Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Mouse Model
2.2. Immunohistochemistry
2.3. Evaluation of Pulmonary Vascular Structure
2.4. Evaluation of Alveolar Development
2.5. Evaluation of Right Ventricular Hypertrophy
2.6. Protein Expression
2.7. SOD Activity Assay
2.8. Statistical Analysis
3. Results
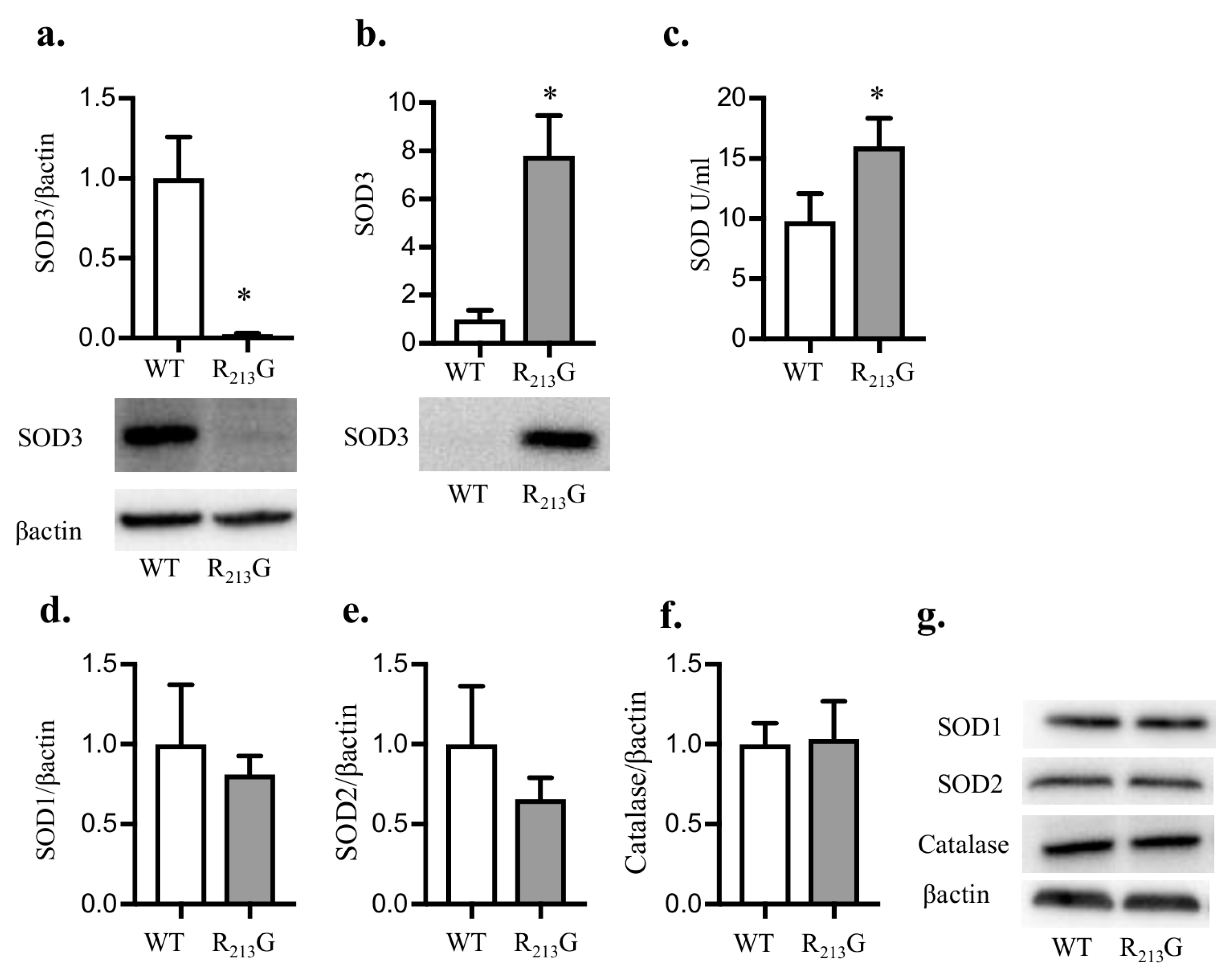
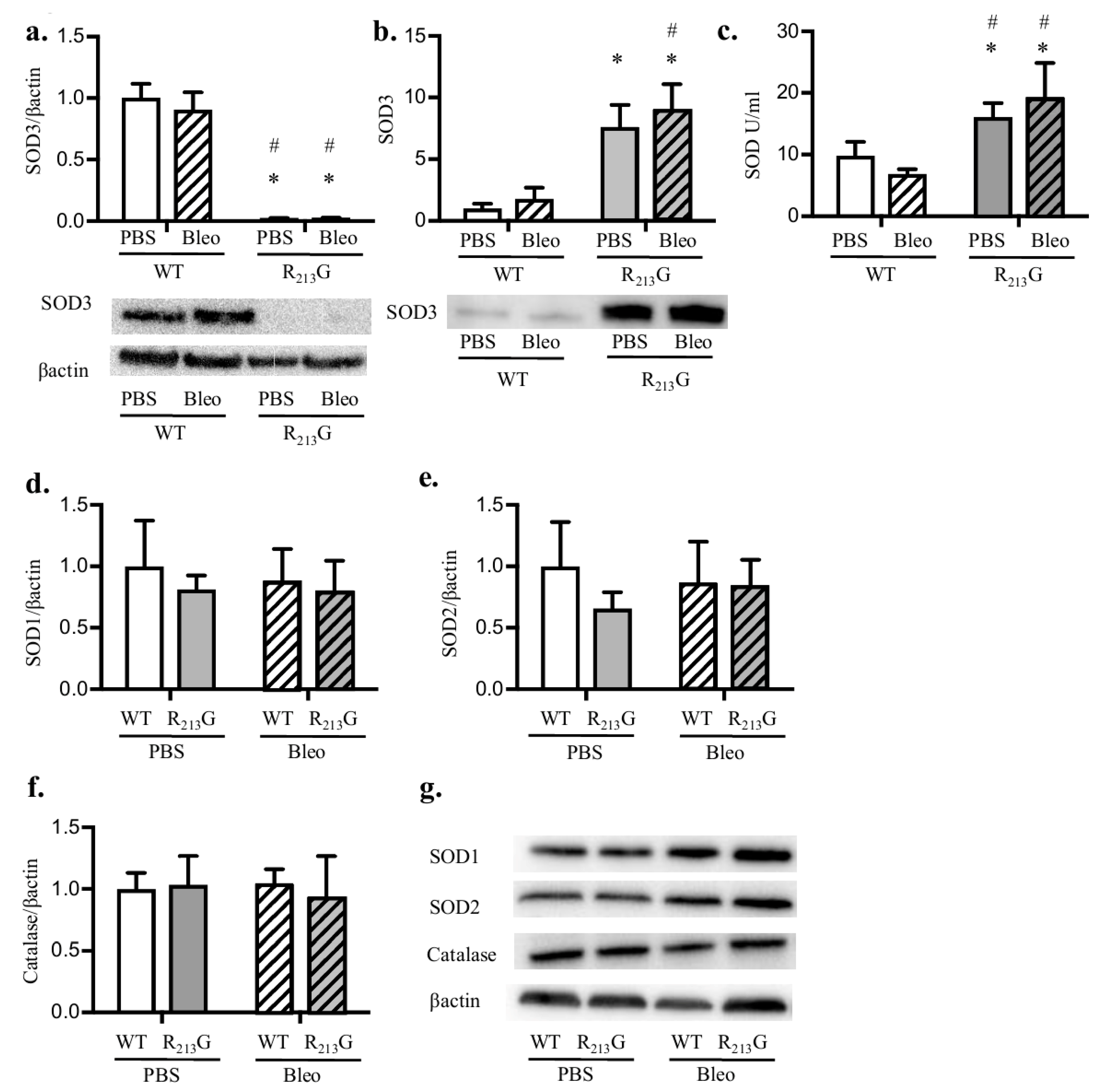
3.1. The R213G SNP Redistributed SOD3 from The Lung to The Extracellular Fluid in Neonatal Mice
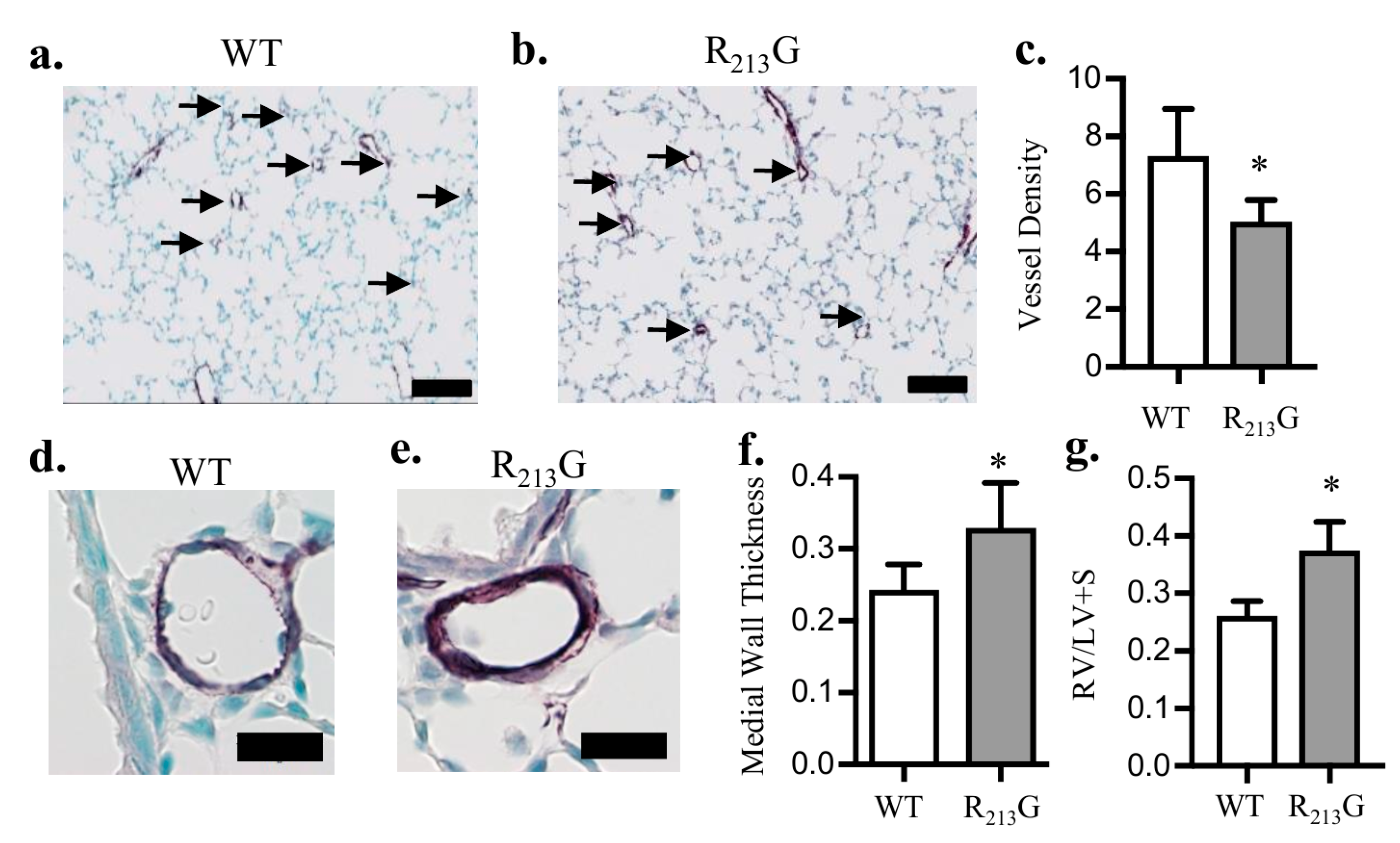
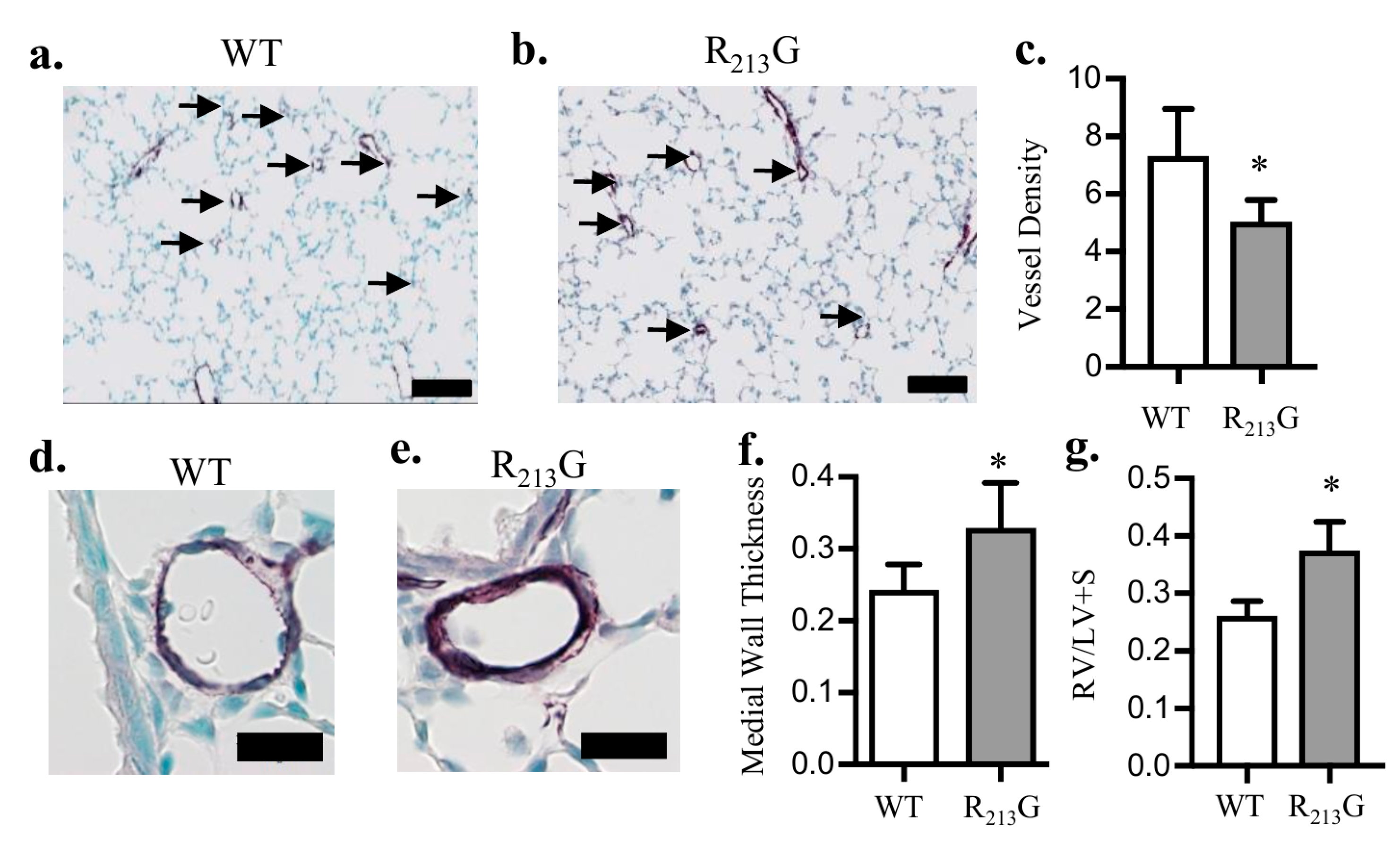
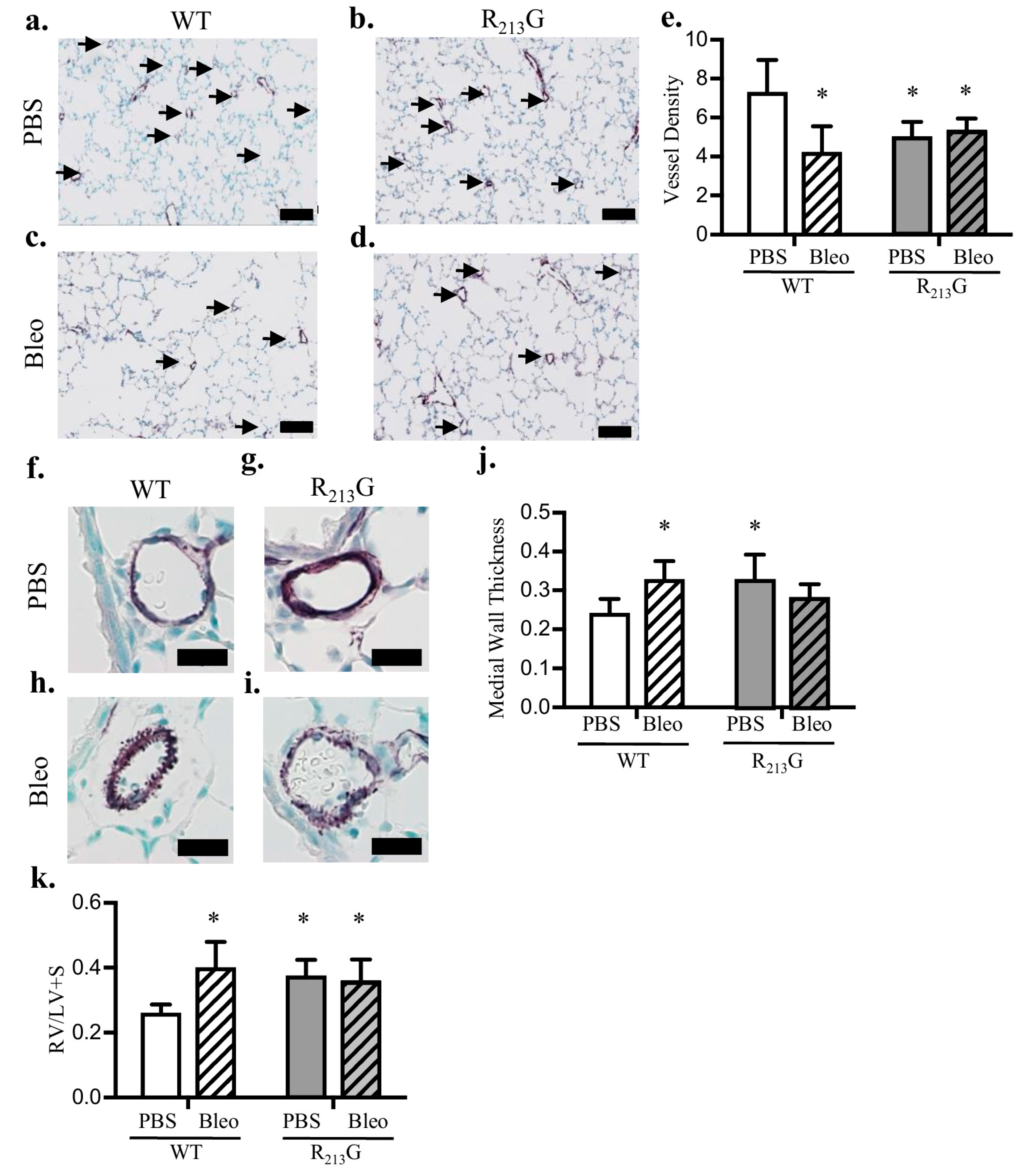
3.2. Mice Expressing the R213G SNP Demonstrated Impaired Pulmonary Vascular Development and PH Under Control Conditions
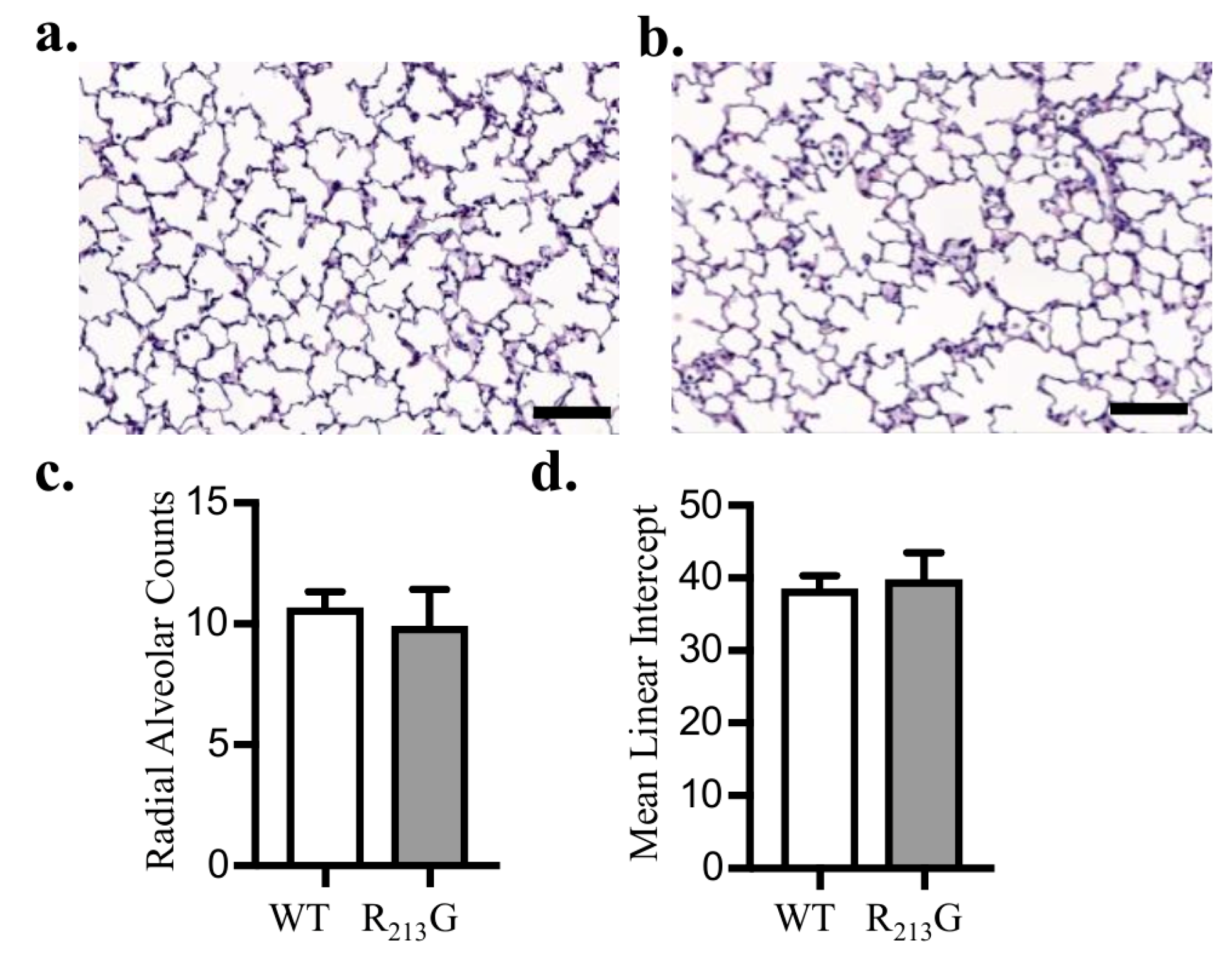
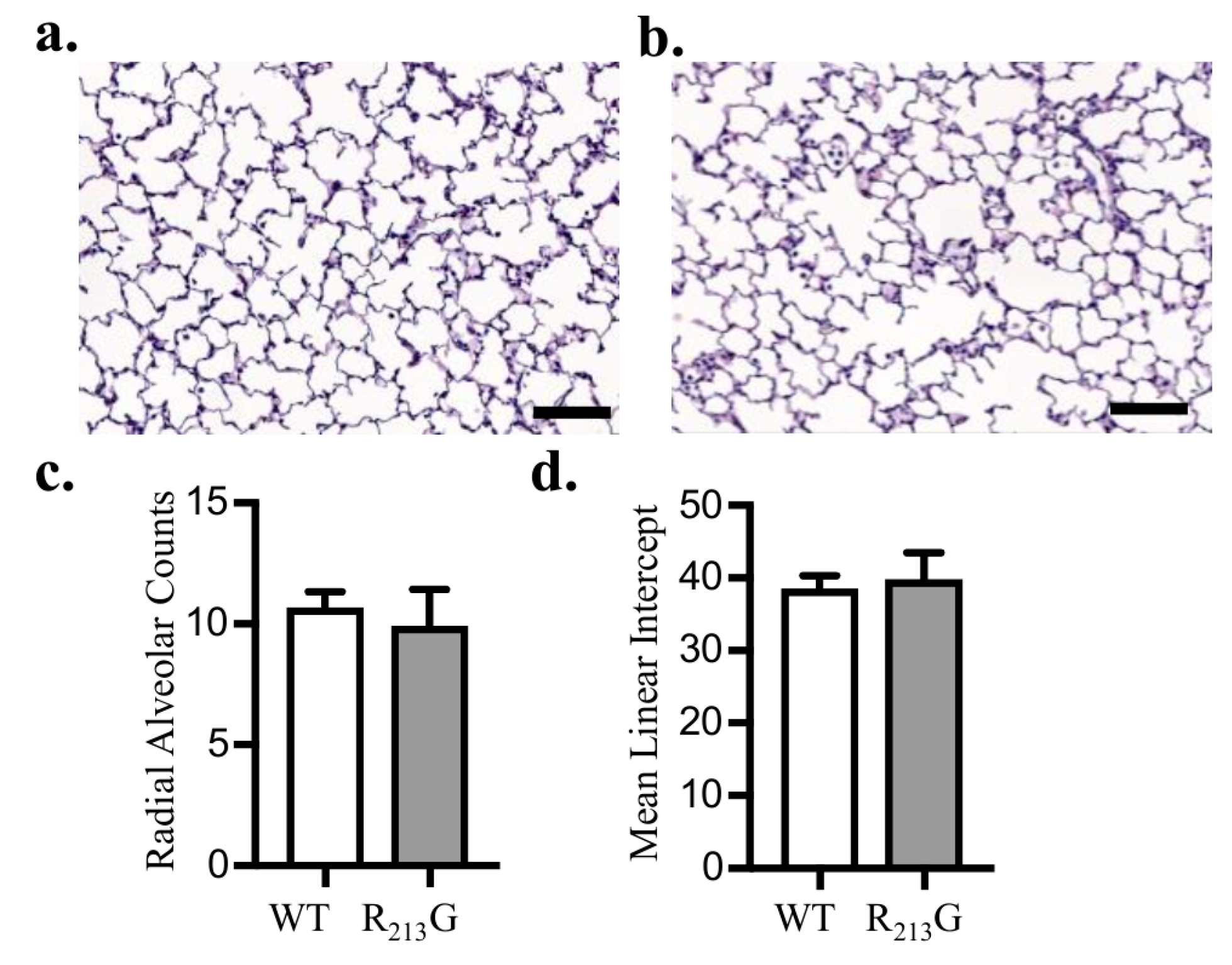
3.3. Alveolar Development Was Not Altered in Mice Expressing the R213G SNP
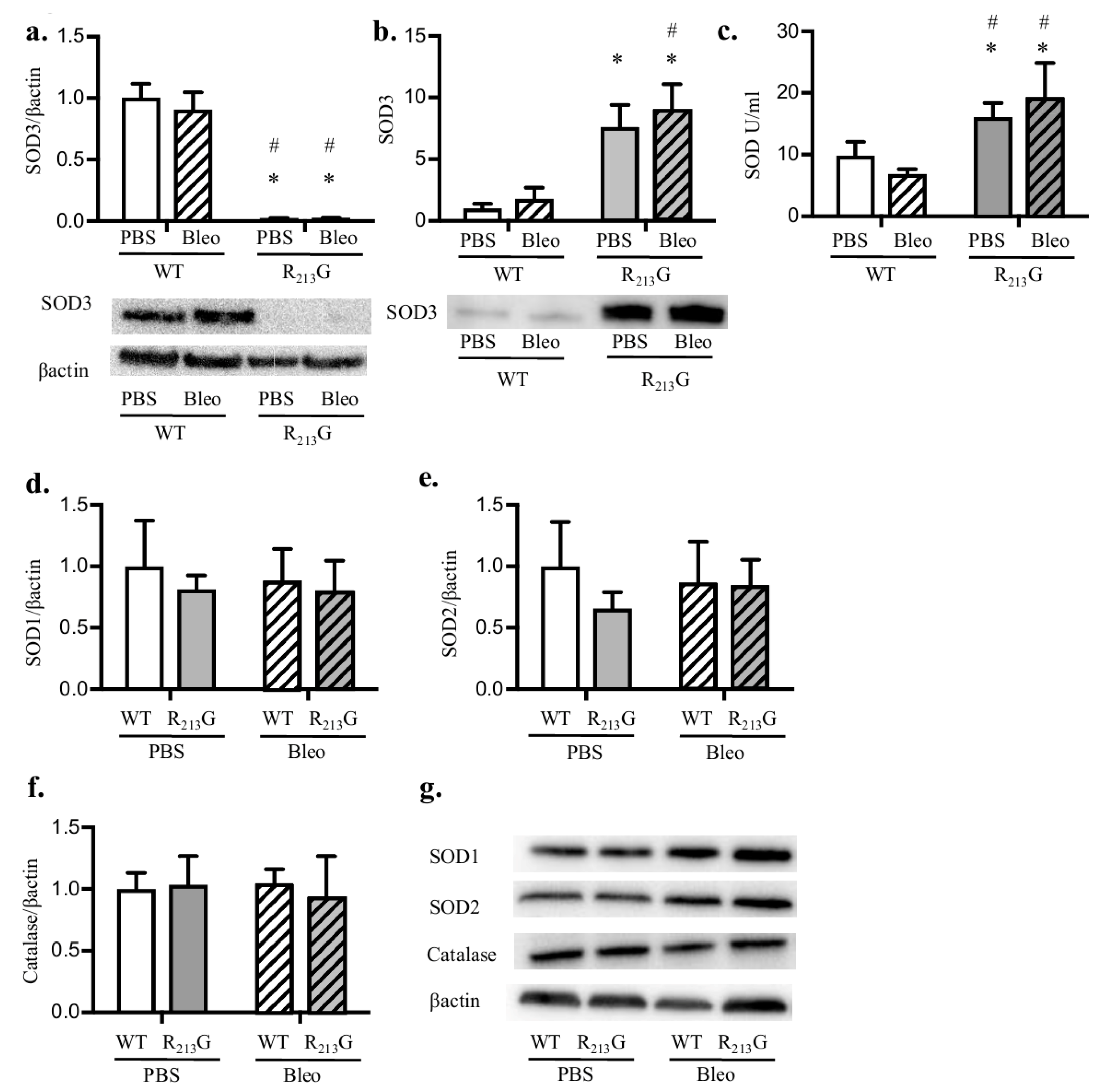
3.4. Bleomycin Did Not Alter Antioxidant Enzyme Expression in the Lung or Plasma in Either Strain
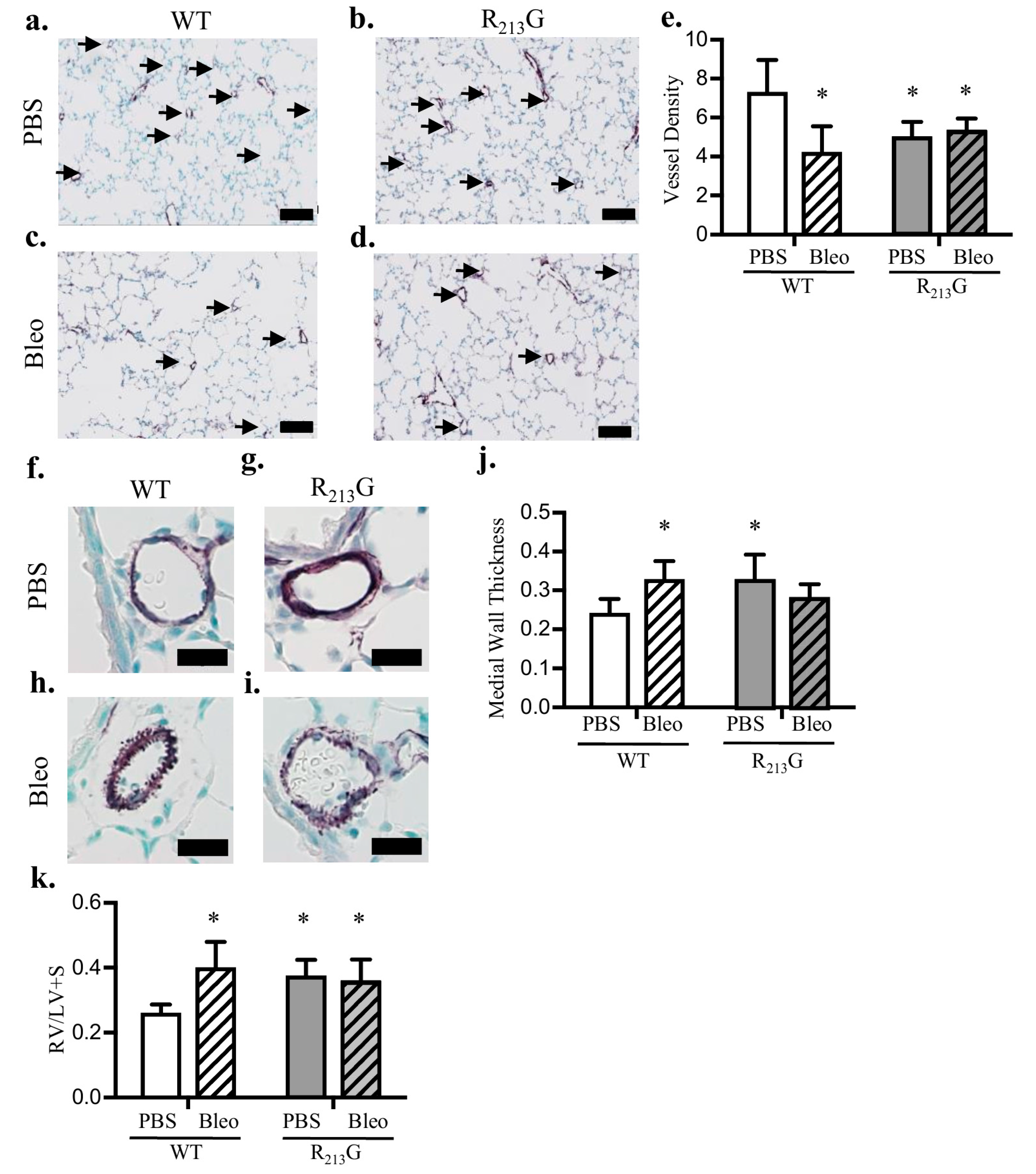
3.5. Mice Expressing the R213G SNP Do Not Have Further Worsening of Pulmonary Vascular Development or PH after Bleomycin Injury
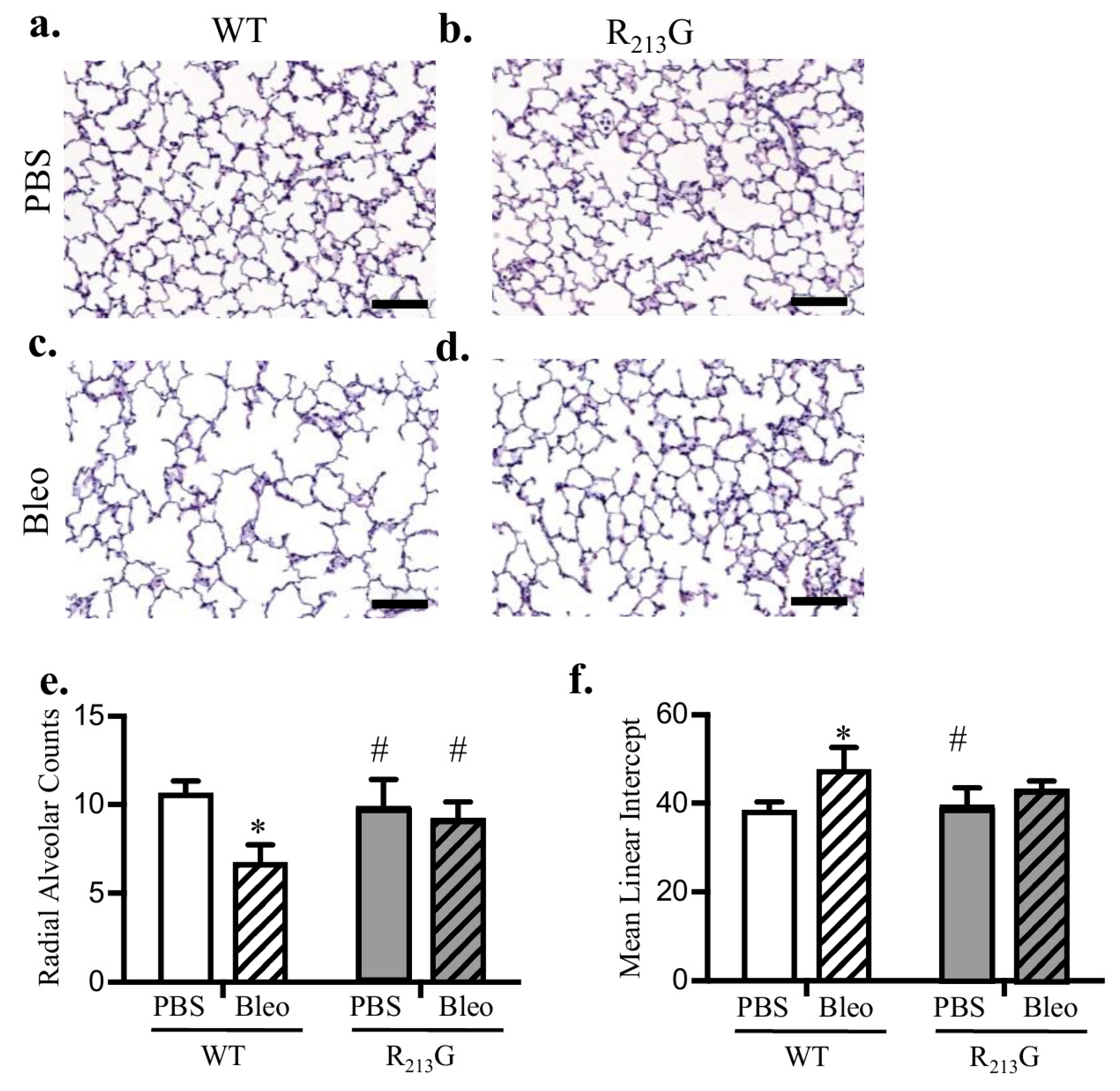
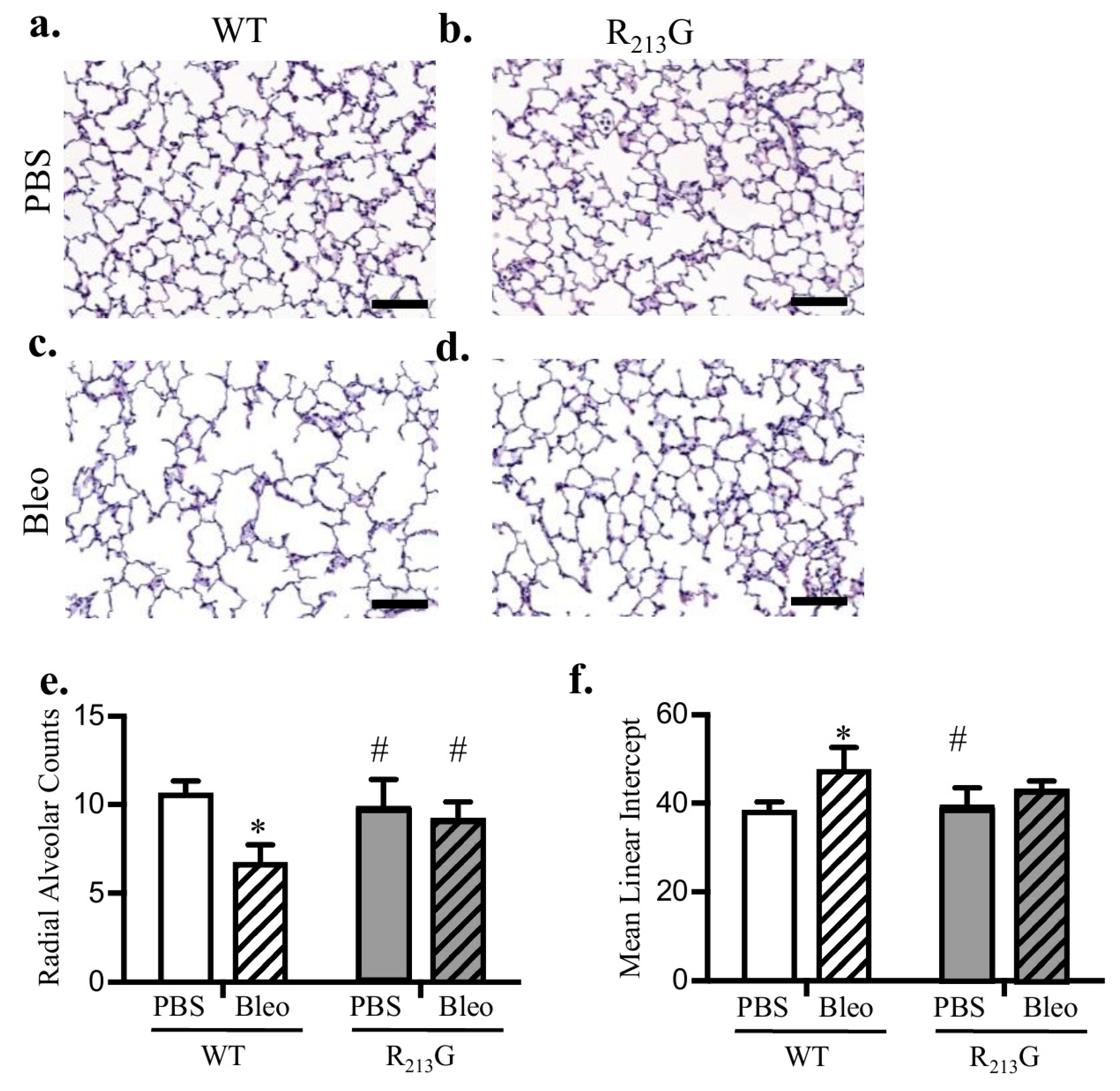
3.6. The R213G SNP Mitigated Bleomycin Induced Alveolar Simplification at 22 Days
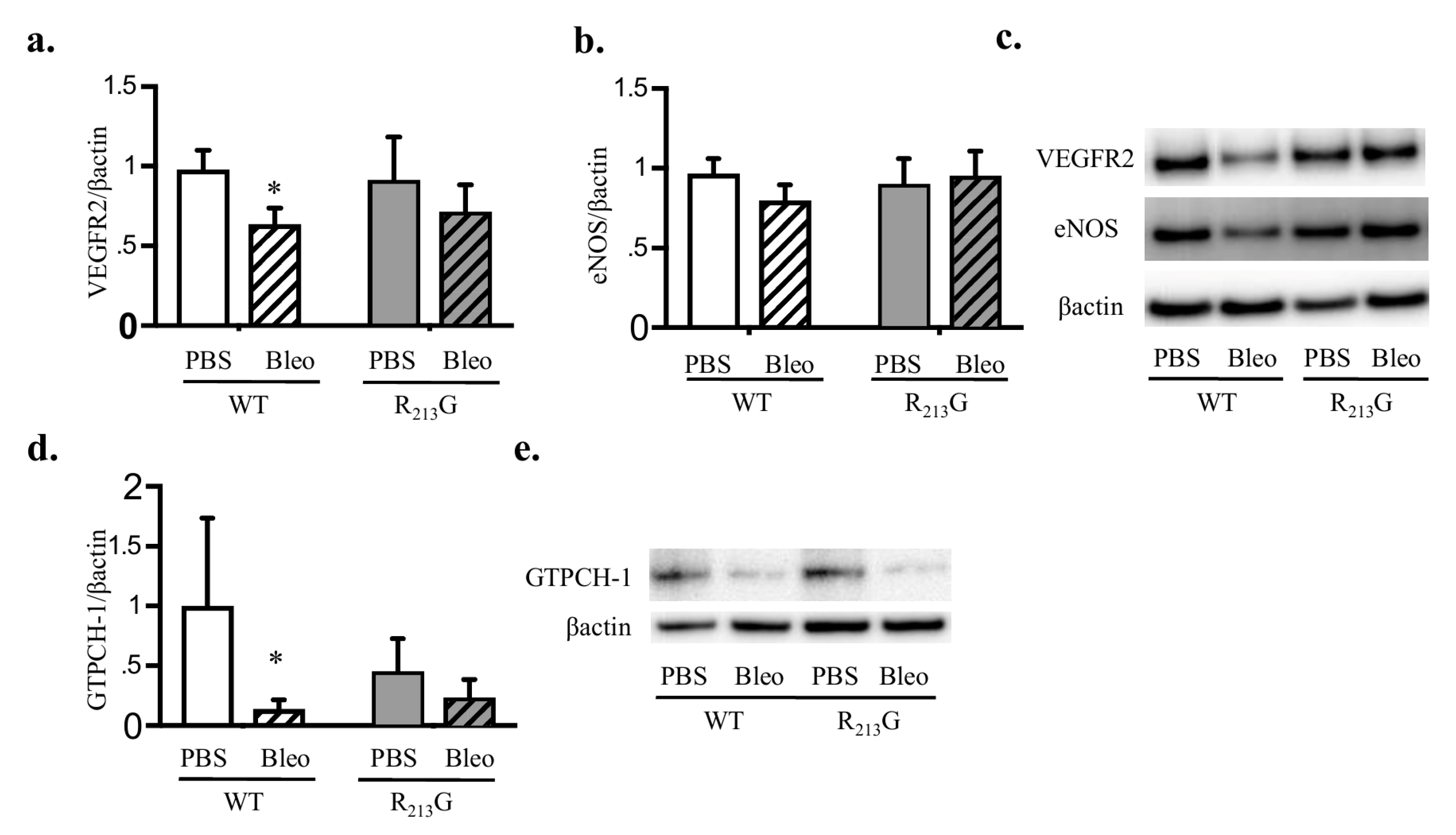
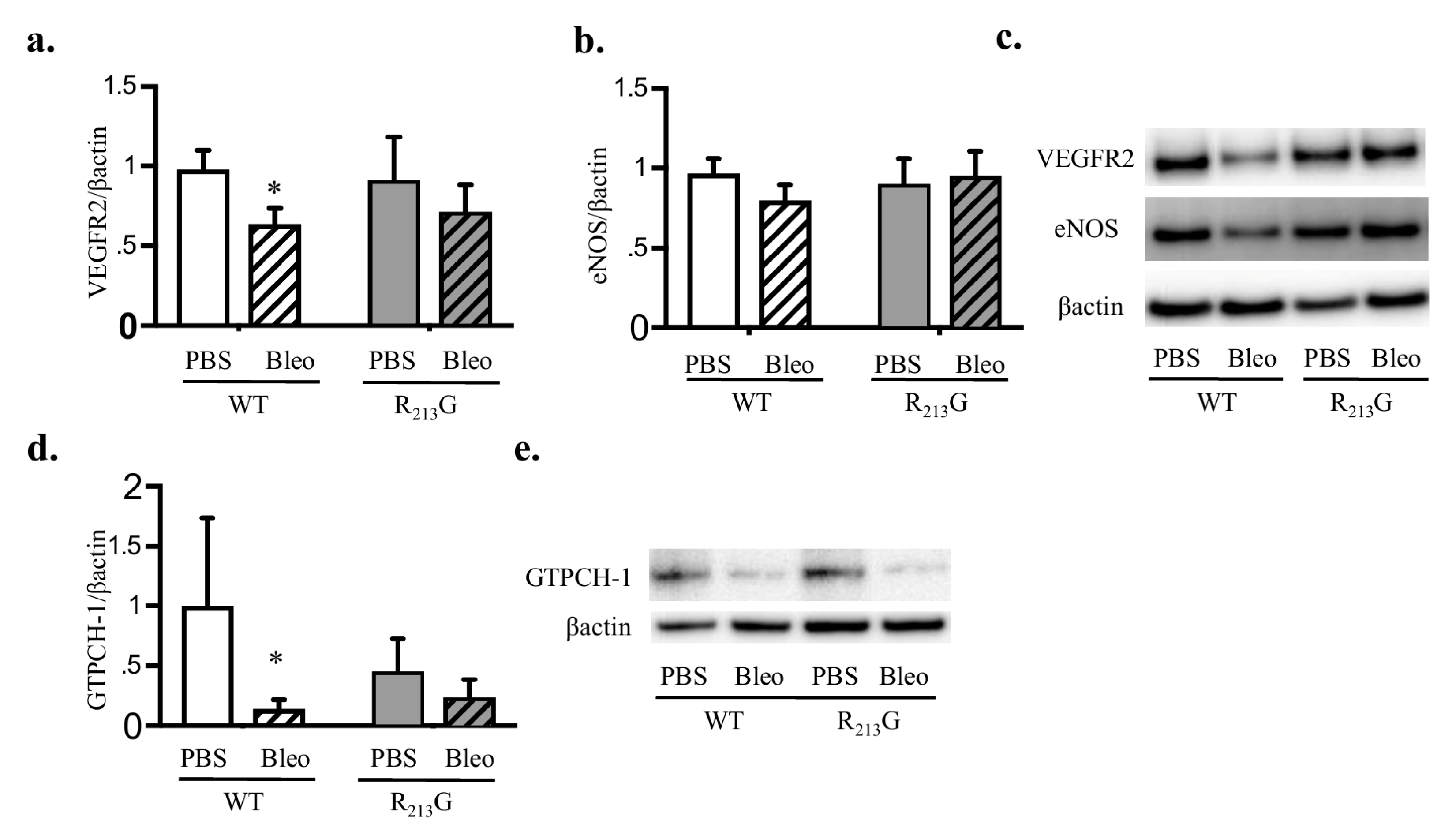
3.7. Bleomycin Only Decreased VEGFR2 and GTPCH-1 Levels in WT Mice
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Islam, J.Y.; Keller, R.L.; Aschner, J.L.; Hartert, T.V.; Moore, P.E. Understanding the short- and long-term respiratory outcomes of prematurity and bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2015, 192, 134–156. [Google Scholar] [CrossRef] [PubMed]
- Stoll, B.J.; Hansen, N.I.; Bell, E.F.; Walsh, M.C.; Carlo, W.A.; Shankaran, S.; Laptook, A.R.; Sanchez, P.J.; Van Meurs, K.P.; Wyckoff, M.; et al. Trends in care practices, morbidity, and mortality of extremely preterm neonates, 1993–2012. JAMA 2015, 314, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Jensen, E.A.; Schmidt, B. Epidemiology of bronchopulmonary dysplasia. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Khemani, E.; McElhinney, D.B.; Rhein, L.; Andrade, O.; Lacro, R.V.; Thomas, K.C.; Mullen, M.P. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: Clinical features and outcomes in the surfactant era. Pediatrics 2007, 120, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Steinhorn, R.H. Role of reactive oxygen species in neonatal pulmonary vascular disease. Antioxid. Redox Signal. 2014, 21, 1926–1942. [Google Scholar] [CrossRef] [PubMed]
- Berkelhamer, S.K.; Farrow, K.N. Developmental regulation of antioxidant enzymes and their impact on neonatal lung disease. Antioxid. Redox Signal. 2014, 21, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Hartney, J.M.; Stidham, T.; Goldstrohm, D.A.; Oberley-Deegan, R.E.; Weaver, M.R.; Valnickova-Hansen, Z.; Scavenius, C.; Benninger, R.K.; Leahy, K.F.; Johnson, R.; et al. A common polymorphism in extracellular superoxide dismutase affects cardiopulmonary disease risk by altering protein distribution. Circ. Cardiovasc. Genet. 2014, 7, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Kodera, T.; Ohta, H.; Hayashi, K.; Hirano, K. The heparin binding site of human extracellular-superoxide dismutase. Arch. Biochem. Biophys. 1992, 297, 155–161. [Google Scholar] [CrossRef]
- Karlsson, K.; Lindahl, U.; Marklund, S.L. Binding of human extracellular superoxide dismutase C to sulphated glycosaminoglycans. Biochem. J. 1988, 256, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Olsen, D.A.; Petersen, S.V.; Oury, T.D.; Valnickova, Z.; Thogersen, I.B.; Kristensen, T.; Bowler, R.P.; Crapo, J.D.; Enghild, J.J. The intracellular proteolytic processing of extracellular superoxide dismutase (EC-SOD) is a two-step event. J. Biol. Chem. 2004, 279, 22152–22157. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.V.; Olsen, D.A.; Kenney, J.M.; Oury, T.D.; Valnickova, Z.; Thogersen, I.B.; Crapo, J.D.; Enghild, J.J. The high concentration of Arg213→Gly extracellular superoxide dismutase (EC-SOD) in plasma is caused by a reduction of both heparin and collagen affinities. Biochem. J. 2005, 385, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Gaurav, R.; Varasteh, J.T.; Weaver, M.R.; Jacobson, S.R.; Hernandez-Lagunas, L.; Liu, Q.; Nozik-Grayck, E.; Chu, H.W.; Alam, R.; Nordestgaard, B.G.; et al. The R213G polymorphism in SOD3 protects against allergic airway inflammation. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Juul, K.; Tybjaerg-Hansen, A.; Marklund, S.; Lange, P.; Nordestgaard, B.G. Genetically increased antioxidative protection and decreased chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 173, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Mouradian, G.C.; Gaurav, R.; Pugliese, S.; El Kasmi, K.; Hartman, B.; Hernandez-Lagunas, L.; Stenmark, K.R.; Bowler, R.P.; Nozik-Grayck, E. Superoxide dismutase 3 R213G single-nucleotide polymorphism blocks murine bleomycin-induced fibrosis and promotes resolution of inflammation. Am. J. Respir. Cell Mol. Biol. 2017, 56, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Juul, K.; Tybjaerg-Hansen, A.; Marklund, S.; Heegaard, N.H.; Steffensen, R.; Sillesen, H.; Jensen, G.; Nordestgaard, B.G. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The copenhagen city heart study. Circulation 2004, 109, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.; Wright, R.H.; Tang, J.R.; Woods, C.; Villegas, L.; Sherlock, L.; Savani, R.C.; Abman, S.H.; Nozik-Grayck, E. Lack of EC-SOD worsens alveolar and vascular development in a neonatal mouse model of bleomycin-induced bronchopulmonary dysplasia and pulmonary hypertension. Pediatric Res. 2015, 78, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Giles, B.L.; Suliman, H.; Mamo, L.B.; Piantadosi, C.A.; Oury, T.D.; Nozik-Grayck, E. Prenatal hypoxia decreases lung extracellular superoxide dismutase expression and activity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L549–L554. [Google Scholar] [CrossRef] [PubMed]
- Poonyagariyagorn, H.K.; Metzger, S.; Dikeman, D.; Mercado, A.L.; Malinina, A.; Calvi, C.; McGrath-Morrow, S.; Neptune, E.R. Superoxide dismutase 3 dysregulation in a murine model of neonatal lung injury. Am. J. Respir. Cell Mol. Biol. 2014, 51, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Auten, R.L.; O’Reilly, M.A.; Oury, T.D.; Nozik-Grayck, E.; Whorton, M.H. Transgenic extracellular superoxide dismutase protects postnatal alveolar epithelial proliferation and development during hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L32–L40. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.N.; Suliman, H.B.; Folz, R.J.; Nozik-Grayck, E.; Golson, M.L.; Mason, S.N.; Auten, R.L. Extracellular superoxide dismutase protects lung development in hyperoxia-exposed newborn mice. Am. J. Respir. Crit. Care Med. 2003, 167, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Min, J.H.; Codipilly, C.N.; Nasim, S.; Miller, E.J.; Ahmed, M.N. Synergistic protection against hyperoxia-induced lung injury by neutrophils blockade and EC-SOD overexpression. Respir. Res. 2012, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Della Latta, V.; Cecchettini, A.; Del Ry, S.; Morales, M.A. Bleomycin in the setting of lung fibrosis induction: From biological mechanisms to counteractions. Pharmacol. Res. 2015, 97, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.M. DNA strand scission by activated bleomycin group antibiotics. Fed. Proc. 1986, 45, 2784–2791. [Google Scholar] [PubMed]
- Sewing, A.C.; Kantores, C.; Ivanovska, J.; Lee, A.H.; Masood, A.; Jain, A.; McNamara, P.J.; Tanswell, A.K.; Jankov, R.P. Therapeutic hypercapnia prevents bleomycin-induced pulmonary hypertension in neonatal rats by limiting macrophage-derived tumor necrosis factor-alpha. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L75–L87. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Dhaliwal, R.; Ivanovska, J.; Kantores, C.; McNamara, P.J.; Scott, J.A.; Belik, J.; Jankov, R.P. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L503–L510. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Dhaliwal, R.; Kantores, C.; Ivanovska, J.; Gosal, K.; McNamara, P.J.; Letarte, M.; Jankov, R.P. Rho-kinase inhibitor prevents bleomycin-induced injury in neonatal rats independent of effects on lung inflammation. Am. J. Respir. Cell Mol. Biol. 2014, 50, 61–73. [Google Scholar] [PubMed]
- Nardiello, C.; Mizikova, I.; Morty, R.E. Looking ahead: Where to next for animal models of bronchopulmonary dysplasia? Cell Tissue Res. 2017, 367, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Van Rheen, Z.; Fattman, C.; Domarski, S.; Majka, S.; Klemm, D.; Stenmark, K.R.; Nozik-Grayck, E. Lung extracellular superoxide dismutase overexpression lessens bleomycin-induced pulmonary hypertension and vascular remodeling. Am. J. Respir. Cell Mol. Biol. 2011, 44, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Cooney, T.P.; Thurlbeck, W.M. The radial alveolar count method of emery and mithal: A reappraisal 2--intrauterine and early postnatal lung growth. Thorax 1982, 37, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Woods, C.; Taylor, J.M.; Benninger, R.K.; Johnson, R.D.; Villegas, L.R.; Stenmark, K.R.; Harrison, D.G.; Majka, S.M.; Irwin, D.; et al. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L868–L876. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Lakshminrusimha, S.; Reda, W.J.; Wedgwood, S.; Czech, L.; Gugino, S.F.; Davis, J.M.; Russell, J.A.; Steinhorn, R.H. Superoxide dismutase restores eNOS expression and function in resistance pulmonary arteries from neonatal lambs with persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L979–L987. [Google Scholar] [CrossRef] [PubMed]
- Oury, T.D.; Day, B.J.; Crapo, J.D. Extracellular superoxide dismutase: A regulator of nitric oxide bioavailability. Lab. Investig. 1996, 75, 617–636. [Google Scholar] [PubMed]
- Langston, W.; Chidlow, J.H., Jr.; Booth, B.A.; Barlow, S.C.; Lefer, D.J.; Patel, R.P.; Kevil, C.G. Regulation of endothelial glutathione by ICAM-1 governs VEGF-A-mediated eNOS activity and angiogenesis. Free Radic. Biol. Med. 2007, 42, 720–729. [Google Scholar] [CrossRef] [PubMed]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Donate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Bachschmid, M.M.; Matsui, R. Regulation of neovascularization by S-glutathionylation via the Wnt5a/sFlt-1 pathway. Biochem. Soc. Trans. 2014, 42, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kim, S.J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, C749–C764. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Sudhahar, V.; Kim, S.J.; Chen, G.F.; McKinney, R.D.; Kojda, G.; Fukai, T.; Ushio-Fukai, M. Critical role of endothelial hydrogen peroxide in post-ischemic neovascularization. PLoS ONE 2013, 8, e57618. [Google Scholar] [CrossRef] [PubMed]
- Kunig, A.M.; Balasubramaniam, V.; Markham, N.E.; Morgan, D.; Montgomery, G.; Grover, T.R.; Abman, S.H. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L529–L535. [Google Scholar] [CrossRef] [PubMed]
- Kunig, A.M.; Balasubramaniam, V.; Markham, N.E.; Seedorf, G.; Gien, J.; Abman, S.H. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1068–L1078. [Google Scholar] [CrossRef] [PubMed]
- Le Cras, T.D.; Markham, N.E.; Tuder, R.M.; Voelkel, N.F.; Abman, S.H. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L555–L562. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Lakshminrusimha, S.; Czech, L.; Schumacker, P.T.; Steinhorn, R.H. Increased p22(phox)/Nox4 expression is involved in remodeling through hydrogen peroxide signaling in experimental persistent pulmonary hypertension of the newborn. Antioxid. Redox Signal. 2013, 18, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Groh, B.S.; Schumacker, P.T.; Lakshminrusimha, S.; Czech, L.; Gugino, S.F.; Russell, J.A.; Steinhorn, R.H. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ. Res. 2008, 102, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Farrow, K.N.; Lakshminrusimha, S.; Czech, L.; Groh, B.S.; Gugino, S.F.; Davis, J.M.; Russell, J.A.; Steinhorn, R.H. SOD and inhaled nitric oxide normalize phosphodiesterase 5 expression and activity in neonatal lambs with persistent pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L109–L116. [Google Scholar] [CrossRef] [PubMed]
- Gien, J.; Tseng, N.; Seedorf, G.; Kuhn, K.; Abman, S.H. Endothelin-1-Rho kinase interactions impair lung structure and cause pulmonary hypertension after bleomycin exposure in neonatal rat pups. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1090–L1100. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.D.; Seedorf, G.J.; Wisniewski, B.L.; Black, C.P.; Ryan, S.L.; Balasubramaniam, V.; Abman, S.H. Endothelial colony-forming cell conditioned media promote angiogenesis in vitro and prevent pulmonary hypertension in experimental bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L73–L81. [Google Scholar] [CrossRef] [PubMed]
- Ee, M.T.; Kantores, C.; Ivanovska, J.; Wong, M.J.; Jain, A.; Jankov, R.P. Leukotriene B4 mediates macrophage influx and pulmonary hypertension in bleomycin-induced chronic neonatal lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L292–L302. [Google Scholar] [CrossRef] [PubMed]
- Tourneux, P.; Markham, N.; Seedorf, G.; Balasubramaniam, V.; Abman, S.H. Inhaled nitric oxide improves lung structure and pulmonary hypertension in a model of bleomycin-induced bronchopulmonary dysplasia in neonatal rats. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1103–L1111. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.; Sherlock, L.; Fisher, S.; Maltzahn, J.K.; Wright, C.J.; Nozik-Grayck, E. Serotonin 2A receptor inhibition protects against the development of pulmonary hypertension and pulmonary vascular remodeling in neonatal mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H. Bronchopulmonary dysplasia: “A vascular hypothesis”. Am. J. Respir. Crit. Care Med. 2001, 164, 1755–1756. [Google Scholar] [CrossRef] [PubMed]
- De Wijs-Meijler, D.P.M.; van Duin, R.W.B.; Duncker, D.J.; Scherrer, U.; Sartori, C.; Reiss, I.K.M.; Merkus, D. Structural and functional changes of the pulmonary vasculature after hypoxia exposure in the neonatal period—A new swine model of pulmonary vascular disease. Am. J. Physiol. Heart Circ. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Wedgwood, S.; Warford, C.; Agvateesiri, S.C.; Thai, P.; Berkelhamer, S.K.; Perez, M.; Underwood, M.A.; Steinhorn, R.H. Postnatal growth restriction augments oxygen-induced pulmonary hypertension in a neonatal rat model of bronchopulmonary dysplasia. Pediatr. Res. 2016, 80, 894–902. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.M.; Herington, J.L.; Stouch, A.N.; Mukherjee, A.B.; Lakhdari, O.; Blackwell, T.S.; Prince, L.S. Ikkbeta activation in the fetal lung mesenchyme alters lung vascular development but not airway morphogenesis. Am. J. Pathol. 2017, 187, 2635–2644. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.D.; Benjamin, J.T.; Kelly, D.R.; Frank, D.B.; Prince, L.S. Chorioamnionitis stimulates angiogenesis in saccular stage fetal lungs via CC chemokines. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L637–L645. [Google Scholar] [CrossRef] [PubMed]
- An, H.S.; Bae, E.J.; Kim, G.B.; Kwon, B.S.; Beak, J.S.; Kim, E.K.; Kim, H.S.; Choi, J.H.; Noh, C.I.; Yun, Y.S. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Korean Circ. J. 2010, 40, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, M.; Beardsmore, C.S.; Owers-Bradley, J.; Dogaru, C.M.; Mada, M.; Ball, I.; Garipov, R.R.; Kuehni, C.E.; Spycher, B.D.; Silverman, M. Catch-up alveolarization in ex-preterm children: Evidence from (3)He magnetic resonance. Am. J. Respir. Crit. Care Med. 2013, 187, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, W.; Wang, L.; Lingappan, K. Sex-specific differences in the modulation of growth differentiation factor 15 (GDF15) by hyperoxia in vivo and in vitro: Role of Hif-1alpha. Toxicol. Appl. Pharmacol. 2017, 332, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Redente, E.F.; Jacobsen, K.M.; Solomon, J.J.; Lara, A.R.; Faubel, S.; Keith, R.C.; Henson, P.M.; Downey, G.P.; Riches, D.W. Age and sex dimorphisms contribute to the severity of bleomycin-induced lung injury and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L510–L518. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherlock, L.G.; Trumpie, A.; Hernandez-Lagunas, L.; McKenna, S.; Fisher, S.; Bowler, R.; Wright, C.J.; Delaney, C.; Nozik-Grayck, E. Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia. Antioxidants 2018, 7, 42. https://doi.org/10.3390/antiox7030042
Sherlock LG, Trumpie A, Hernandez-Lagunas L, McKenna S, Fisher S, Bowler R, Wright CJ, Delaney C, Nozik-Grayck E. Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia. Antioxidants. 2018; 7(3):42. https://doi.org/10.3390/antiox7030042
Chicago/Turabian StyleSherlock, Laurie G., Ashley Trumpie, Laura Hernandez-Lagunas, Sarah McKenna, Susan Fisher, Russell Bowler, Clyde J. Wright, Cassidy Delaney, and Eva Nozik-Grayck. 2018. "Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia" Antioxidants 7, no. 3: 42. https://doi.org/10.3390/antiox7030042