Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid


1
School of Chemical Sciences, The University of Auckland, Auckland 1142, New Zealand
2
School of Mathematical and Physical Sciences, University of Technology Sydney, Sydney, Broadway NSW 2007, Australia
*
Authors to whom correspondence should be addressed.
Membranes 2018, 8(2), 20; https://doi.org/10.3390/membranes8020020
Submission received: 27 March 2018
/
Revised: 12 April 2018
/
Accepted: 12 April 2018
/
Published: 18 April 2018
(This article belongs to the Special Issue Polymeric Membranes for Gas Separation)
Abstract
:Carboxylate-functionalised polymers of intrinsic microporosity (C-PIMs) are highly desirable materials for membrane separation applications. The recently reported method to afford C-PIMs was via an extensive base hydrolysis process requiring 360 h. Herein, a novel and effective method to convert PIM-CONH2 to C-PIM using nitrous acid was studied. The chemical structure of C-PIM was characterised by 1H NMR, 13C NMR, FTIR, elemental analysis, UV-Vis, TGA and TGA-MS. Complete conversion from amide to carboxylic acid groups was confirmed. Decarboxylation of C-PIM was also successfully studied by TGA-MS for the first time, with a loss of m/z 44 amu (CO2) observed at the first degradation stage. TGA also revealed decreased thermal stability of C-PIM relative to PIM-CONH2 under both N2 and air atmosphere. Gel permeation chromatography (GPC) analysis showed continuous molecular weight degradation of C-PIM with extended reaction time. Aromatic nitration was also observed as a side reaction in some cases.
1. Introduction
The unprecedented energy consumption of the past century has led to dramatic increases in atmospheric CO2 concentration [1,2,3]. Because of this, there is a growing demand for efficient and cost-effective CO2 separation and capture technology [1,2,3]. In addition to the current chemical and thermal separation processes such as absorption, cryogenic distillation and pressure swing adsorption (PSA), membrane separation is slowly gaining ground in oil refineries and petrochemical plants due to its low energy consumption and environmental friendliness [4,5,6,7]. In the past three decades, polymeric membrane separations have exhibited strong growth rates and now have been installed for N2/O2 and CO2/CH4 separations plus H2 recovery from HCs, etc. Polymeric membranes enjoy low cost fabrication, easy scale-up, flexibility and mild operation conditions [8].
A unique class of microporous organic material (i.e., Polymers of Intrinsic Microporosity (PIMs)) was first developed by Budd and McKeown in 2004 [9]. Since the chemical structures and physical properties of the membrane materials influence their corresponding permeability and permselectivity, various new PIM structures and synthetics methods have been reported, in order to improve the gas separation performance [10,11,12,13,14,15]. The nitrile (–CN) group of PIM-1 is the most popular target for transformation, and has been converted to various CO2-philic functionalities such as nitrogen-containing groups [16,17,18,19,20,21,22] and carboxylic acid-based functional groups [23,24,25,26,27,28,29,30]. Carboxylate functionalised PIMs (C-PIMs) were inspired by the well-known carboxylic acid-containing polyimide based on the 6FDA-DABA, which exhibits superior gas separation performance after decarboxylation and/or crosslinking [31]. The first synthetic attempt toward C-PIM was reported in 2009 [24]. The synthesis was based on hydrolysis of the nitrile group under alkaline conditions. The resultant polymer showed an overall increase in permselectivity and decreased permeability [24]. However, in 2014, Satilmis et al. pointed out that the base-hydrolysed PIM-1 materials contained a mixture of amide, carboxylic acid, ammonium carboxylate and sodium carboxylate functional groups where the actual percentage of carboxylic acid was less than 20% [32]. Even after extensive base-hydrolysis, only 51% carboxylation was achieved [32]. In 2017, Santoso et al. reported a critical update on the characterisation of C-PIM [33]. Based on the spectroscopic data obtained from FT-IR, 1H NMR, 13C NMR, and 1H-15N HSQC, the amide and carboxylic acid groups were unambiguously distinguishable. The multiple proton signals between 7.0–8.5 ppm in 1H NMR were correctly assigned to the active amide protons (–C(=O)-NH2) whereas the carboxylic acid proton (–C(=O)-OH) is located further downfield in the range of 13.0–14.0 ppm. Moreover, the carbonyl stretch vibration at 1666 cm−1 observed by FTIR belongs to the amide carbonyl, whereas the carboxylic acid carbonyl stretch is located at 1720 cm−1 [33].
Most recently, Jeon et al. successfully obtained a fully converted C-PIM (>92 mol % COOH content) after an exhaustive 360 h (15 days) base-hydrolysis [34]. The resulting C-PIM film exhibited outstanding selectivity of 53.6 for CO2/N2 gas pair [34]. However, this extremely slow reaction rate is not ideal and could be a barrier to large scale industrial implementation. During the base hydrolysis process, the nitrile to amide conversion is relatively fast. The bottleneck appears to be the slow conversion from the amide to carboxylic acid [35]. The hydroxide (OH−) attacks the carbonyl carbon of the benzamide to give a tetrahedral intermediate. Since NH2− is a stronger base and a less favourable leaving group, the tetrahedral intermediate can either revert to the amide starting material or form the carboxylic acid product.
Alternatively, conversion of nitrile to carboxylic acid via acidic hydrolysis route was published by Weng et al. in 2015. The reported reaction time was 48 h [23]. However, our group failed to reproduce the original experimental results.
A new synthesis of C-PIM via a fast and efficient reaction route is therefore required. As summarised in Table 1, herein we report an effective and fast method to convert amide-containing PIM-1 (i.e., PIM-CONH2) to C-PIM via reaction with nitrous acid. A combination of FTIR, 1H, 13C NMR, elemental analysis, UV-Vis, GPC, TGA, and TGA-MS were used to characterise the resulting C-PIM.
2. Materials and Methods
All chemicals and solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA). 3,3,3′,3′-Tetramethyl-1,1′-spirobiindane-5,5′,6,6′-tetraol (Sigma-Aldrich, 96%) and 2,3,5,6-tetrafluoroterephthalonitrile (Sigma-Aldrich, 99%) were recrystallised before use and other chemicals and solvents were used as received without further treatment. However, caution is required: sodium nitrite needs to be handled with care due to high acute toxicity.
NMR spectra were recorded on Bruker DRX 400 MHz NMR spectrometers at 25 °C. The measurement frequencies for 1H NMR was 399.89 and 400.13 MHz. The chemical shifts for the NMR solvents reported for 1H and 13C in parts per million (ppm) are referenced to 7.26 ppm and 77.0 ppm for CDCl3, 2.5 ppm and 39.5 ppm for DMSO-d6, 1.7 ppm with 3.6 ppm and 67.2 ppm with 25.3 ppm for THF-d8, respectively. Broadband decoupled mode was used for recording 13C NMR spectra.
Infrared spectra were recorded on Nicolet iS50 FT-IR Spectrometer with a Universal ATR Sampling Accessory. Each sample was scanned 32 times at resolution of 4 cm−1.
UV-Vis spectra were recorded on a Shimadzu UV-2700 spectrophotometer. Pure solvents were used for baseline correction. For these measurements, PIM-CONH2 and C-PIM were dissolved in DMSO and THF at a concentration of 0.1 mg/mL.
Elemental analysis was performed in the Campbell Microanalytical Laboratory located at University of Otago. All measured microanalysis results have an uncertainty of ±0.4%.
Thermogravimetric analysis (TGA) was performed using a TG 5000 instrument. For polymer thermal degradation analysis, samples were heated from room temperature to 100 °C at a rate of 10 °C/min followed by isotherm for 60 min to remove water and solvents and then heated to 800 °C at a rate of 10 °C/min. Identical conditions were used for TGA measurements under nitrogen flow and air flow. For the thermal decarboxylation experiment, C-PIM was heated from room temperature to 100 °C at a rate of 10 °C/min followed by isotherm for 60 min and then heated to 350 °C at a rate of 10 °C/min followed by isotherm for 60 min. The leftover material was further characterised by FTIR.
Thermogravimetric analysis was coupled to a gas chromatography mass spectrometer (TG-GC-MS), which was used to analyse the gases evolved during pyrolysis. TG measurement was carried out using a Netzsch STA449 F5 Jupiter® instrument. The experiments were carried out in a helium atmosphere flowing at 60 mL/min (40 mL/min directly to the furnace +20 mL to the furnace via the mass balance). High purity Helium 5.0 gas (99.999%) was supplied by Coregas. The sample insertion temperature was 40 °C followed by isotherm for 10 min. Samples were then heated to 1000 °C at a rate of 10 °C/min followed by isotherm for 20 min. Crucible cleaning was achieved by influx of 10 mL/min oxygen in the purge gas and cooling to 40 °C. The GC-MS configuration measurement range from 5 to 100 m/z was analysed using an Agilent 7890 GC with Agilent MSD 5977 EI.
PIM-1 is the polycondensation product of 3,3,3′,3′-Tetramethyl-1,1′-spirobiindane-5,5′,6,6′-tetraol and 2,3,5,6-tetrafluoroterephthalonitrile and was synthesised following the procedures reported by Budd and McKeown [9].
We previously reported the synthesis of PIM-CONH2 [36]. Typically, dimethyl sulfoxide (DMSO) (30.0 mL) was added to fine PIM-1 powder (0.500 g) and stirred at moderate speed for 1 h at R.T. Potassium carbonate (K2CO3) (0.500 g, 3.62 mmol) was then added to the stirring mixture to give a slightly basic pH 9–10 solution, followed by drop-wise addition of 25% hydrogen peroxide (H2O2) (5.0 mL, 32.64 mmol). The reaction was performed at RT for 24 h. Water (250 mL) was added at the end of the reaction and was further stirred rapidly overnight. A light-yellow polymer was collected by vacuum filtration and washed with water and methanol. The solid was dried in oven at 25 °C for 24 h to give PIM-CONH2 (0.48 g, yield > 80%). 1H NMR (400 MHz, DMSO-d6): δ ppm 7.94–7.57 (br, 2H), 6.80 (s, 2H), 6.18 (s, 2H), 2.21–2.07 (br, 4H), 1.29–1.21 (br, 12H). 13C NMR (100.57 MHz, DMSO-d6 ): δ ppm 162.0, 148.2, 145.5, 140.2, 140.0, 133.5, 115.7, 111.0, 110.0, 58.5, 56.7, 43.1, 31.1, 29.6.
Synthesis of C-PIM using nitrous acid: In a pressure tube, PIM-CONH2 (50 mg) was suspended in a stirred mixture of MeCN (0.8 mL) and 2M aqueous H2SO4 (0.6 mL, 1.2 mmol). NaNO2 (0.150 g, 2.4 mmol) was added portion-wise at 0 °C. After addition, the cap was screwed tightly, and the reaction vessel immersed in oil at 90 °C for 6 h. The reaction mixture was then diluted with water (4 mL) and extracted with ethyl acetate (8 mL). The organic layer was then concentrated in vacuo. The crude product was then dissolved in THF, filtered and solution concentrated in vacuo and dried in a vacuum oven at 70 °C overnight. The crude polymer was re-dissolved in THF, precipitated by addition of MeOH and then dried in vacuum to give C-PIM (35 mg, yield = 70%) as a dark yellow powder which was fluorescent under UV. 1H NMR (400 MHz, DMSO-d6): δ ppm 13.84 (br, 2H), 6.87 (s, 2H), 6.29 (s, 2H), 2.26–2.11 (br, 4H), 1.33–1.25 (br, 12H). 13C NMR (100.57 MHz, DMSO-d6 ): δ ppm 162.3, 148.6, 145.7, 139.8, 133.6, 113.4, 111.3, 110.1, 58.3, 56.7, 43.1, 31.0, 29.5.
3. Results
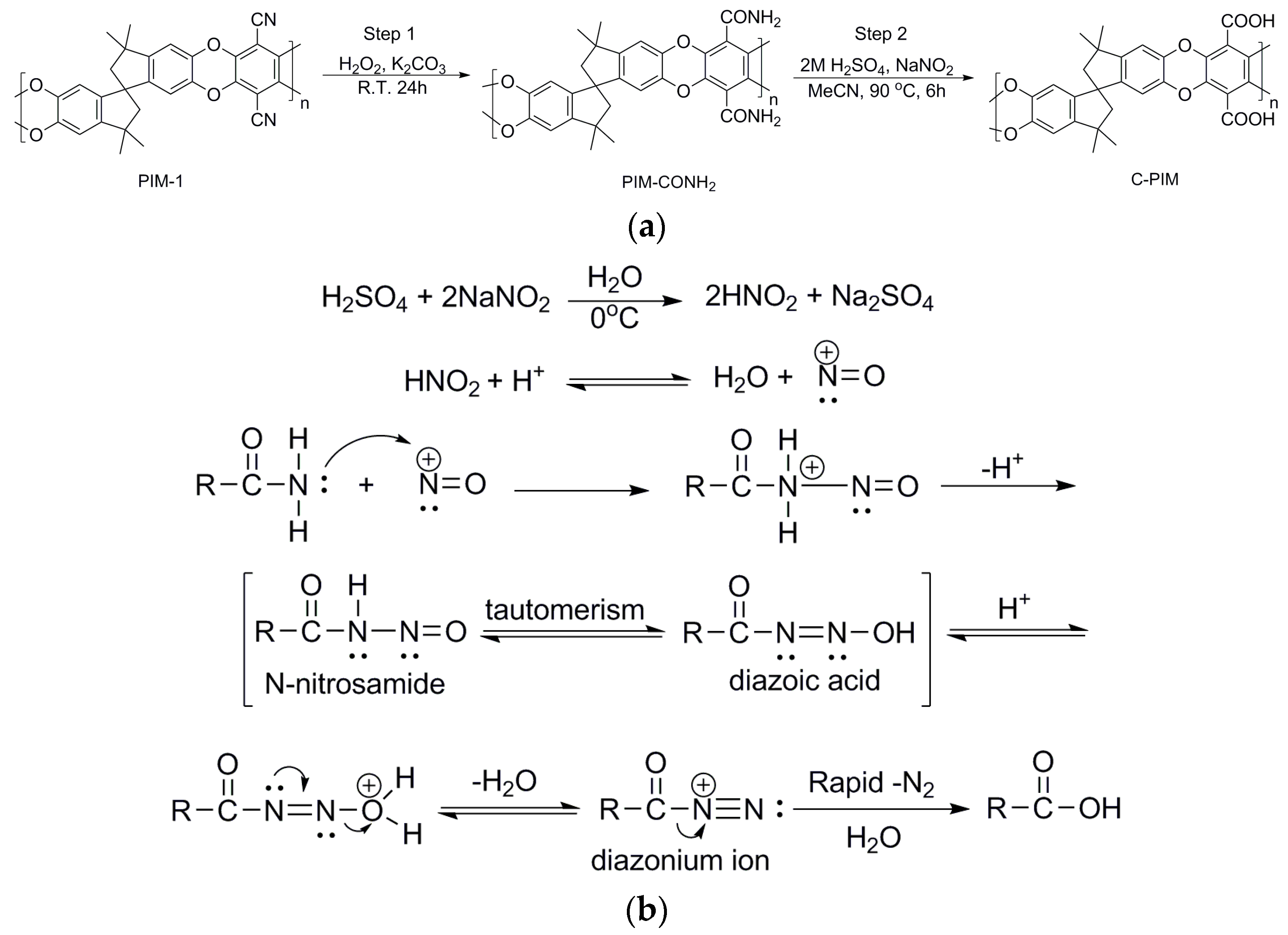
Previously reported post-polymerisation modifications of PIM-1 to C-PIM have utilised either acid or base-catalysed hydrolysis procedures [23,24,32,33,34,37]. In both pathways, PIM-CONH2 is formed as an intermediate. This most likely results in the co-existence of two distinct functional groups (amide and carboxylic acid) on a single polymer chain. Furthermore, when using the basic conditions reported by Jeon et al. the C-PIM is obtained after an extremely long reaction time (15 days) [34]. Based on these issues, we have developed a two-step approach to C-PIM as shown in Scheme 1a. PIM-1 is first converted to PIM-CONH2 using hydrogen peroxide. Pure PIM-CONH2 is purified and isolated. In the second step, PIM-CONH2 undergoes reaction with nitrous acid (HNO2) to afford C-PIM. Since nitrous acid is unstable, it is prepared in situ by the reaction of sodium nitrite and dilute acid in the absence of heat. We have previously reported the detailed synthesis of PIM-CONH2 [36]. Here we only focus on the second reaction. Optimisation of reaction solvent, the choice of the type of acid and acidity, and reaction time are discussed in detail below.
Pioneered by George Olah et al., nitrous acid has found use as an alternative method to convert amide compounds that are resistant to standard hydrolysis to the corresponding carboxylic acids [38]. In 1965, Olah et al. reported the conversion sterically hindered benzamides and benzenesulfonamides to the corresponding acids by treatment with nitrosonium tetrafluoroborate at 50 °C in acetonitrile, in high yield (>80%) [38]. The amide can react with the nitrous acid to give the corresponding carboxylic acid and nitrogen gas. Scheme 1b gives the proposed reaction mechanism. The key reactive species is the highly electrophilic nitrosonium (NO+) cation, which is readily added to amide nitrogen, resulting in the formation of the corresponding N-nitrosamide. The N-nitrosamide tautomerises to the diazoic acid which forms a diazonium cation by losing a molecule of water. The diazonium cation decomposes rapidly through evolution of nitrogen and forms acyl carbocation. Finally, the acyl carbocation is quenched by water to give the carboxylic acid product.
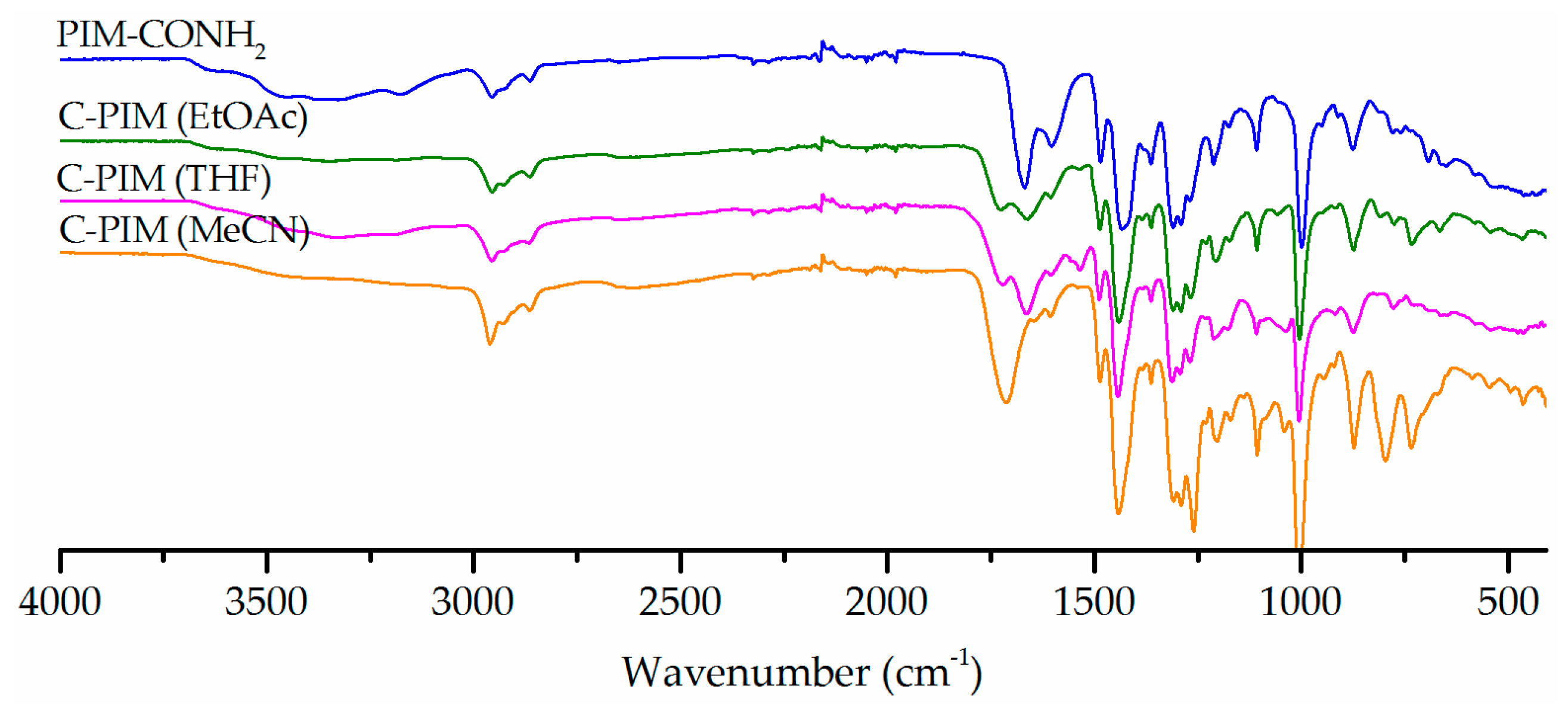
First, three common organic solvents: ethyl acetate (EtOAc), tetrahydrofuran (THF) and acetonitrile (MeCN) were tested as reaction media. The reaction temperature was adjusted with the solvent’s boiling point. Therefore, the temperature was set at 70 °C for THF, 80 °C for EtOAc and 90 °C for MeCN. At the beginning of the reaction, PIM-CONH2 was not soluble in any of these solvents. As the reaction proceeded, the reaction mixtures became partially soluble in EtOAc and MeCN and fully soluble in THF. Figure 1 shows the effect of solvent on the reaction of PIM-CONH2 with NaNO2 and 2M H2SO4 for 6 h. Conversion was observed in all three solvents as assessed by FTIR which exhibited a carboxylic acid carbonyl stretching peak at 1720 cm−1. However, the experiments carried out in EtOAc and THF still showed the peak at 1666 cm−1 which corresponds to the stretch vibration of amide carbonyl. Furthermore, a new peak at 1545 cm−1 was present in these two solvent systems. In prior literature, this type of signal was found in nitrated polyphenylene oxide (PPO-NO2) reported by Bhole et al. [39] and nitrated poly (diphenylacetylene) reported by Sakaguchi et al. [40]. Their FTIR showed absorption peaks at 1532 cm−1 and 1364 cm−1, which corresponded to the aromatic –NO2 antisymmetric stretch and –NO2 symmetrical stretch, respectively. This is therefore strong evidence that nitration of PIM-CONH2 has also occurred in EtOAc and THF. On the basis of these results, MeCN is best suited for the present reaction, in which the product gave the most intense carboxylic acid carbonyl stretching peak, with minimal amide carbonyl stretching. Notably no nitration side reaction occurred in MeCN.
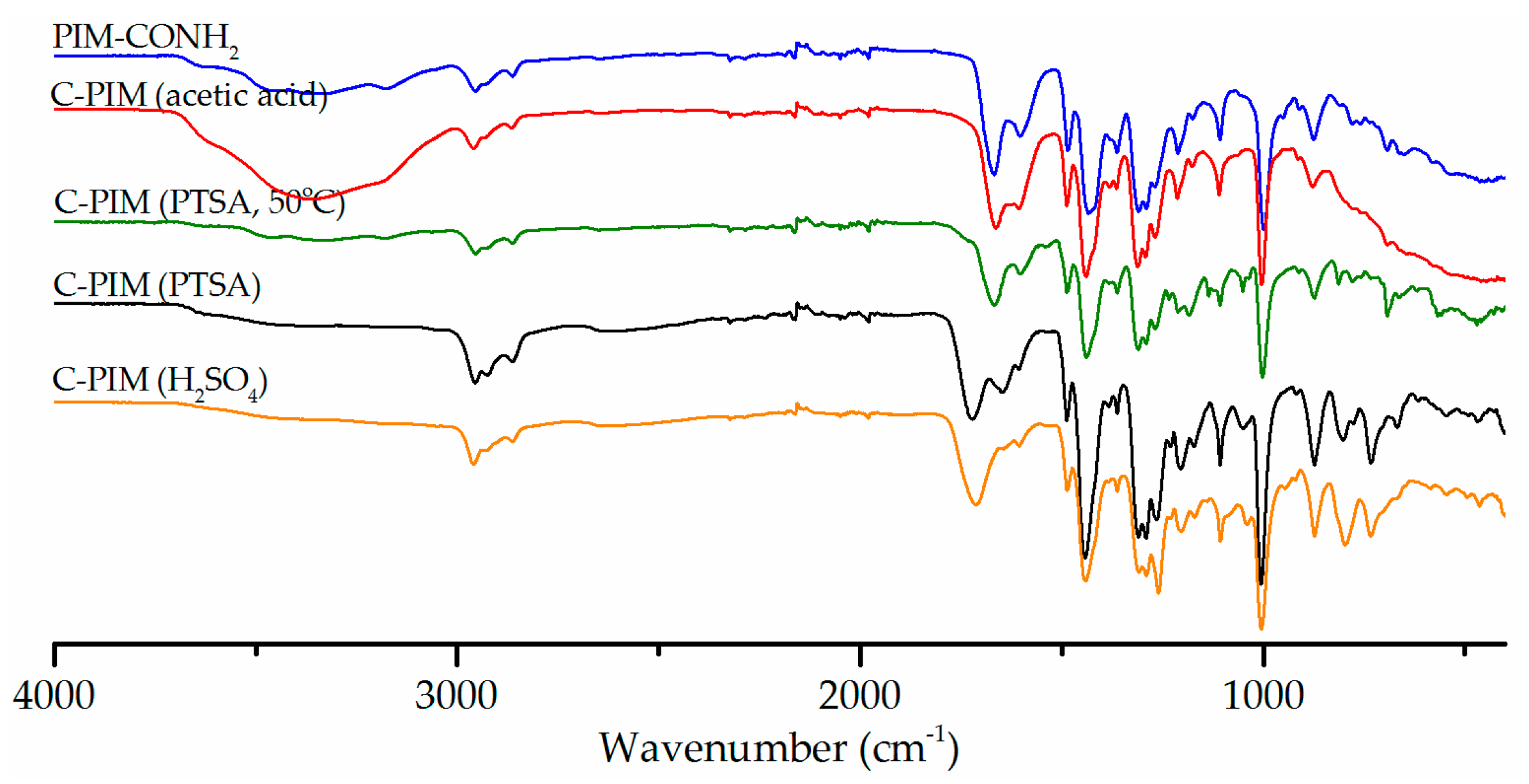
Secondly, besides the combination of sodium nitrite with sulfuric acid, organic acids such as acetic acid and p-toluenesulfonic acid (PTSA) with sodium nitrite [41,42,43] were tested. The use of organic acids was expected to be more compatible with reaction solvent (MeCN) while still facilitating the generation of nitrous acid. Figure 2 shows the effect of the acid on the reaction outcome using NaNO2 in MeCN. All acids were made into the same concentration of 2M and performed under otherwise identical reaction conditions (90 °C for 6 h). A lower temperature reaction at 50 °C was also performed for PTSA in accordance with literature precedent [43]. The acetic acid system showed no conversion, as the parent amide absorption peak at 1666 cm−1 was still intact and no carboxylic acid carbonyl absorption peak at 1720 cm−1 was observed. This result was not surprising since when H2SO4 and PTSA were mixed with sodium nitrite, nitrogen gas was evolved which confirmed that nitrous acid had been produced, but this was not the case with acetic acid. This is likely due to the lower strength of acetic acid compared to the other acids. The temperature dependence of the reaction was also observed in the reaction performed using PTSA. The reaction with PTSA at 50 °C is not as effective as the one at 90 °C. At 50 °C, the amide stretch at 1666 cm−1 was still the dominant absorption in the IR spectrum, with a very weak carboxylic acid carbonyl absorption peak at 1720 cm−1. Comparing H2SO4 and PTSA, the former resulted in a higher conversion rate.
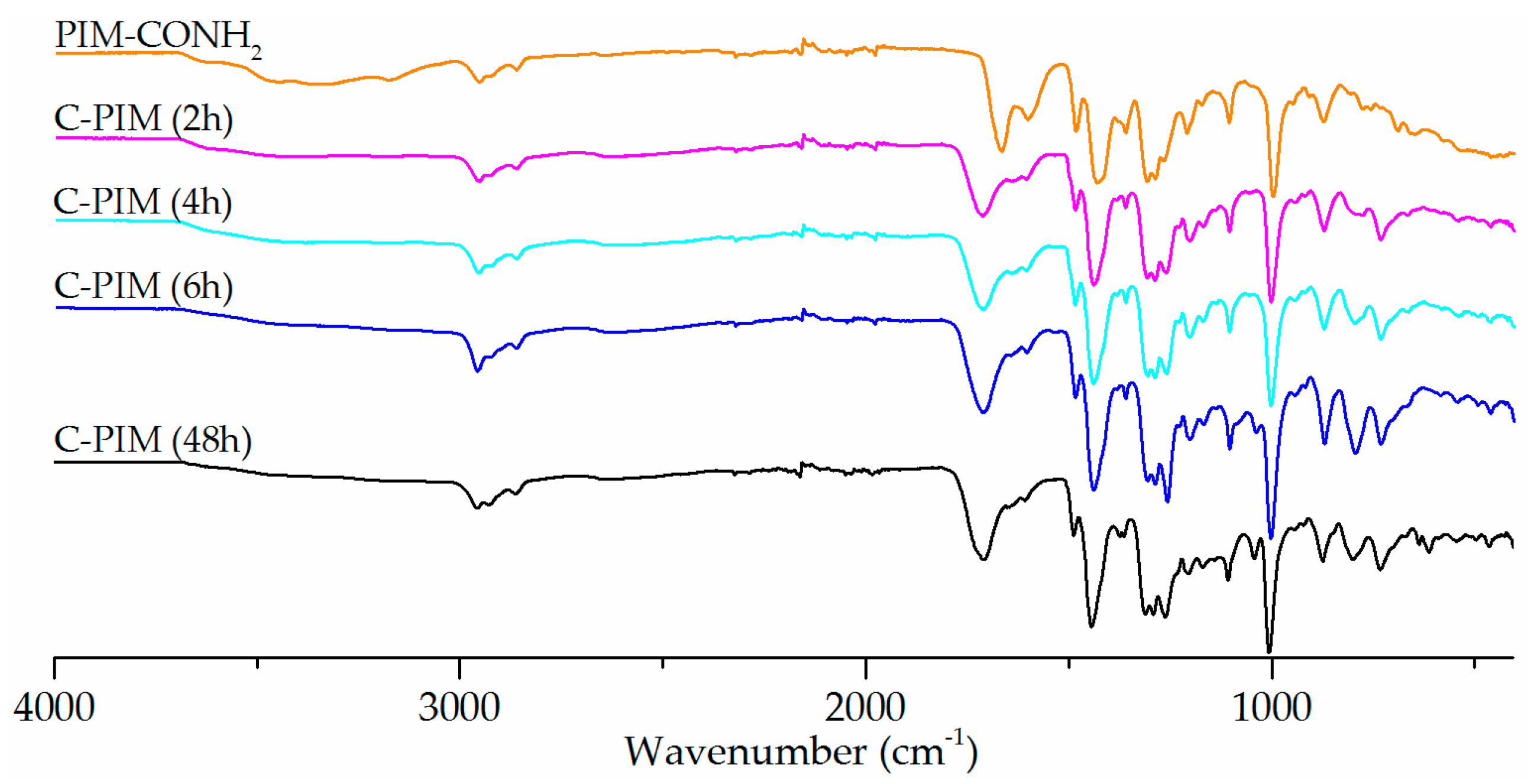
After identifying the best reaction solvent and acid, the reaction kinetics were monitored by FTIR. Figure 3 shows the reaction progression between 2 to 48 h. Remarkably, under the optimised reaction conditions (MeCN, NaNO2 and H2SO4 at 90 °C) the conversion from amide to carboxylic acid was pleasingly fast. Within 2 h, the amide band at 1666 cm−1 had nearly disappeared and was taken over by the carboxylic acid peak at 1720 cm−1. There was no obvious change in the intensity of the C=O band of carboxylic acid after 6 h. Clearly, the reaction kinetics for the nitrous acid reaction are much faster than acid or base catalysed hydrolysis.
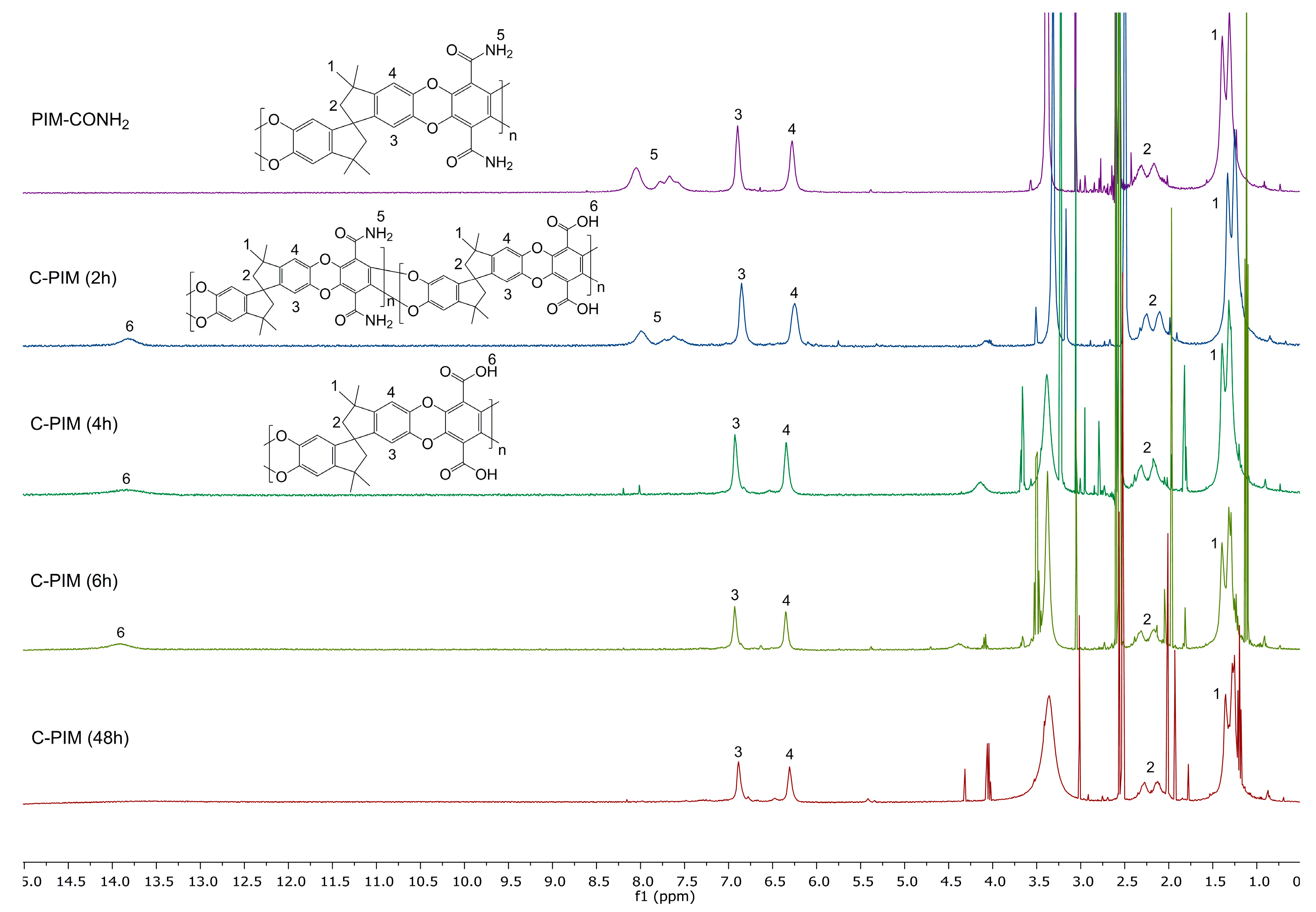
The 1H NMR spectra were also used to probe the reaction kinetics. Figure 4 compares the 1H NMR spectra of PIM-CONH2, C-PIM (2 h), C-PIM (4 h), C-PIM (6 h) and C-PIM (48 h) samples. For PIM-CONH2, multiple peaks are observed at 7.0–8.5 ppm which are due to the four active amide (–C(=O)NH2) protons. For the C-PIM (2 h) sample, both amide and carboxylic acid (–C(=O)OH) proton resonances in the 13.0–14.0 ppm region were present. As the reaction progressed to 4 h, the amide proton region (7.0–8.5 ppm) turned to flat baseline. After 6 h reaction time, there were no obvious changes in 1H NMR spectra, consistent with the FTIR results. We therefore concluded that the conversion was complete after 6 h.
The conversion of the amide to the carboxylic acid was also verified by elemental analysis (Table 2). Increased percentages of hydrogen atoms (4.47% to 5.33%) and oxygen atoms (14.69% to 23.90%) were observed from the conversion of PIM-1 to PIM-CONH2, where four additional hydrogen atoms and two additional oxygen atoms were gained per repeat unit. From PIM-CONH2 to C-PIM, the percentage of hydrogen atoms was decreased (5.33% to 4.80%) with an increase in oxygen atoms (23.90% to 30.74%), where two hydrogen atoms were lost and two more oxygen atoms were gained for each repeat unit in C-PIM polymer. Most significantly, the decrease of nitrogen content from 5.24% in PIM-CONH2 to 0.91% in C-PIM indicated successful conversion of the amide groups to carboxylic acid functionalities.
The solubility of C-PIM in common organic solvents is very important for applications involving membrane casting. In Table 3, the solubility of PIM-1, PIM-CONH2, and C-PIM are compared. It was well known that PIM-1 was readily soluble in tetrahydrofuran (THF), dichloromethane (DCM), and chloroform (CHCl3), but insoluble in polar aprotic solvents such as dimethyl formamide (DMF), dimethyl acetamide (DMAc), and N-methyl pyrrolidone (NMP). In contrast to PIM-1, PIM-CONH2 showed good solubility in polar aprotic solvents such as DMF, DMAc, NMP, and DMSO. The C-PIM prepared in this work exhibited wide solubility in many common solvents except less polar ones such as DCM and CHCl3. Interestingly, C-PIM was even soluble in acetone. The higher solubility of C-PIM than PIM-CONH2 was attributed to favourable interactions between the carboxylic acid groups of C-PIM with the solvent molecules.
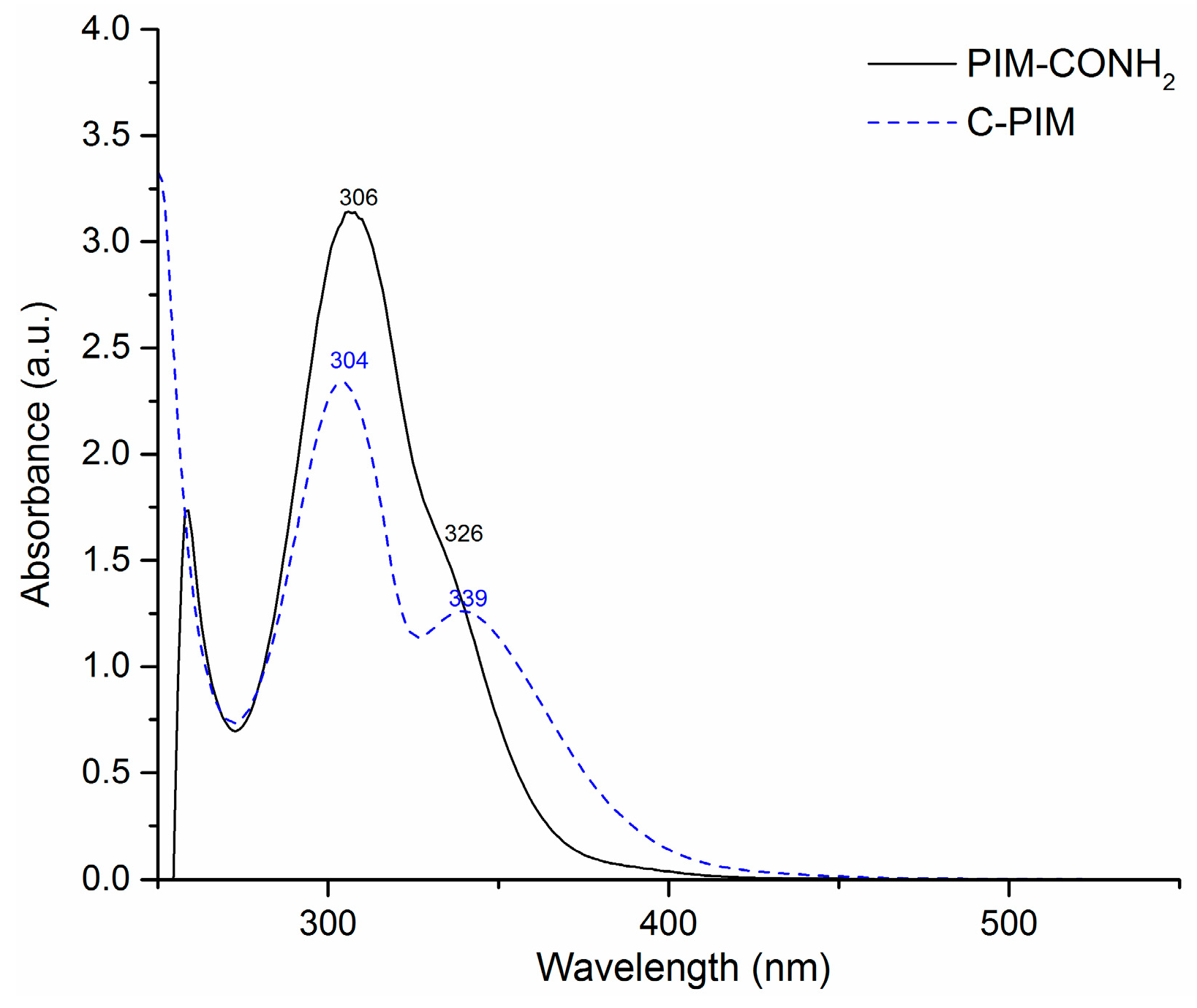
The UV-Vis spectra of PIM-CONH2 and C-PIM with same concentration (0.1 mg/mL) are shown in Figure 5. For the starting polymer PIM-CONH2, the absorption maximum (λmax) was at 306 nm corresponding to the π-π* transition while the shoulder peak at 326 nm corresponds to the n-π* transition of the carbonyl group. For the product polymer C-PIM, λmax at 304 nm corresponds to the π-π* transition and λmax at 339 nm arise from the n to π* transition. A red shift of about 13 nm was observed for the n-π* transition (–C=O) of C-PIM compared to that of PIM-CONH2. This indicated that the energy gap between n orbital and π* orbital for C-PIM was smaller than that of PIM-CONH2.
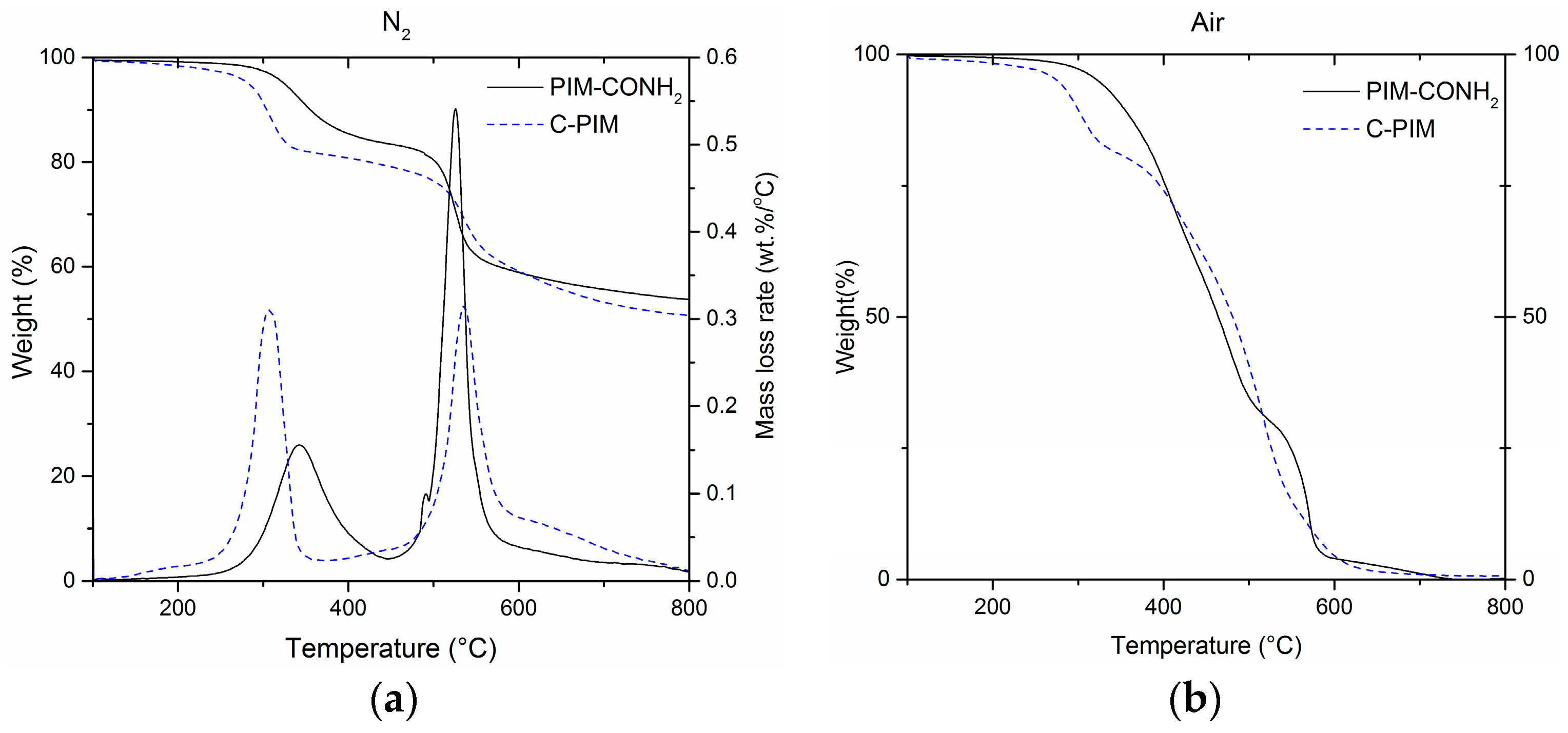
The thermal stability of C-PIM and PIM-CONH2 were investigated by TGA under nitrogen and air, respectively. Figure 6a shows the pyrolysis behaviour of PIM-CONH2 and C-PIM under a nitrogen atmosphere. Both C-PIM and PIM-CONH2 clearly showed two distinct stages of thermal degradation. Subsequently, two main peaks can be clearly resolved from the differential TGA curve (DTG). In PIM-CONH2, the first degradation stage is associated with a mass loss of ~15% in the range of 270–450 °C. This was attributed to the release of the labile pendant group (i.e., amide). During the second degradation stage, a mass loss of ~25% was observed in the range of 450–600 °C which was ascribed to decomposition of the PIM backbone. The C-PIM gave a similar TGA thermogram with two degradation stages. However, the conversion of the amide group to the carboxylic acid group decreased in thermal stability. Under nitrogen, the onset temperature for decarboxylation was measured to be 250 °C, with a major loss of CO2 at 307 °C. A total mass loss of 16% was observed in the range of 250–370 °C corresponding to the decarboxylation. As the theoretical value for full removal of the side chain is 18%, this means that near-total decarboxylation had occurred. This decarboxylation result is in agreement with the decarboxylation of polythiophene ester reported by Søndergaard et al. [44]. The second degradation stage of C-PIM occurred between 450–600 °C, and was associated with a mass loss of 20%.
Moreover, as analysed from the DTG curves, in the first stage of degradation (loss of pendant group), the maximum mass loss rate (rmax) of the C-PIM was rmax = 0.31 %/°C at 307 °C which was twice the value of PIM-CONH2 with rmax = 0.16 %/°C at 342 °C. This suggests that the rate of decarboxylation is faster than the de-amidation rate. In the second stage of degradation (main chain breakdown), C-PIM has rmax = 0.32 %/°C at 536 °C while PIM-CONH2 has a very large rmax = 0.54 %/°C at 526 °C. This interesting DTG data might reveal that the C-PIM polymer after decarboxylation stage is relatively more stable as a result of decarboxylation-induced aryl radical homo-coupling (i.e., cross-linking).
Figure 6b shows the pyrolysis behaviour of PIM-CONH2 and C-PIM under an air atmosphere. The C-PIM still showed decarboxylation in the range of 270–350 °C. This indicates that decarboxylation of C-PIM was not affected by the oxidative environment. For PIM-CONH2, there was no obvious first stage degradation. Both C-PIM and PIM-CONH2 decomposed completely as 100% weight loss was observed at the end of pyrolysis at 800 °C, whereas under the N2 atmosphere, ~50% char yields were obtained.
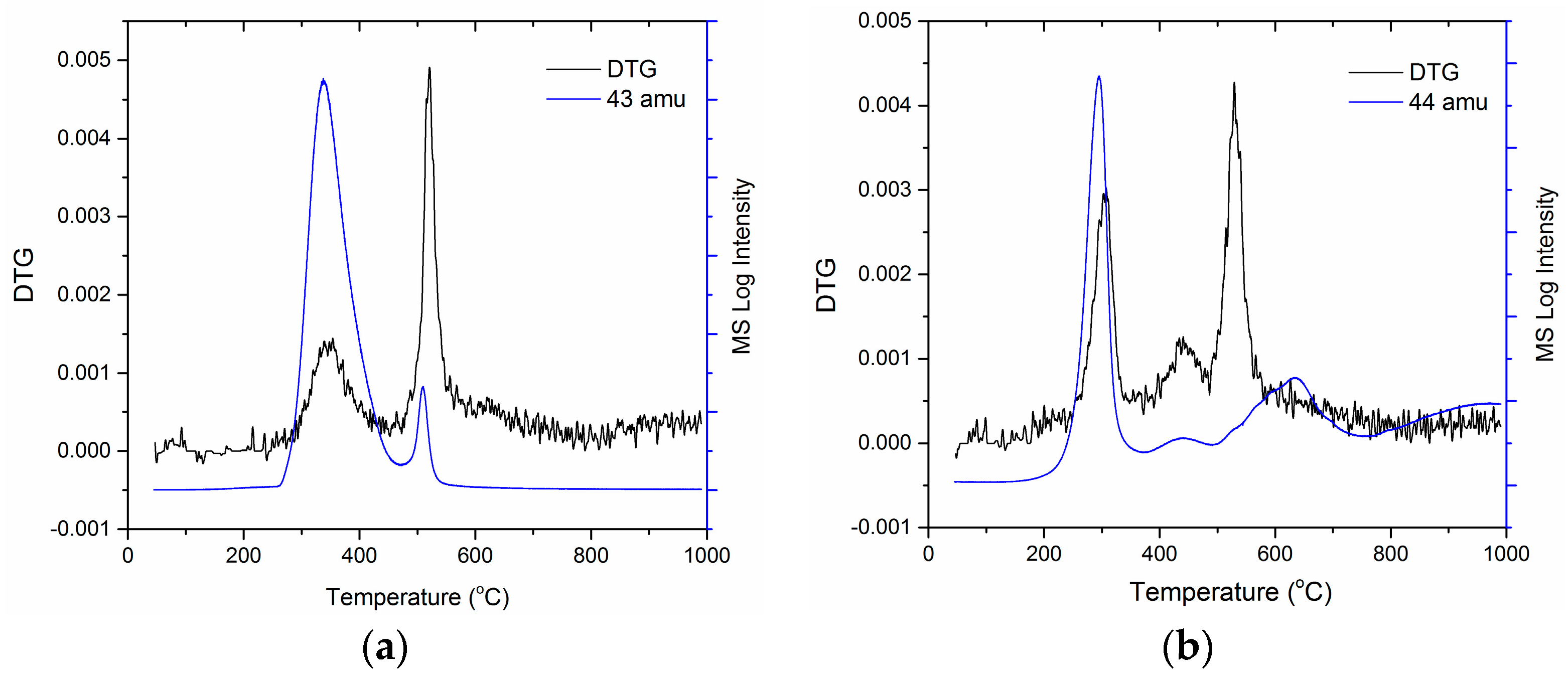
For the first time, TGA-MS was used to analyse the composition of the evolved gases from C-PIM and PIM-CONH2 during TGA scans, focusing on their first degradation stages (decarboxylation and de-amidation, respectively). In Figure 7a, the DTG curve of PIM-CONH2 is overlaid with MS signal for the molecular ion with m/z 43 amu evolved during the TGA scan. A mass loss of 43 amu (CONH) showed the best match with the PIM-CONH2 first DTG peak at 350 °C. This confirmed that the first mass loss peak resulted from release of the amide group. In Figure 7b, the DTG curve of C-PIM is overlaid with the MS signal for the molecular ion of m/z 44 amu evolved during the TGA scan. The MS peak associated with the molecular weight of 44 (CO2) showed the perfect match with the C-PIM first DTG peak at 300 °C, which confirms that the first mass loss peak is due to the decarboxylation of C-PIM.
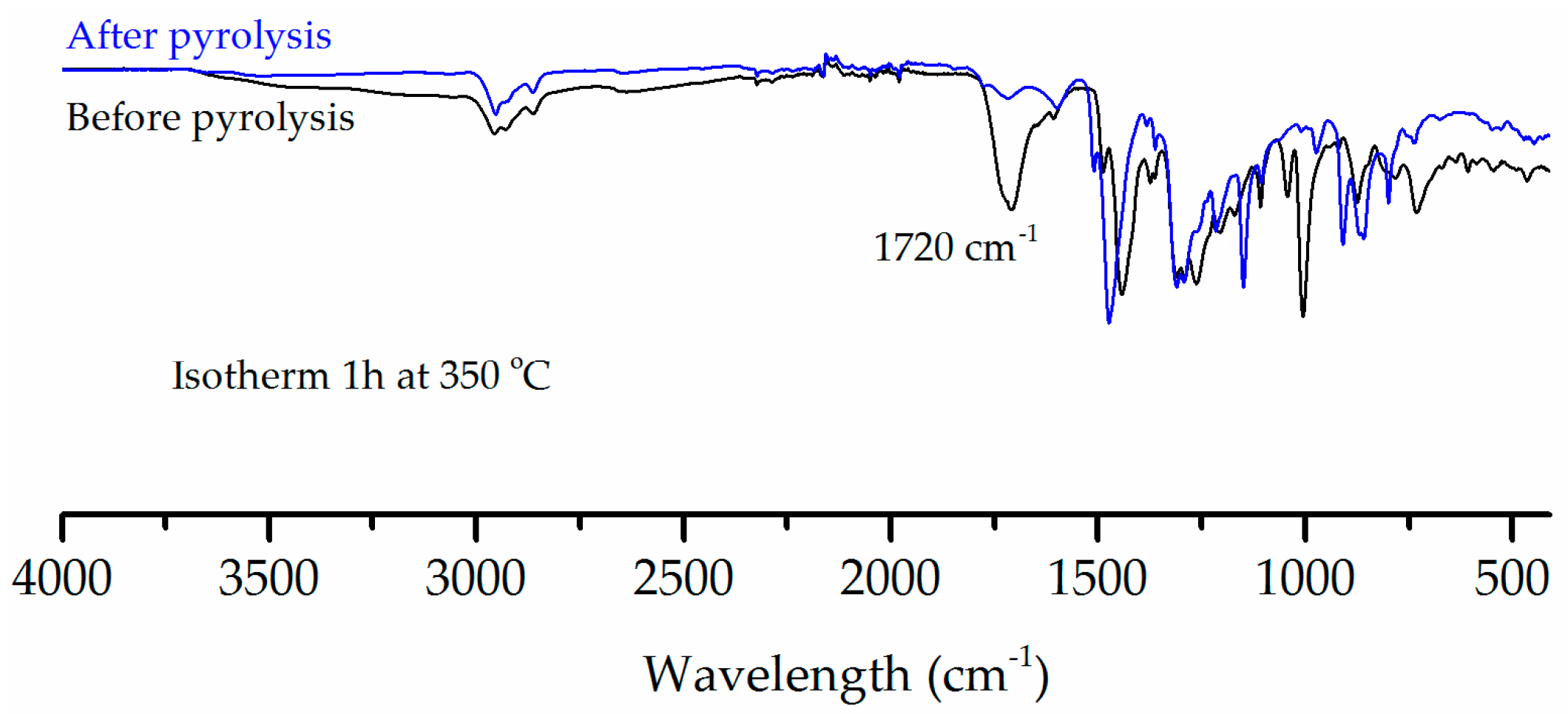
Figure 8 compares the FTIR of C-PIM samples before and after pyrolysis. The pyrolysed C-PIM sample was held isothermally at 350 °C for 60 min under nitrogen. The pyrolysis residue clearly shows the disappearance of the original carboxylic acid C=O stretch at 1720 cm−1. This further confirmed that the first degradation stage is the decarboxylation of C-PIM.
4. Discussion
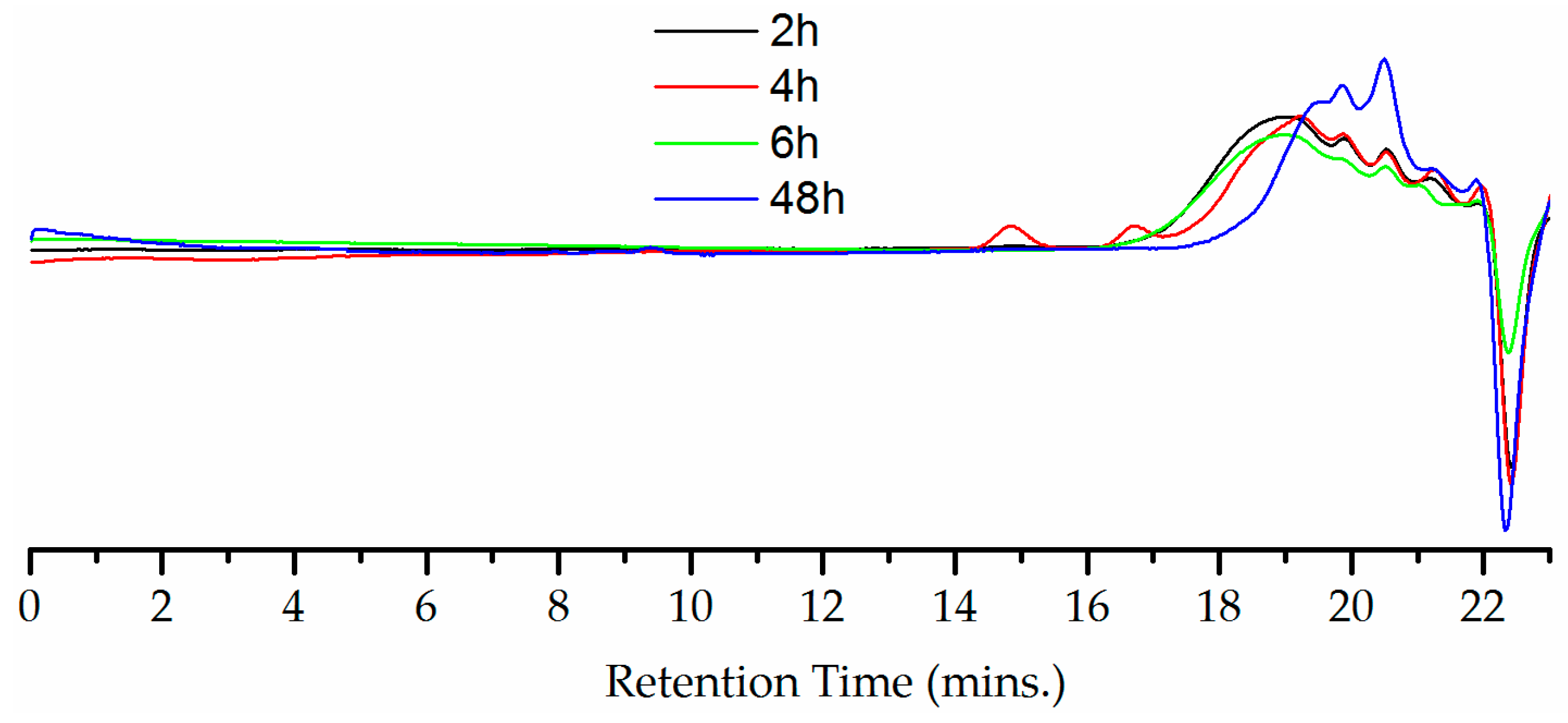
The film forming ability and mechanical properties of PIMs are important for membrane applications. The C-PIM produced via acid hydrolysis reported by Weng et al. [23] and the base hydrolysis reported by Jeon et al. [34] had the ability to form free standing isotropic films. Film casting of C-PIM from this work was attempted. But unfortunately, the C-PIM produced via nitrous acid route was unable to form a flexible film. It appears likely that the main reason for this was that the starting material PIM-CONH2 was unable to form a film, so the C-PIM carried on this disadvantage. Moreover, the GPC analysis of C-PIM in THF (Figure 9) revealed that molecular weight degradation had occurred during the nitrous acid reaction. It was observed that longer reaction times, were associated with decreases in molecular weight.
In our approach, another side reaction is the aromatic ring nitration. In the early discussion of the reaction solvent choice, aromatic nitration was observed for the reactions conducted in both THF and EtOAc. Aromatic nitration is caused by the nitronium ion (NO2+) in the reaction system. A typical example of nitration of polystyrene is reported by Philippides et al. where they used nitric acid and sulfuric acid mixture [45]. In the nitrous acid reaction, it seems that nitric acid (HNO3) was also inevitably produced, which in turn resulted in the formation of NO2+.
5. Conclusions
A novel and effective method to convert PIM-1 to C-PIM was studied. The C-PIM was successfully prepared from PIM-CONH2 using nitrous acid as a key reagent. The optimum reaction conditions were NaNO2, H2SO4 in MeCN at 90 °C for 6 h. The resulting C-PIM was soluble in various organic solvents such as THF, acetone, DMSO and NMP. The chemical structure of C-PIM was characterised by 1H NMR, 13C NMR, FTIR, elemental analysis, UV-Vis, TGA and TGA-MS. These measurements confirmed complete conversion of the amide groups to carboxylic acids. Thermal analysis by TGA showed that C-PIM has two degradation stages where the first stage was decarboxylation and second stage was backbone decomposition. For the first time, the decarboxylation product was confirmed by TGA-MS, in which a loss of m/z 44 amu (CO2) at the first degradation stage was found. TGA also showed that the thermal stability of C-PIM was less than PIM-CONH2 under both N2 and air atmosphere. GPC analysis showed continuous molecular weight degradation of C-PIM with extended reaction times. Aromatic nitration was observed as a side-reaction in EtOAc and THF. Further optimisation of this reaction is currently underway.
Acknowledgments
This work was financially supported by the Science for Technological Innovation (SfTI) seed fund, New Zealand National Science Challenges (NSC) and Product Accelerator program from the Ministry of Business, Innovation and Employment (MBIE). Wei-Hsuan Wu would also like to thank her parents for funding her study at The University of Auckland.
Author Contributions
Wei-Hsuan Wu performed the experiments and the data analysis; Paul Thomas conducted TGA-MS experiment. Paul Hume conceived and designed the synthetic experiments; Jianyong Jin is the project leader, supervised the study and provided scientific discussion. All the authors contributed to the writing of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pachauri, R.K.; Allen, M.R.; Barros, V.R.; Broome, J.; Cramer, W.; Christ, R.; Church, J.A.; Clarke, L.; Dahe, Q.; Dasgupta, P.; et al. Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2014. [Google Scholar]
- Sokolov, A.P.; Stone, P.H.; Forest, C.E.; Prinn, R.; Sarofim, M.C.; Webster, M.; Paltsev, S.; Schlosser, C.A.; Kicklighter, D.; Dutkiewicz, S.; et al. Probabilistic forecast for twenty-first-century climate based on uncertainties in emissions (without policy) and climate parameters. J. Clim. 2009, 22, 5175–5204. [Google Scholar] [CrossRef]
- Agiaye, E.O.; Othman, M. CO2 Capture & Utilization: Harnessing the CO2 Content in Natural Gas for Environmental and Economic Gains. In Proceedings of the SPE Nigeria Annual International Conference and Exhibition, Lagos, Nigeria, 4–6 August 2015; Society of Petroleum Engineers: Richardson, TX, USA, 2015. [Google Scholar]
- Chakravarti, S.; Gupta, A.; Hunek, B. Advanced Technology for the Capture of Carbon Dioxide from Flue Gases. In Proceedings of the First National Conference on Carbon Sequestration, Washington, DC, USA, 15–17 May 2001; Praxair, Inc.: Tonawanda, NY, USA, 2001; pp. 15–17. [Google Scholar]
- Pires, J.; Martins, F.; Alvim-Ferraz, M.; Simões, M. Recent developments on carbon capture and storage: An overview. Chem. Eng. Res. Des. 2011, 89, 1446–1460. [Google Scholar] [CrossRef]
- Riemer, P.W. The Capture of Carbon Dioxide from Fossil Fuel Fired Power Stations; IEA Greenhouse Gas R & D Programme: Cheltenham, UK, 1993. [Google Scholar]
- Pfaff, I.; Kather, A. Comparative thermodynamic analysis and integration issues of CCS steam power plants based on oxy-combustion with cryogenic or membrane based air separation. Energy Procedia 2009, 1, 495–502. [Google Scholar] [CrossRef]
- Scholes, C.A.; Kentish, S.E.; Stevens, G.W. Effects of minor components in carbon dioxide capture using polymeric gas separation membranes. Sep. Purif. Rev. 2009, 38, 1–44. [Google Scholar] [CrossRef]
- Budd, P.M.; Ghanem, B.S.; Makhseed, S.; McKeown, N.B.; Msayib, K.J.; Tattershall, C.E. Polymers of intrinsic microporosity (PIMs): Robust, solution-processable, organic nanoporous materials. Chem. Commun. 2004, 230–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jin, J.; Cooney, R.; Fu, Q.; Qiao, G.G.; Thomas, S.; Merkel, T.C. Synthesis of perfectly alternating copolymers for polymers of intrinsic microporosity. Polym. Chem. 2015, 6, 5003–5008. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, J.; Cooney, R.; Zhang, S. Synthesis of polymers of intrinsic microporosity using an ab-type monomer. Polymer 2015, 57, 45–50. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, J.; Cooney, R.; Zhang, S. Fluoride-mediated polycondensation for the synthesis of polymers of intrinsic microporosity. Polymer 2015, 76, 168–172. [Google Scholar] [CrossRef]
- Zhang, J.; Kang, H.; Martin, J.; Zhang, S.; Thomas, S.; Merkel, T.C.; Jin, J. The enhancement of chain rigidity and gas transport performance of polymers of intrinsic microporosity via intramolecular locking of the spiro-carbon. Chem. Commun. 2016, 52, 6553–6556. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.R.; Jin, J.; Freeman, B.D.; Paul, D.R. Physical aging, CO2 sorption and plasticization in thin films of polymer with intrinsic microporosity (PIM-1). J. Membr. Sci. 2017, 537, 362–371. [Google Scholar] [CrossRef]
- Starannikova, L.; Belov, N.; Shantarovich, V.; Zhang, J.; Jin, J.; Yampolskii, Y. Effective increase in permeability and free volume of PIM copolymers containing ethanoanthracene unit and comparison between the alternating and random copolymers. J. Membr. Sci. 2018, 548, 593–597. [Google Scholar] [CrossRef]
- Du, N.; Park, H.B.; Robertson, G.P.; Dal-Cin, M.M.; Visser, T.; Scoles, L.; Guiver, M.D. Polymer nanosieve membranes for CO2-capture applications. Nat. Mater. 2011, 10, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Robertson, G.P.; Dal-Cin, M.M.; Scoles, L.; Guiver, M.D. Polymers of intrinsic microporosity (PIMs) substituted with methyl tetrazole. Polymer 2012, 53, 4367–4372. [Google Scholar] [CrossRef]
- Patel, H.A.; Yavuz, C.T. Noninvasive functionalization of polymers of intrinsic microporosity for enhanced CO2 capture. Chem. Commun. 2012, 48, 9989–9991. [Google Scholar] [CrossRef] [PubMed]
- Swaidan, R.; Ghanem, B.S.; Litwiller, E.; Pinnau, I. Pure-and mixed-gas CO2/CH4 separation properties of PIM-1 and an amidoxime-functionalized PIM-1. J. Membr. Sci. 2014, 457, 95–102. [Google Scholar] [CrossRef]
- Mason, C.R.; Maynard-Atem, L.; Al-Harbi, N.M.; Budd, P.M.; Bernardo, P.; Bazzarelli, F.; Clarizia, G.; Jansen, J.C. Polymer of intrinsic microporosity incorporating thioamide functionality: Preparation and gas transport properties. Macromolecules 2011, 44, 6471–6479. [Google Scholar] [CrossRef]
- Mason, C.R.; Maynard-Atem, L.; Heard, K.W.; Satilmis, B.; Budd, P.M.; Friess, K.; Lanč, M.; Bernardo, P.; Clarizia, G.; Jansen, J.C. Enhancement of CO2 affinity in a polymer of intrinsic microporosity by amine modification. Macromolecules 2014, 47, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Satilmis, B.; Alnajrani, M.N.; Budd, P.M. Hydroxyalkylaminoalkylamide PIMs: Selective adsorption by ethanolamine-and diethanolamine-modified PIM-1. Macromolecules 2015, 48, 5663–5669. [Google Scholar] [CrossRef]
- Weng, X.; Baez, J.E.; Khiterer, M.; Hoe, M.Y.; Bao, Z.; Shea, K.J. Chiral polymers of intrinsic microporosity: Selective membrane permeation of enantiomers. Angew. Chem. Int. Ed. 2015, 54, 11214–11218. [Google Scholar] [CrossRef] [PubMed]
- Du, N.; Robertson, G.P.; Song, J.; Pinnau, I.; Guiver, M.D. High-performance carboxylated polymers of intrinsic microporosity (PIMs) with tunable gas transport properties. Macromolecules 2009, 42, 6038–6043. [Google Scholar] [CrossRef] [Green Version]
- Du, N.; Dal-Cin, M.M.; Robertson, G.P.; Guiver, M.D. Decarboxylation-induced cross-linking of polymers of intrinsic microporosity (PIMs) for membrane gas separation. Macromolecules 2012, 45, 5134–5139. [Google Scholar] [CrossRef]
- Yong, W.F.; Chung, T.-S. Miscible blends of carboxylated polymers of intrinsic microporosity (cPIM-1) and Matrimid. Polymer 2015, 59, 290–297. [Google Scholar] [CrossRef]
- Yong, W.F.; Li, F.Y.; Chung, T.S.; Tong, Y.W. Molecular interaction, gas transport properties and plasticization behavior of cPIM-1/Torlon blend membranes. J. Membr. Sci. 2014, 462, 119–130. [Google Scholar] [CrossRef]
- Salehian, P.; Yong, W.F.; Chung, T.-S. Development of high performance carboxylated PIM-1/P84 blend membranes for pervaporation dehydration of isopropanol and CO2/CH4 separation. J. Membr. Sci. 2016, 518, 110–119. [Google Scholar] [CrossRef]
- Liao, K.-S.; Lai, J.-Y.; Chung, T.-S. Metal ion modified PIM-1 and its application for propylene/propane separation. J. Membr. Sci. 2016, 515, 36–44. [Google Scholar] [CrossRef]
- Zhao, H.; Xie, Q.; Ding, X.; Chen, J.; Hua, M.; Tan, X.; Zhang, Y. High performance post-modified polymers of intrinsic microporosity (PIM-1) membranes based on multivalent metal ions for gas separation. J. Membr. Sci. 2016, 514, 305–312. [Google Scholar] [CrossRef]
- Wind, J.D.; Staudt-Bickel, C.; Paul, D.R.; Koros, W.J. The effects of crosslinking chemistry on CO2 plasticization of polyimide gas separation membranes. Ind. Eng. Chem. Res. 2002, 41, 6139–6148. [Google Scholar] [CrossRef]
- Satilmis, B.; Budd, P.M. Base-catalysed hydrolysis of PIM-1: Amide versus carboxylate formation. RSC Adv. 2014, 4, 52189–52198. [Google Scholar] [CrossRef]
- Santoso, B.; Yanaranop, P.; Kang, H.; Leung, I.K.; Jin, J. A Critical Update on the Synthesis of Carboxylated Polymers of Intrinsic Microporosity (C-PIMs). Macromolecules 2017, 50, 3043–3050. [Google Scholar] [CrossRef]
- Jeon, J.W.; Kim, D.-G.; Sohn, E.-H.; Yoo, Y.; Kim, Y.S.; Kim, B.G.; Lee, J.-C. Highly Carboxylate-Functionalized Polymers of Intrinsic Microporosity for CO2-Selective Polymer Membranes. Macromolecules 2017, 50, 8019–8027. [Google Scholar] [CrossRef]
- McMurry, J. Organic Chemistry, 7th ed.; Thomson Brooks Cole: Pacific Grove, CA, USA, 2008. [Google Scholar]
- Yanaranop, P.; Santoso, B.; Etzion, R.; Jin, J. Facile conversion of nitrile to amide on polymers of intrinsic microporosity (PIM-1). Polymer 2016, 98, 244–251. [Google Scholar] [CrossRef]
- Satilmis, B.; Budd, P.M.; Uyar, T. Systematic hydrolysis of PIM-1 and electrospinning of hydrolyzed PIM-1 ultrafine fibers for an efficient removal of dye from water. React. Funct. Polym. 2017, 121, 67–75. [Google Scholar] [CrossRef]
- Olah, G.A.; Olah, J.A. Reactions of Amides and Sulfonamides with Nitrosonium Salts. J. Org. Chem. 1965, 30, 2386–2387. [Google Scholar] [CrossRef]
- Bhole, Y.S. Investigations on Gas Permeation and Related Physical Properties of structurally Architectured Aromatic Polymers (Polyphenylene Oxides and Polyarylates), Polyarlate-Clay Nanocomposites and Poly (Ionic Liquid). Ph.D. Thesis, CSIR-National Chemical Laboratory, Pune, India, 2007. [Google Scholar]
- Sakaguchi, T.; Yumoto, K.; Shiotsuki, M.; Sanda, F.; Yoshikawa, M.; Masuda, T. Synthesis of poly (diphenylacetylene) membranes by desilylation of various precursor polymers and their properties. Macromolecules 2005, 38, 2704–2709. [Google Scholar] [CrossRef]
- Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. A new, one-step, effective protocol for the iodination of aromatic and heterocyclic compounds via aprotic diazotization of amines. Synthesis 2007, 2007, 81–84. [Google Scholar] [CrossRef]
- Filimonov, V.D.; Trusova, M.; Postnikov, P.; Krasnokutskaya, E.A.; Lee, Y.M.; Hwang, H.Y.; Kim, H.; Chi, K.-W. Unusually stable, versatile, and pure arenediazonium tosylates: Their preparation, structures, and synthetic applicability. Org. Lett. 2008, 10, 3961–3964. [Google Scholar] [CrossRef] [PubMed]
- Borikar, S.P.; Paul, V. N-Nitrosation of Secondary Amines Using p-TSA-NaNO2 as a Novel Nitrosating Agent Under Mild Conditions. Synth. Commun. 2010, 40, 654–660. [Google Scholar] [CrossRef]
- Søndergaard, R.R.; Norrman, K.; Krebs, F.C. Low-temperature side-chain cleavage and decarboxylation of polythiophene esters by acid catalysis. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1127–1132. [Google Scholar] [CrossRef]
- Philippides, A.; Budd, P.M.; Price, C.; Cuncliffe, A.V. The nitration of polystyrene. Polymer 1993, 34, 3509–3513. [Google Scholar] [CrossRef]
Scheme 1.
(a) Two-step synthesis of C-PIM via PIM-CONH2. (b) Proposed reaction mechanism for the conversion of amides to carboxylic acid using nitrous acid [38].
Scheme 1.
(a) Two-step synthesis of C-PIM via PIM-CONH2. (b) Proposed reaction mechanism for the conversion of amides to carboxylic acid using nitrous acid [38].
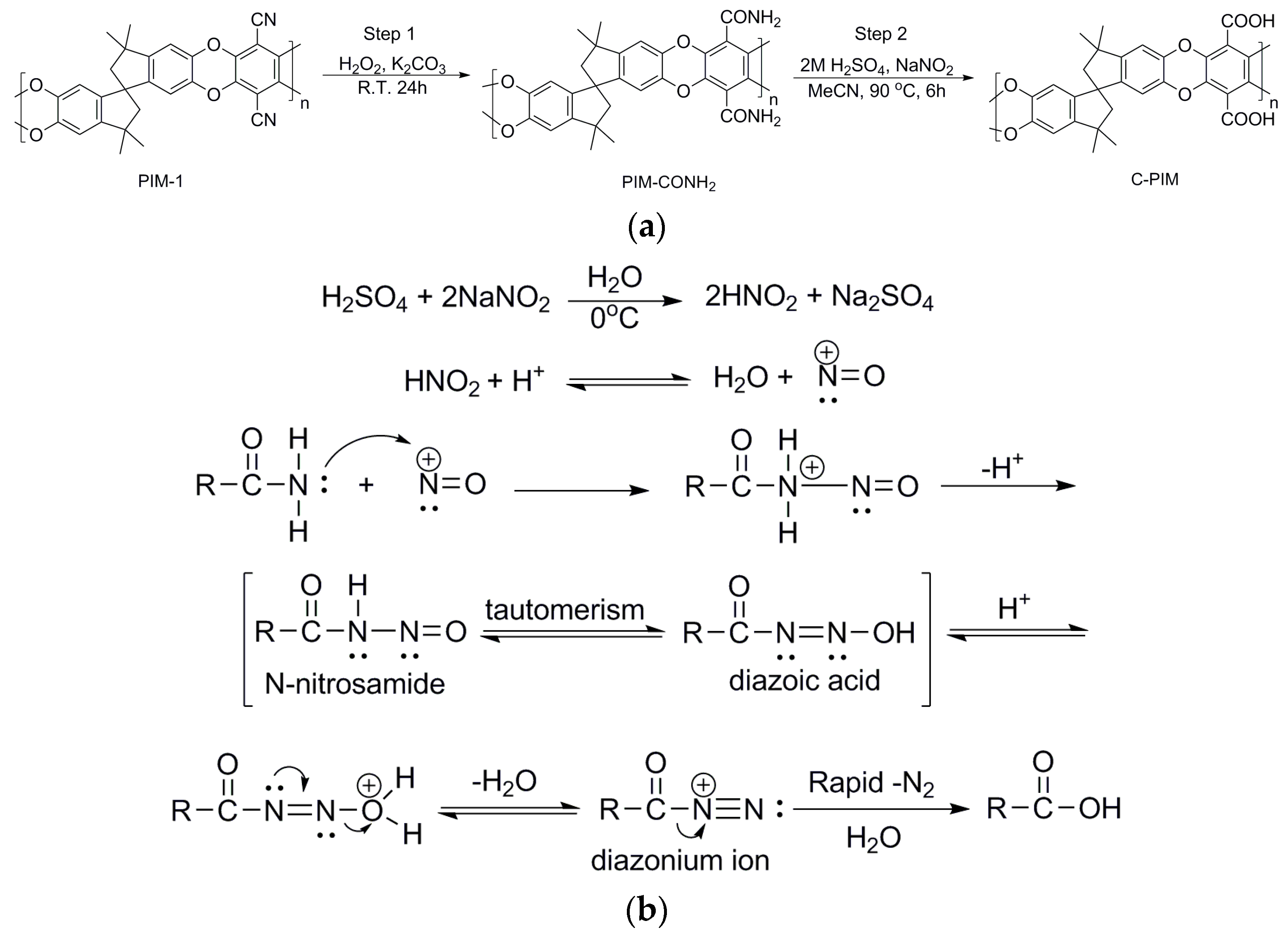
Figure 1.
FTIR spectra of the reaction products in three solvents (EtOAc, THF and MeCN) with NaNO2 and 2M H2SO4 for 6 h.
Figure 1.
FTIR spectra of the reaction products in three solvents (EtOAc, THF and MeCN) with NaNO2 and 2M H2SO4 for 6 h.
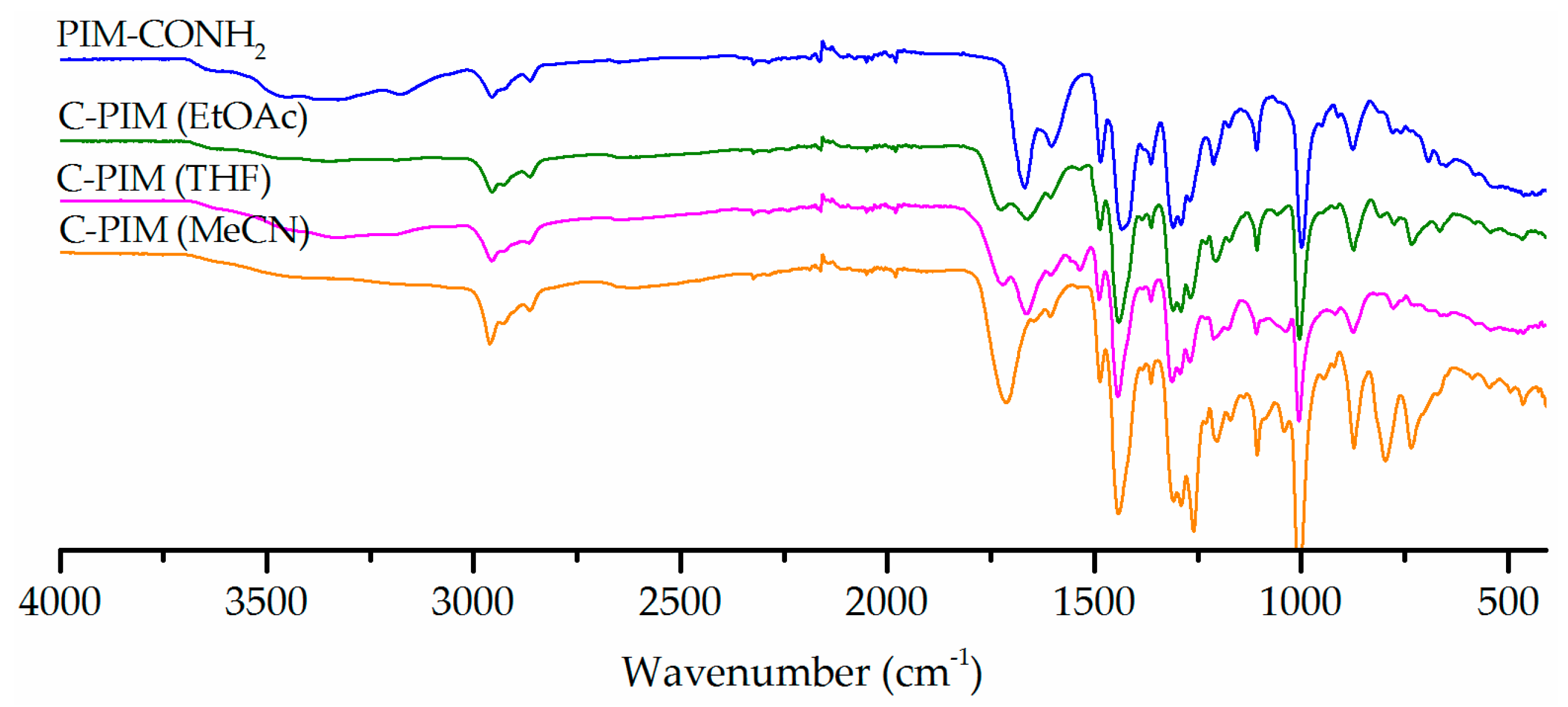
Figure 2.
FTIR spectra comparison of the reaction products in three acid systems (acetic acid, PTSA, H2SO4) with NaNO2 in MeCN at 90 °C for 6 h.
Figure 2.
FTIR spectra comparison of the reaction products in three acid systems (acetic acid, PTSA, H2SO4) with NaNO2 in MeCN at 90 °C for 6 h.
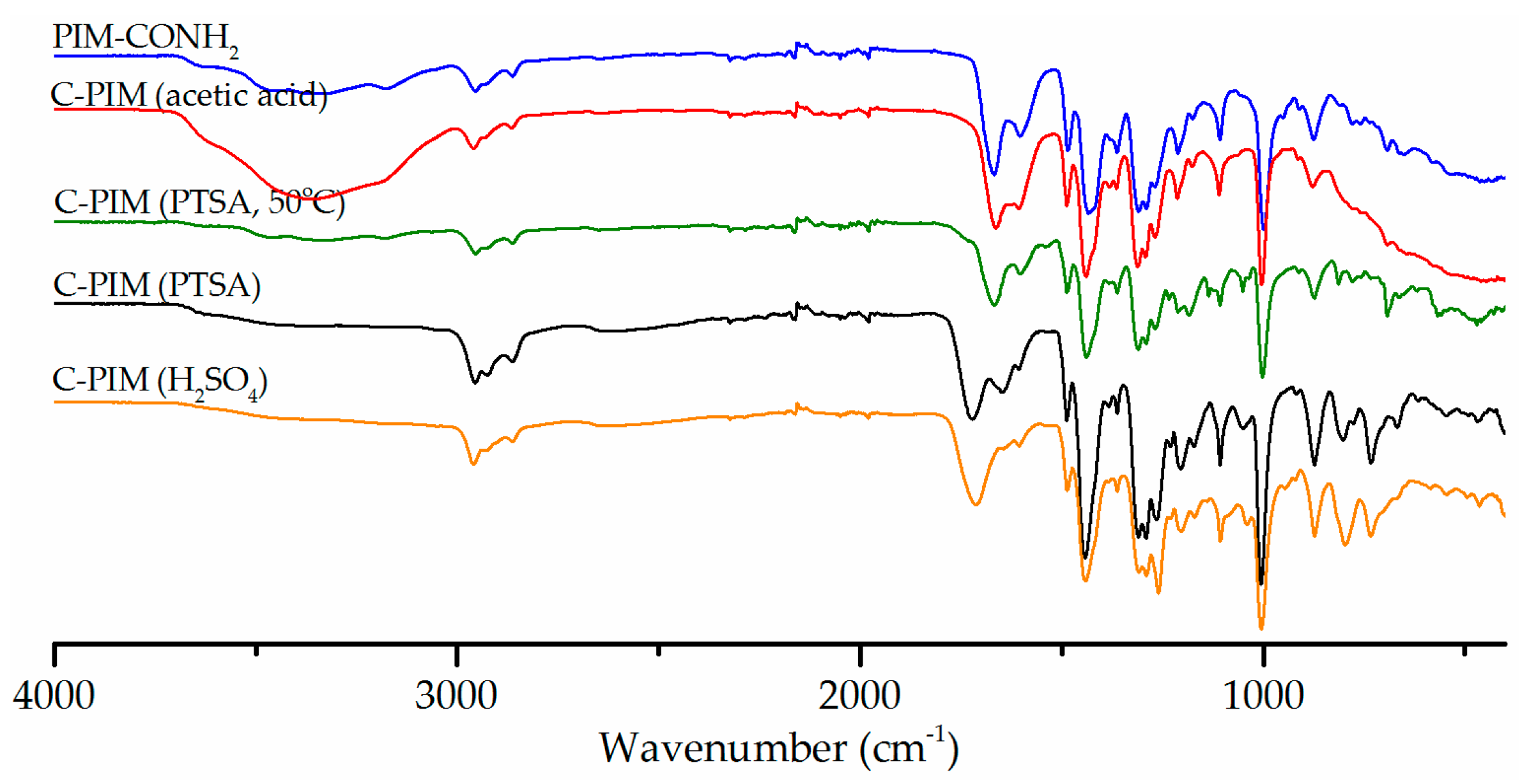
Figure 3.
FTIR spectra of reaction of PIM-CONH2 with NaNO2 and H2SO4 in MeCN at 90 °C at reaction times from 2–48 h.
Figure 3.
FTIR spectra of reaction of PIM-CONH2 with NaNO2 and H2SO4 in MeCN at 90 °C at reaction times from 2–48 h.
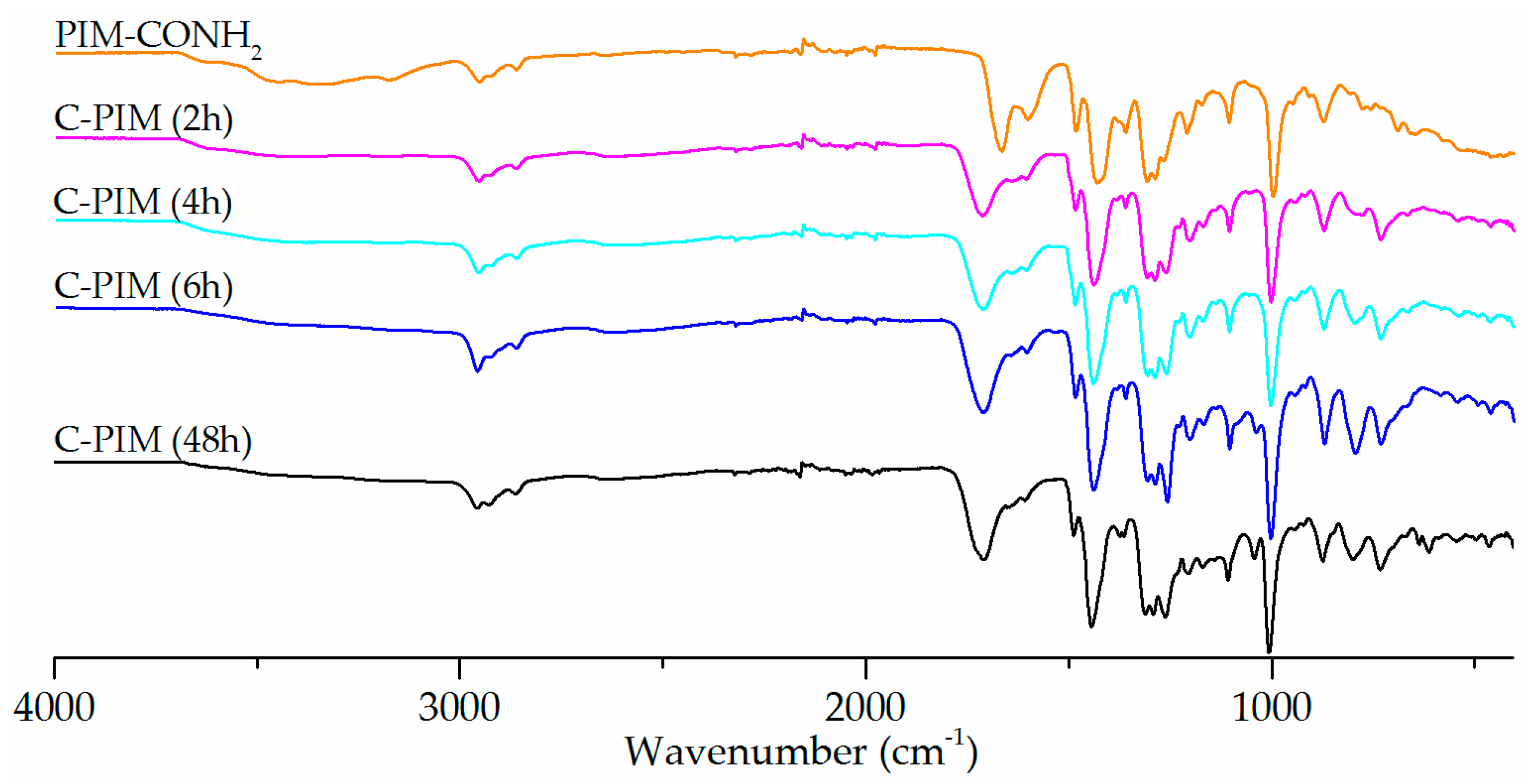
Figure 4.
1H NMR spectra of C-PIM at reaction times from 2 h to 48 h.
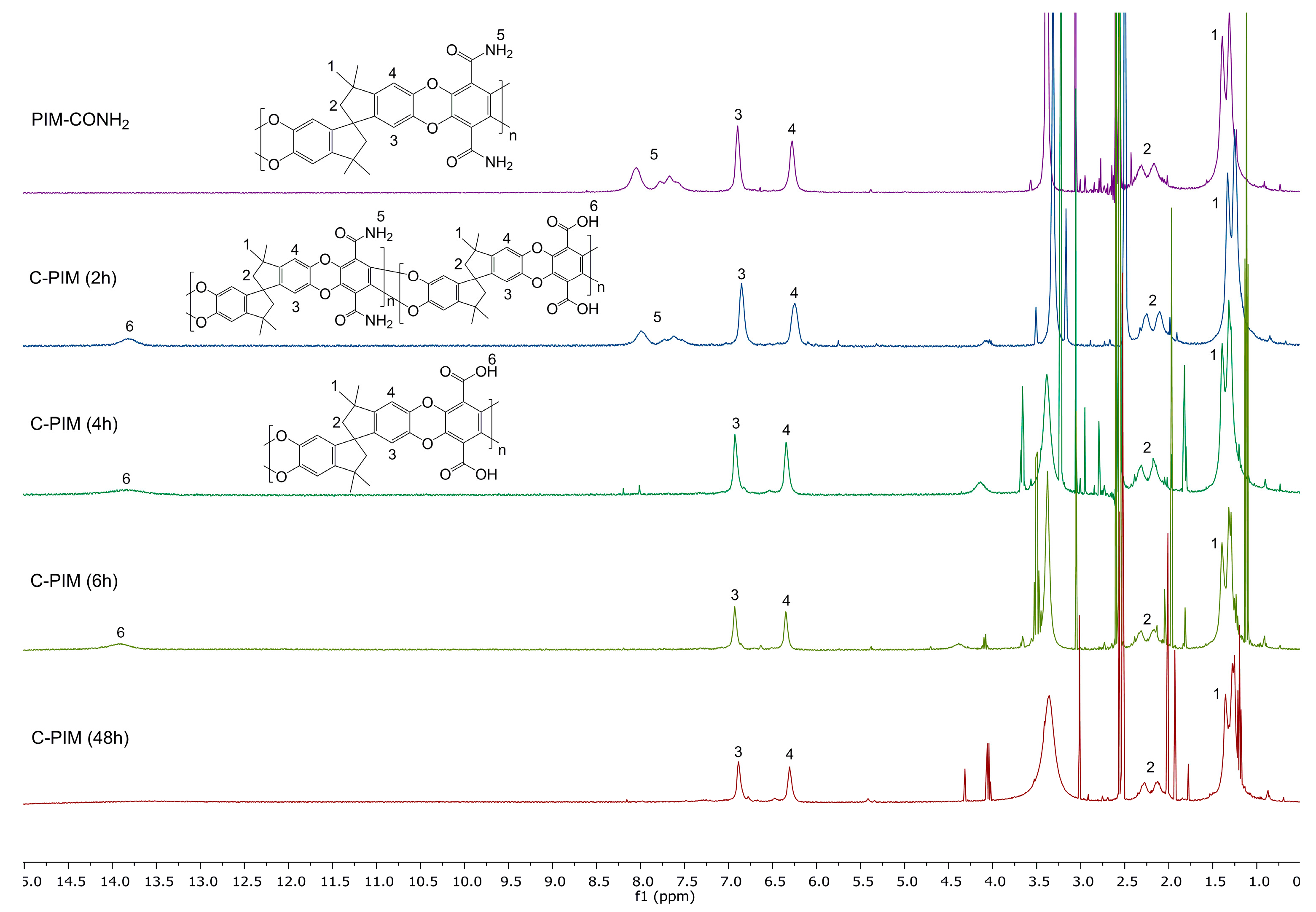
Figure 5.
UV-Vis spectra of PIM-CONH2 in DMSO and C-PIM in THF.
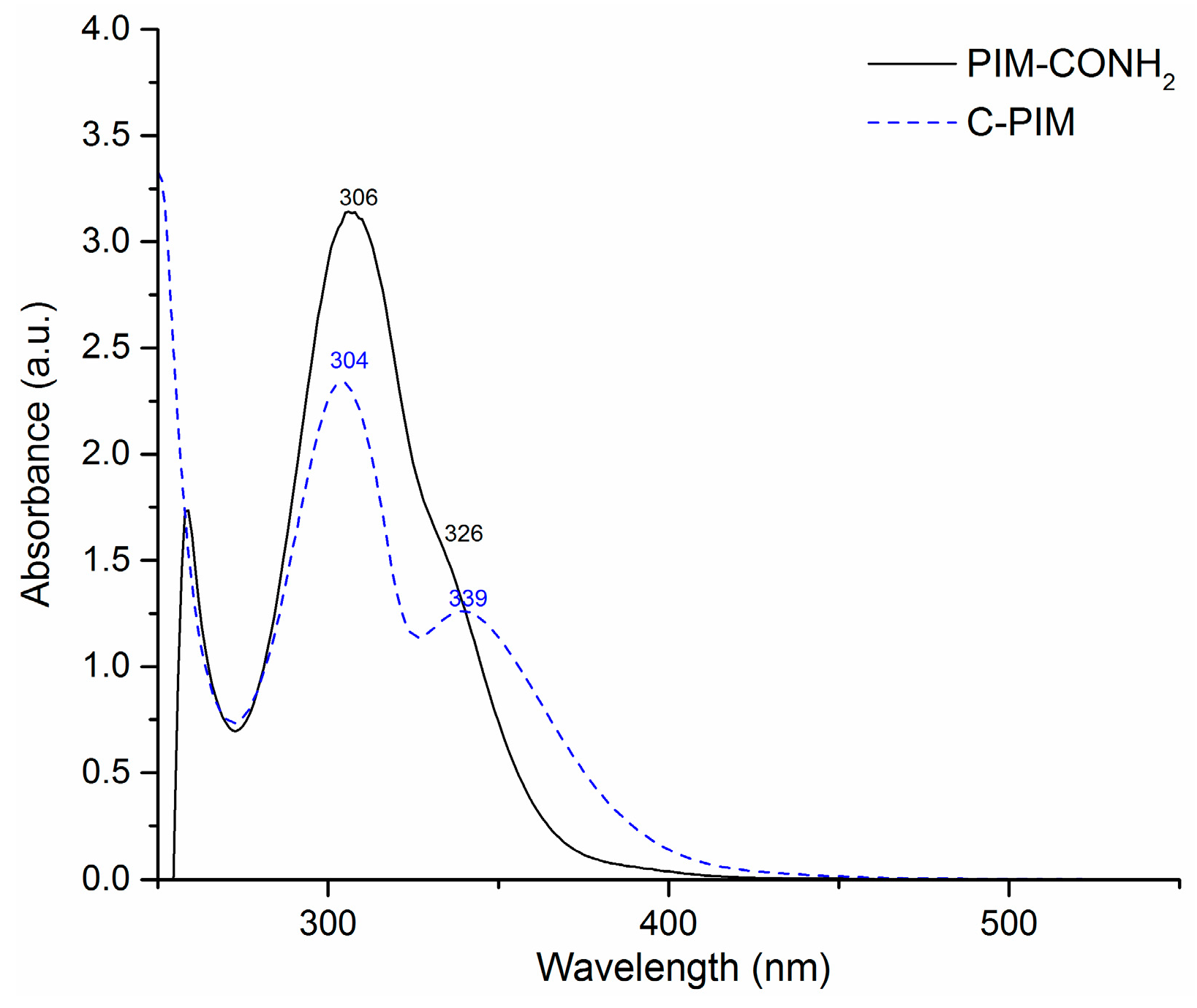
Figure 6.
(a) Thermogravimetric analysis of PIM-CONH2 and C-PIM at 10 °C/min under N2 atmosphere. The left axis marks weight loss change in % and the right axis marks mass loss rate in wt. %/°C; (b) Thermogravimetric analysis of PIM-CONH2 and C-PIM at 10 °C/min under air atmosphere.
Figure 6.
(a) Thermogravimetric analysis of PIM-CONH2 and C-PIM at 10 °C/min under N2 atmosphere. The left axis marks weight loss change in % and the right axis marks mass loss rate in wt. %/°C; (b) Thermogravimetric analysis of PIM-CONH2 and C-PIM at 10 °C/min under air atmosphere.
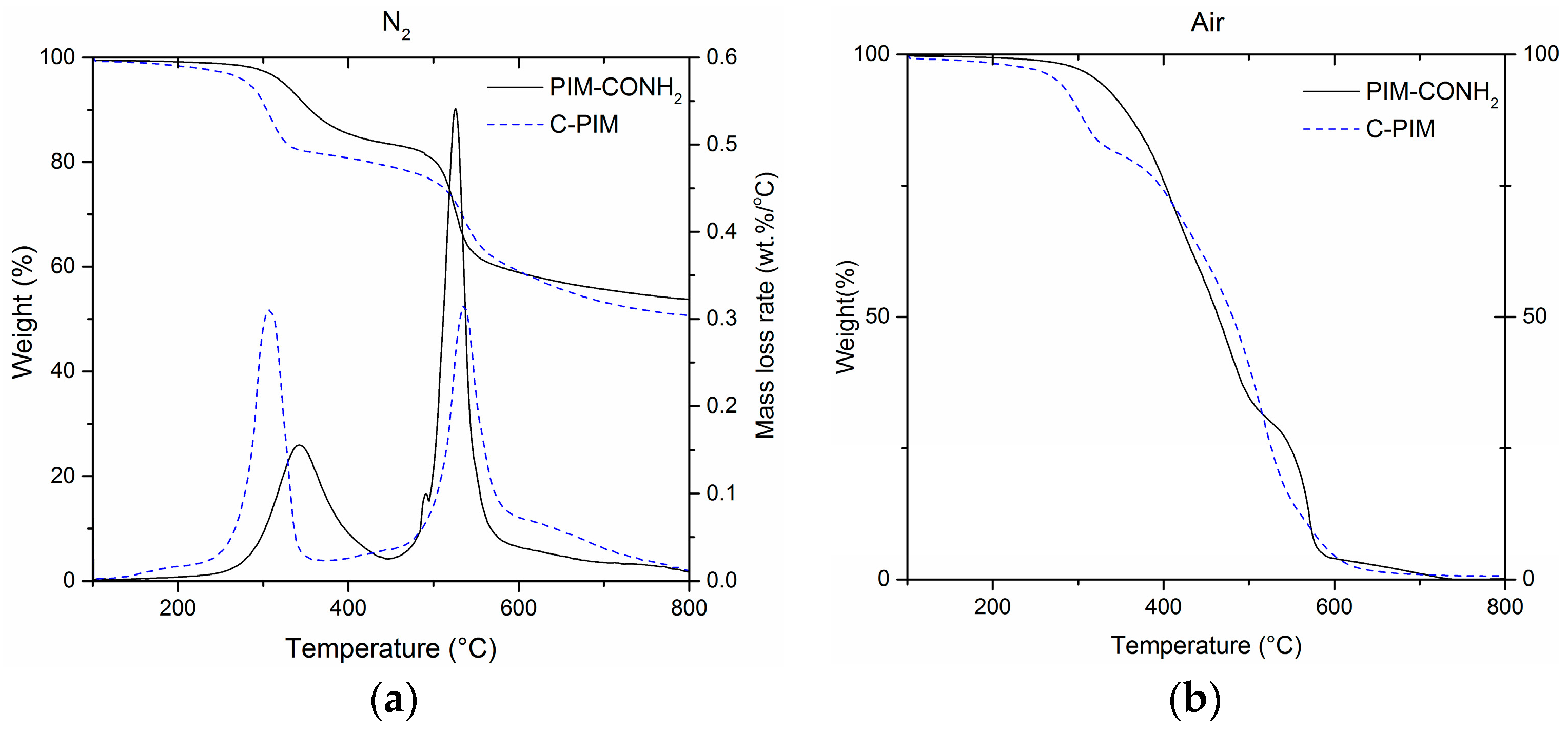
Figure 7.
Thermogravimetric analysis combined with mass spectroscopy (TGA-MS). (a) DTG curve of PIM-CONH2 overlaid with MS trace of m/z 43 amu and (b) DTG curve of C-PIM overlaid with MS trace of m/z 44 amu.
Figure 7.
Thermogravimetric analysis combined with mass spectroscopy (TGA-MS). (a) DTG curve of PIM-CONH2 overlaid with MS trace of m/z 43 amu and (b) DTG curve of C-PIM overlaid with MS trace of m/z 44 amu.
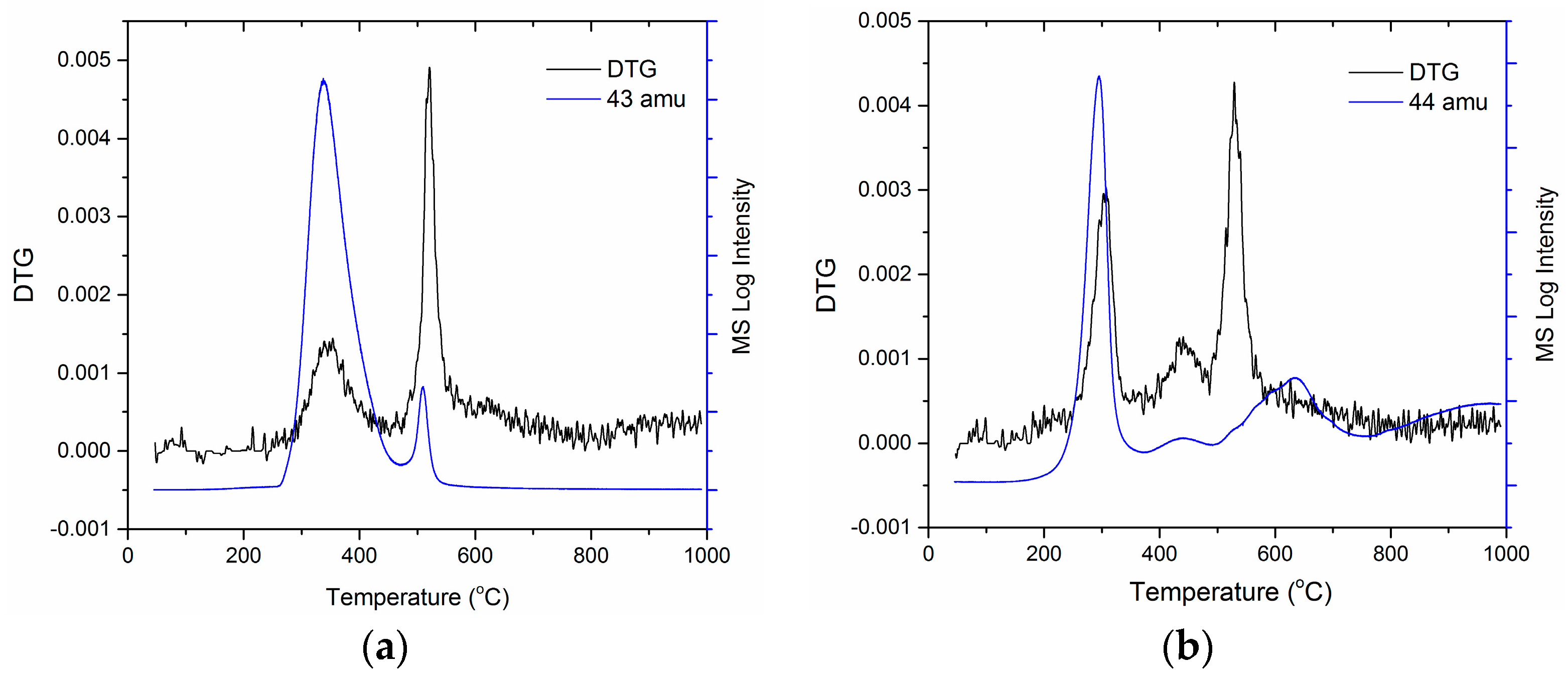
Figure 8.
FTIR spectra comparison of the C-PIM samples before and after decarboxylation via isothermal pyrolysis at 350 °C for 60 min.
Figure 8.
FTIR spectra comparison of the C-PIM samples before and after decarboxylation via isothermal pyrolysis at 350 °C for 60 min.
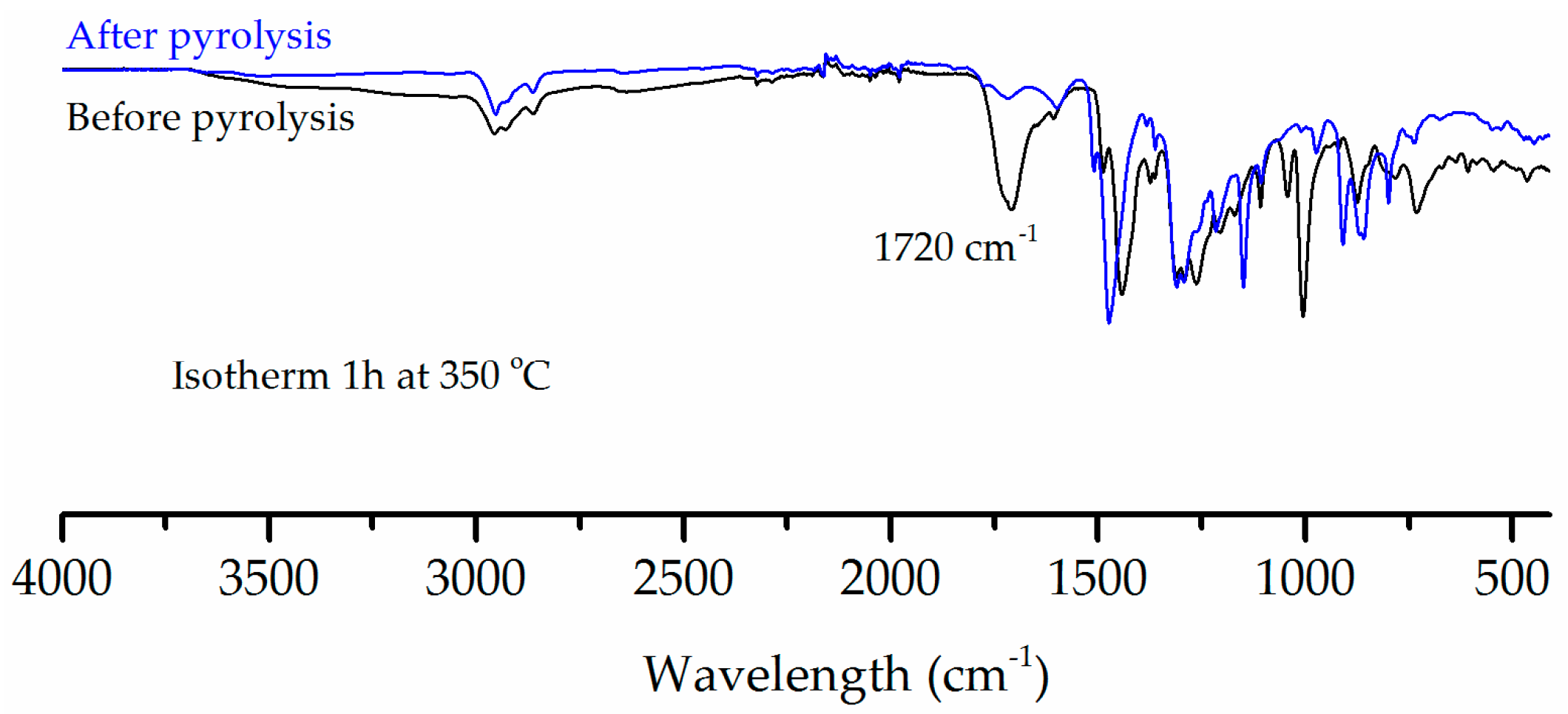
Figure 9.
GPC chromatograph showing molecular weight change of C-PIM at different reaction times from 2 h to 48 h using THF as eluent.
Figure 9.
GPC chromatograph showing molecular weight change of C-PIM at different reaction times from 2 h to 48 h using THF as eluent.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of C-PIM synthesis methods.
| Methods | Base Hydrolysis | Acid Hydrolysis | This Work |
|---|---|---|---|
| Starting Material | PIM-1 | PIM-1 | PIM-CONH2 |
| Reaction Conditions | 20% NaOH at 120 °C, 5 h | H2SO4, H2O and Acetic acid at 105 °C, 48 h | NaNO2, H2SO4 and MeCN at 90 °C, 6 h |
Table 2.
Elemental analysis results for PIM-1, PIM-CONH2, and C-PIM.
| Samples | C% | H% | N% | O% * |
|---|---|---|---|---|
| PIM-1 | 74.65 | 4.47 | 6.19 | 14.69 |
| PIM-CONH2 | 65.53 | 5.33 | 5.24 | 23.90 |
| C-PIM | 63.55 | 4.80 | 0.91 | 30.74 |
* O% = 100–(C% + H% + N%).
Table 3.
Solubility comparison of PIM-1, PIM-CONH2 and C-PIM in organic solvents.
| Solvent | PIM-1 | PIM-CONH2 | C-PIM Obtained in This Work |
|---|---|---|---|
| DCM | soluble | insoluble | insoluble |
| EtOAc | insoluble | insoluble | partially soluble |
| CHCl3 | soluble | insoluble | insoluble |
| THF | soluble | partially soluble | soluble |
| DMF | insoluble | soluble | soluble |
| DMAc | insoluble | soluble | soluble |
| NMP | insoluble | soluble | soluble |
| MeOH | insoluble | partially soluble | partially soluble |
| MeCN | insoluble | insoluble | partially soluble |
| EtOH | insoluble | insoluble | partially soluble |
| Acetone | insoluble | insoluble | soluble |
| DMSO | insoluble | soluble | soluble |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wu, W.-H.; Thomas, P.; Hume, P.; Jin, J. Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid. Membranes 2018, 8, 20. https://doi.org/10.3390/membranes8020020
AMA Style
Wu W-H, Thomas P, Hume P, Jin J. Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid. Membranes. 2018; 8(2):20. https://doi.org/10.3390/membranes8020020
Chicago/Turabian StyleWu, Wei-Hsuan, Paul Thomas, Paul Hume, and Jianyong Jin. 2018. "Effective Conversion of Amide to Carboxylic Acid on Polymers of Intrinsic Microporosity (PIM-1) with Nitrous Acid" Membranes 8, no. 2: 20. https://doi.org/10.3390/membranes8020020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.