
TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis

 , ,
, ,
Abstract
:1. Introduction
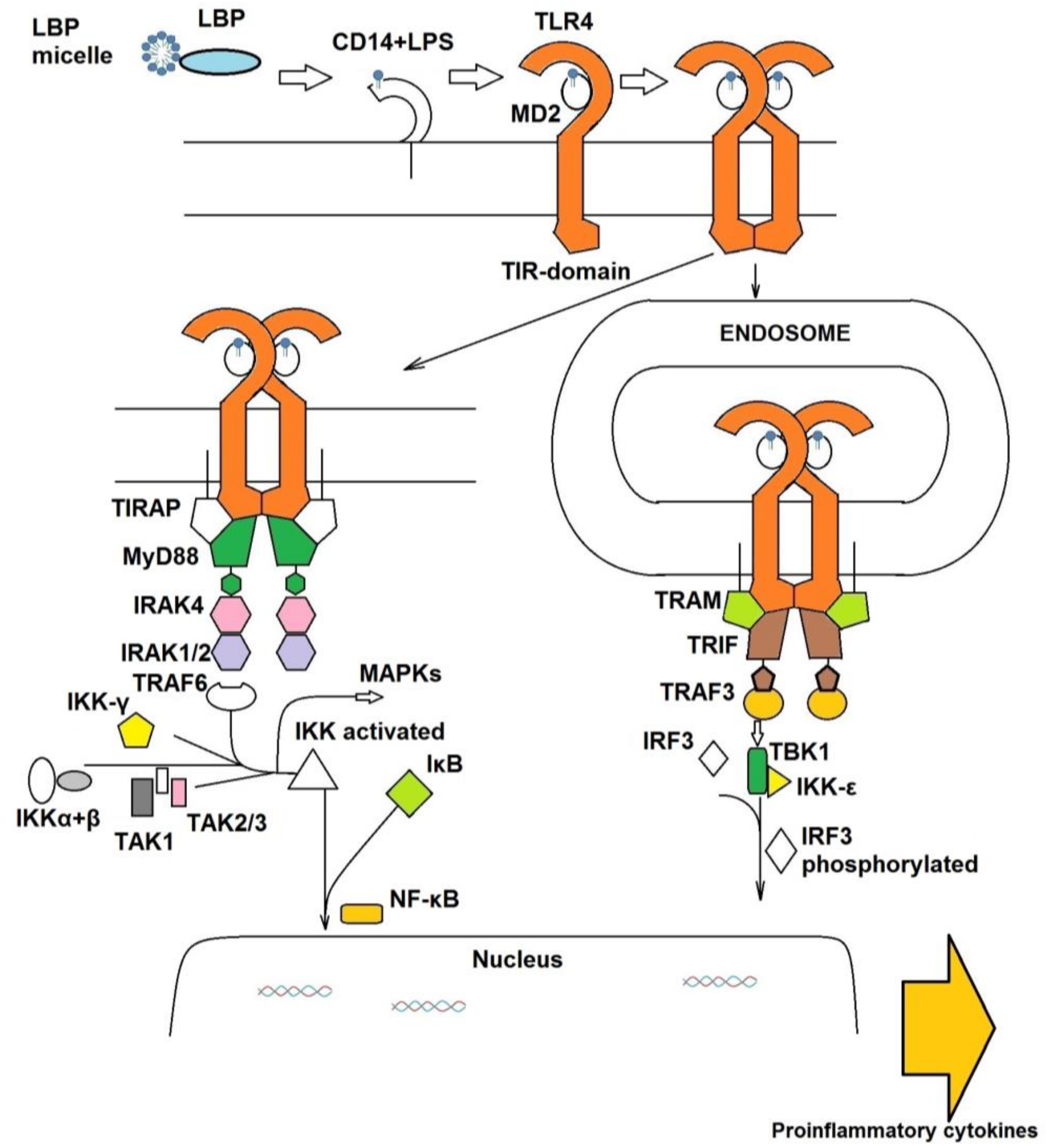
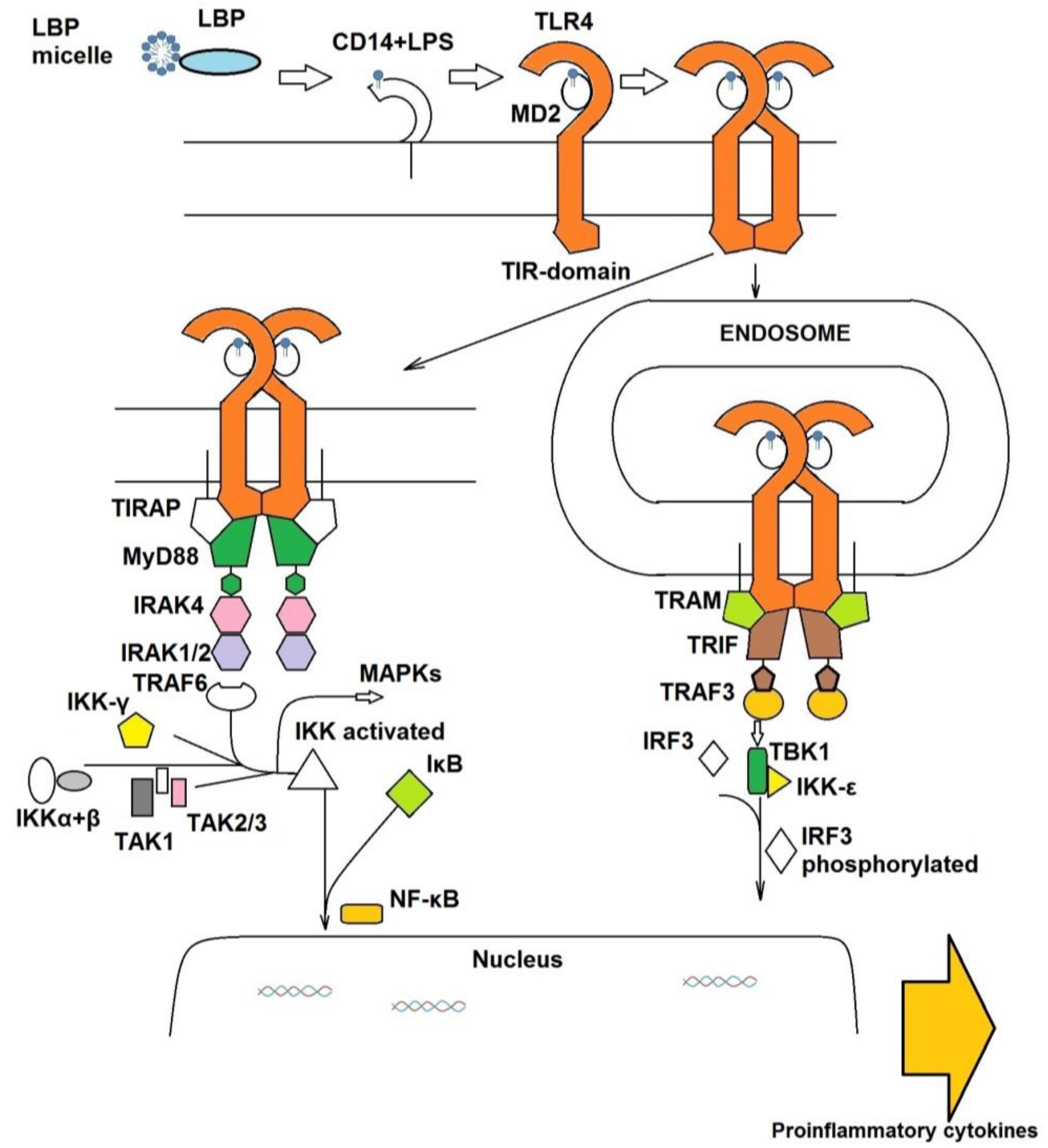
1.1. The Extracellular TLR4 Receptor System
1.2. The Intracellular Signal Cascade
2. Pathologies Related to TLR4 Signaling
2.1. Sepsis and Septic Shock
2.2. Animal Models of Sepsis
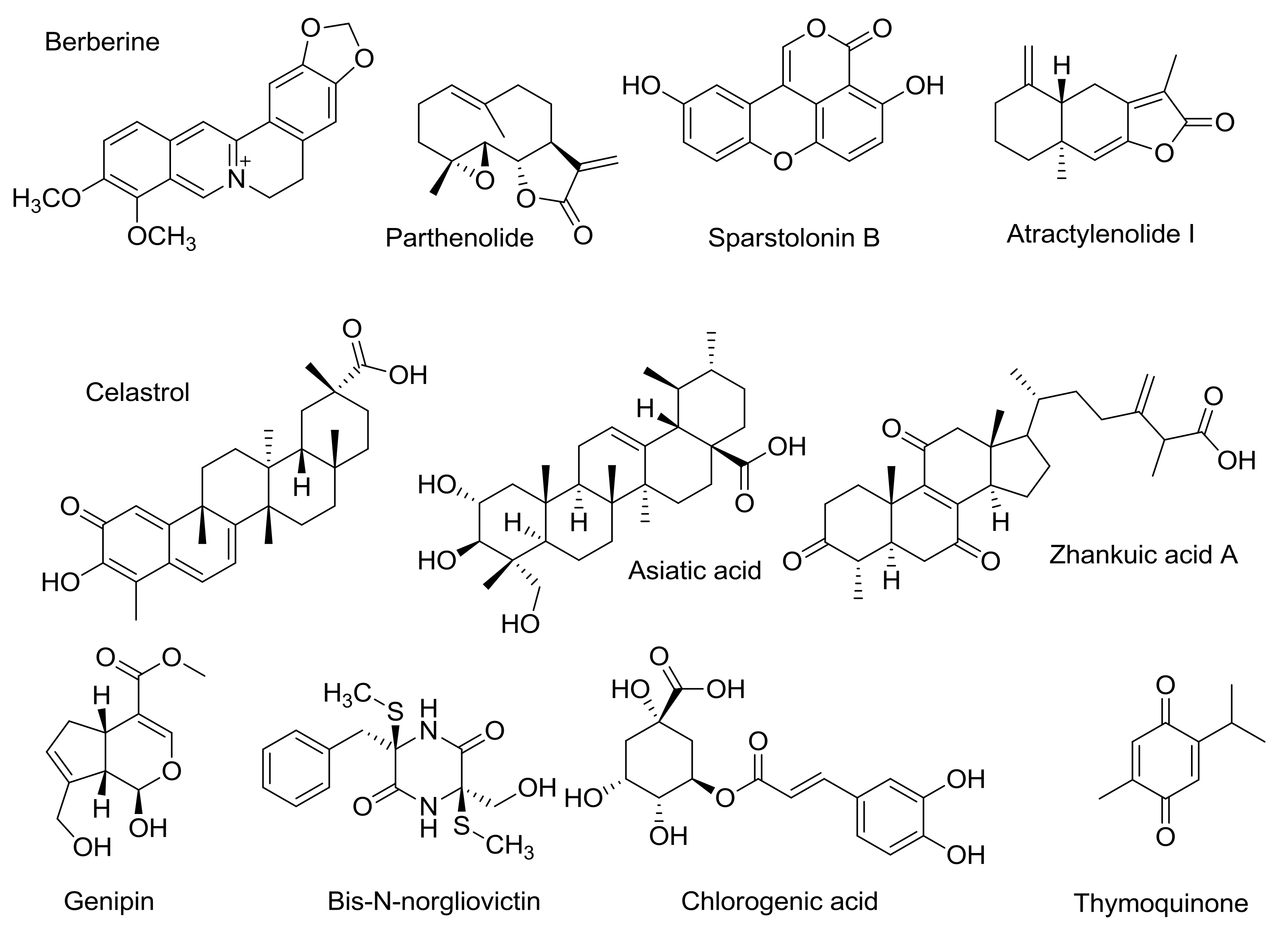
3. TLR4 Antagonists from Natural Sources
4. Synthetic TLR4 Antagonists
4.1. TAK-242 and Eritoran: Clinical Trials
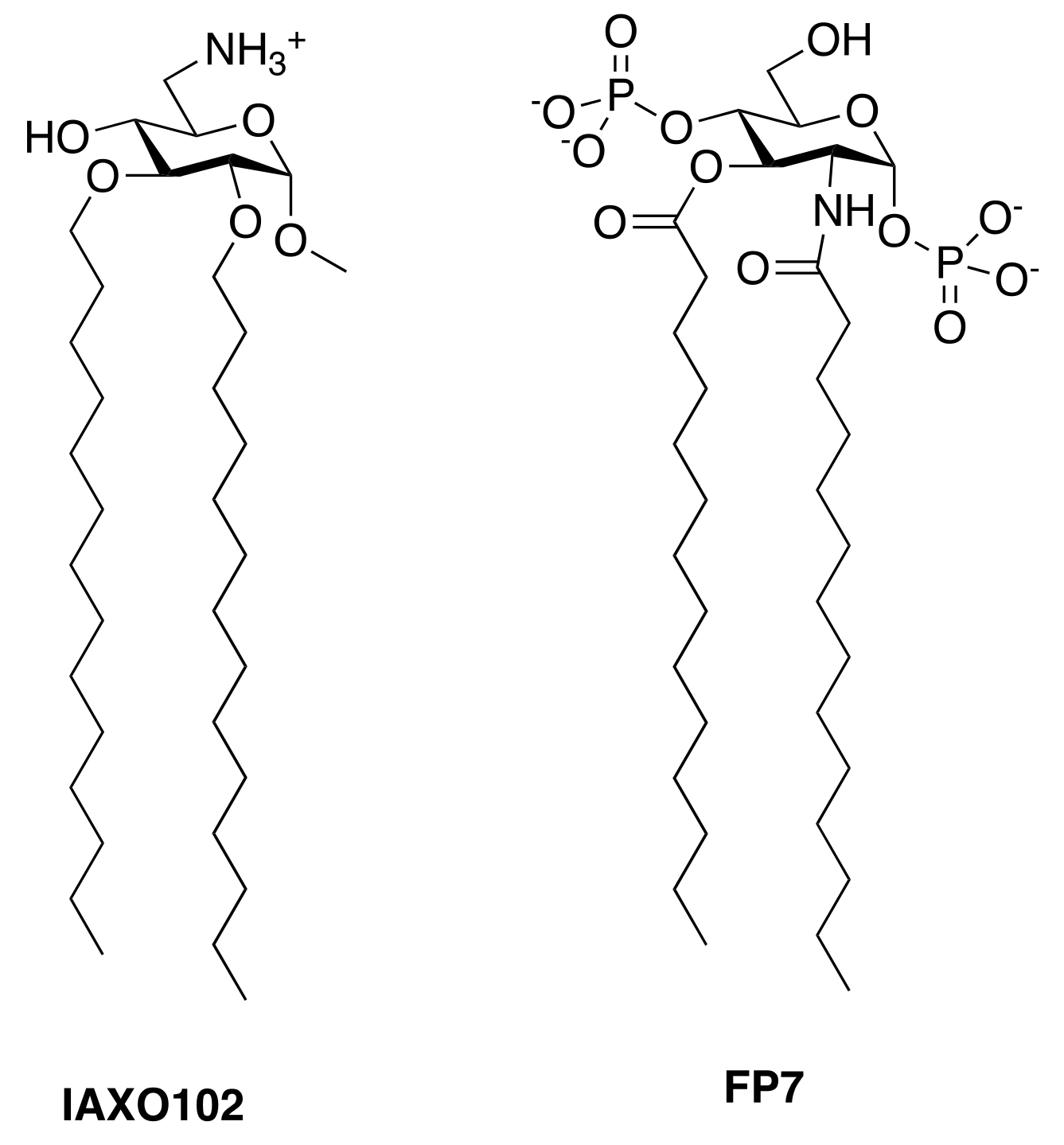
4.2. Synthetic Cationic and Anionic Amphiphiles
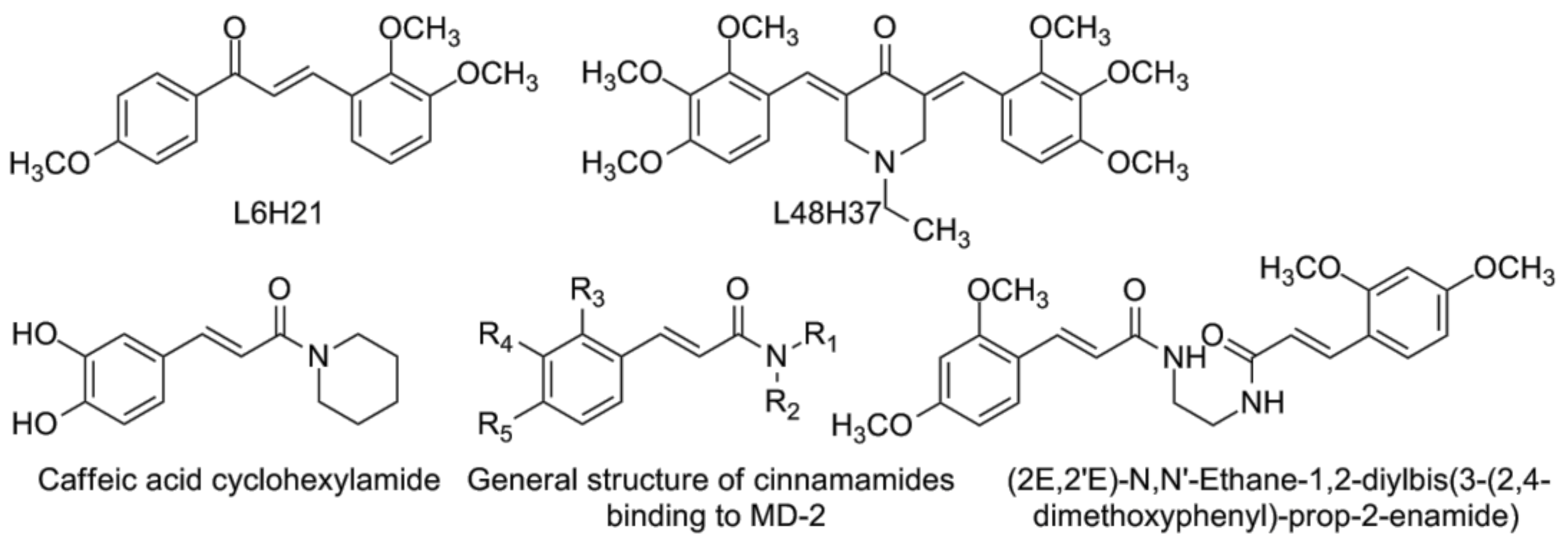
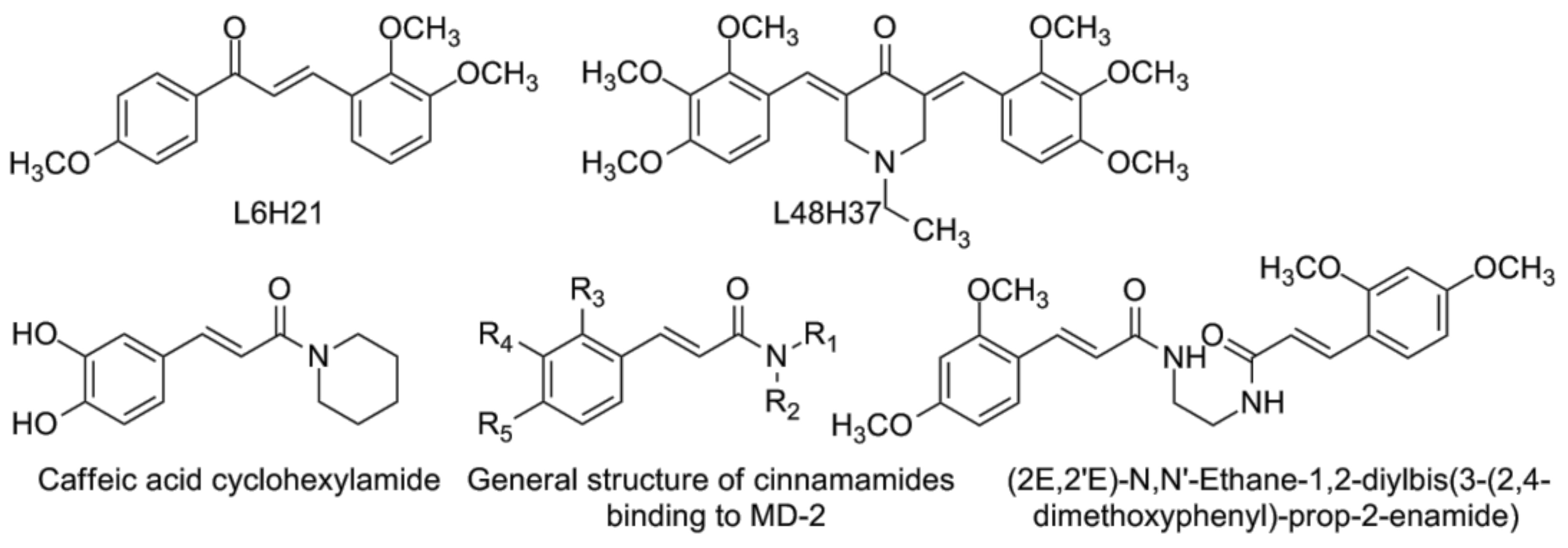
4.3. Chalcone Derivatives and Curcumin Analogues
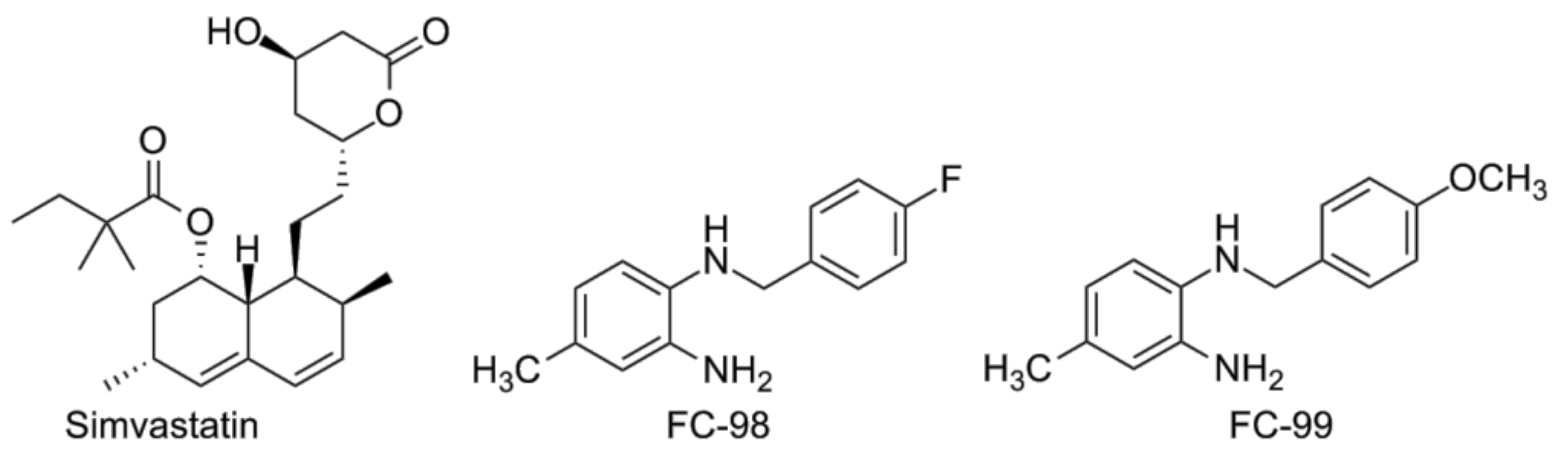
4.4. Other Compounds
4.5. In Silico Studies
5. Peptide TLR4 Modulators
5.1. Peptides that Disrupt the TLR4/MD-2 Interaction
5.2. Peptides that Disrupt TIR/TIR Interactions
6. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HEJ and C57BL/10SCCR mice: Mutations in TLR4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. TLR4 as the mammalian endotoxin sensor. Curr. Top Microbiol. Immunol. 2002, 270, 109–120. [Google Scholar] [PubMed]
- Beutler, B.; Du, X.; Poltorak, A. Identification of toll-like receptor 4 (TLR4) as the sole conduit for LPS signal transduction: Genetic and evolutionary studies. J. Endotoxin Res. 2001, 7, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Kobe, B.; Deisenhofer, J. The leucine-rich repeat: A versatile binding motif. Trends Biochem. Sci. 1994, 19, 415–421. [Google Scholar] [CrossRef]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. Md-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Gioannini, T.; Teghanemt, A.; Zhang, D.; Levis, E.; Weiss, J. Monomeric endotoxin: Protein complexes are essential for TLR4-dependent cell activation. J. Endotoxin Res. 2005, 11, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Gioannini, T.; Teghanemt, A.; Zhang, D.; Coussens, N.; Dockstader, W.; Ramaswamy, S.; Weiss, J. Isolation of an endotoxin-MD-2 complex that produces toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. USA 2004, 101, 4186–4191. [Google Scholar] [CrossRef] [PubMed]
- Park, B.; Song, D.; Kim, H.; Choi, B.; Lee, H.; Lee, J. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, H.M. Dynamic lipopolysaccharide transfer cascade to TLR4/MD2 complex via LBP and CD14. BMB Rep. 2017, 50, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Kim, S.J.; Rah, S.H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.O.; Park, B.S.; Yoon, T.Y.; et al. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Kitchens, R. Role of CD14 in cellular recognition of bacterial lipopolysaccharides. Chem. Immunol. 2000, 74, 61–82. [Google Scholar] [PubMed]
- O’Neill, L.A.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Guven-Maiorov, E.; Keskin, O.; Gursoy, A.; VanWaes, C.; Chen, Z.; Tsai, C.J.; Nussinov, R. The architecture of the TIR domain signalosome in the toll-like receptor-4 signaling pathway. Sci. Rep. 2015, 5, 13128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, D.W.; Heo, K.H.; Kim, Y.K.; Sim, E.J.; Kang, T.B.; Choi, J.W.; Sim, D.W.; Cheong, S.H.; Lee, S.H.; Bang, J.K.; et al. Anti-inflammatory action of an antimicrobial model peptide that suppresses the TRIF-dependent signalling pathway via inhibition of toll-like receptor 4 endocytosis in lipopolysaccharide-stimulated macrophages. PLoS ONE 2015, 10, e0126871. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 controls the LPS-induced endocytosis of toll-like receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Valkov, E.; Stamp, A.; Dimaio, F.; Baker, D.; Verstak, B.; Roversi, P.; Kellie, S.; Sweet, M.J.; Mansell, A.; Gay, N.J.; et al. Crystal structure of toll-like receptor adaptor mal/tirap reveals the molecular basis for signal transduction and disease protection. Proc. Natl. Acad. Sci. USA 2011, 108, 14879–14884. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Lu, J.; Zhou, W.; Shen, Y. Structural insights into TIR domain specificity of the bridging adaptor mal in TLR4 signaling. PLoS ONE 2012, 7, e34202. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1r signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, R.; Zhou, H.; Shan, Y.; Liu, Q.; Li, Q.; Shaw, D.E.; Li, X.; Wu, H. Irak4 dimerization and trans-autophosphorylation are induced by myddosome assembly. Mol. Cell 2014, 55, 891–903. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, M. The TAK1-TRAF6 signalling pathway. Int. J. Biochem. Cell Biol. 2010, 42, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Enokizono, Y.; Kumeta, H.; Funami, K.; Horiuchi, M.; Sarmiento, J.; Yamashita, K.; Standley, D.M.; Matsumoto, M.; Seya, T.; Inagaki, F. Structures and interface mapping of the tir domain-containing adaptor molecules involved in interferon signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 19908–19913. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Symmons, M.F.; Gangloff, M.; Bryant, C.E. Assembly and localization of toll-like receptor signalling complexes. Nat. Rev. Immunol. 2014, 14, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Funami, K.; Sasai, M.; Oshiumi, H.; Seya, T.; Matsumoto, M. Homo-oligomerization is essential for toll/interleukin-1 receptor domain-containing adaptor molecule-1-mediated NF-kappaB and interferon regulatory factor-3 activation. J. Biol. Chem. 2008, 283, 18283–18291. [Google Scholar] [CrossRef] [PubMed]
- Heipertz, E.L.; Harper, J.; Walker, W.E. Sting and trif contribute to mouse sepsis, depending on severity of the disease model. Shock 2017, 47, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins mavs, sting, and trif induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Seong, S.Y. Partial role of TLR4 as a receptor responding to damage-associated molecular pattern. Immunol. Lett. 2009, 125, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.; Lotze, M.; Yang, H.; Li, J.; Tracey, K.; Geller, D.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S. TLR4 and HMGB1: Partners in crime? Kidney Int. 2011, 80, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C. The roles of toll-like receptors in atherosclerosis. J. Innate Immun. 2009, 1, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi-Roodsaz, S.; Joosten, L.A.; Roelofs, M.F.; Radstake, T.R.; Matera, G.; Popa, C.; van der Meer, J.W.; Netea, M.G.; van den Berg, W.B. Inhibition of toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007, 56, 2957–2967. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Tanga, F.; Deleo, J. The contributing role of cd14 in toll-like receptor 4 dependent neuropathic pain. Neuroscience 2009, 158, 896–903. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, Y.; Levy, R.M.; Fan, J.J.; Hackam, D.J.; Vodovotz, Y.; Yang, H.; Tracey, K.J.; Billiar, T.R.; Wilson, M.A. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: Role of HMGB1-TLR4 signaling. J. Immunol. 2007, 178, 6573–6580. [Google Scholar] [CrossRef] [PubMed]
- Bachtell, R.; Hutchinson, M.R.; Wang, X.; Rice, K.C.; Maier, S.F.; Watkins, L.R. Targeting the toll of drug abuse: The translational potential of Toll-like receptor 4. CNS Neurol. Disord. Drug Targets 2015, 14, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Buisman-Pijlman, F.; Hutchinson, M.R. Toll-like receptor 4: Innate immune regulator of neuroimmune and neuroendocrine interactions in stress and major depressive disorder. Front. Neurosci. 2014, 8, 309. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.K.; Huang, T.L.; Huang, K.W.; Huang, Y.L.; Hung, Y.Y. Association between toll-like receptor 4 expression and symptoms of major depressive disorder. Neuropsychiatr. Dis. Treat. 2015, 11, 1853–1857. [Google Scholar] [PubMed]
- De Paola, M.; Sestito, S.E.; Mariani, A.; Memo, C.; Fanelli, R.; Freschi, M.; Bendotti, C.; Calabrese, V.; Peri, F. Synthetic and natural small molecule TLR4 antagonists inhibit motoneuron death in cultures from ALS mouse model. Pharmacol. Res. 2016, 103, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.K.; Saini, J.; Mahajan, K.; Singh, D.; Jayswal, D.P.; Mishra, S.; Bishayee, A.; Sethi, G.; Kunnumakkara, A.B. Therapeutic implications of toll-like receptors in peripheral neuropathic pain. Pharmacol. Res. 2017, 115, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Bettoni, I.; Comelli, F.; Rossini, C.; Granucci, F.; Giagnoni, G.; Peri, F.; Costa, B. Glial TLR4 receptor as new target to treat neuropathic pain: Efficacy of a new receptor antagonist in a model of peripheral nerve injury in mice. Glia 2008, 56, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Fink, M.P.; Marshall, J.C.; Abraham, E.; Angus, D.; Cook, D.; Cohen, J.; Opal, S.M.; Vincent, J.L.; Ramsay, G.; et al. 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Intensive Care Med. 2003, 29, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, C.; Scherag, A.; Adhikari, N.K.; Hartog, C.S.; Tsaganos, T.; Schlattmann, P.; Angus, D.C.; Reinhart, K. International Forum of Acute Care Trialists. Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am. J. Respir. Crit. Care Med. 2016, 193, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Suarez De La Rica, A.; Gilsanz, F.; Maseda, E. Epidemiologic trends of sepsis in western countries. Ann. Transl. Med. 2016, 4, 325. [Google Scholar] [CrossRef] [PubMed]
- Keynan, Y.; Fowke, K.R.; Ball, T.B.; Meyers, A.F.A. Toll-like receptors dysregulation after influenza virus infection: Insights into pathogenesis of subsequent bacterial pneumonia. ISRN Pulmonol. 2011, 2011. [Google Scholar] [CrossRef]
- Shah, N.S.; Greenberg, J.A.; McNulty, M.C.; Gregg, K.S.; Riddell, J.T.; Mangino, J.E.; Weber, D.M.; Hebert, C.L.; Marzec, N.S.; Barron, M.A.; et al. Bacterial and viral co-infections complicating severe influenza: Incidence and impact among 507 U.S. Patients, 2013–14. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2016, 80, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Crum-Cianflone, N.F. Invasive aspergillosis associated with severe influenza infections. Open Forum Infect. Dis. 2016, 3, ofw171. [Google Scholar] [CrossRef] [PubMed]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 antagonist eritoran protects mice from lethal influenza infection. Nature 2013, 497, 498–502. [Google Scholar] [CrossRef] [PubMed]
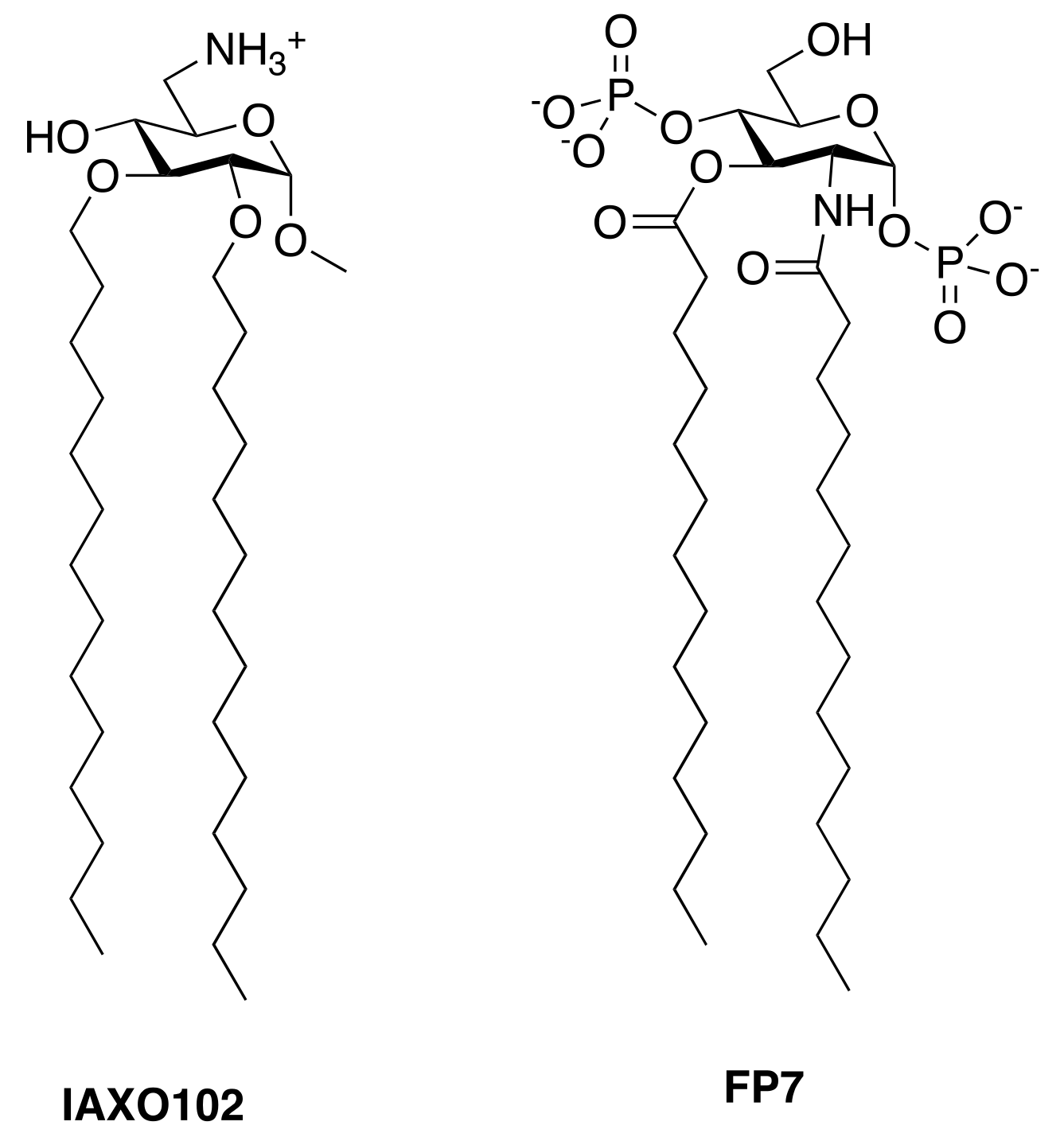
- Perrin-Cocon, L.; Aublin-Gex, A.; Sestito, S.E.; Shirey, K.A.; Patel, M.C.; André, P.; Blanco, J.C.; Vogel, S.N.; Peri, F.; Lotteau, V. TLR4 antagonist FP7 inhibits LPS-induced cytokine production and glycolytic reprogramming in dendritic cells, and protects mice from lethal influenza infection. Sci. Rep. 2017, 7, 40791. [Google Scholar] [CrossRef] [PubMed]
- Decker, T. Sepsis: Avoiding its deadly toll. J. Clin. Investig. 2004, 113, 1387–1389. [Google Scholar] [CrossRef] [PubMed]
- Lakshmikanth, C.L.; Jacob, S.P.; Chaithra, V.H.; de Castro-Faria-Neto, H.C.; Marathe, G.K. Sepsis: In search of cure. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2016, 65, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.M.; Davis, E.T.; Jones, S.W. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology 2017, 56, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Maor, Y.; Nanau, R.M.; Melzer, E.; Mell, H.; Opris, M.; Cohen, L.; Malnick, S. Alcoholic liver disease: Role of cytokines. Biomolecules 2015, 5, 2023–2034. [Google Scholar] [CrossRef] [PubMed]
- Wittebole, X.; Castanares-Zapatero, D.; Laterre, P.F. Toll-like receptor 4 modulation as a strategy to treat sepsis. Mediat. Inflamm. 2010, 2010, 568396. [Google Scholar] [CrossRef] [PubMed]
- Savva, A.; Roger, T. Targeting toll-like receptors: Promising therapeutic strategies for the management of sepsis-associated pathology and infectious diseases. Front. Immunol. 2013, 4, 387. [Google Scholar] [CrossRef] [PubMed]
- Stortz, J.A.; Raymond, S.L.; Mira, J.C.; Moldawer, L.L.; Mohr, A.M.; Efron, P.A. Murine models of sepsis and trauma: Can we bridge the gap? ILAR J. 2017, 58, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Deitch, E.A. Animal models of sepsis and shock: A review and lessons learned. Shock 1998, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal models of sepsis: Setting the stage. Nat. Rev. Drug Dis. 2005, 4, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Seemann, S.; Zohles, F.; Lupp, A. Comprehensive comparison of three different animal models for systemic inflammation. J. Biomed. Sci. 2017, 24, 60. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P.; Heard, S.O. Laboratory models of sepsis and septic shock. J. Surg. Res. 1990, 49, 186–196. [Google Scholar] [CrossRef]
- Dejager, L.; Pinheiro, I.; Dejonckheere, E.; Libert, C. Cecal ligation and puncture: The gold standard model for polymicrobial sepsis? Trends Microbiol. 2011, 19, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, W.J.; Choudhry, M.; Schwacha, M.G.; Kerby, J.D.; Rue, L.W., III; Bland, K.I.; Chaudry, I.H. Cecal ligation and puncture. Shock 2005, 24, S52–S57. [Google Scholar] [CrossRef]
- Pinsky, M.R.; Vincent, J.L.; Deviere, J.; Alegre, M.; Kahn, R.J.; Dupont, E. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest 1993, 103, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Wichterman, K.A.; Baue, A.E.; Chaudry, I.H. Sepsis and septic shock—A review of laboratory models and a proposal. J. Surg. Res. 1980, 29, 189–201. [Google Scholar] [CrossRef]
- Chu, M.; Ding, R.; Chu, Z.Y.; Zhang, M.B.; Liu, X.Y.; Xie, S.H.; Zhai, Y.J.; Wang, Y.D. Role of berberine in anti-bacterial as a high-affinity lps antagonist binding to TLR4/MD-2 receptor. BMC Complement. Altern. Med. 2014, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Sung, B.; Kunnumakkara, A.B.; Sethi, G.; Chaturvedi, M.M.; Aggarwal, B.B. Berberine modifies cysteine 179 of Ikappabalpha kinase, suppresses nuclear factor-kappab-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 2008, 68, 5370–5379. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gao, X.; Wu, X.; Wu, Z.; Cheng, L.; Zhu, L.; Shen, D.; Tong, X. Parthenolide inhibits LPS-induced inflammatory cytokines through the toll-like receptor 4 signal pathway in THP-1 cells. Acta Biochim. Biophys. Sin. 2015, 47, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.J.; Lee, D.H.; Lee, M.S.; Lee, C.S. Sesquiterpene lactone parthenolide attenuates production of inflammatory mediators by suppressing the toll-like receptor-4-mediated activation of the AKT, mTOR, and NF-kappaB pathways. Naunyn-Schmiedeberg Arch. Pharmacol. 2015, 388, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Saadane, A.; Masters, S.; DiDonato, J.; Li, J.; Berger, M. Parthenolide inhibits IkappaB kinase, NF-kappaB activation, and inflammatory response in cystic fibrosis cells and mice. Am. J. Respir. Cell Mol. Biol. 2007, 36, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Shin, H.J.; Youn, H.S. Parthenolide inhibits TRIF-dependent signaling pathway of toll-like receptors in raw264.7 macrophages. Mol. Cells 2011, 31, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, M.; Wong, H.R.; Hake, P.W.; Malhotra, V.; O’Connor, M.; Zingarelli, B. Parthenolide, an inhibitor of the nuclear factor-kappab pathway, ameliorates cardiovascular derangement and outcome in endotoxic shock in rodents. Mol. Pharmacol. 2002, 61, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cui, X.; Li, Y.; Fitz, Y.; Hsu, L.; Eichacker, P.Q. Parthenolide has limited effects on nuclear factor-kappa beta increases and worsens survival in lipopolysaccharide-challenged C57BL/6J mice. Cytokine 2006, 33, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.C.; Zhang, J.Q.; Zeng, C.; Chen, W.L.; Ren, W.X.; Yan, G.X.; Wang, H.X.; Li, Q.B.; Chen, Z.C. Inhibitory effects of parthenolide on the activity of NF-kappaB in multiple myeloma via targeting TRAF6. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wu, Q.; Jiang, J.; Duan, J.; Wang, C.; Smith, M.D.; Lu, H.; Wang, Q.; Nagarkatti, P.; Fan, D. Characterization of sparstolonin b, a Chinese herb-derived compound, as a selective toll-like receptor antagonist with potent anti-inflammatory properties. J. Biol. Chem. 2011, 286, 26470–26479. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Dong, S.; Lei, L.; Liu, J.; Zhang, J.; Li, J.; Duan, J.; Fan, D. Protective effects of sparstolonin b, a selective TLR2 and TLR4 antagonist, on mouse endotoxin shock. Cytokine 2015, 75, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xiu, L.; Diao, J.; Wei, L.; Sun, J. Sparstolonin b inhibits lipopolysaccharide-induced inflammation in 3T3-L1 adipocytes. Eur. J. Pharmacol. 2015, 769, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Yu, F.; Cui, X.; Duan, J.; Wu, Q.; Nagarkatti, P.; Fan, D. Sparstolonin b suppresses lipopolysaccharide-induced inflammation in human umbilical vein endothelial cells. Arch. Pharmacal Res. 2013, 36, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Chen, R.; Zheng, J. Atractylenolide i inhibits lipopolysaccharide-induced inflammatory responses via mitogen-activated protein kinase pathways in raw264.7 cells. Immunopharmacol. Immunotoxicol. 2014, 36, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Huang, W.M.; Zeng, Q.Y. Atractylenolide I protects mice from lipopolysaccharide-induced acute lung injury. Eur. J. Pharmacol. 2015, 765, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Xiao, Z.; Zhou, L.; Zhang, J.; Li, X.; He, Q. The protective effect of atractylenolide I on systemic inflammation in the mouse model of sepsis created by cecal ligation and puncture. Pharm. Biol. 2016, 54, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; He, L.C.; Jin, J.Q. Atractylenolide I and atractylenolide III inhibit lipopolysaccharide-induced TNF-alpha and no production in macrophages. Phytother. Res. PTR 2007, 21, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Shiau, A.L.; Wang, S.H.; Yang, J.S.; Chang, S.J.; Wu, C.L.; Wu, T.S. Zhankuic acid a isolated from taiwanofungus camphoratus is a novel selective TLR4/MD-2 antagonist with anti-inflammatory properties. J. Immunol. 2014, 192, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular, extensible docking program: Dock 5. J. Comput.-Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput.-Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, B.H.; Kim, N.D.; Lee, J.Y. Celastrol blocks binding of lipopolysaccharides to a toll-like receptor4/myeloid differentiation factor 2 complex in a thiol-dependent manner. J. Ethnopharmacol. 2015, 172, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Veerappan, K.; Natarajan, S.; Ethiraj, P.; Vetrivel, U.; Samuel, S. Inhibition of Ikkbeta by celastrol and its analogues—An in silico and in vitro approach. Pharm. Biol. 2017, 55, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xiao, X.; Yang, M. Asiatic acid inhibits lipopolysaccharide-induced acute lung injury in mice. Inflammation 2016, 39, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.R.; Mohapatra, P.; Patel, H.M.; Goyal, S.N.; Ojha, S.; Kundu, C.N.; Patil, C.R. Pentacyclic triterpenoids inhibit Ikkbeta mediated activation of NF-kappab pathway: In silico and in vitro evidences. PLoS ONE 2015, 10, e0125709. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Yoon, S.J.; Lee, S.M. Genipin attenuates sepsis by inhibiting toll-like receptor signaling. Mol. Med. 2012, 18, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Dou, H.; Gong, W.; Liu, X.; Yu, Z.; Li, E.; Tan, R.; Hou, Y. Bis-n-norgliovictin, a small-molecule compound from marine fungus, inhibits LPS-induced inflammation in macrophages and improves survival in sepsis. Eur. J. Pharmacol. 2013, 705, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, S.J.; Lee, S.M. Genipin attenuates sepsis-induced immunosuppression through inhibition of T lymphocyte apoptosis. Int. Immunopharmacol. 2015, 27, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Baek, S.I.; Yun, J.; Lee, S.; Yoon, D.Y.; Jung, J.K.; Jung, S.H.; Hwang, B.Y.; Hong, J.T.; Han, S.B.; et al. IRAK4 as a molecular target in the amelioration of innate immunity-related endotoxic shock and acute liver injury by chlorogenic acid. J. Immunol. 2015, 194, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Roh, E.; Kim, H.S.; Baek, S.I.; Choi, N.S.; Kim, N.; Hwang, B.Y.; Han, S.B.; Kim, Y. Inhibition of IRAK-4 activity for rescuing endotoxin LPS-induced septic mortality in mice by lonicerae flos extract. Biochem. Biophys. Res. Commun. 2013, 442, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bae, G.S.; Jo, I.J.; Choi, S.B.; Kim, D.G.; Shin, J.Y.; Lee, S.K.; Kim, M.J.; Shin, S.; Song, H.J.; et al. Loganin protects against pancreatitis by inhibiting NF-kappaB activation. Eur. J. Pharmacol. 2015, 765, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Hossen, M.J.; Yang, W.S.; Kim, D.; Aravinthan, A.; Kim, J.H.; Cho, J.Y. Thymoquinone: An IRAK1 inhibitor with in vivo and in vitro anti-inflammatory activities. Sci. Rep. 2017, 7, 42995. [Google Scholar] [CrossRef] [PubMed]
- Alkharfy, K.M.; Ahmad, A.; Raish, M.; Vanhoutte, P.M. Thymoquinone modulates nitric oxide production and improves organ dysfunction of sepsis. Life Sci. 2015, 143, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Alkharfy, K.M.; Al-Daghri, N.M.; Al-Attas, O.S.; Alokail, M.S. The protective effect of thymoquinone against sepsis syndrome morbidity and mortality in mice. Int. Immunopharmacol. 2011, 11, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Li, X. IRAK4 in TLR/IL-1r signaling: Possible clinical applications. Eur. J. Immunol. 2008, 38, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Seganish, W.M. Inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4): A patent review (2012-2015). Expert Opin. Ther. Pat. 2016, 26, 917–932. [Google Scholar] [CrossRef] [PubMed]
- Tumey, L.N.; Boschelli, D.H.; Bhagirath, N.; Shim, J.; Murphy, E.A.; Goodwin, D.; Bennett, E.M.; Wang, M.; Lin, L.L.; Press, B.; et al. Identification and optimization of indolo[2,3-c]quinoline inhibitors of IRAK4. Bioorg. Med. Chem. Lett. 2014, 24, 2066–2072. [Google Scholar] [CrossRef] [PubMed]
- Patra, M.C.; Choi, S. Recent progress in the molecular recognition and therapeutic importance of interleukin-1 receptor-associated kinase 4. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, D.; Robinson, S.; Romero, D.L. Recent advances in the discovery of small molecule inhibitors of interleukin-1 receptor-associated kinase 4 (IRAK4) as a therapeutic target for inflammation and oncology disorders. J. Med. Chem. 2015, 58, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; Romero, D.L.; Yang, Y.; Shaffer, A.L., III; Chaudhary, D.; Robinson, S.; Miao, W.; Rui, L.; Westlin, W.F.; Kapeller, R.; et al. Selective interleukin-1 receptor-associated kinase 4 inhibitors for the treatment of autoimmune disorders and lymphoid malignancy. J. Exp. Med. 2015, 212, 2189–2201. [Google Scholar] [CrossRef] [PubMed]
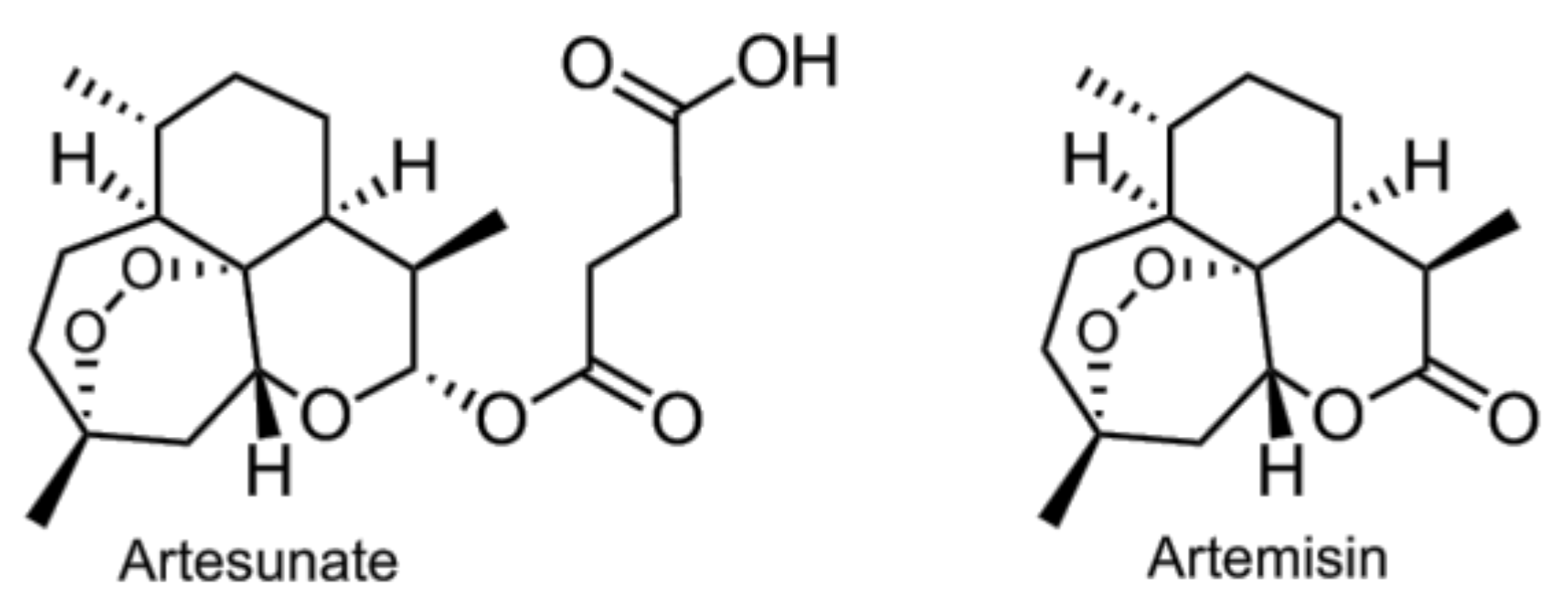
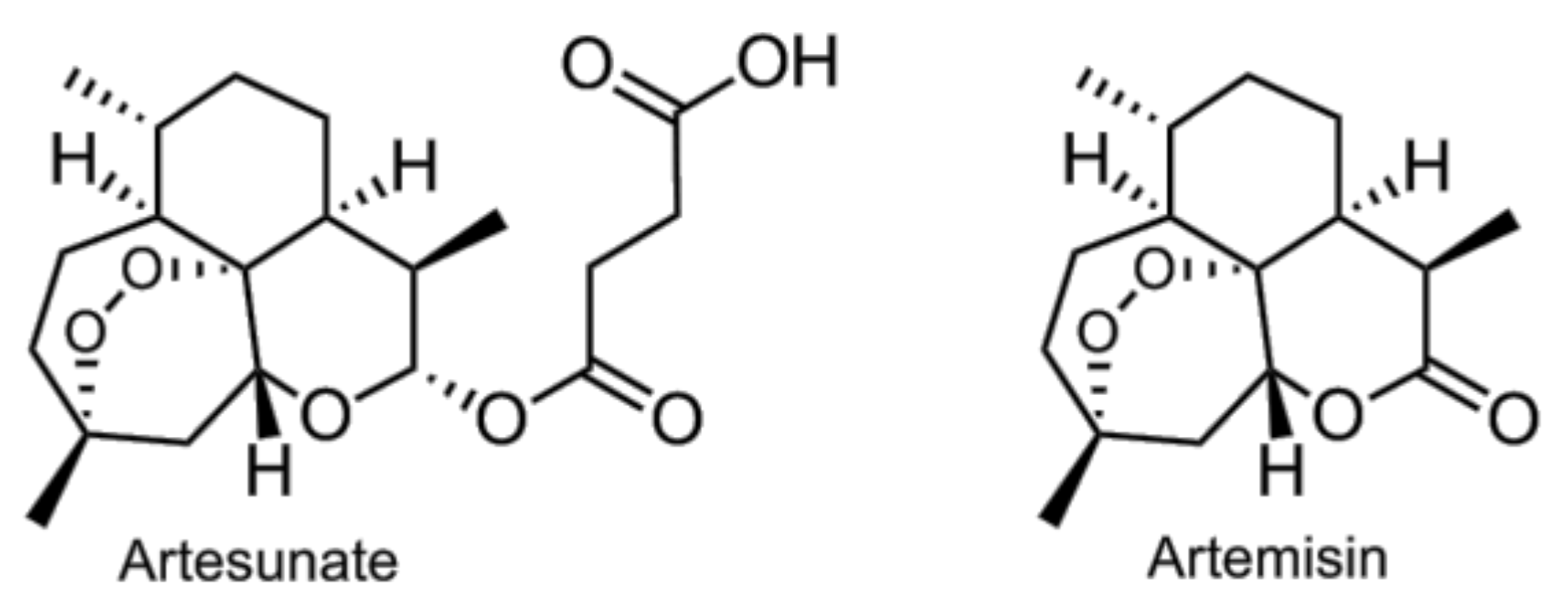
- Li, B.; Zhang, R.; Li, J.; Zhang, L.; Ding, G.; Luo, P.; He, S.; Dong, Y.; Jiang, W.; Lu, Y.; et al. Antimalarial artesunate protects sepsis model mice against heat-killed escherichia coli challenge by decreasing TLR4, TLR9 mRNA expressions and transcription factor NF-kappa B activation. Int. Immunopharmacol. 2008, 8, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shi, J.; Lv, S.; Xu, W.; Li, J.; Ge, W.; Xiao, C.; Geng, D.; Liu, Y. Artesunate attenuates lipopolysaccharide-stimulated proinflammatory responses by suppressing TLR4, MyD88 expression, and NF-kappaB activation in microglial cells. Inflammation 2015, 38, 1925–1932. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhou, H.; Zheng, J.; Cheng, J.; Liu, W.; Ding, G.; Wang, L.; Luo, P.; Lu, Y.; Cao, H.; et al. The antimalarial artemisinin synergizes with antibiotics to protect against lethal live escherichia coli challenge by decreasing proinflammatory cytokine release. Antimicrob. Agents Chemother. 2006, 50, 2420–2427. [Google Scholar] [CrossRef] [PubMed]
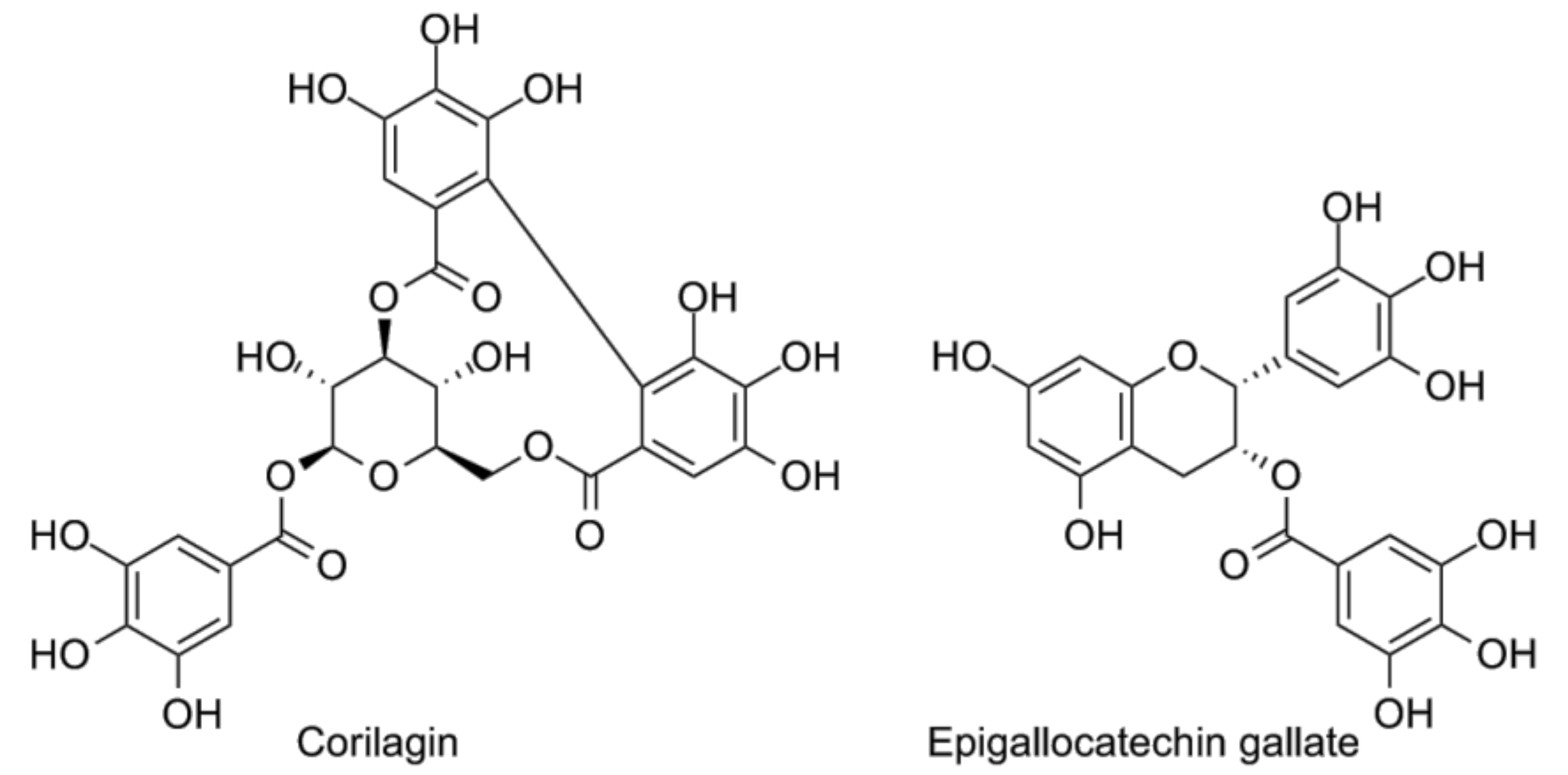
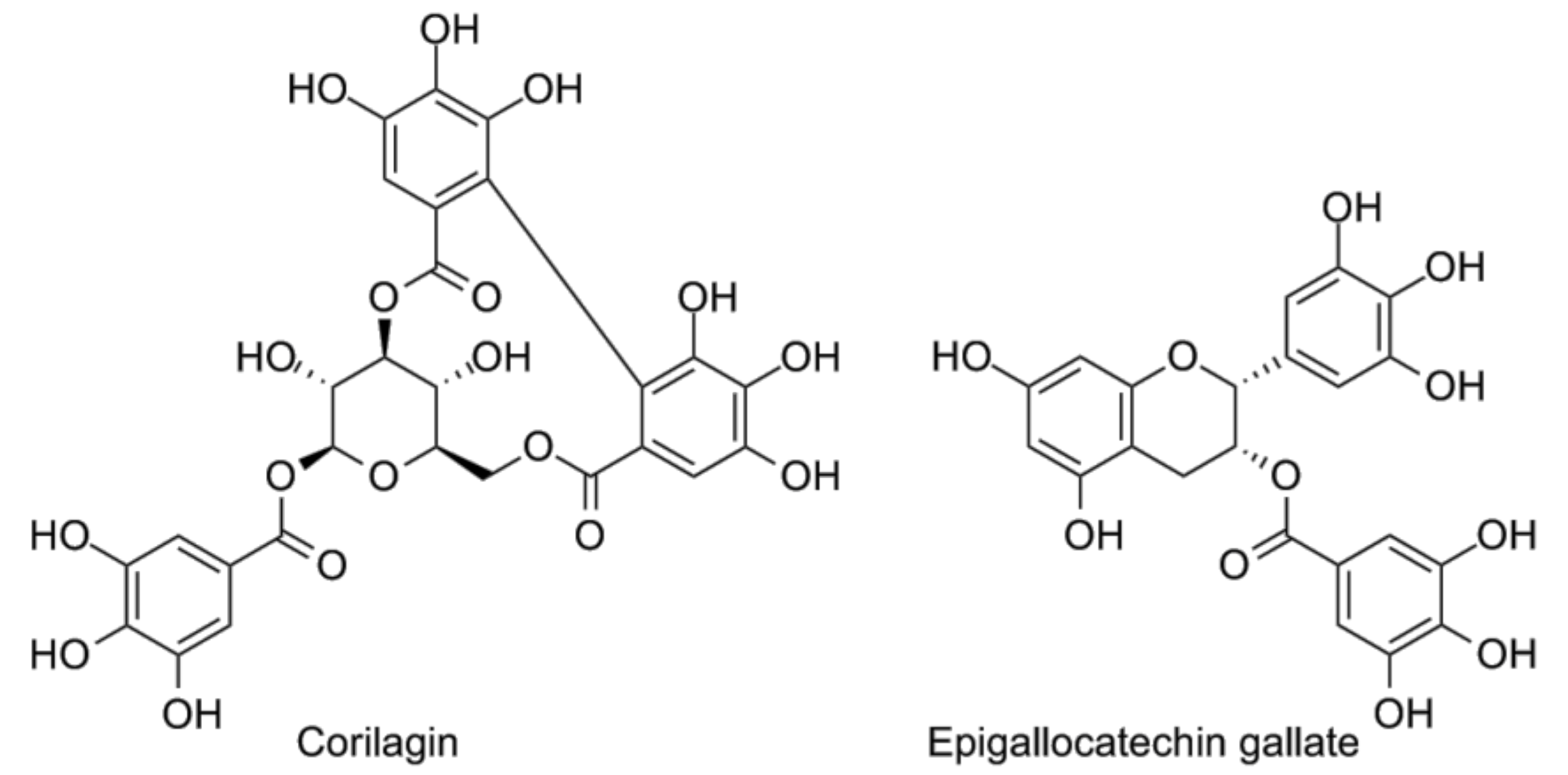
- Li, H.R.; Liu, J.; Zhang, S.L.; Luo, T.; Wu, F.; Dong, J.H.; Guo, Y.J.; Zhao, L. Corilagin ameliorates the extreme inflammatory status in sepsis through TLR4 signaling pathways. BMC Complement. Altern. Med. 2017, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Umar, S.; Riegsecker, S.; Chourasia, M.; Ahmed, S. Regulation of transforming growth factor beta-activated kinase activation by epigallocatechin-3-gallate in rheumatoid arthritis synovial fibroblasts: Suppression of k(63) -linked autoubiquitination of tumor necrosis factor receptor-associated factor 6. Arthritis Rheumatol. 2016, 68, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.S.; Lahni, P.M.; Hake, P.W.; Denenberg, A.G.; Wong, H.R.; Snead, C.; Catravas, J.D.; Zingarelli, B. The green tea polyphenol epigallocatechin-3-gallate improves systemic hemodynamics and survival in rodent models of polymicrobial sepsis. Shock 2007, 28, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Peri, F.; Calabrese, V. Toll-like receptor 4 (TLR4) modulation by synthetic and natural compounds: An update. J. Med. Chem. 2014, 57, 3612–3622. [Google Scholar] [CrossRef] [PubMed]
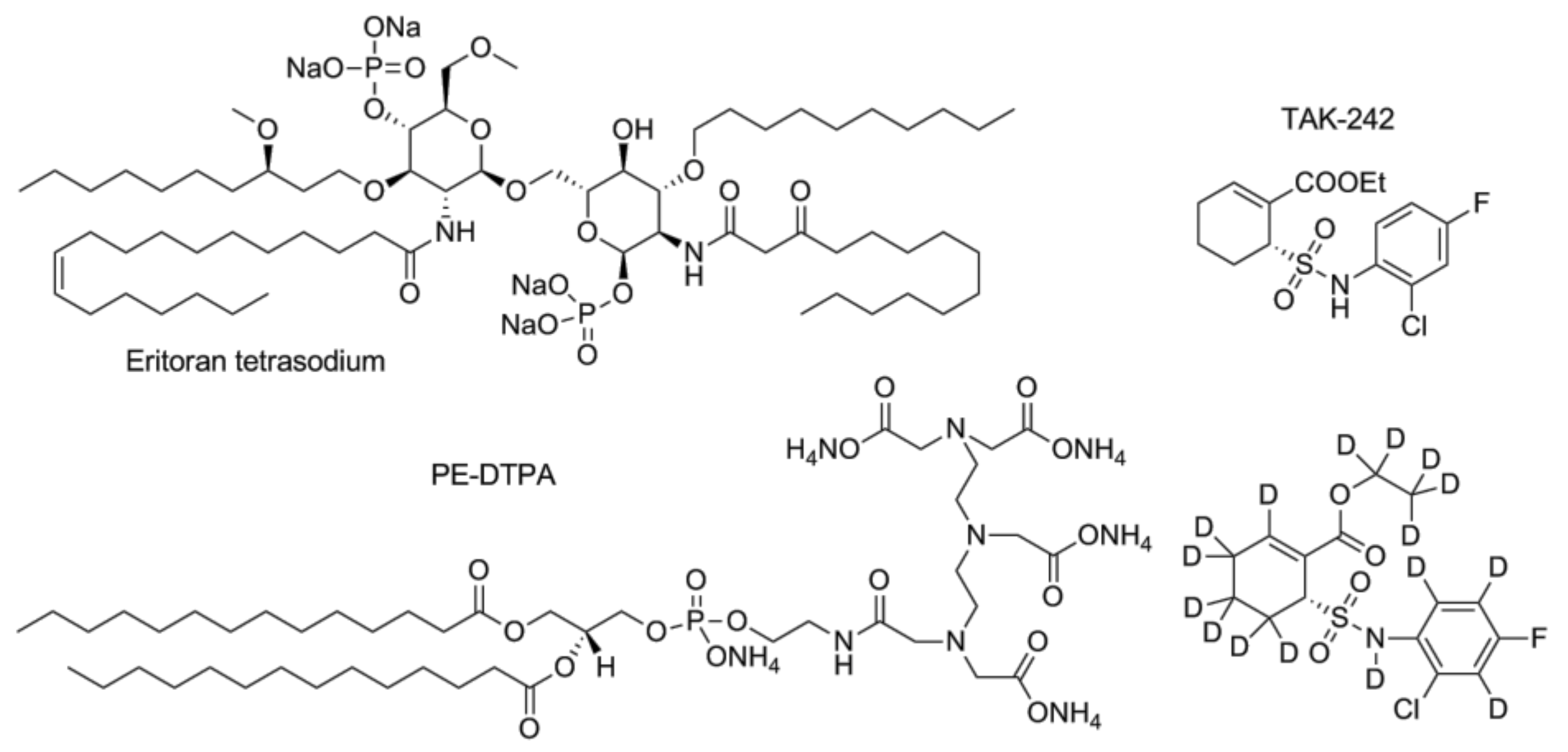
- Takashima, K.; Matsunaga, N.; Yoshimatsu, M.; Hazeki, K.; Kaisho, T.; Uekata, M.; Hazeki, O.; Akira, S.; Iizawa, Y.; Ii, M. Analysis of binding site for the novel small-molecule TLR4 signal transduction inhibitor TAK-242 and its therapeutic effect on mouse sepsis model. Br. J. Pharmacol. 2009, 157, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sha, T.; Sunamoto, M.; Kitazaki, T.; Sato, J.; Ii, M.; Iizawa, Y. Therapeutic effects of TAK-242, a novel selective toll-like receptor 4 signal transduction inhibitor, in mouse endotoxin shock model. Eur. J. Pharmacol. 2007, 571, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Sha, T.; Iizawa, Y.; Ii, M. Combination of imipenem and TAK-242, a toll-like receptor 4 signal transduction inhibitor, improves survival in a murine model of polymicrobial sepsis. Shock 2011, 35, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Tang, H.; Wang, J.; Prunty, M.C.; Hua, X.; Sayeed, I.; Stein, D.G. TAK-242, an antagonist for toll-like receptor 4, protects against acute cerebral ischemia/reperfusion injury in mice. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Fenhammar, J.; Rundgren, M.; Hultenby, K.; Forestier, J.; Taavo, M.; Kenne, E.; Weitzberg, E.; Eriksson, S.; Ozenci, V.; Wernerson, A.; et al. Renal effects of treatment with a TLR4 inhibitor in conscious septic sheep. Crit. Care 2014, 18, 488. [Google Scholar] [CrossRef] [PubMed]
- Kuno, M.; Nemoto, K.; Ninomiya, N.; Inagaki, E.; Kubota, M.; Matsumoto, T.; Yokota, H. The novel selective toll-like receptor 4 signal transduction inhibitor TAK-242 prevents endotoxaemia in conscious guinea-pigs. Clin. Exp. Pharmacol. Physiol. 2009, 36, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Garate, I.; Garcia-Bueno, B.; Madrigal, J.L.; Caso, J.R.; Alou, L.; Gomez-Lus, M.L.; Leza, J.C. Toll-like 4 receptor inhibitor TAK-242 decreases neuroinflammation in rat brain frontal cortex after stress. J. Neuroinflamm. 2014, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Rice, T.W.; Wheeler, A.P.; Bernard, G.R.; Vincent, J.L.; Angus, D.C.; Aikawa, N.; Demeyer, I.; Sainati, S.; Amlot, N.; Cao, C.; et al. A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis. Crit. Care Med. 2010, 38, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.G.S.S.C.Z. Preparation of Deuterated Cyclohexenes as Modulators of TLR4 Signaling for Disease Treatment. U.S. Patent No. US 20090022706 A1, 2009. [Google Scholar]
- Kim, H.; Park, B.; Kim, J.; Kim, S.; Lee, J.; Oh, S.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Bennett-Guerrero, E.; Grocott, H.P.; Levy, J.H.; Stierer, K.A.; Hogue, C.W.; Cheung, A.T.; Newman, M.F.; Carter, A.A.; Rossignol, D.P.; Collard, C.D. A phase II, double-blind, placebo-controlled, ascending-dose study of eritoran (e5564), a lipid a antagonist, in patients undergoing cardiac surgery with cardiopulmonary bypass. Anesth. Analg. 2007, 104, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Barochia, A.; Solomon, S.; Cui, X.; Natanson, C.; Eichacker, P.Q. Eritoran tetrasodium (e5564) treatment for sepsis: Review of preclinical and clinical studies. Expert Opin. Drug Metab. Toxicol. 2011, 7, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Tidswell, M.; LaRosa, S.P. Toll-like receptor-4 antagonist eritoran tetrasodium for severe sepsis. Expert Rev. Anti-Infect. Ther. 2011, 9, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.P.; Wong, N.; Noveck, R.; Lynn, M. Continuous pharmacodynamic activity of eritoran tetrasodium, a TLR4 antagonist, during intermittent intravenous infusion into normal volunteers. Innate Immun. 2008, 14, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Opal, S.M.; Laterre, P.F.; Francois, B.; LaRosa, S.P.; Angus, D.C.; Mira, J.P.; Wittebole, X.; Dugernier, T.; Perrotin, D.; Tidswell, M.; et al. Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: The access randomized trial. JAMA 2013, 309, 1154–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tidswell, M.; Tillis, W.; Larosa, S.P.; Lynn, M.; Wittek, A.E.; Kao, R.; Wheeler, J.; Gogate, J.; Opal, S.M.; Eritoran Sepsis Study, G. Phase 2 trial of eritoran tetrasodium (e5564), a toll-like receptor 4 antagonist, in patients with severe sepsis. Crit. Care Med. 2010, 38, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; An, H.J.; Kim, J.L.; Lee, H.; Paik, S.G. Inhibitory effect of a phosphatidyl ethanolamine derivative on LPS-induced sepsis. Mol. Cells 2009, 27, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.; Rossini, C.; Della Fiorentina, S.; Pozzi, C.; Comelli, F.; Bettoni, I.; Fusi, P.; Costa, B.; Peri, F. Glycolipids and benzylammonium lipids as novel antisepsis agents: Synthesis and biological characterization. J. Med. Chem. 2009, 52, 1209–1213. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.; Yu, L.; Teghanemt, A.; Gioannini, T.; Weiss, J.; Peri, F. Evidence of a specific interaction between new synthetic antisepsis agents and CD14. Biochemistry 2009, 48, 12337–12344. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Lavado, J.; Sestito, S.E.; Cighetti, R.; Aguilar Moncayo, E.M.; Oblak, A.; Lainšček, D.; Jiménez Blanco, J.L.; García Fernández, J.M.; Ortiz Mellet, C.; Jerala, R.; et al. Trehalose- and glucose-derived glycoamphiphiles: Small-molecule and nanoparticle toll-like receptor 4 (TLR4) modulators. J. Med. Chem. 2014, 57, 9105–9123. [Google Scholar] [CrossRef] [PubMed]
- Cighetti, R.; Ciaramelli, C.; Sestito, S.E.; Zanoni, I.; Kubik, Ł.; Ardá-Freire, A.; Calabrese, V.; Granucci, F.; Jerala, R.; Martín-Santamaría, S.; et al. Modulation of cd14 and TLR4·MD-2 activities by a synthetic lipid a mimetic. Chembiochem 2014, 15, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shan, X.; Chen, G.; Jiang, L.; Wang, Z.; Fang, Q.; Liu, X.; Wang, J.; Zhang, Y.; Wu, W.; et al. MD-2 as the target of a novel small molecule, l6h21, in the attenuation of LPS-induced inflammatory response and sepsis. Br. J. Pharmacol. 2015, 172, 4391–4405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, J.; Ying, S.; Chen, G.; Wu, B.; Xu, T.; Liu, Z.; Liu, X.; Huang, L.; Shan, X.; et al. Discovery of new MD2 inhibitor from chalcone derivatives with anti-inflammatory effects in LPS-induced acute lung injury. Sci. Rep. 2016, 6, 25130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shan, X.; Dai, Y.; Jiang, L.; Chen, G.; Zhang, Y.; Wang, Z.; Dong, L.; Wu, J.; Guo, G.; et al. Curcumin analog l48h37 prevents lipopolysaccharide-induced TLR4 signaling pathway activation and sepsis via targeting MD2. J. Pharmacol. Exp. Ther. 2015, 353, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.; Zhu, R.; Liu, Q.; Fei, J.; Wang, S. Anti-inflammatory activity of curcumin-loaded solid lipid nanoparticles in IL-1beta transgenic mice subjected to the lipopolysaccharide-induced sepsis. Biomaterials 2015, 53, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Youn, H.S.; Saitoh, S.I.; Miyake, K.; Hwang, D.H. Inhibition of homodimerization of toll-like receptor 4 by curcumin. Biochem. Pharmacol. 2006, 72, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Gradisar, H.; Keber, M.M.; Pristovsek, P.; Jerala, R. MD-2 as the target of curcumin in the inhibition of response to LPS. J. Leukoc. Biol. 2007, 82, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P.; Simental-Mendia, L.E.; Bo, S.; Sahebkar, A. Effect of curcumin on circulating interleukin-6 concentrations: A systematic review and meta-analysis of randomized controlled trials. Pharmacol. Res. 2016, 111, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, Y.; Liu, X.; Fang, Q.; Wang, Z.; Fu, L.; Liu, Z.; Wang, Y.; Zhao, Y.; Li, X.; et al. Discovery of a new inhibitor of myeloid differentiation 2 from cinnamamide derivatives with anti-inflammatory activity in sepsis and acute lung injury. J. Med. Chem. 2016, 59, 2436–2451. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Park, S.H.; Jung, J.K.; Cho, W.J.; Ahn, B.; Yun, C.Y.; Choi, Y.P.; Yeo, J.H.; Lee, H.; Hong, J.T.; et al. Caffeic acid cyclohexylamide rescues lethal inflammation in septic mice through inhibition of Ikappab kinase in innate immune process. Sci. Rep. 2017, 7, 41180. [Google Scholar] [CrossRef] [PubMed]
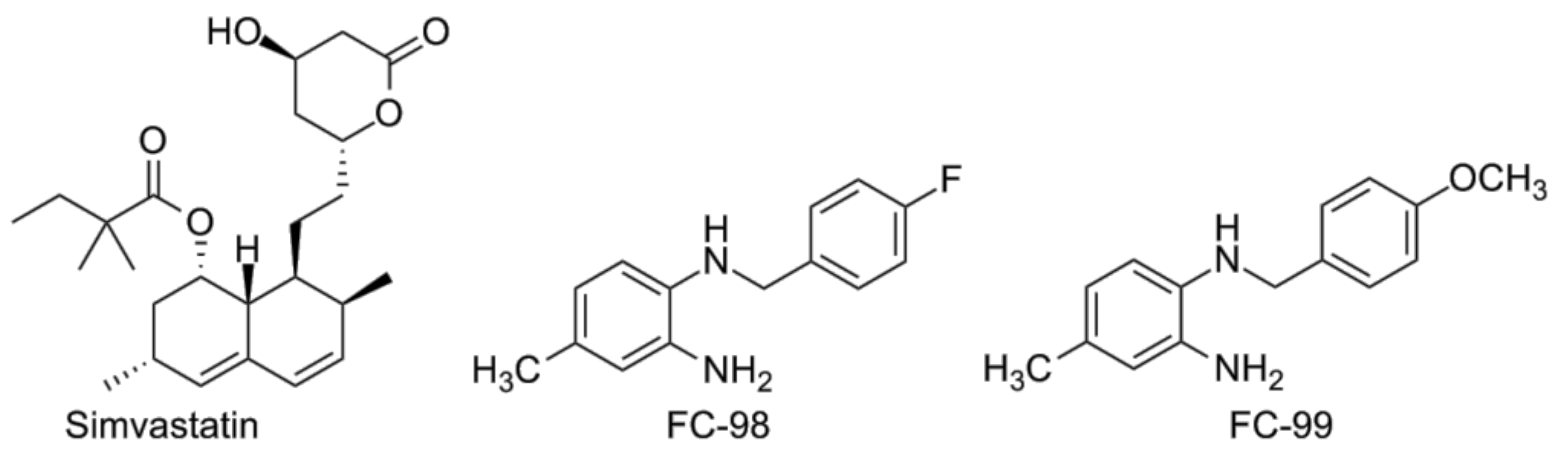
- Niessner, A.; Steiner, S.; Speidl, W.S.; Pleiner, J.; Seidinger, D.; Maurer, G.; Goronzy, J.J.; Weyand, C.M.; Kopp, C.W.; Huber, K.; et al. Simvastatin suppresses endotoxin-induced upregulation of toll-like receptors 4 and 2 in vivo. Atherosclerosis 2006, 189, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Dou, H.; Song, Y.; Liu, X.; Yang, L.; Jiang, N.; Chen, D.; Li, E.; Tan, R.; Hou, Y. A novel benzenediamine derivate rescued mice from experimental sepsis by attenuating proinflammatory mediators via IRAK4. Am. J. Respir. Cell Mol. Biol. 2014, 51, 191–200. [Google Scholar] [PubMed]
- Song, Y.; Liu, X.; Yue, H.; Ji, J.; Dou, H.; Hou, Y. Anti-inflammatory effects of benzenediamine derivate FC-98 on sepsis injury in mice via suppression of JNK, NF-kappaB and IRF3 signaling pathways. Mol. Immunol. 2015, 67, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the toll-like receptor-dependent and -independent antiviral response. Nature 2006, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.N.; Sodhi, C.P.; Hackam, D.J. Novel TLR4 inhibitors for the treatment of human infectious and inflammatory disorders. PCT International Application No. PCT/ US2011/053293 Publication No. WO 2012040719 A2, 29 May 2015. [Google Scholar]
- Billod, J.M.; Lacetera, A.; Guzman-Caldentey, J.; Martin-Santamaria, S. Computational approaches to toll-like receptor 4 modulation. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Klett, J.; Reeves, J.; Oberhauser, N.; Perez-Regidor, L.; Martin-Santamaria, S. Modulation of toll-like receptor 4. Insights from X-ray crystallography and molecular modeling. Curr. Top. Med. Chem. 2014, 14, 2672–2683. [Google Scholar] [CrossRef] [PubMed]
- Perez-Regidor, L.; Zarioh, M.; Ortega, L.; Martin-Santamaria, S. Virtual screening approaches towards the discovery of toll-like receptor modulators. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Murgueitio, M.S.; Rakers, C.; Frank, A.; Wolber, G. Balancing inflammation: Computational design of small-molecule toll-like receptor modulators. Trends Pharmacol. Sci. 2017, 38, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Slivka, P.; Shridhar, M.; Lee, G.; Sammond, D.; Hutchinson, M.; Martinko, A.; Buchanan, M.; Sholar, P.; Kearney, J.; Harrison, J.; et al. A peptide antagonist of the TLR4-MD2 interaction. Chembiochem 2009, 10, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kim, H.J.; Lee, S.C.; Hong, S.; Park, K.; Jeon, Y.H.; Kim, D.; Cheong, H.K.; Kim, H.S. Structure-based rational design of a toll-like receptor 4 (TLR4) decoy receptor with high binding affinity for a target protein. PLoS ONE 2012, 7, e30929. [Google Scholar] [CrossRef] [PubMed]
- Couture, L.A.; Piao, W.; Ru, L.W.; Vogel, S.N.; Toshchakov, V.Y. Targeting toll-like receptor (TLR) signaling by toll/interleukin-1 receptor (TIR) domain-containing adapter protein/myd88 adapter-like (TIRAP/MAL)-derived decoy peptides. J. Biol. Chem. 2012, 287, 24641–24648. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tao, X.; Shen, B.; Horng, T.; Medzhitov, R.; Manley, J.L.; Tong, L. Structural basis for signal transduction by the toll/interleukin-1 receptor domains. Nature 2000, 408, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Dunne, A.; Ejdeback, M.; Ludidi, P.L.; O’Neill, L.A.; Gay, N.J. Structural complementarity of toll/interleukin-1 receptor domains in toll-like receptors and the adaptors mal and myd88. J. Biol. Chem. 2003, 278, 41443–41451. [Google Scholar] [CrossRef] [PubMed]
- Nunez Miguel, R.; Wong, J.; Westoll, J.F.; Brooks, H.J.; O’Neill, L.A.; Gay, N.J.; Bryant, C.E.; Monie, T.P. A dimer of the toll-like receptor 4 cytoplasmic domain provides a specific scaffold for the recruitment of signalling adaptor proteins. PLoS ONE 2007, 2, e788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piao, W.; Vogel, S.N.; Toshchakov, V.Y. Inhibition of TLR4 signaling by tram-derived decoy peptides in vitro and in vivo. J. Immunol. 2013, 190, 2263–2272. [Google Scholar] [CrossRef] [PubMed]
- Toshchakov, V.Y.; Szmacinski, H.; Couture, L.A.; Lakowicz, J.R.; Vogel, S.N. Targeting TLR4 signaling by TLR4 Toll/IL-1 receptor domain-derived decoy peptides: Identification of the TLR4 Toll/IL-1 receptor domain dimerization interface. J. Immunol. 2011, 186, 4819–4827. [Google Scholar] [CrossRef] [PubMed]
- Piao, W.; Ru, L.W.; Piepenbrink, K.H.; Sundberg, E.J.; Vogel, S.N.; Toshchakov, V.Y. Recruitment of tlr adapter TRIF to TLR4 signaling complex is mediated by the second helical region of TRIF TIR domain. Proc. Natl. Acad. Sci. USA 2013, 110, 19036–19041. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Li, W.; Wang, Y.; Yang, M.; Guo, J.; Zhan, S.; Du, X.; Wang, Z.; Yang, M.; Li, J.; et al. Inhibition of TLR4 signaling by brucella TIR-containing protein TCPB-derived decoy peptides. Int. J. Med. Microbiol. 2016, 306, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed]
- Newman, R.M.; Salunkhe, P.; Godzik, A.; Reed, J.C. Identification and characterization of a novel bacterial virulence factor that shares homology with mammalian toll/interleukin-1 receptor family proteins. Infect. Immun. 2006, 74, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Patot, S.; Imbert, P.R.; Baude, J.; Martins Simoes, P.; Campergue, J.B.; Louche, A.; Nijland, R.; Bes, M.; Tristan, A.; Laurent, F.; et al. The tir homologue lies near resistance genes in staphylococcus aureus, coupling modulation of virulence and antimicrobial susceptibility. PLoS Pathog. 2017, 13, e1006092. [Google Scholar] [CrossRef]
- Cirl, C.; Miethke, T. Microbial toll/interleukin 1 receptor proteins: A new class of virulence factors. Int. J. Med. Microbiol. 2010, 300, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Rana, R.R.; Zhang, M.; Spear, A.M.; Atkins, H.S.; Byrne, B. Bacterial TIR-containing proteins and host innate immune system evasion. Med. Microbiol. Immunol. 2013, 202, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Tian, Y.; Wang, T.; Zhang, W.; Wang, W.; Gao, X.; Qu, S.; Cao, Y.; Zhang, N. Tram-derived decoy peptides inhibits the inflammatory response in mouse mammary epithelial cells and a mastitis model in mice. Eur. J. Pharmacol. 2015, 764, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Hines, D.J.; Choi, H.B.; Hines, R.M.; Phillips, A.G.; MacVicar, B.A. Prevention of LPS-induced microglia activation, cytokine production and sickness behavior with TLR4 receptor interfering peptides. PLoS ONE 2013, 8, e60388. [Google Scholar] [CrossRef] [PubMed]
- Lysakova-Devine, T.; Keogh, B.; Harrington, B.; Nagpal, K.; Halle, A.; Golenbock, D.T.; Monie, T.; Bowie, A.G. Viral inhibitory peptide of TLR4, a peptide derived from vaccinia protein a46, specifically inhibits TLR4 by directly targeting Myd88 adaptor-like and TRIF-related adaptor molecule. J. Immunol. 2010, 185, 4261–4271. [Google Scholar] [CrossRef] [PubMed]
- Loiarro, M.; Sette, C.; Gallo, G.; Ciacci, A.; Fanto, N.; Mastroianni, D.; Carminati, P.; Ruggiero, V. Peptide-mediated interference of TIR domain dimerization in Myd88 inhibits interleukin-1-dependent activation of NF-{kappa}B. J. Biol. Chem. 2005, 280, 15809–15814. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Qin, J. Modulation of toll-interleukin 1 receptor mediated signaling. J. Mol. Med. 2005, 83, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Srivastava, M.; Saqib, U.; Liu, D.; Faisal, S.M.; Sugathan, S.; Bishnoi, S.; Baig, M.S. Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways. Int. Immunopharmacol. 2016, 40, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Vaure, C.; Liu, Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Marcuzzi, A.; Secchiero, P.; Crovella, S.; Zauli, G. Trail administration down-modulated the acute systemic inflammatory response induced in a mouse model by muramyldipeptide or lipopolysaccharide. Cytokine 2012, 60, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Medicines and Healthcare products Regulatory Agency. Supplementary chapters sci basis of pharmacopeial requirements. In British Pharmacopeia; Medicines and Healthcare products Regulatory Agency: London, UK, 2012; Volume 5. [Google Scholar]
- Muzio, M.; Bosisio, D.; Polentarutti, N.; D’Amico, G.; Stoppacciaro, A.; Mancinelli, R.; van’t Veer, C.; Penton-Rol, G.; Ruco, L.P.; Allavena, P.; et al. Differential expression and regulation of toll-like receptors (tlr) in human leukocytes: Selective expression of TLR3 in dendritic cells. J. Immunol. 2000, 164, 5998–6004. [Google Scholar] [CrossRef] [PubMed]
- Lichte, P.; Grigoleit, J.S.; Steiner, E.M.; Kullmann, J.S.; Schedlowski, M.; Oberbeck, R.; Kobbe, P. Low dose lps does not increase TLR4 expression on monocytes in a human in vivo model. Cytokine 2013, 63, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Iwami, K.I.; Matsuguchi, T.; Masuda, A.; Kikuchi, T.; Musikacharoen, T.; Yoshikai, Y. Cutting edge: Naturally occurring soluble form of mouse toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol. 2000, 165, 6682–6686. [Google Scholar] [CrossRef] [PubMed]
- Akashi, S.; Shimazu, R.; Ogata, H.; Nagai, Y.; Takeda, K.; Kimoto, M.; Miyake, K. Cutting edge: Cell surface expression and lipopolysaccharide signaling via the toll-like receptor 4-MD-2 complex on mouse peritoneal macrophages. J. Immunol. 2000, 164, 3471–3475. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.X.; Souza, D.G.; Amaral, F.A.; Fagundes, C.T.; Rodrigues, I.P.; Alves-Filho, J.C.; Kosco-Vilbois, M.; Ferlin, W.; Shang, L.; Elson, G.; et al. Therapeutic effects of treatment with anti-TLR2 and anti-TLR4 monoclonal antibodies in polymicrobial sepsis. PLoS ONE 2015, 10, e0132336. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.H.; Dudics, S.; Astry, B.; Moudgil, K.D. Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathog. Dis. 2016, 74. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Liu, D.; Zhang, Y.; Qian, Y.; Zhang, H.; Guo, S.; Sunagawa, M.; Hisamitsu, T.; Liu, Y. Celastrol inhibits lipopolysaccharide-stimulated rheumatoid fibroblast-like synoviocyte invasion through suppression of tlr4/nf-kappab-mediated matrix metalloproteinase-9 expression. PLoS ONE 2013, 8, e68905. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Lan, Y.; Zhou, H.; Li, W.; Xiang, M. Total coumarins from urtica dentata hand prevent murine autoimmune diabetes via suppression of the TLR4-signaling pathways. J. Ethnopharmacol. 2013, 146, 379–392. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance name | Mechanism of action | Clinical trial, design and results | Reference |
|---|---|---|---|
| TAK-242 (Resatorvid) | Binds covalently to Cys747 of TLR4-TIR domain and blocks TLR4/TIRAP and TLR4/TRAM interactions | Randomized, double-blind, placebo-controlled phase 2 trial (274 patients). Failed to suppress cytokine levels in patients with sepsis and shock or respiratory failure. | [124] |
| Eritoran (E5564) | Lipid A mimic, binds to MD-2 | Phase 2, double-blind, placebo-controlled, ascending-dose study (152 patients). Eritoran was well tolerated but did not attenuated systemic inflammation significantly. | [127] |
| Randomized, double-blind, placebo-controlled, multinational phase 3 trial (1961 patient). Treatment did not improve 28-day survival. | [131] | ||
| Prospective, randomized, double-blind, placebo-controlled, multicenter, ascending-dose phase II trial (293 patients). Eritoran was well tolerated but the mortality was not decreased significantly. | [132] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. https://doi.org/10.3390/vaccines5040034
Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, Peri F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines. 2017; 5(4):34. https://doi.org/10.3390/vaccines5040034
Chicago/Turabian StyleKuzmich, Nikolay N., Konstantin V. Sivak, Vladimir N. Chubarev, Yuri B. Porozov, Tatiana N. Savateeva-Lyubimova, and Francesco Peri. 2017. "TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis" Vaccines 5, no. 4: 34. https://doi.org/10.3390/vaccines5040034