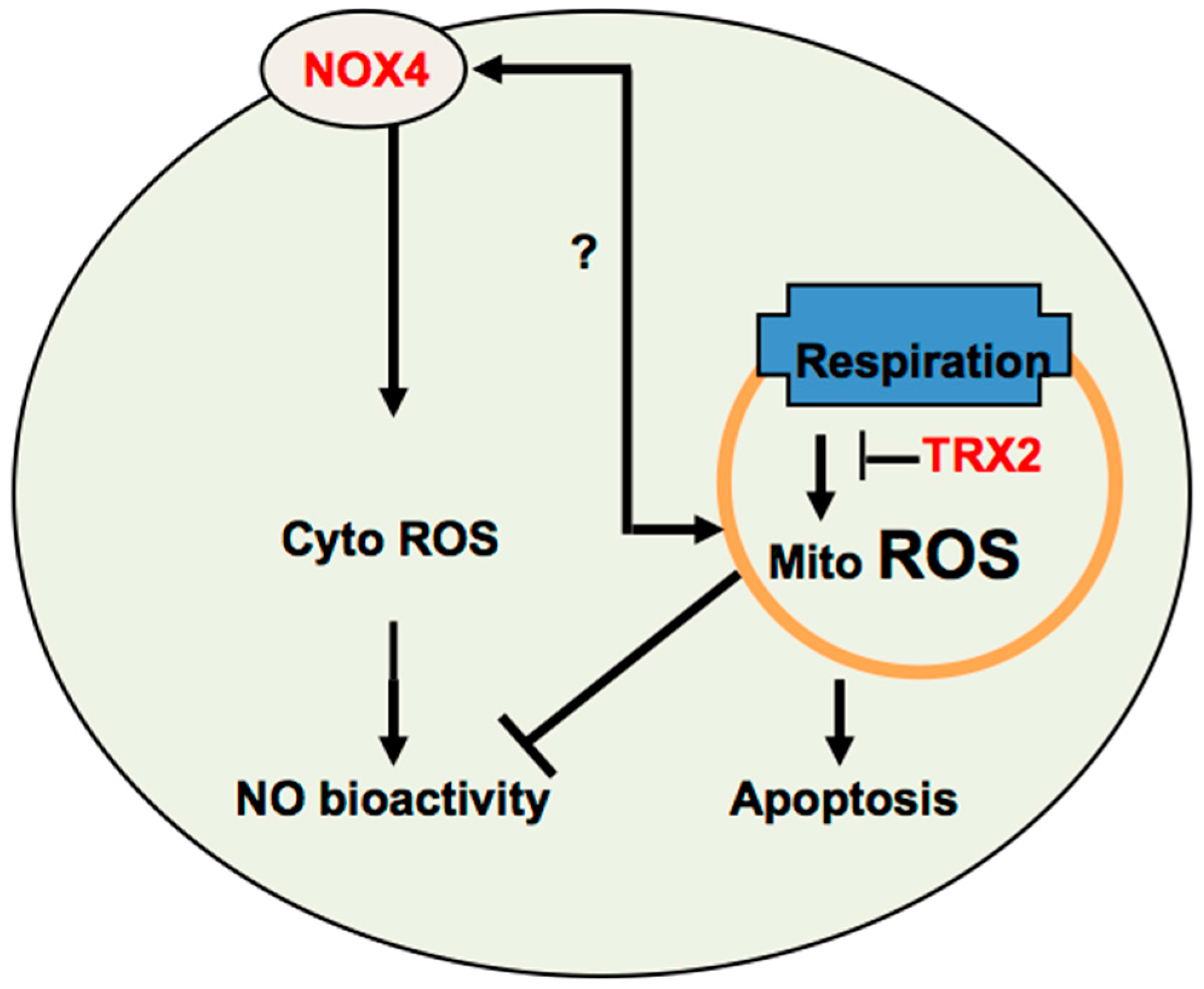
Figure 3.
Cross-talk between NOX4 and TRX2-mediated ROS signaling. It is plausible that cytosolic ROS enhance, while mitochondrial ROS inhibit angiogenesis. NOX-generated cytosolic ROS induce angiogenesis, in part, by increasing NO activity. TRX2 prevents mitochondrial ROS-mediated NO dysfunction and EC apoptosis to increase angiogenesis. It is unknown how NOX4 and TRX2 cross-talk. A low level of mitochondrial ROS may upregulate NOX4, which in turn is translocated into mitochondria to induce more ROS production in mitochondria.
Figure 3.
Cross-talk between NOX4 and TRX2-mediated ROS signaling. It is plausible that cytosolic ROS enhance, while mitochondrial ROS inhibit angiogenesis. NOX-generated cytosolic ROS induce angiogenesis, in part, by increasing NO activity. TRX2 prevents mitochondrial ROS-mediated NO dysfunction and EC apoptosis to increase angiogenesis. It is unknown how NOX4 and TRX2 cross-talk. A low level of mitochondrial ROS may upregulate NOX4, which in turn is translocated into mitochondria to induce more ROS production in mitochondria.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, C.; Li, L.; Zhou, H.J.; Min, W. The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk. Antioxidants 2017, 6, 42. https://doi.org/10.3390/antiox6020042
AMA Style
Chen C, Li L, Zhou HJ, Min W. The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk. Antioxidants. 2017; 6(2):42. https://doi.org/10.3390/antiox6020042
Chicago/Turabian StyleChen, Chaofei, Li Li, Huanjiao Jenny Zhou, and Wang Min. 2017. "The Role of NOX4 and TRX2 in Angiogenesis and Their Potential Cross-Talk" Antioxidants 6, no. 2: 42. https://doi.org/10.3390/antiox6020042
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.