
Co-Administration of Lipid Nanoparticles and Sub-Unit Vaccine Antigens Is Required for Increase in Antigen-Specific Immune Responses in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Adjuvants and Vaccine Antigens
2.3. Antigen-Specific Antibody Titers
2.4. ELISpot Assays
2.5. T-Cell Intracellular Staining (ICS)
2.6. Statistical Analysis
3. Results
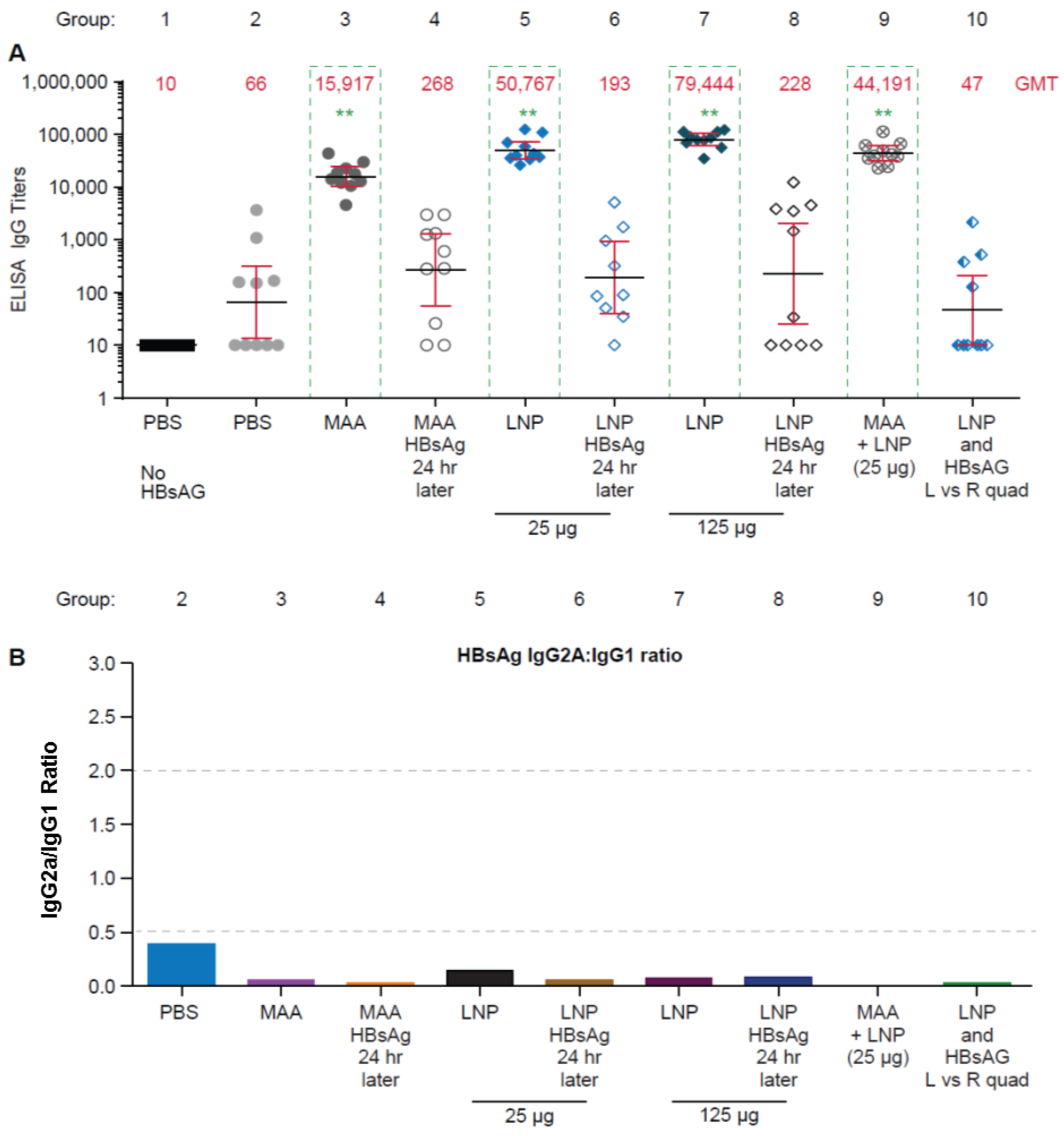
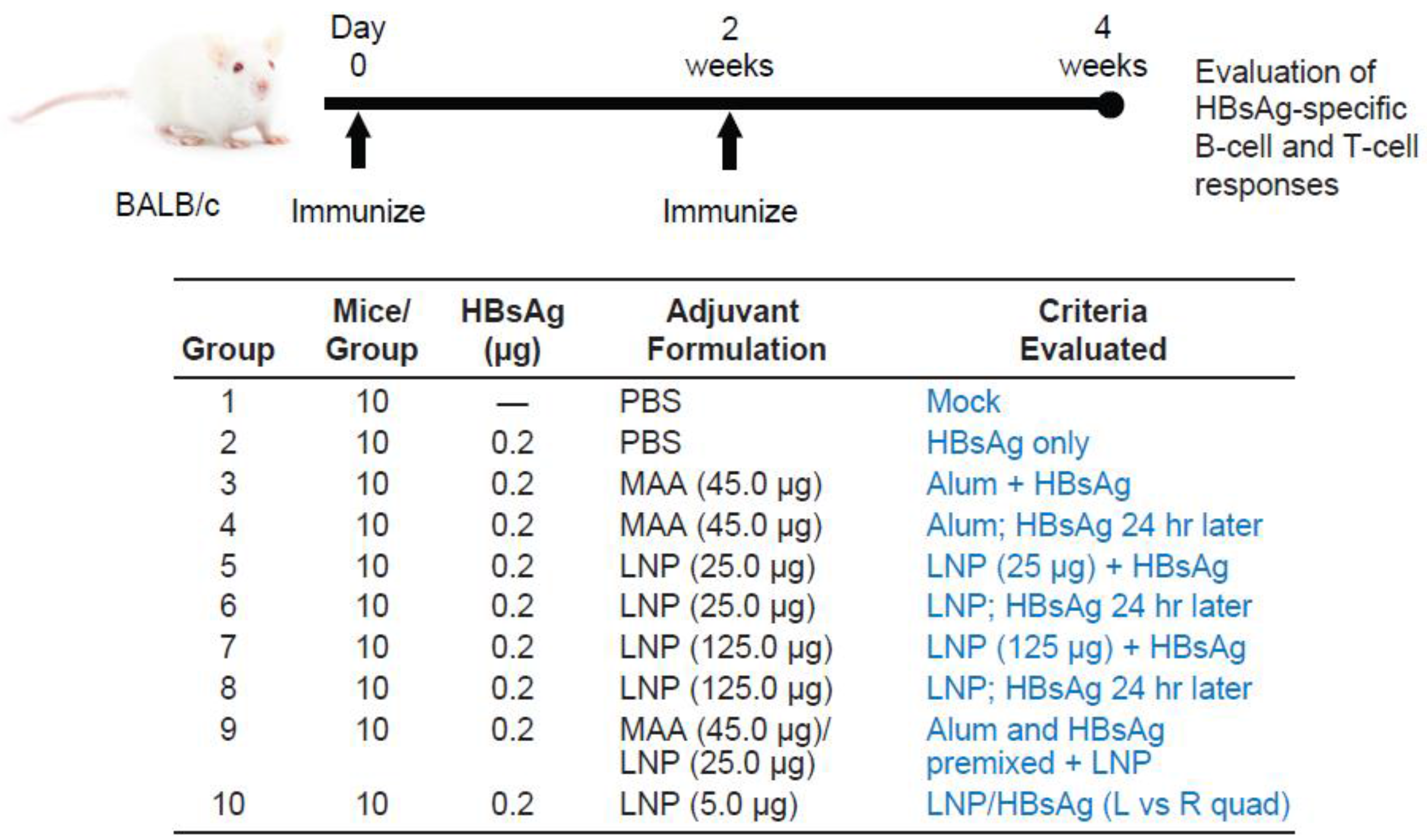
3.1. Requirement for Coadministration of HBsAg with LNP for Antigen-Specific B-Cell Responses
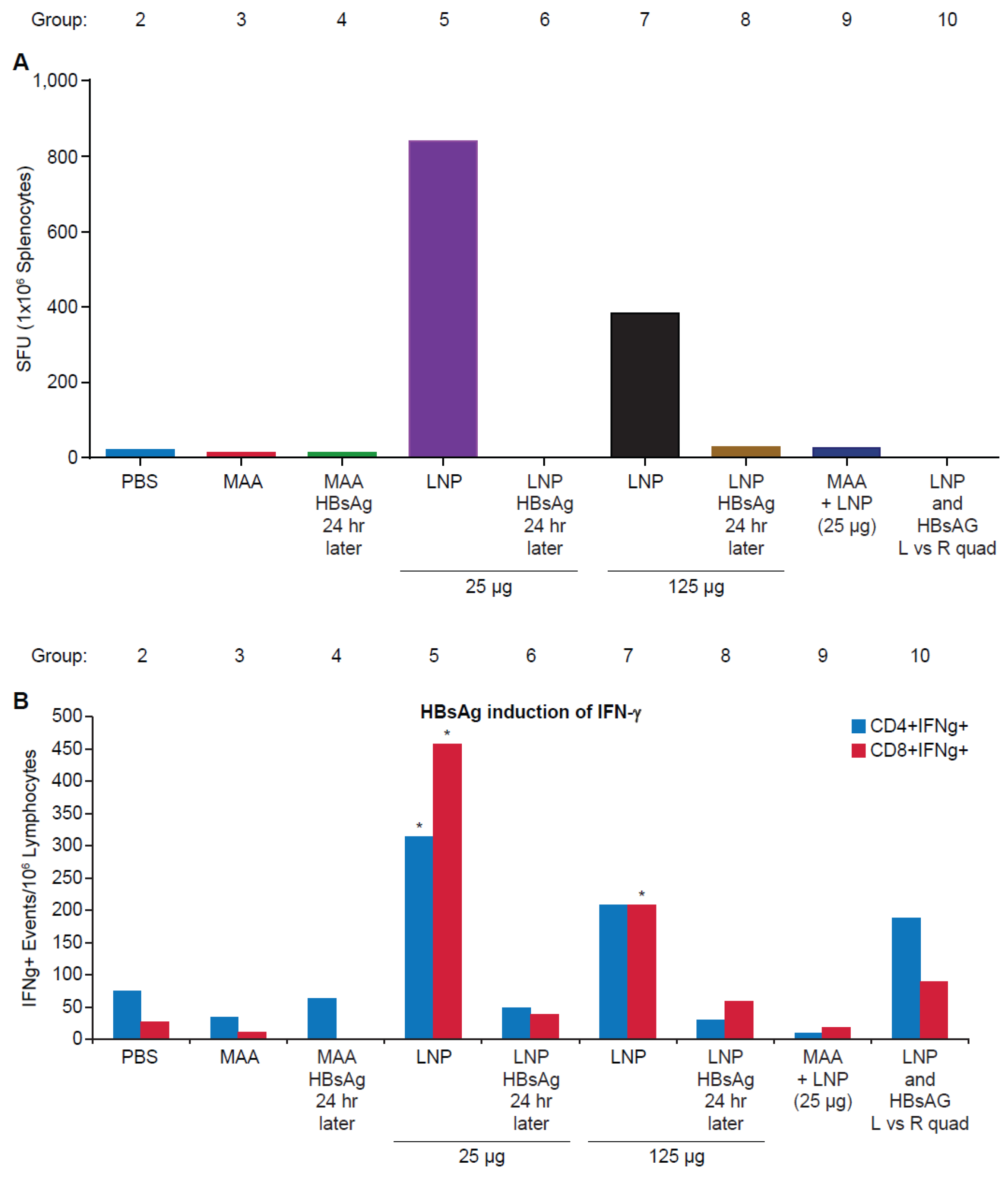
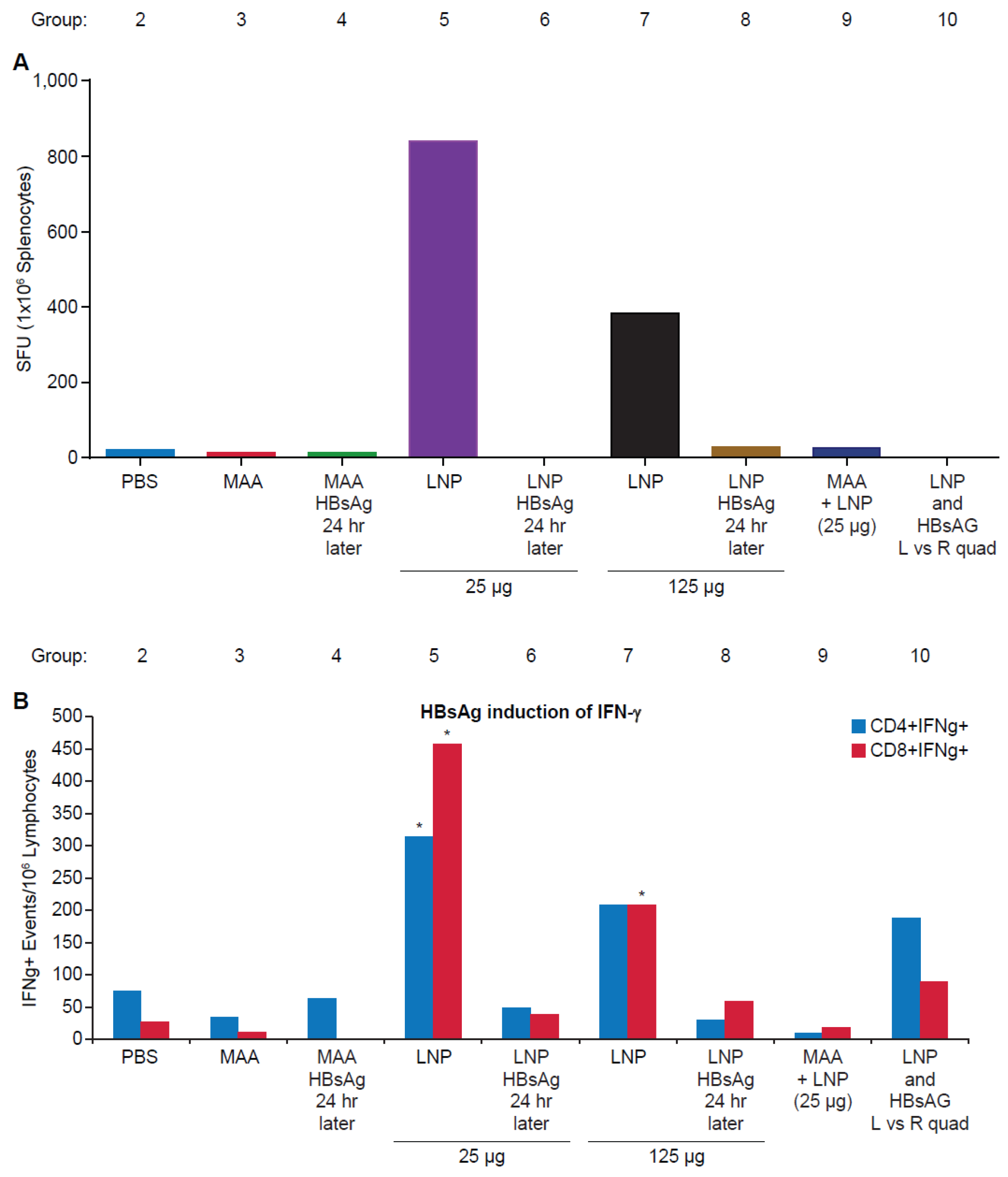
3.2. Requirement for Co-Administration of HBsAg with LNP for Antigen-Specific T-Cell Responses
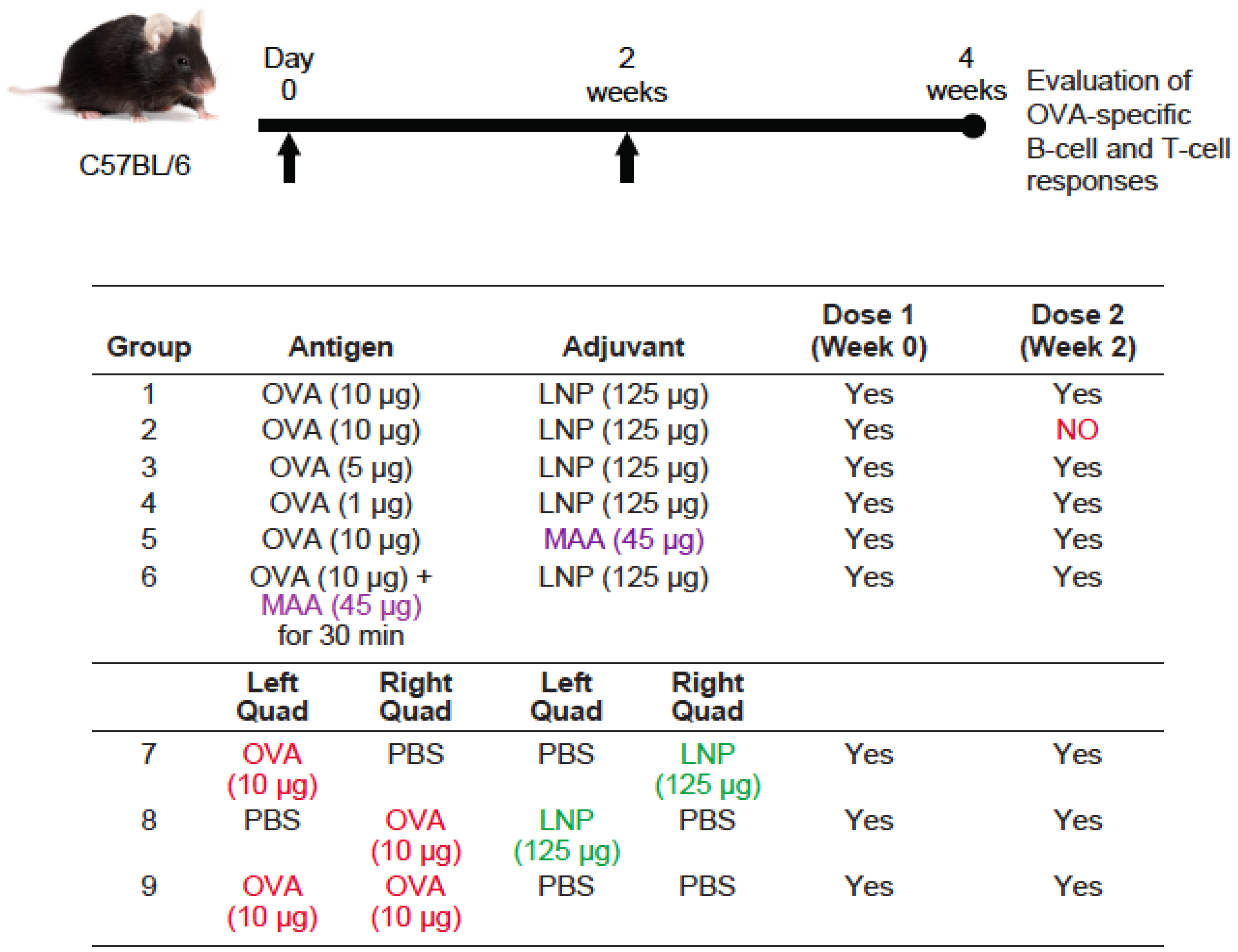
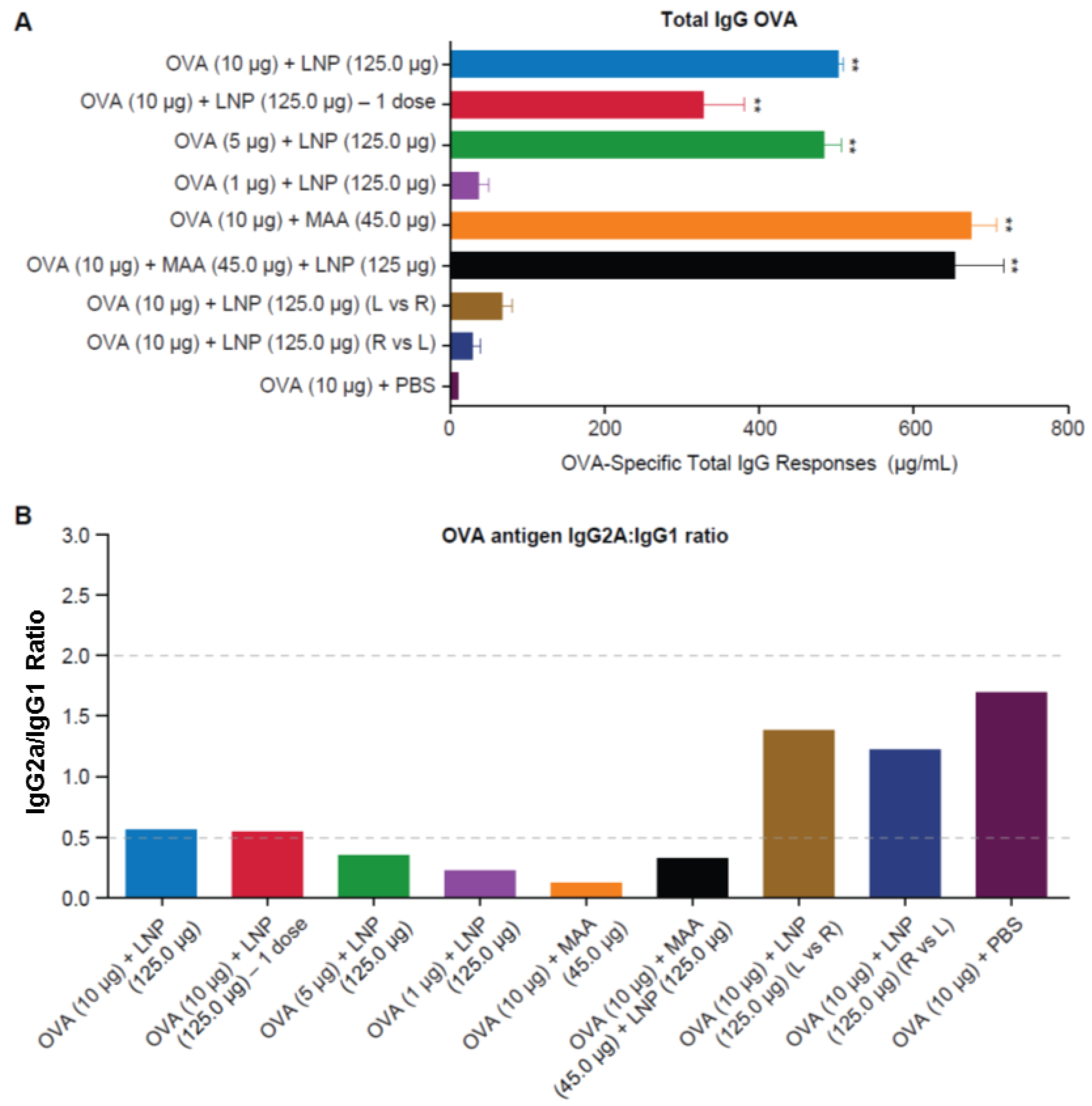
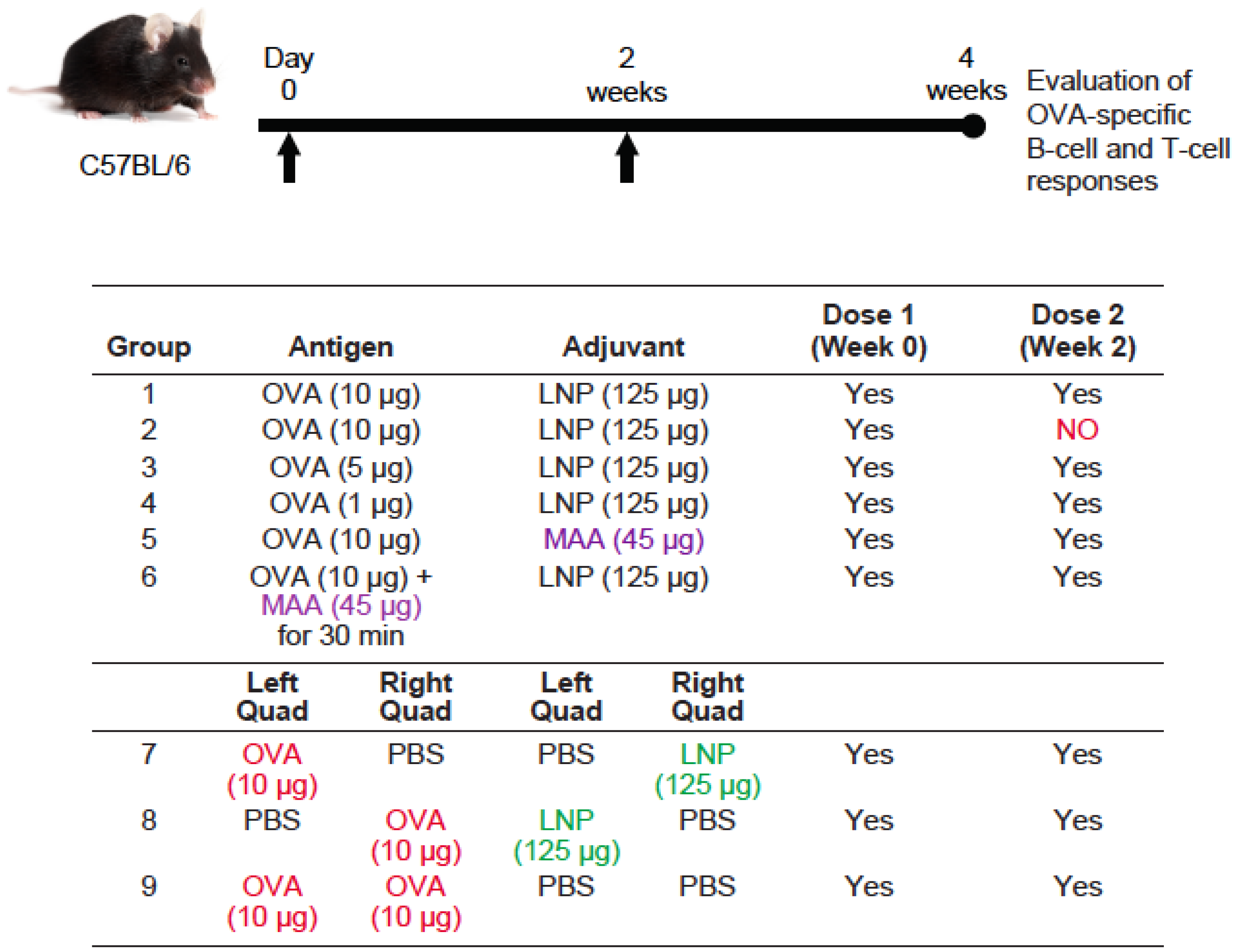
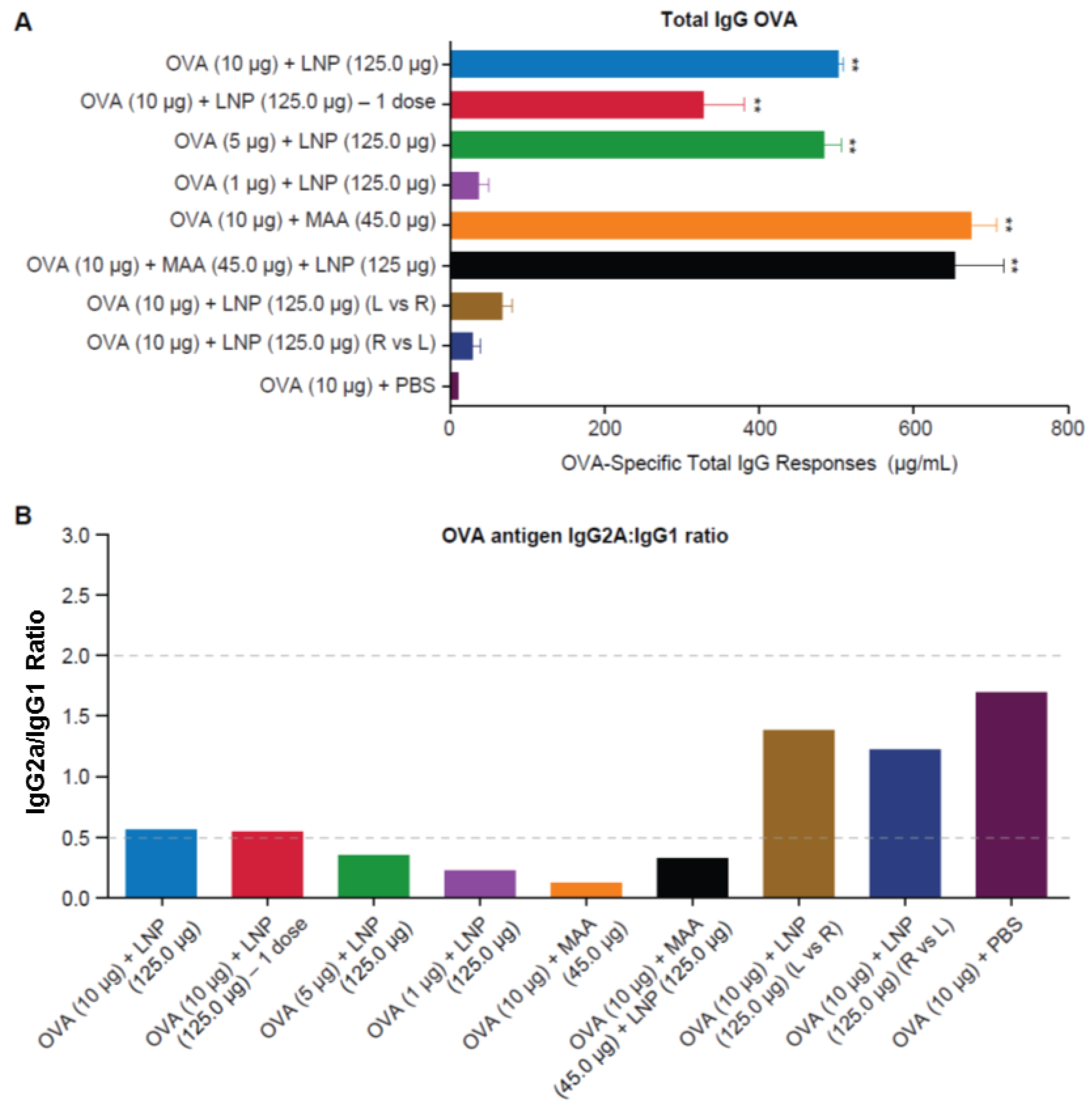
3.3. Requirement for Co-Administration of OVA with LNP for Antigen-Specific B-Cell Responses
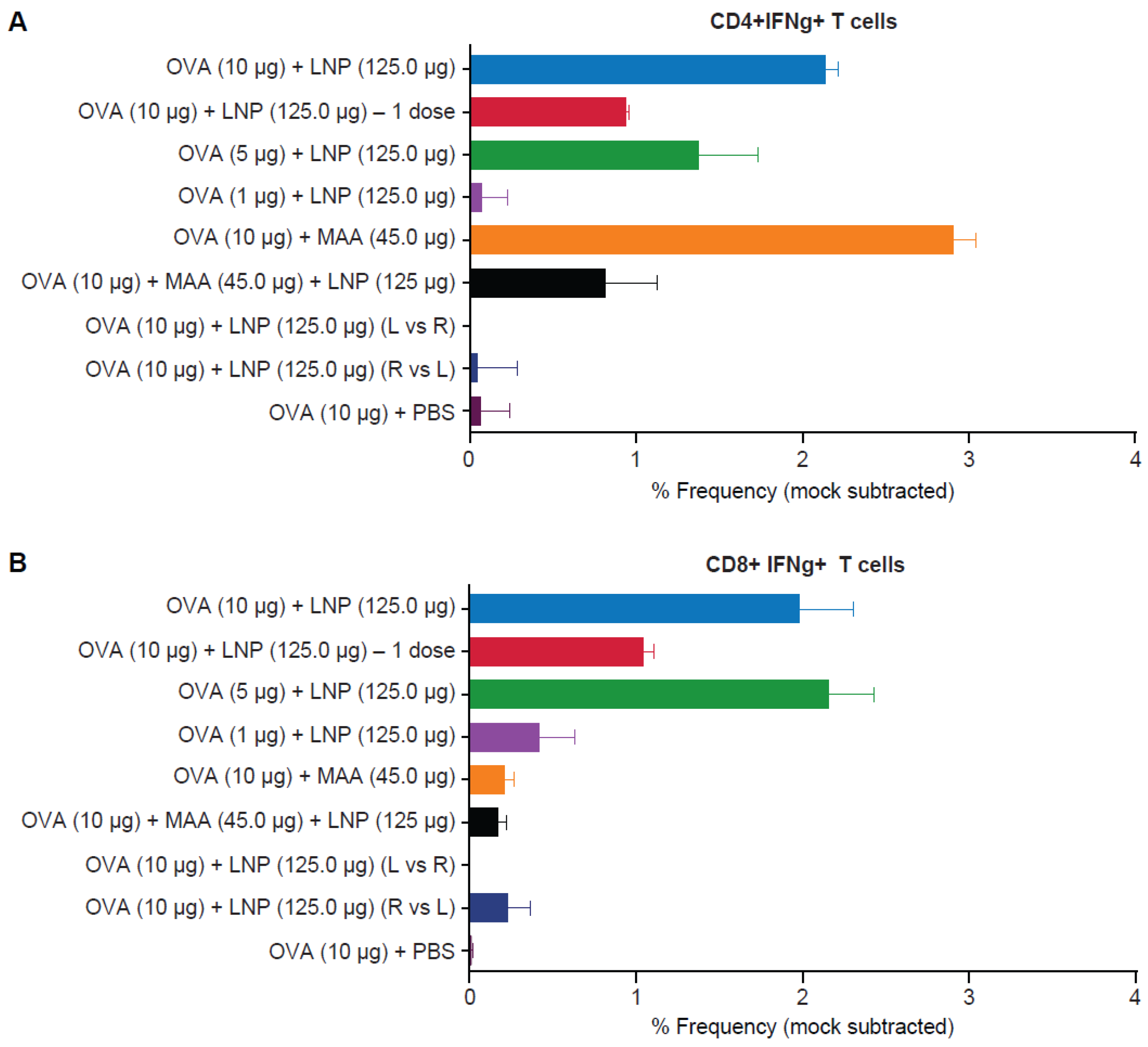
3.4. Requirement for Co-Administration of OVA with LNP for Antigen-Specific T-Cell Responses
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A
References
- Demana, P.H.; Davies, N.M.; Hook, S.; Rades, T. Quil a-lipid powder formulations releasing iscoms and related colloidal structures upon hydration. J. Control Release 2005, 103, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Vangala, A.; Kirby, D.; Rosenkrands, I.; Agger, E.M.; Andersen, P.; Perrie, Y. A comparative study of cationic liposome and niosome-based adjuvant systems for protein subunit vaccines: Characterisation, environmental scanning electron microscopy and immunisation studies in mice. J. Pharm. Pharmacol. 2006, 58, 787–799. [Google Scholar] [PubMed]
- Guy, B. The perfect mix: Recent progress in adjuvant research. Nat. Rev. Microbiol 2007, 5, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Perrie, Y.; Mohammed, A.R.; Kirby, D.J.; McNeil, S.E.; Bramwell, V.W. Vaccine adjuvant systems: Enhancing the efficacy of sub-unit protein antigens. Int. J. Pharm. 2008, 364, 272–280. [Google Scholar] [CrossRef]
- Hansen, B.; Sokolovska, A.; HogenEsch, H.; Hem, S.L. Relationship between the strength of antigen adsorption to an aluminum-containing adjuvant and the immune response. Vaccine 2007, 25, 6618–6624. [Google Scholar] [CrossRef] [PubMed]
- Apostolico Jde, S.; Lunardelli, V.A.; Coirada, F.C.; Boscardin, S.B.; Rosa, D.S. Adjuvants: Classification, modus operandi, and licensing. J. Immunol. Res. 2016. [Google Scholar] [CrossRef]
- Olafsdottir, T.; Lindqvist, M.; Harandi, A.M. Molecular signatures of vaccine adjuvants. Vaccine 2015, 33, 5302–5307. [Google Scholar] [CrossRef] [PubMed]
- Leroux-Roels, G. Unmet needs in modern vaccinology: Adjuvants to improve the immune response. Vaccine 2010, 28, C25–C36. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Yadav, S.K. Cellular interactions of therapeutically delivered nanoparticles. Expert Opin. Drug Deliv. 2011, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Hadinoto, K.; Sundaresan, A.; Cheow, W.S. Lipid-polymer hybrid nanoparticles as a new generation therapeutic delivery platform: A review. Eur. J. Pharm. Biopharm. 2013, 85, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Wilson, K.; Day, S.; Fuchsberger, M.; Plebanski, M. Methods of effective conjugation of antigens to nanoparticles as non-inflammatory vaccine carriers. Methods 2013, 60, 232–241. [Google Scholar] [CrossRef]
- Rosalia, R.A.; Cruz, L.J.; van Duikeren, S.; Tromp, A.T.; Silva, A.L.; Jiskoot, W.; de Gruijl, T.; Lowik, C.; Oostendorp, J.; van der Burg, S.H.; et al. Cd40-targeted dendritic cell delivery of plga-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials 2015, 40, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, K.; Aoshi, T.; Narita, H.; Kuroda, E.; Hayashi, M.; Tetsutani, K.; Koyama, S.; Mochizuki, S.; Sakurai, K.; Katakai, Y.; et al. Nonagonistic dectin-1 ligand transforms cpg into a multitask nanoparticulate tlr9 agonist. Proc. Natl. Acad. Sci. USA 2014, 111, 3086–3091. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.J.; Swartz, M.A.; Szeto, G.L. Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 2013, 12, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Zeelenberg, I.S.; Cruz, L.J.; van Hout-Kuijer, M.A.; van de Glind, G.; Fokkink, R.G.; Lambeck, A.J.; Figdor, C.G. Targeted delivery of tlr ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 2011, 118, 6836–6844. [Google Scholar] [CrossRef]
- Katare, Y.K.; Panda, A.K. Influences of excipients on in vitro release and in vivo performance of tetanus toxoid loaded polymer particles. Eur. J. Pharm. Sci. 2006, 28, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Thoryk, E.A.; Cox, K.S.; Meschino, S.; Dubey, S.A.; Vora, K.A.; Celano, R.; Gindy, M.; Casimiro, D.R.; Bett, A.J. A novel lipid nanoparticle adjuvant significantly enhances B cell and t cell responses to sub-unit vaccine antigens. Vaccine 2016, 34, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chiu, Y.C.; Tostanoski, L.H.; Jewell, C.M. Polyelectrolyte multilayers assembled entirely from immune signals on gold nanoparticle templates promote antigen-specific T cell response. ACS Nano 2015, 9, 6465–6477. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, A.; Blanchette, C.; Boone, T.; Corzett, M.; Fischer, N.; Hoeprich, P.; Driks, A.; Rasley, A. Co-delivery of adjuvant and subunit antigens via a nanoparticle platform induces tissue-associated and systemic adaptive immune responses (P4409). J. Immunol. 2013, 190, 205–214. [Google Scholar]
- Gindy, M.E.; Feuston, B.; Glass, A.; Arrington, L.; Haas, R.M.; Schariter, J.; Stirdivant, S.M. Stabilization of ostwald ripening in low molecular weight amino lipid nanoparticles for systemic delivery of sirna therapeutics. Mol. Pharm. 2014, 11, 4143–4153. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, D.; Antonello, J.M.; Bett, A.J.; Medi, M.B.; Casimiro, D.R.; ter Meulen, J. Immunogenicity of a reduced-dose whole killed rabies vaccine is significantly enhanced by ISCOMATRIX adjuvant, merck amorphous aluminum hydroxylphosphate sulfate (MAA) or a synthetic tlr9 agonist in rhesus macaques. Vaccine 2013, 31, 4888–4893. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Thoryk, E.A.; Cox, K.S.; Smith, J.S.; Wolf, J.J.; Gindy, M.E.; Casimiro, D.R.; Bett, A.J. A tetravalent sub-unit dengue vaccine formulated with ionizable cationic lipid nanoparticle induces significant immune responses in rodents and non-human primates. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Miyahira, Y.; Murata, K.; Rodriguez, D.; Rodriguez, J.R.; Esteban, M.; Rodrigues, M.M.; Zavala, F. Quantification of antigen specific CD8+ T cells using an elispot assay. J. Immunol. Methods 1995, 181, 45–54. [Google Scholar] [CrossRef]
- Fan, Y.; Moon, J.J. Nanoparticle drug delivery systems designed to improve cancer vaccines and immunotherapy. Vaccines 2015, 3, 662–685. [Google Scholar] [PubMed]
- Hogenesch, H. Mechanism of immunopotentiation and safety of aluminum adjuvants. Front. Immunol. 2012. [Google Scholar] [CrossRef] [PubMed]
- HogenEsch, H. Mechanisms of stimulation of the immune response by aluminum adjuvants. Vaccine 2002, 20, S34–S39. [Google Scholar] [CrossRef]
- Noe, S.M.; Green, M.A.; HogenEsch, H.; Hem, S.L. Mechanism of immunopotentiation by aluminum-containing adjuvants elucidated by the relationship between antigen retention at the inoculation site and the immune response. Vaccine 2010, 28, 3588–3594. [Google Scholar] [PubMed]
- Didierlaurent, A.M.; Morel, S.; Lockman, L.; Giannini, S.L.; Bisteau, M.; Carlsen, H.; Kielland, A.; Vosters, O.; Vanderheyde, N.; Schiavetti, F.; et al. As04, an aluminum salt- and tlr4 agonist-based adjuvant system, induces a transient localized innate immune response leading to enhanced adaptive immunity. J. Immunol. 2009, 183, 6186–6197. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Fierens, K.; Lambrecht, B.N. Alum adjuvant: Some of the tricks of the oldest adjuvant. J. Med. Microbiol. 2012, 61, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Saade, F.; Honda-Okubo, Y.; Trec, S.; Petrovsky, N. A novel hepatitis b vaccine containing advax, a polysaccharide adjuvant derived from delta inulin, induces robust humoral and cellular immunity with minimal reactogenicity in preclinical testing. Vaccine 2013, 31, 1999–2007. [Google Scholar]
- Martin, R.M.; Brady, J.L.; Lew, A.M. The need for IgG2c specific antiserum when isotyping antibodies from C57bl/6 and NOD mice. J. Immunol. Methods 1998, 212, 187–192. [Google Scholar] [CrossRef]
- Romero Mendez, I.Z.; Shi, Y.; HogenEsch, H.; Hem, S.L. Potentiation of the immune response to non-adsorbed antigens by aluminum-containing adjuvants. Vaccine 2007, 25, 825–833. [Google Scholar] [CrossRef]
- Hansen, B.; Belfast, M.; Soung, G.; Song, L.; Egan, P.M.; Capen, R.; Hogenesch, H.; Mancinelli, R.; Hem, S.L. Effect of the strength of adsorption of hepatitis b surface antigen to aluminum hydroxide adjuvant on the immune response. Vaccine 2009, 27, 888–892. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, S.; Benson, R.A.; Gibson, V.B.; Pollock, A.H.; Garside, P.; Brewer, J.M. Antigen depot is not required for alum adjuvanticity. FASEB J. 2012, 26, 1272–1279. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thoryk, E.A.; Swaminathan, G.; Meschino, S.; Cox, K.S.; Gindy, M.; Casimiro, D.R.; Bett, A.J. Co-Administration of Lipid Nanoparticles and Sub-Unit Vaccine Antigens Is Required for Increase in Antigen-Specific Immune Responses in Mice. Vaccines 2016, 4, 47. https://doi.org/10.3390/vaccines4040047
Thoryk EA, Swaminathan G, Meschino S, Cox KS, Gindy M, Casimiro DR, Bett AJ. Co-Administration of Lipid Nanoparticles and Sub-Unit Vaccine Antigens Is Required for Increase in Antigen-Specific Immune Responses in Mice. Vaccines. 2016; 4(4):47. https://doi.org/10.3390/vaccines4040047
Chicago/Turabian StyleThoryk, Elizabeth A., Gokul Swaminathan, Steven Meschino, Kara S. Cox, Marian Gindy, Danilo R. Casimiro, and Andrew J. Bett. 2016. "Co-Administration of Lipid Nanoparticles and Sub-Unit Vaccine Antigens Is Required for Increase in Antigen-Specific Immune Responses in Mice" Vaccines 4, no. 4: 47. https://doi.org/10.3390/vaccines4040047