p53-Mediated Oxidative Stress Enhances Indirubin-3′-Monoxime-Induced Apoptosis in HCT116 Colon Cancer Cells by Upregulating Death Receptor 5 and TNF-Related Apoptosis-Inducing Ligand Expression


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. Western Blotting Analysis
2.5. Flow Cytometry Analysis
2.6. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.7. Transient Knockdown of p53 and CHOP
2.8. Fluorescence Microscopy
2.9. Statistical Analysis
3. Results
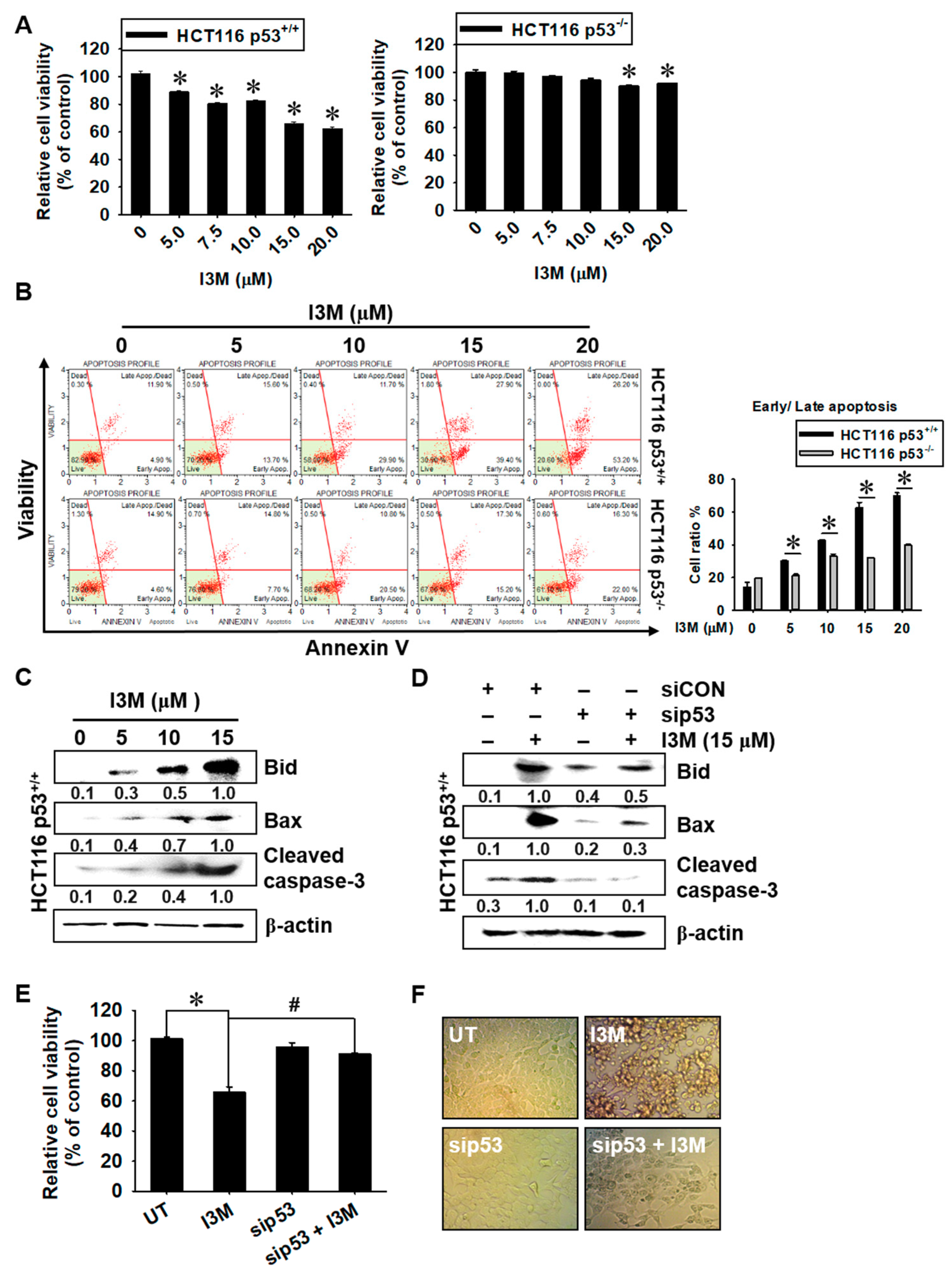
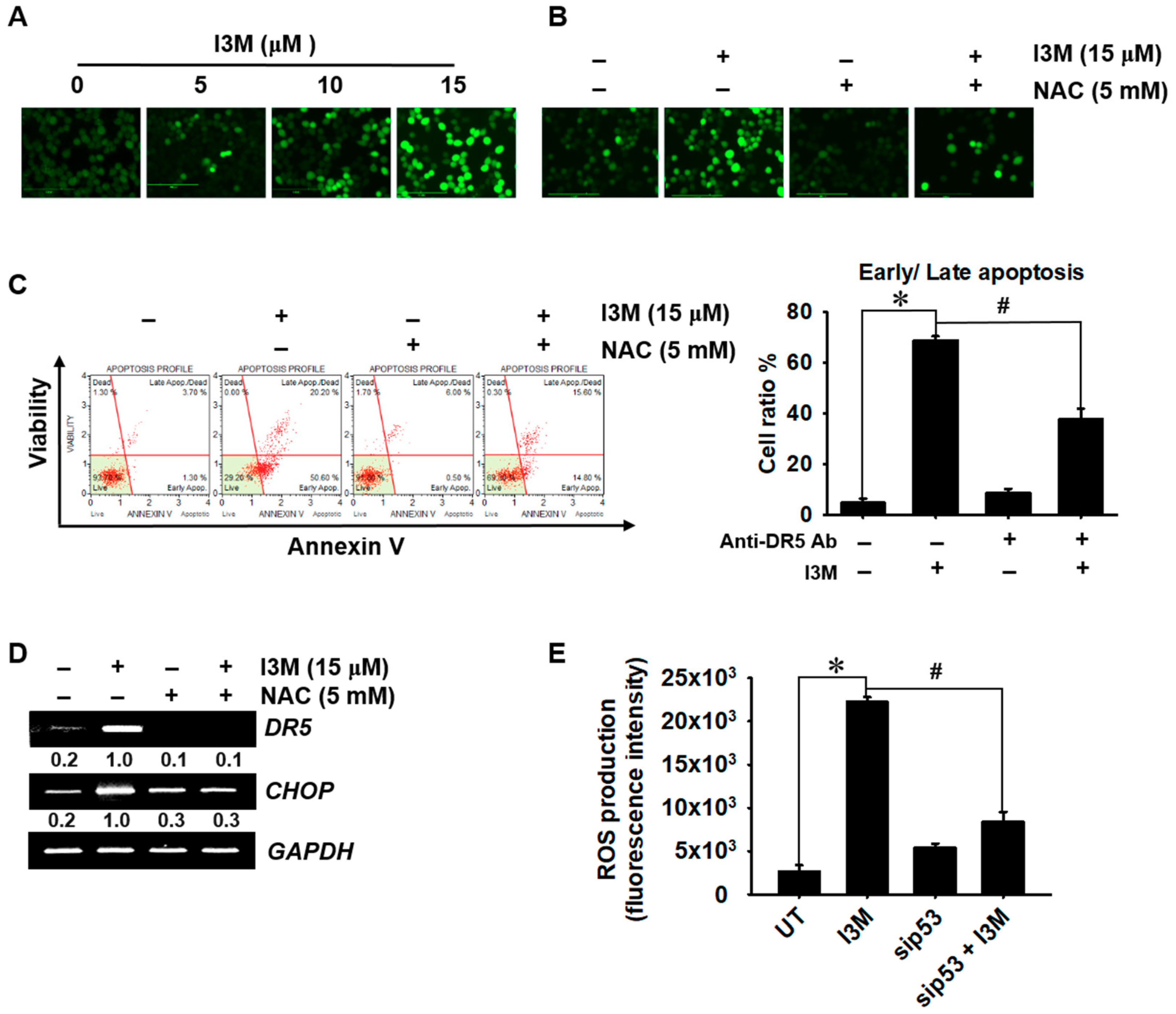
3.1. I3M-Induced HCT116 Apoptosis Is Dependent on p53 Expression
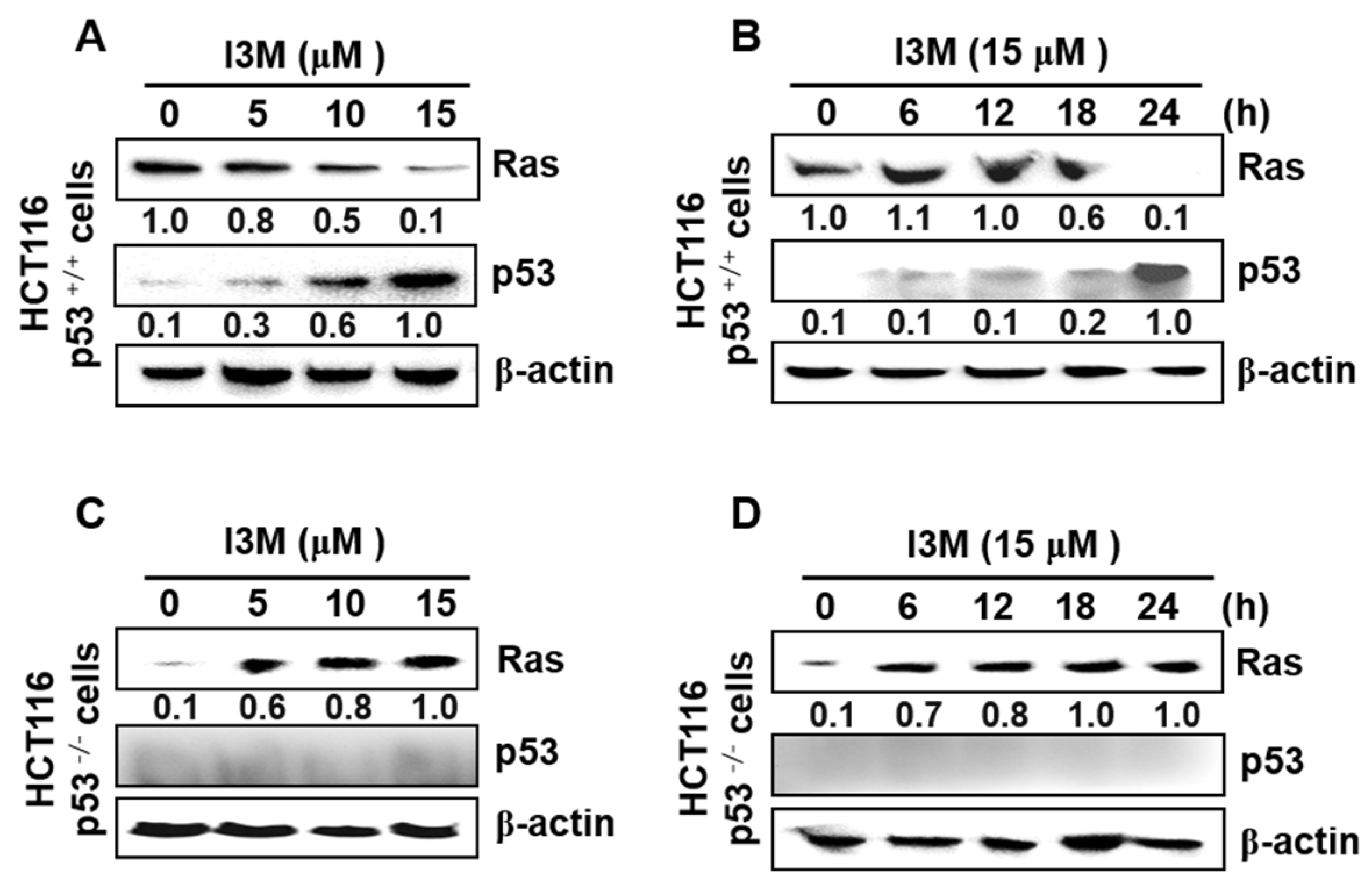
3.2. I3M Enhances p53 Expression in HCT116 p53+/+ Cells, Accompanied by Ras Downregulation
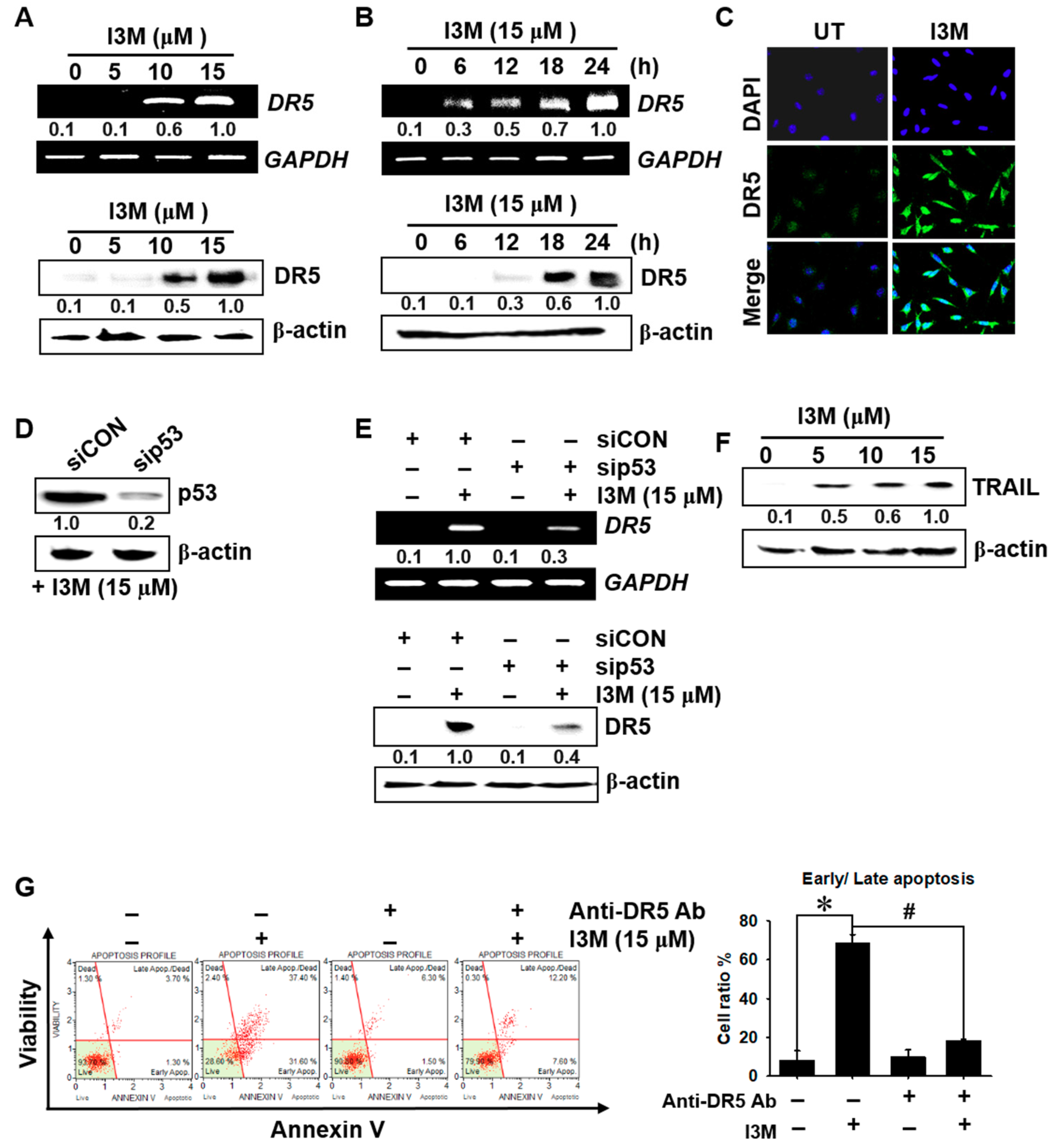
3.3. I3M-Induced DR5 Expression Depends on p53 Expression
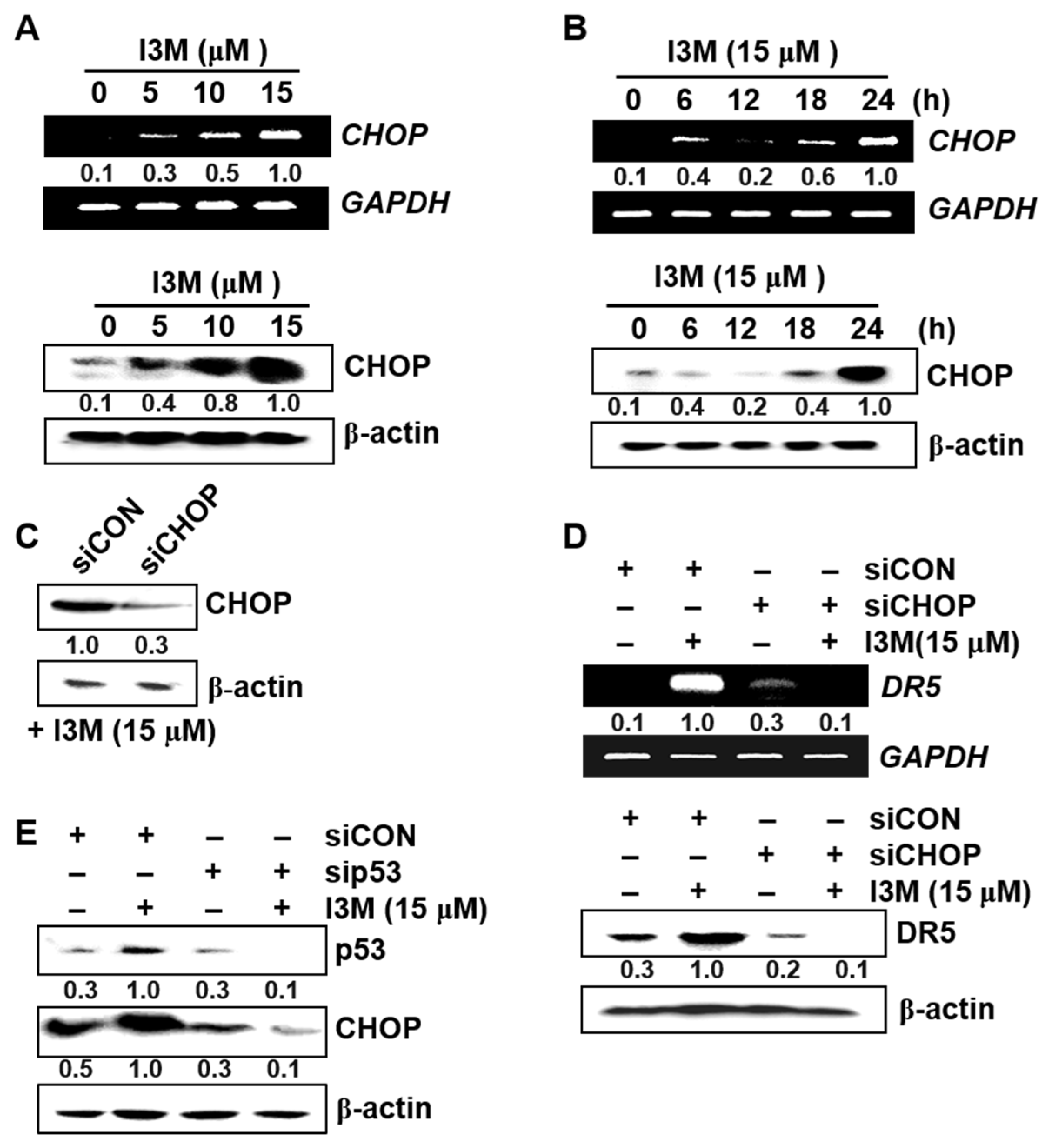
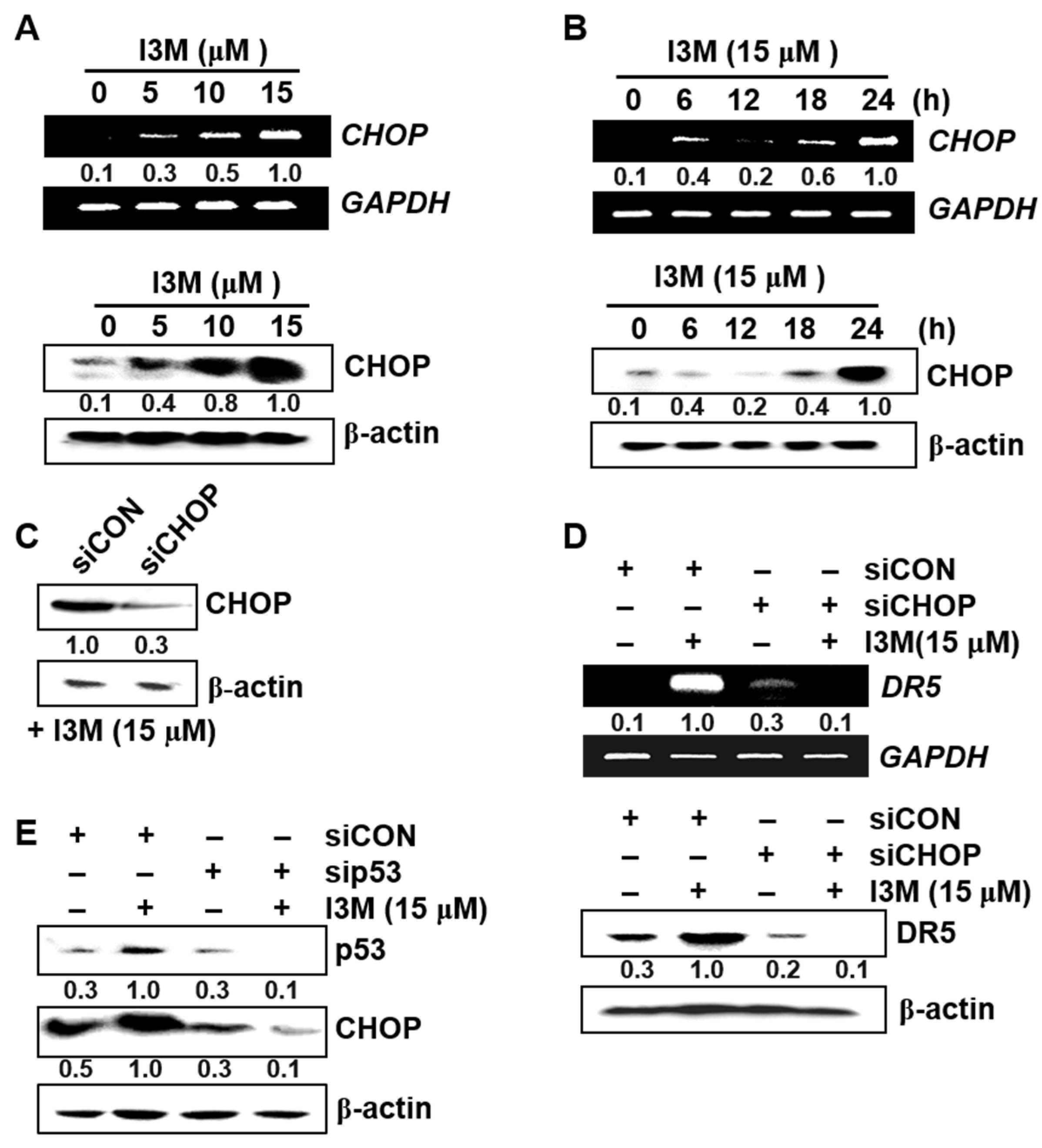
3.4. I3M-Induced DR5 Upregulation Is Mediated via CHOP Induction
3.5. I3M-Mediated p53 Increases Oxidative Stress-Mediated Apoptosis
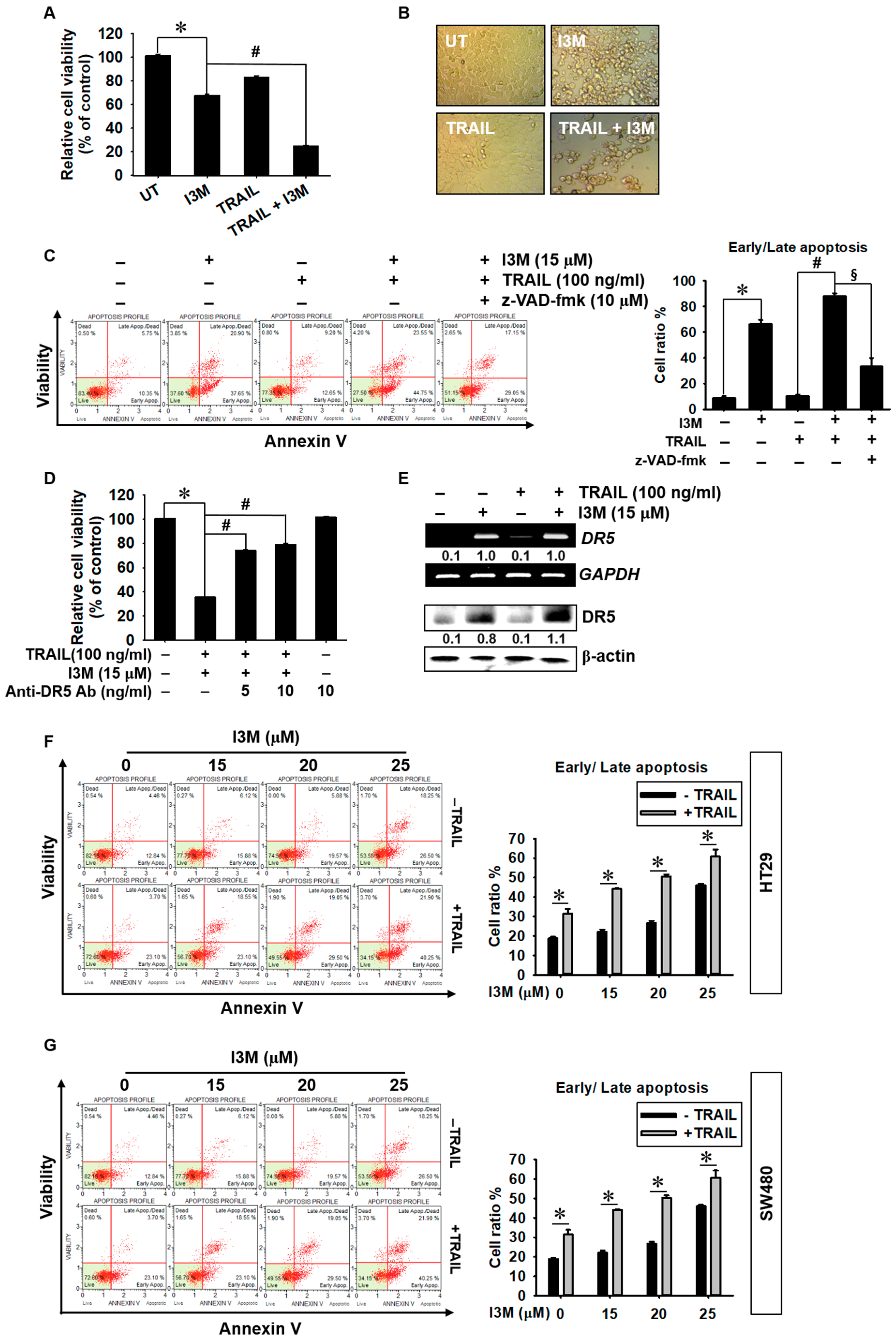
3.6. Co-Treatment with I3M and TRAIL Potentiates Apoptosis in HCT116, HT 29, and SW480 Colon Cancer Cells
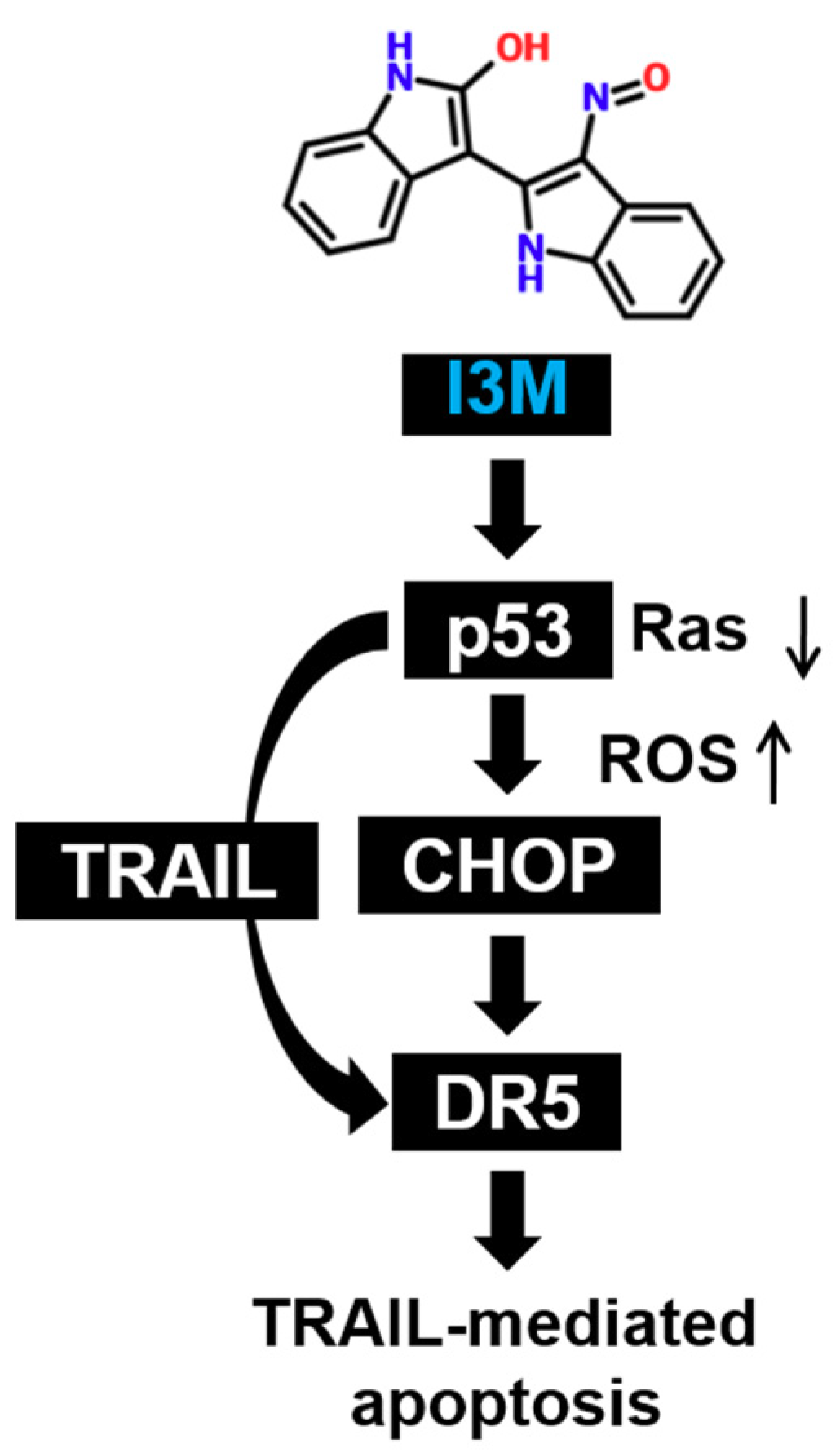
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Simpson, E.R.; Brown, K.A. p53: Protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 2015, 75, 5001–5007. [Google Scholar] [CrossRef] [PubMed]
- Essmann, F.; Schulze-Osthoff, K. Translational approaches targeting the p53 pathway for anti-cancer therapy. Br. J. Pharmacol. 2012, 165, 328–344. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hickman, J.H.; Wang, S.J.; Gu, W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 2015, 14, 2881–2885. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, Y.; St., Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Lu, Y.; Shen, H.M. Targeting p53 as a therapeutic strategy in sensitizing TRAIL-induced apoptosis in cancer cells. Cancer Lett. 2012, 314, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.Z.; Yao, B.Y. Progress in indirubin treatment of chronic myelocytic leukemia. J. Tradit. Chin. Med. 1983, 3, 245–248. [Google Scholar] [PubMed]
- Ahn, M.Y.; Kim, T.H.; Kwon, S.M.; Yoon, H.E.; Kim, H.S.; Kim, J.I.; Kim, Y.C.; Kang, K.W.; Ahn, S.G.; Yoon, J.H. 5-Nitro-5′-hydroxy-indirubin-3′-oxime (AGM130), an indirubin-3′-oxime derivative, inhibits tumor growth by inducing apoptosis against non-small cell lung cancer in vitro and in vivo. Eur. J. Pharm. Sci. 2015, 79, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Ichimaru, Y.; Kurita, M.; Hayashi, E.; Homma, T.; Saito, H.; Masuda, S.; Nemoto, N.; Hemmi, A.; Suzuki, T.; et al. Induction of cell death in pancreatic ductal adenocarcinoma by indirubin 3′-oxime and 5-methoxyindirubin 3′-oxime in vitro and in vivo. Cancer Lett. 2017, 397, 72–82. [Google Scholar] [CrossRef]
- Perabo, F.G.; Landwehrs, G.; Frossler, C.; Schmidt, D.H.; Mueller, S.C. Antiproliferative and apoptosis inducing effects of indirubin-3′-monoxime in renal cell cancer cells. Urol. Oncol. 2011, 29, 815–820. [Google Scholar] [CrossRef]
- Damiens, E.; Baratte, B.; Marie, D.; Eisenbrand, G.; Meijer, L. Anti-mitotic properties of indirubin-3′-monoxime, a CDK/GSK-3 inhibitor: Induction of endoreplication following prophase arrest. Oncogene 2001, 20, 3786–3797. [Google Scholar] [CrossRef] [PubMed]
- Ries, S.; Biederer, C.; Woods, D.; Shifman, O.; Shirasawa, S.; Sasazuki, T.; McMahon, M.; Oren, M.; McCormick, F. Opposing effects of Ras on p53: Transcriptional activation of mdm2 and induction of p19ARF. Cell 2000, 103, 321–330. [Google Scholar] [CrossRef]
- Takimoto, R.; El-Deiry, W.S. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 2000, 19, 1735–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL resistance: Novel targets for TRAIL sensitization in cancer cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef]
- Kramer, T.; Schmidt, B.; Lo Monte, F. Small-molecule inhibitors of GSK-3: Structural insights and their application to Alzheimer’s disease models. Int. J. Alzheimers Dis. 2012, 2012, 381029. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Bush, A.I.; Adlard, P.A. GSK-3 in neurodegenerative diseases. Int. J. Alzheimers Dis. 2011, 2011, 189246. [Google Scholar] [CrossRef]
- Blazevic, T.; Heiss, E.H.; Atanasov, A.G.; Breuss, J.M.; Dirsch, V.M.; Uhrin, P. Indirubin and indirubin derivatives for counteracting proliferative diseases. Evid. Based Complement. Altern. Med. 2015, 2015, 654098. [Google Scholar] [CrossRef]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef]
- Dimberg, L.Y.; Anderson, C.K.; Camidge, R.; Behbakht, K.; Thorburn, A.; Ford, H.L. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene 2013, 32, 1341–1350. [Google Scholar] [CrossRef]
- Kuribayashi, K.; Krigsfeld, G.; Wang, W.; Xu, J.; Mayes, P.A.; Dicker, D.T.; Wu, G.S.; El-Deiry, W.S. TNFSF10 (TRAIL), a p53 target gene that mediates p53-dependent cell death. Cancer Biol. Ther. 2008, 7, 2034–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willms, A.; Schittek, H.; Rahn, S.; Sosna, J.; Mert, U.; Adam, D.; Trauzold, A. Impact of p53 status on TRAIL-mediated apoptotic and non-apoptotic signaling in cancer cells. PLoS ONE 2019, 14, e0214847. [Google Scholar] [CrossRef] [PubMed]
- Solomon, H.; Brosh, R.; Buganim, Y.; Rotter, V. Inactivation of the p53 tumor suppressor gene and activation of the Ras oncogene: Cooperative events in tumorigenesis. Discov. Med. 2010, 9, 448–454. [Google Scholar] [PubMed]
- Park, E.J.; Choi, K.S.; Yoo, Y.H.; Kwon, T.K. Nutlin-3, a small-molecule MDM2 inhibitor, sensitizes Caki cells to TRAIL-induced apoptosis through p53-mediated PUMA upregulation and ROS-mediated DR5 upregulation. Anticancer Drugs 2013, 24, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Yang, J.S.; Tsai, F.J.; Chiang, N.N.; Lu, C.C.; Huang, Y.S.; Chen, C.; Chen, F.A. Kaempferol induces ATM/p53-mediated death receptor and mitochondrial apoptosis in human umbilical vein endothelial cells. Int. J. Oncol. 2016, 48, 2007–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotliarova, S.; Pastorino, S.; Kovell, L.C.; Kotliarov, Y.; Song, H.; Zhang, W.; Bailey, R.; Maric, D.; Zenklusen, J.C.; Lee, J.; et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 2008, 68, 6643–6651. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilshara, M.G.; Molagoda, I.M.N.; Jayasooriya, R.G.P.T.; Choi, Y.H.; Park, C.; Lee, K.T.; Lee, S.; Kim, G.-Y. p53-Mediated Oxidative Stress Enhances Indirubin-3′-Monoxime-Induced Apoptosis in HCT116 Colon Cancer Cells by Upregulating Death Receptor 5 and TNF-Related Apoptosis-Inducing Ligand Expression. Antioxidants 2019, 8, 423. https://doi.org/10.3390/antiox8100423
Dilshara MG, Molagoda IMN, Jayasooriya RGPT, Choi YH, Park C, Lee KT, Lee S, Kim G-Y. p53-Mediated Oxidative Stress Enhances Indirubin-3′-Monoxime-Induced Apoptosis in HCT116 Colon Cancer Cells by Upregulating Death Receptor 5 and TNF-Related Apoptosis-Inducing Ligand Expression. Antioxidants. 2019; 8(10):423. https://doi.org/10.3390/antiox8100423
Chicago/Turabian StyleDilshara, Matharage Gayani, Ilandarage Menu Neelaka Molagoda, Rajapaksha Gedara Prasad Tharanga Jayasooriya, Yung Hyun Choi, Cheol Park, Kyoung Tae Lee, Seungheon Lee, and Gi-Young Kim. 2019. "p53-Mediated Oxidative Stress Enhances Indirubin-3′-Monoxime-Induced Apoptosis in HCT116 Colon Cancer Cells by Upregulating Death Receptor 5 and TNF-Related Apoptosis-Inducing Ligand Expression" Antioxidants 8, no. 10: 423. https://doi.org/10.3390/antiox8100423