Molecular Mechanisms of Retinoid Receptors in Diabetes-Induced Cardiac Remodeling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
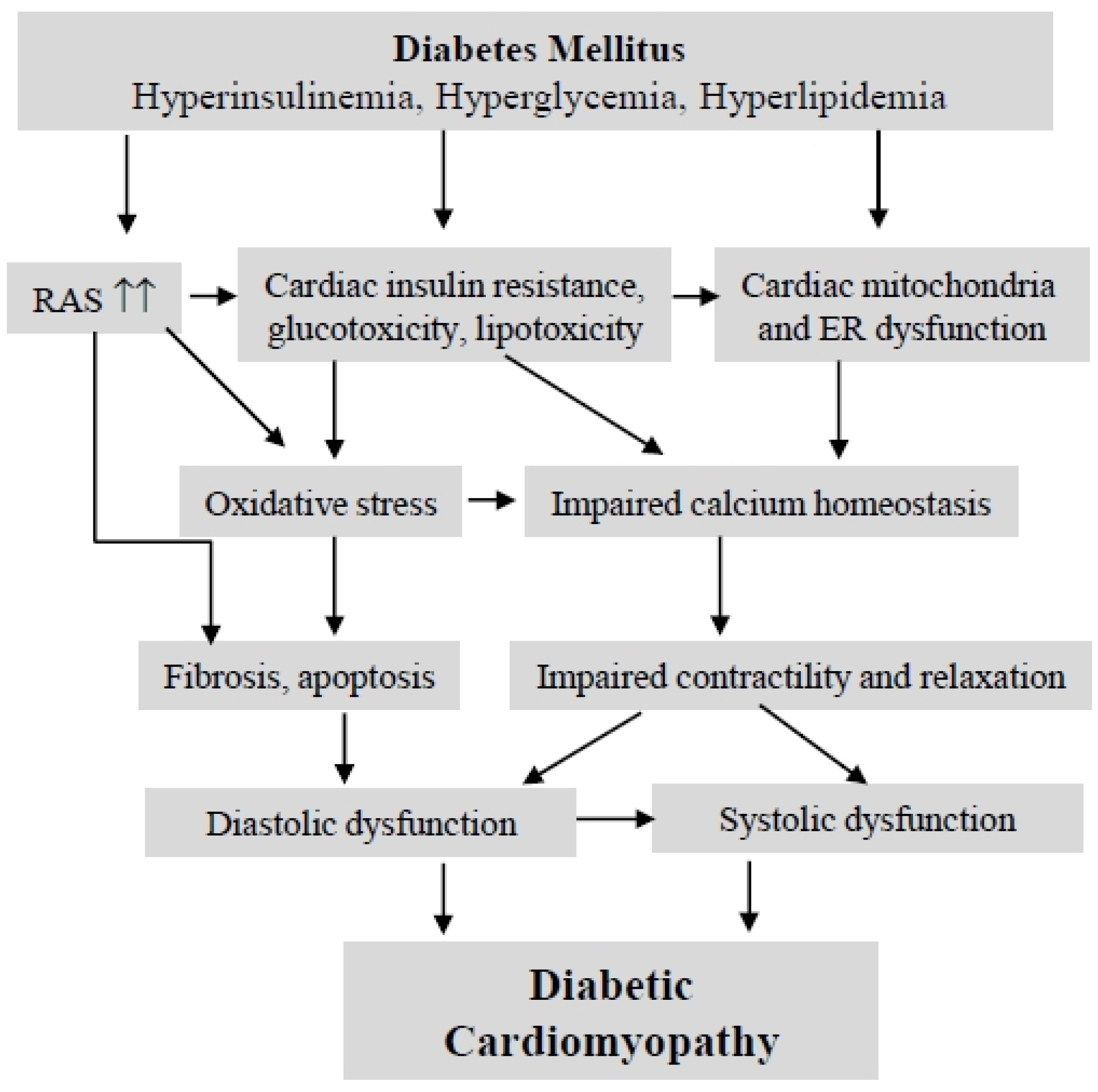
2. Diabetic Cardiomyopathy

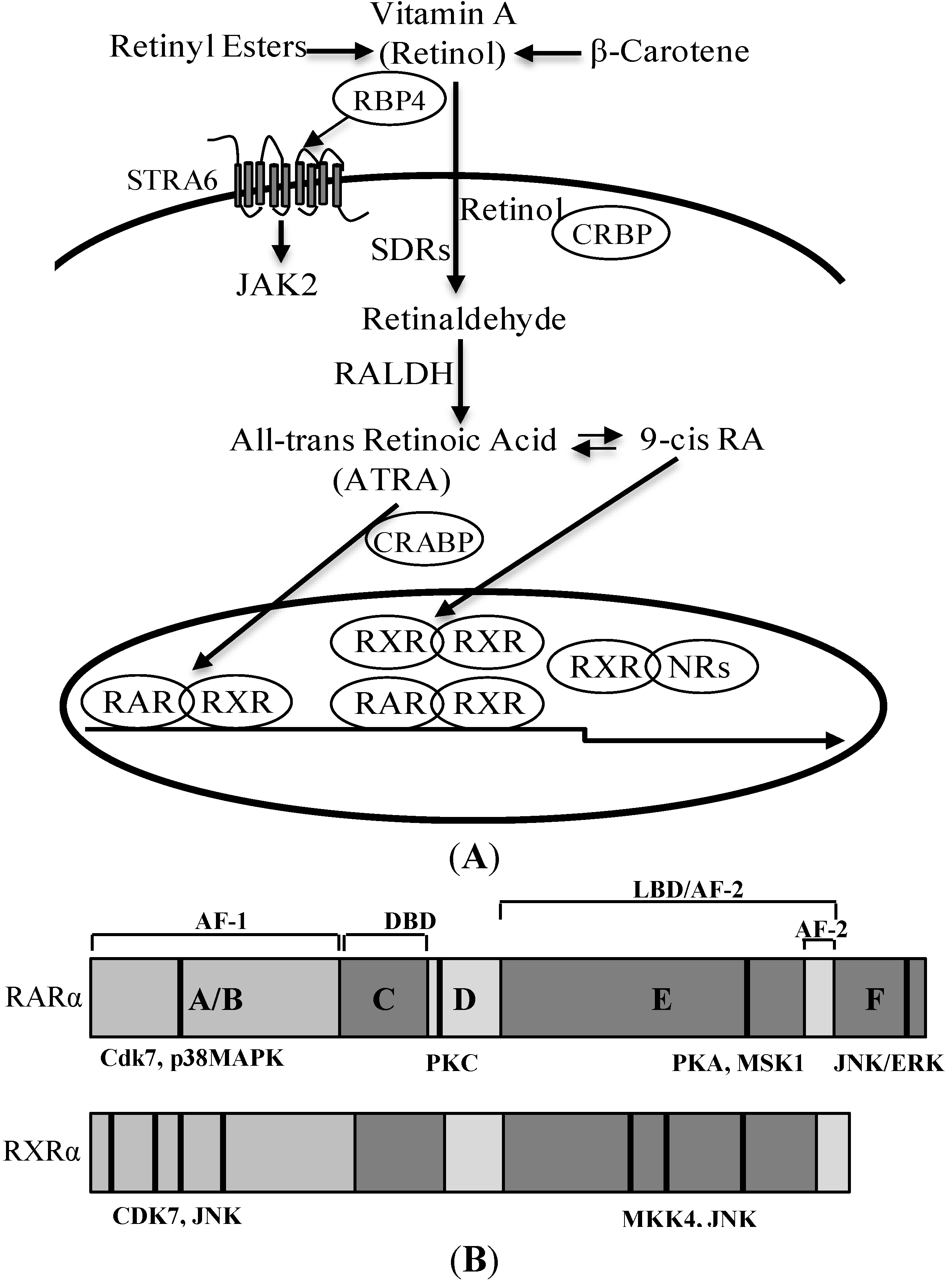
3. Retinoic Acid and Retinoid Receptor Signaling

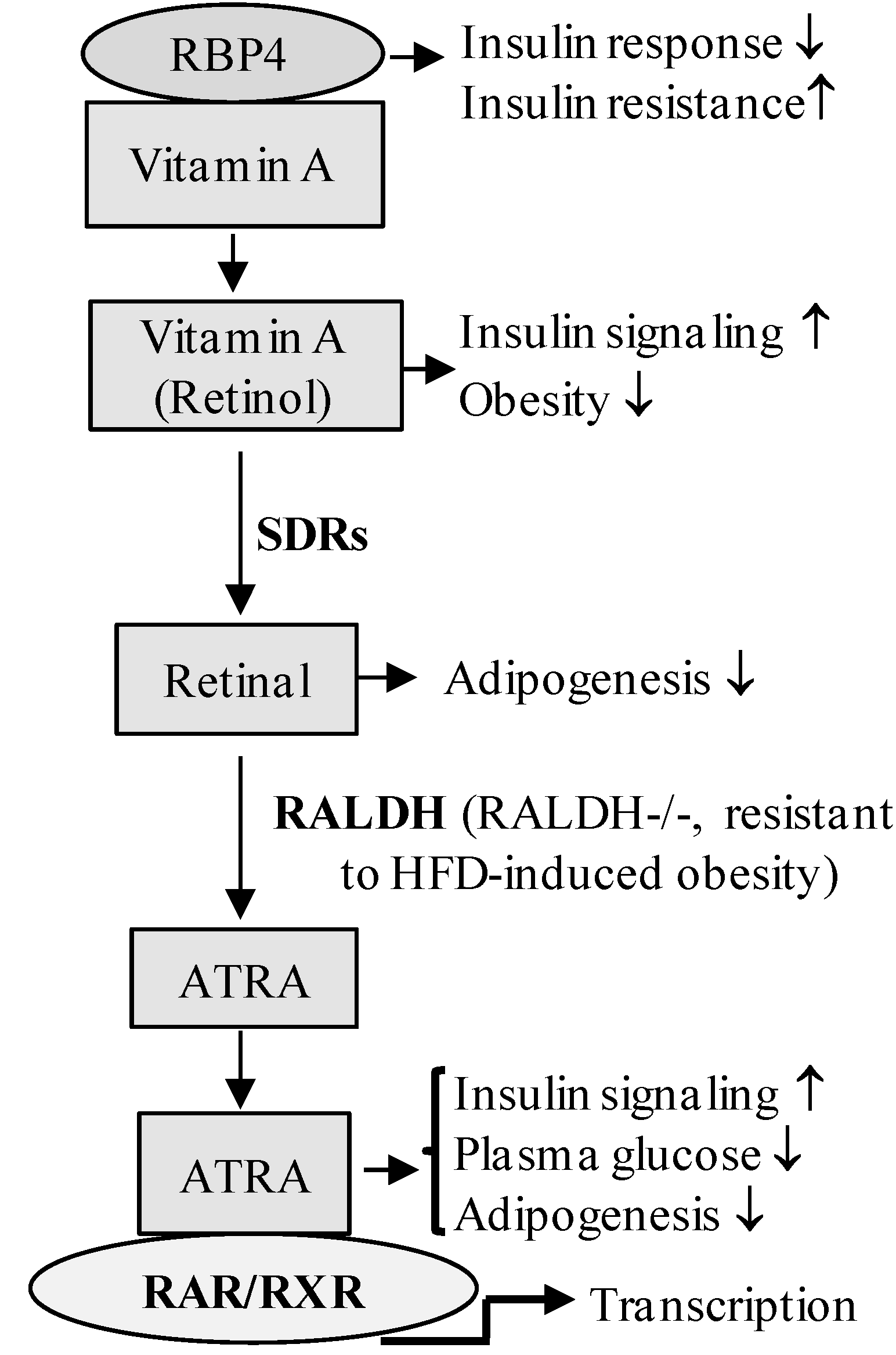
4. Retinoic Acid Signaling Is Involved in the Development of Diabetes

5. Retinoid Receptor-Mediated Signaling and Diabetic Cardiomyopathy
5.1. Retinoid Receptor-Mediated Signaling in Cardiac Remodeling
5.2. Mechanisms Involved in Retinoid Receptor Signaling Regulated Diabetic Cardiomyopathy
5.2.1. Activation of RAR/RXR Signaling Improves Cardiac Insulin Signaling and Glucose/Lipid Metabolism
5.2.2. RAR/RXR Signaling in the Regulation of Cardiac NF-κB-Mediated Inflammatory Responses
5.2.3. RAR/RXR Signaling in the Regulation of the Renin-Angiotensin System
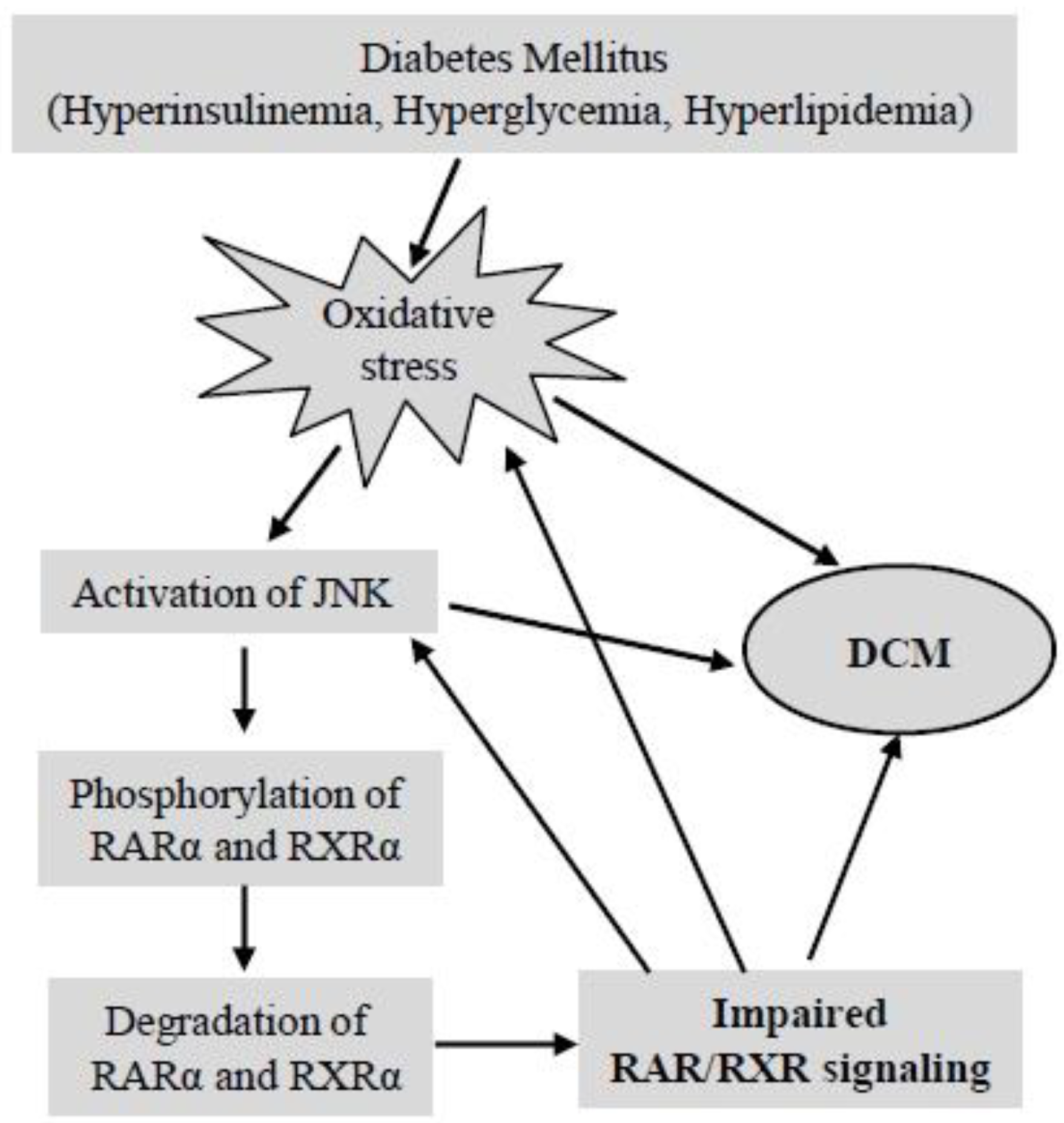
5.2.4. Does Impaired RAR/RXR Signaling Contribute to the Development of DCM?
6. Conclusions

Acknowledgments
Conflicts of Interest
References
- Battiprolu, P.K.; Gillette, T.G.; Wang, Z.V.; Lavandero, S.; Hill, J.A. Diabetic Cardiomyopathy: Mechanisms, Therapeutic Targets. Drug Discov. Today Dis. Mech. 2010, 7, e135–e143. [Google Scholar] [CrossRef]
- Murarka, S.; Movahed, M.R. Diabetic cardiomyopathy. J. Card. Fail. 2010, 16, 971–979. [Google Scholar] [CrossRef]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-κB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc. Res. 2010, 85, 473–483. [Google Scholar] [CrossRef]
- Westermann, D.; Rutschow, S.; van Linthout, S.; Linderer, A.; Bucker-Gartner, C.; Sobirey, M.; Riad, A.; Pauschinger, M.; Schultheiss, H.P.; Tschope, C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia 2006, 49, 2507–2513. [Google Scholar] [CrossRef]
- Li, C.J.; Lv, L.; Li, H.; Yu, D.M. Cardiac fibrosis and dysfunction in experimental diabetic cardiomyopathy are ameliorated by α-lipoic acid. Cardiovasc. Diabetol. 2012, 11. [Google Scholar] [CrossRef]
- Nizamutdinova, I.T.; Guleria, R.S.; Singh, A.B.; Kendall, J.A., Jr.; Baker, K.M.; Pan, J. Retinoic acid protects cardiomyocytes from high glucose-induced apoptosis via inhibition of sustained activation of NF-κB signaling. J. Cell. Physiol. 2013, 228, 380–392. [Google Scholar] [CrossRef]
- Guleria, R.S.; Singh, A.B.; Nizamutdinova, I.T.; Souslova, T.; Mohammad, A.A.; Kendall, J.A., Jr.; Baker, K.M.; Pan, J. Activation of retinoid receptor-mediated signaling ameliorates diabetes-induced cardiac dysfunction in, Zucker diabetic rats. J. Mol. Cell. Cardiol. 2013, 57, 106–118. [Google Scholar] [CrossRef]
- Guleria, R.S.; Choudhary, R.; Tanaka, T.; Baker, K.M.; Pan, J. Retinoic acid receptor-mediated signaling protects cardiomyocytes from hyperglycemia induced apoptosis: Role of the renin-angiotensin system. J. Cell. Physiol. 2011, 226, 1292–1307. [Google Scholar] [CrossRef]
- Frey, S.K.; Vogel, S. Vitamin A metabolism and adipose tissue biology. Nutrients 2011, 3, 27–39. [Google Scholar] [CrossRef]
- Norseen, J.; Hosooka, T.; Hammarstedt, A.; Yore, M.M.; Kant, S.; Aryal, P.; Kiernan, U.A.; Phillips, D.A.; Maruyama, H.; Kraus, B.J.; et al. Retinol-binding protein 4 inhibits insulin signaling in adipocytes by inducing proinflammatory cytokines in macrophages through a c-Jun N-terminal kinase- and toll-like receptor 4-dependent and retinol-independent mechanism. Mol. Cell. Biol. 2012, 32, 2010–2019. [Google Scholar] [CrossRef]
- Raghow, R. Metabolic balancing acts of vitamin A in type-2 diabetes and obesity. World J. Diabetes 2012, 3, 174–177. [Google Scholar] [CrossRef]
- Kiefer, F.W.; Vernochet, C.; O’Brien, P.; Spoerl, S.; Brown, J.D.; Nallamshetty, S.; Zeyda, M.; Stulnig, T.M.; Cohen, D.E.; Kahn, C.R.; et al. Retinaldehyde dehydrogenase 1 regulates a thermogenic program in white adipose tissue. Nat. Med. 2012, 18, 918–925. [Google Scholar] [CrossRef]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef]
- Ziouzenkova, O.; Orasanu, G.; Sharlach, M.; Akiyama, T.E.; Berger, J.P.; Viereck, J.; Hamilton, J.A.; Tang, G.; Dolnikowski, G.G.; Vogel, S.; et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 2007, 13, 695–702. [Google Scholar] [CrossRef]
- Berry, D.C.; DeSantis, D.; Soltanian, H.; Croniger, C.M.; Noy, N. Retinoic acid upregulates preadipocyte genes to block adipogenesis and suppress diet-induced obesity. Diabetes 2012, 61, 1112–1121. [Google Scholar] [CrossRef]
- Shaw, J.E.; Sicree, R.A.; Zimmet, P.Z. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res. Clin. Pract. 2010, 87, 4–14. [Google Scholar] [CrossRef]
- Grundy, S.M.; Benjamin, I.J.; Burke, G.L.; Chait, A.; Eckel, R.H.; Howard, B.V.; Mitch, W.; Smith, S.C., Jr.; Sowers, J.R. Diabetes and cardiovascular disease: A statement for healthcare professionals from the American Heart Association. Circulation 1999, 100, 1134–1146. [Google Scholar] [CrossRef]
- Kannel, W.B. Incidence and epidemiology of heart failure. Heart Fail. Rev. 2000, 5, 167–173. [Google Scholar] [CrossRef]
- Liu, L.; Eisen, H.J. Epidemiology of heart failure and scope of the problem. Cardiol. Clin. 2014, 32, 1–8. [Google Scholar] [CrossRef]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Executive summary: Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation 2013, 127, 143–152. [Google Scholar] [CrossRef]
- Galderisi, M.; Anderson, K.M.; Wilson, P.W.; Levy, D. Echocardiographic evidence for the existence of a distinct diabetic cardiomyopathy (the Framingham Heart Study). Am. J. Cardiol. 1991, 68, 85–89. [Google Scholar] [CrossRef]
- Rutter, M.K.; Parise, H.; Benjamin, E.J.; Levy, D.; Larson, M.G.; Meigs, J.B.; Nesto, R.W.; Wilson, P.W.; Vasan, R.S. Impact of glucose intolerance and insulin resistance on cardiac structure and function: Sex-related differences in the Framingham Heart Study. Circulation 2003, 107, 448–454. [Google Scholar] [CrossRef]
- Battiprolu, P.K.; Lopez-Crisosto, C.; Wang, Z.V.; Nemchenko, A.; Lavandero, S.; Hill, J.A. Diabetic cardiomyopathy and metabolic remodeling of the heart. Life Sci. 2013, 92, 609–615. [Google Scholar] [CrossRef]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Patil, M.B.; Burji, N.P. Echocardiographic evaluation of diastolic dysfunction in asymptomatic type 2 diabetes mellitus. J. Assoc. Physicians India 2012, 60, 23–26. [Google Scholar]
- Paillole, C.; Dahan, M.; Paycha, F.; Solal, A.C.; Passa, P.; Gourgon, R. Prevalence and significance of left ventricular filling abnormalities determined by Doppler echocardiography in young type I (insulin-dependent) diabetic patients. Am. J. Cardiol. 1989, 64, 1010–1016. [Google Scholar] [CrossRef]
- Van Heerebeek, L.; Somsen, A.; Paulus, W.J. The failing diabetic heart: Focus on diastolic left ventricular dysfunction. Curr. Diab. Rep. 2009, 9, 79–86. [Google Scholar] [CrossRef]
- Mandavia, C.H.; Aroor, A.R.; Demarco, V.G.; Sowers, J.R. Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci. 2013, 92, 601–608. [Google Scholar] [CrossRef]
- Tillquist, M.N.; Maddox, T.M. Update on diabetic cardiomyopathy: Inches forward, miles to go. Curr. Diab. Rep. 2012, 12, 305–313. [Google Scholar]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Boyer, J.K.; Thanigaraj, S.; Schechtman, K.B.; Perez, J.E. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. Am. J. Cardiol. 2004, 93, 870–875. [Google Scholar] [CrossRef]
- Galderisi, M. Diastolic dysfunction and diabetic cardiomyopathy: Evaluation by Doppler echocardiography. J. Am. Coll. Cardiol. 2006, 48, 1548–1551. [Google Scholar] [CrossRef]
- Khouri, S.J.; Maly, G.T.; Suh, D.D.; Walsh, T.E. A practical approach to the echocardiographic evaluation of diastolic function. J. Am. Soc. Echocardiogr. 2004, 17, 290–297. [Google Scholar] [CrossRef]
- Ng, A.C.; Delgado, V.; Bertini, M.; van der Meer, R.W.; Rijzewijk, L.J.; Shanks, M.; Nucifora, G.; Smit, J.W.; Diamant, M.; Romijn, J.A.; et al. Findings from left ventricular strain and strain rate imaging in asymptomatic patients with type 2 diabetes mellitus. Am. J. Cardiol. 2009, 104, 1398–1401. [Google Scholar] [CrossRef]
- Ernande, L.; Bergerot, C.; Rietzschel, E.R.; de Buyzere, M.L.; Thibault, H.; Pignonblanc, P.G.; Croisille, P.; Ovize, M.; Groisne, L.; Moulin, P.; et al. Diastolic dysfunction in patients with type 2 diabetes mellitus: Is it really the first marker of diabetic cardiomyopathy? J. Am. Soc. Echocardiogr. 2011, 24, 1268–1275. [Google Scholar] [CrossRef]
- Jeudy, J.; White, C.S. Cardiac magnetic resonance imaging: Techniques and principles. Semin. Roentgenol. 2008, 43, 173–182. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Varughese, G.I.; Sriraman, R.; Arunagirinathan, G. Diabetic cardiomyopathy: Pathophysiology, diagnostic evaluation and management. World J. Diabetes 2013, 4, 177–189. [Google Scholar]
- Li, G.; Hu, Y.; Yang, W.; Jiang, Y.; Wang, J.; Xiao, J.; Hu, Z.; Pan, X.; Howard, B.V.; Bennett, P.H. Effects of insulin resistance and insulin secretion on the efficacy of interventions to retard development of type 2 diabetes mellitus: The DA Qing IGT.; Diabetes Study. Diabetes Res. Clin. Pract. 2002, 58, 193–200. [Google Scholar] [CrossRef]
- Aguiar, E.J.; Morgan, P.J.; Collins, C.E.; Plotnikoff, R.C.; Callister, R. Efficacy of interventions that include diet, aerobic and resistance training components for type 2 diabetes prevention: A systematic review with meta-analysis. Int. J. Behav. Nutr. Phys. Act. 2014, 11. [Google Scholar] [CrossRef]
- Ismail-Beigi, F.; Craven, T.; Banerji, M.A.; Basile, J.; Calles, J.; Cohen, R.M.; Cuddihy, R.; Cushman, W.C.; Genuth, S.; Grimm, R.H., Jr.; et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: An analysis of the ACCORD randomised trial. Lancet 2010, 376, 419–430. [Google Scholar] [CrossRef]
- Hemmingsen, B.; Lund, S.S.; Gluud, C.; Vaag, A.; Almdal, T.P.; Hemmingsen, C.; Wetterslev, J. Targeting intensive glycaemic control versus targeting conventional glycaemic control for type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2013, 11. [Google Scholar] [CrossRef]
- Gerstein, H.C.; Miller, M.E.; Byington, R.P.; Goff, D.C., Jr.; Bigger, J.T.; Buse, J.B.; Cushman, W.C.; Genuth, S.; Ismail-Beigi, F.; Grimm, R.H., Jr.; et al. Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 2008, 358, 2545–2559. [Google Scholar] [CrossRef]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef]
- Briel, M.; Ferreira-Gonzalez, I.; You, J.J.; Karanicolas, P.J.; Akl, E.A.; Wu, P.; Blechacz, B.; Bassler, D.; Wei, X.; Sharman, A.; et al. Association between change in high density lipoprotein cholesterol and cardiovascular disease morbidity and mortality: Systematic review and meta-regression analysis. BMJ 2009, 338. [Google Scholar] [CrossRef]
- Robinson, J.G.; Wang, S.; Jacobson, T.A. Meta-analysis of comparison of effectiveness of lowering apolipoprotein B versus low-density lipoprotein cholesterol and nonhigh-density lipoprotein cholesterol for cardiovascular risk reduction in randomized trials. Am. J. Cardiol. 2012, 110, 1468–1476. [Google Scholar] [CrossRef]
- Huynh, K.; Bernardo, B.C.; McMullen, J.R.; Ritchie, R.H. Diabetic cardiomyopathy: Mechanisms and new treatment strategies targeting antioxidant signaling pathways. Pharmacol. Ther. 2014, 142, 375–415. [Google Scholar] [CrossRef]
- Voulgari, C.; Papadogiannis, D.; Tentolouris, N. Diabetic cardiomyopathy: From the pathophysiology of the cardiac myocytes to current diagnosis and management strategies. Vasc. Health Risk Manag. 2010, 6, 883–903. [Google Scholar]
- Shekelle, P.G.; Rich, M.W.; Morton, S.C.; Atkinson, C.S.; Tu, W.; Maglione, M.; Rhodes, S.; Barrett, M.; Fonarow, G.C.; Greenberg, B.; et al. Efficacy of angiotensin-converting enzyme inhibitors and β-blockers in the management of left ventricular systolic dysfunction according to race, gender, and diabetic status: A meta-analysis of major clinical trials. J. Am. Coll. Cardiol. 2003, 41, 1529–1538. [Google Scholar] [CrossRef]
- Kawasaki, D.; Kosugi, K.; Waki, H.; Yamamoto, K.; Tsujino, T.; Masuyama, T. Role of activated renin-angiotensin system in myocardial fibrosis and left ventricular diastolic dysfunction in diabetic patients—Reversal by chronic angiotensin II type 1A receptor blockade. Circ. J. 2007, 71, 524–529. [Google Scholar] [CrossRef]
- Riccioni, G. The role of direct renin inhibitors in the treatment of the hypertensive diabetic patient. Ther. Adv. Endocrinol. Metab. 2013, 4, 139–145. [Google Scholar] [CrossRef]
- Parving, H.H.; Brenner, B.M.; McMurray, J.J.; de Zeeuw, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Persson, F.; Desai, A.S.; Nicolaides, M.; et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N. Engl. J. Med. 2012, 367, 2204–2213. [Google Scholar] [CrossRef]
- Wu, M.T.; Tung, S.C.; Hsu, K.T.; Lee, C.T. Aliskiren add-on therapy effectively reduces proteinuria in chronic kidney disease: An open-label prospective trial. J. Renin-Angiotensin-Aldosterone Syst. 2012. [Google Scholar] [CrossRef]
- Fonarow, G.C. A review of evidence-based β-blockers in special populations with heart failure. Rev. Cardiovasc. Med. 2008, 9, 84–95. [Google Scholar]
- Grossman, E.; Messerli, F.H. Calcium antagonists. Prog. Cardiovasc. Dis. 2004, 47, 34–57. [Google Scholar] [CrossRef]
- Rhinn, M.; Dolle, P. Retinoic acid signalling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef]
- Naylor, H.M.; Newcomer, M.E. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry 1999, 38, 2647–2653. [Google Scholar] [CrossRef]
- Noy, N.; Xu, Z.J. Interactions of retinol with binding proteins: Implications for the mechanism of uptake by cells. Biochemistry 1990, 29, 3878–3883. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar] [CrossRef]
- Berry, D.C.; Jacobs, H.; Marwarha, G.; Gely-Pernot, A.; O’Byrne, S.M.; DeSantis, D.; Klopfenstein, M.; Feret, B.; Dennefeld, C.; Blaner, W.S.; et al. The STRA6 receptor is essential for retinol-binding protein-induced insulin resistance but not for maintaining vitamin A homeostasis in tissues other than the eye. J. Biol. Chem. 2013, 288, 24528–24539. [Google Scholar] [CrossRef]
- Terra, R.; Wang, X.; Hu, Y.; Charpentier, T.; Lamarre, A.; Zhong, M.; Sun, H.; Mao, J.; Qi, S.; Luo, H.; et al. To investigate the necessity of STRA6 upregulation in T cells during T cell immune responses. PLoS One 2013, 8, e82808. [Google Scholar] [CrossRef]
- Duester, G.; Mic, F.A.; Molotkov, A. Cytosolic retinoid dehydrogenases govern ubiquitous metabolism of retinol to retinaldehyde followed by tissue-specific metabolism to retinoic acid. Chem. Biol. Interact. 2003, 143–144, 201–210. [Google Scholar] [CrossRef]
- Pares, X.; Farres, J.; Kedishvili, N.; Duester, G. Medium- and short-chain dehydrogenase/reductase gene and protein families: Medium-chain and short-chain dehydrogenases/reductases in retinoid metabolism. Cell. Mol. Life Sci. 2008, 65, 3936–3949. [Google Scholar] [CrossRef]
- Farjo, K.M.; Moiseyev, G.; Nikolaeva, O.; Sandell, L.L.; Trainor, P.A.; Ma, J.X. RDH10 is the primary enzyme responsible for the first step of embryonic vitamin A metabolism and retinoic acid synthesis. Dev. Biol. 2011, 357, 347–355. [Google Scholar] [CrossRef]
- Napoli, J.L. Physiological insights into all-trans-retinoic acid biosynthesis. Biochim. Biophys. Acta 2012, 1821, 152–167. [Google Scholar]
- Napoli, J.L. Retinoic acid biosynthesis and metabolism. FASEB J. 1996, 10, 993–1001. [Google Scholar]
- Wolf, G. Cellular retinoic acid-binding protein II: A coactivator of the transactivation by the retinoic acid receptor complex, RAR.RXR. Nutr. Rev. 2000, 58, 151–153. [Google Scholar] [CrossRef]
- Kedishvili, N.Y. Enzymology of retinoic acid biosynthesis and degradation. J. Lipid Res. 2013, 54, 1744–1760. [Google Scholar] [CrossRef]
- Davidovici, B.B.; Tuzun, Y.; Wolf, R. Retinoid receptors. Dermatol. Clin. 2007, 25, 525–530. [Google Scholar] [CrossRef]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Rochette-Egly, C.; Oulad-Abdelghani, M.; Staub, A.; Pfister, V.; Scheuer, I.; Chambon, P.; Gaub, M.P. Phosphorylation of the retinoic acid receptor-α by protein kinase, A. Mol. Endocrinol. 1995, 9, 860–871. [Google Scholar]
- Rochette-Egly, C.; Adam, S.; Rossignol, M.; Egly, J.M.; Chambon, P. Stimulation of RARα activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell 1997, 90, 97–107. [Google Scholar] [CrossRef]
- Delmotte, M.H.; Tahayato, A.; Formstecher, P.; Lefebvre, P. Serine 157, a retinoic acid receptor α residue phosphorylated by protein kinase C in vitro, is involved in RXR·RARα heterodimerization and transcriptional activity. J. Biol. Chem. 1999, 274, 38225–38231. [Google Scholar] [CrossRef]
- Srinivas, H.; Xia, D.; Moore, N.L.; Uray, I.P.; Kim, H.; Ma, L.; Weigel, N.L.; Brown, P.H.; Kurie, J.M. Akt phosphorylates and suppresses the transactivation of retinoic acid receptor α. Biochem. J. 2006, 395, 653–662. [Google Scholar] [CrossRef]
- Lee, H.Y.; Suh, Y.A.; Robinson, M.J.; Clifford, J.L.; Hong, W.K.; Woodgett, J.R.; Cobb, M.H.; Mangelsdorf, D.J.; Kurie, J.M. Stress pathway activation induces phosphorylation of retinoid X receptor. J. Biol. Chem. 2000, 275, 32193–32199. [Google Scholar] [CrossRef]
- Matsushima-Nishiwaki, R.; Okuno, M.; Adachi, S.; Sano, T.; Akita, K.; Moriwaki, H.; Friedman, S.L.; Kojima, S. Phosphorylation of retinoid X receptor α at serine 260 impairs its metabolism and function in human hepatocellular carcinoma. Cancer Res. 2001, 61, 7675–7682. [Google Scholar]
- Yamazaki, K.; Shimizu, M.; Okuno, M.; Matsushima-Nishiwaki, R.; Kanemura, N.; Araki, H.; Tsurumi, H.; Kojima, S.; Weinstein, I.B.; Moriwaki, H. Synergistic effects of RXRα and PPARγ ligands to inhibit growth in human colon cancer cells—Phosphorylated RXRα is a critical target for colon cancer management. Gut 2007, 56, 1557–1563. [Google Scholar] [CrossRef]
- Kam, R.K.; Deng, Y.; Chen, Y.; Zhao, H. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci. 2012, 2. [Google Scholar] [CrossRef]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; van Veldhuizen, P.; et al. Retinoic acid signaling pathways in development and diseases. Bioorg. Med. Chem. 2014, 22, 673–683. [Google Scholar]
- Connolly, R.M.; Nguyen, N.K.; Sukumar, S. Molecular pathways: Current role and future directions of the retinoic acid pathway in cancer prevention and treatment. Clin. Cancer Res. 2013, 19, 1651–1659. [Google Scholar] [CrossRef]
- Brun, P.J.; Yang, K.J.; Lee, S.A.; Yuen, J.J.; Blaner, W.S. Retinoids: Potent regulators of metabolism. Biofactors 2013, 39, 151–163. [Google Scholar] [CrossRef]
- Zhao, S.; Li, R.; Li, Y.; Chen, W.; Zhang, Y.; Chen, G. Roles of vitamin A status and retinoids in glucose and fatty acid metabolism. Biochem. Cell Biol. 2012, 90, 142–152. [Google Scholar] [CrossRef]
- Mongan, N.P.; Gudas, L.J. Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation 2007, 75, 853–870. [Google Scholar] [CrossRef]
- Park, H.; Green, M.H.; Shaffer, M.L. Association between serum retinol-binding protein 4 concentrations and clinical indices in subjects with type 2 diabetes: A meta-analysis. J. Hum. Nutr. Diet. 2012, 25, 300–310. [Google Scholar] [CrossRef]
- Basu, T.K.; Tze, W.J.; Leichter, J. Serum vitamin A and retinol-binding protein in patients with insulin-dependent diabetes mellitus. Am. J. Clin. Nutr. 1989, 50, 329–331. [Google Scholar]
- Kiefer, F.W.; Orasanu, G.; Nallamshetty, S.; Brown, J.D.; Wang, H.; Luger, P.; Qi, N.R.; Burant, C.F.; Duester, G.; Plutzky, J. Retinaldehyde dehydrogenase 1 coordinates hepatic gluconeogenesis and lipid metabolism. Endocrinology 2012, 153, 3089–3099. [Google Scholar] [CrossRef]
- Wierdsma, N.J.; van Bokhorst-de van der Schueren, M.A.; Berkenpas, M.; Mulder, C.J.; van Bodegraven, A.A. Vitamin and mineral deficiencies are highly prevalent in newly diagnosed celiac disease patients. Nutrients 2013, 5, 3975–3992. [Google Scholar] [CrossRef]
- Noy, N. The one-two punch: Retinoic acid suppresses obesity both by promoting energy expenditure and by inhibiting adipogenesis. Adipocyte 2013, 2, 184–187. [Google Scholar] [CrossRef]
- Bonet, M.L.; Ribot, J.; Palou, A. Lipid metabolism in mammalian tissues and its control by retinoic acid. Biochim. Biophys. Acta 2012, 1821, 177–189. [Google Scholar]
- Amengual, J.; Ribot, J.; Bonet, M.L.; Palou, A. Retinoic acid treatment enhances lipid oxidation and inhibits lipid biosynthesis capacities in the liver of mice. Cell. Physiol. Biochem. 2010, 25, 657–666. [Google Scholar] [CrossRef]
- Basu, T.K.; Basualdo, C. Vitamin A homeostasis and diabetes mellitus. Nutrition 1997, 13, 804–806. [Google Scholar] [CrossRef]
- Basualdo, C.G.; Wein, E.E.; Basu, T.K. Vitamin A (retinol) status of first nation adults with non-insulin-dependent diabetes mellitus. J. Am. Coll. Nutr. 1997, 16, 39–45. [Google Scholar] [CrossRef]
- Baena, R.M.; Campoy, C.; Bayes, R.; Blanca, E.; Fernandez, J.M.; Molina-Font, J.A. Vitamin A retinol binding protein and lipids in type 1 diabetes mellitus. Eur. J. Clin. Nutr. 2002, 56, 44–50. [Google Scholar] [CrossRef]
- Tuitoek, P.J.; Ziari, S.; Tsin, A.T.; Rajotte, R.V.; Suh, M.; Basu, T.K. Streptozotocin-induced diabetes in rats is associated with impaired metabolic availability of vitamin A (retinol). Br. J. Nutr. 1996, 75, 615–622. [Google Scholar] [CrossRef]
- Zunino, S.J.; Storms, D.H.; Stephensen, C.B. Diets rich in polyphenols and vitamin A inhibit the development of type I autoimmune diabetes in nonobese diabetic mice. J. Nutr. 2007, 137, 1216–1221. [Google Scholar]
- Van, Y.H.; Lee, W.H.; Ortiz, S.; Lee, M.H.; Qin, H.J.; Liu, C.P. All-trans retinoic acid inhibits type 1 diabetes by T regulatory (Treg)-dependent suppression of interferon-γ-producing T-cells without affecting Th17 cells. Diabetes 2009, 58, 146–155. [Google Scholar] [CrossRef]
- Cabre, A.; Lazaro, I.; Girona, J.; Manzanares, J.; Marimon, F.; Plana, N.; Heras, M.; Masana, L. Retinol-binding protein 4 as a plasma biomarker of renal dysfunction and cardiovascular disease in type 2 diabetes. J. Intern. Med. 2007, 262, 496–503. [Google Scholar] [CrossRef]
- Rocha, M.; Banuls, C.; Bellod, L.; Rovira-Llopis, S.; Morillas, C.; Sola, E.; Victor, V.M.; Hernandez-Mijares, A. Association of serum retinol binding protein 4 with atherogenic dyslipidemia in morbid obese patients. PLoS One 2013, 8, e78670. [Google Scholar] [CrossRef]
- Cho, Y.M.; Youn, B.S.; Lee, H.; Lee, N.; Min, S.S.; Kwak, S.H.; Lee, H.K.; Park, K.S. Plasma retinol-binding protein-4 concentrations are elevated in human subjects with impaired glucose tolerance and type 2 diabetes. Diabetes Care 2006, 29, 2457–2461. [Google Scholar] [CrossRef]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef]
- Manolescu, D.C.; Sima, A.; Bhat, P.V. All-trans retinoic acid lowers serum retinol-binding protein 4 concentrations and increases insulin sensitivity in diabetic mice. J. Nutr. 2010, 140, 311–316. [Google Scholar] [CrossRef]
- Berry, D.C.; Jin, H.; Majumdar, A.; Noy, N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. USA 2011, 108, 4340–4345. [Google Scholar] [CrossRef]
- Mukherjee, R.; Davies, P.J.; Crombie, D.L.; Bischoff, E.D.; Cesario, R.M.; Jow, L.; Hamann, L.G.; Boehm, M.F.; Mondon, C.E.; Nadzan, A.M.; et al. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature 1997, 386, 407–410. [Google Scholar] [CrossRef]
- Shen, Q.; Cline, G.W.; Shulman, G.I.; Leibowitz, M.D.; Davies, P.J. Effects of rexinoids on glucose transport and insulin-mediated signaling in skeletal muscles of diabetic (db/db) mice. J. Biol. Chem. 2004, 279, 19721–19731. [Google Scholar]
- Jeyakumar, S.M.; Vijaya Kumar, P.; Giridharan, N.V.; Vajreswari, A. Vitamin A improves insulin sensitivity by increasing insulin receptor phosphorylation through protein tyrosine phosphatase 1B regulation at early age in obese rats of WNIN/Ob strain. Diabetes Obes. Metab. 2011, 13, 955–958. [Google Scholar] [CrossRef]
- Mercader, J.; Ribot, J.; Murano, I.; Felipe, F.; Cinti, S.; Bonet, M.L.; Palou, A. Remodeling of white adipose tissue after retinoic acid administration in mice. Endocrinology 2006, 147, 5325–5332. [Google Scholar] [CrossRef]
- Amengual, J.; Ribot, J.; Bonet, M.L.; Palou, A. Retinoic acid treatment increases lipid oxidation capacity in skeletal muscle of mice. Obesity (Silver Spring) 2008, 16, 585–591. [Google Scholar] [CrossRef]
- Ribot, J.; Felipe, F.; Bonet, M.L.; Palou, A. Changes of adiposity in response to vitamin A status correlate with changes of PPARγ 2 expression. Obes. Res. 2001, 9, 500–509. [Google Scholar] [CrossRef]
- Bonet, M.L.; Oliver, J.; Pico, C.; Felipe, F.; Ribot, J.; Cinti, S.; Palou, A. Opposite effects of feeding a vitamin A-deficient diet and retinoic acid treatment on brown adipose tissue uncoupling protein 1 (UCP1), UCP2 and leptin expression. J. Endocrinol. 2000, 166, 511–517. [Google Scholar] [CrossRef]
- Mercader, J.; Madsen, L.; Felipe, F.; Palou, A.; Kristiansen, K.; Bonet, M.L. All-trans retinoic acid increases oxidative metabolism in mature adipocytes. Cell. Physiol. Biochem. 2007, 20, 1061–1072. [Google Scholar] [CrossRef]
- Molotkov, A.; Duester, G. Genetic evidence that retinaldehyde dehydrogenase Raldh1 (Aldh1a1) functions downstream of alcohol dehydrogenase Adh1 in metabolism of retinol to retinoic acid. J. Biol. Chem. 2003, 278, 36085–36090. [Google Scholar] [CrossRef]
- Salazar, J.; Guardiola, M.; Ferre, R.; Coll, B.; Alonso-Villaverde, C.; Winklhofer-Roob, B.M.; Rock, E.; Fernandez-Ballart, J.D.; Civeira, F.; Pocovi, M.; et al. Association of a polymorphism in the promoter of the cellular retinoic acid-binding protein II gene (CRABP2) with increased circulating low-density lipoprotein cholesterol. Clin. Chem. Lab. Med. 2007, 45, 615–620. [Google Scholar]
- Kastner, P.; Messaddeq, N.; Mark, M.; Wendling, O.; Grondona, J.M.; Ward, S.; Ghyselinck, N.; Chambon, P. Vitamin A deficiency and mutations of, RXRα, RXRβ and RARα lead to early differentiation of embryonic ventricular cardiomyocytes. Development 1997, 124, 4749–4758. [Google Scholar]
- Subbarayan, V.; Mark, M.; Messadeq, N.; Rustin, P.; Chambon, P.; Kastner, P. RXRα overexpression in cardiomyocytes causes dilated cardiomyopathy but fails to rescue myocardial hypoplasia in RXRα-null fetuses. J. Clin. Invest. 2000, 105, 387–394. [Google Scholar] [CrossRef]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar]
- Zile, M.H. Vitamin A—Not for your eyes only: Requirement for heart formation begins early in embryogenesis. Nutrients 2010, 2, 532–550. [Google Scholar] [CrossRef]
- Noy, N. Between death and survival: Retinoic acid in regulation of apoptosis. Annu. Rev. Nutr. 2010, 30, 201–217. [Google Scholar] [CrossRef]
- Ahuja, H.S.; Szanto, A.; Nagy, L.; Davies, P.J. The retinoid X receptor and its ligands: Versatile regulators of metabolic function, cell differentiation and cell death. J. Biol. Regul. Homeost. Agents 2003, 17, 29–45. [Google Scholar]
- Zhou, M.D.; Sucov, H.M.; Evans, R.M.; Chien, K.R. Retinoid-dependent pathways suppress myocardial cell hypertrophy. Proc. Natl. Acad. Sci. USA 1995, 92, 7391–7395. [Google Scholar]
- Wu, J.; Garami, M.; Cheng, T.; Gardner, D.G. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J. Clin. Invest. 1996, 97, 1577–1588. [Google Scholar] [CrossRef]
- Wang, H.J.; Zhu, Y.C.; Yao, T. Effects of all-trans retinoic acid on angiotensin II-induced myocyte hypertrophy. J. Appl. Physiol. 2002, 92, 2162–2168. [Google Scholar]
- Palm-Leis, A.; Singh, U.S.; Herbelin, B.S.; Olsovsky, G.D.; Baker, K.M.; Pan, J. Mitogen-activated protein kinases and mitogen-activated protein kinase phosphatases mediate the inhibitory effects of all-trans retinoic acid on the hypertrophic growth of cardiomyocytes. J. Biol. Chem. 2004, 279, 54905–54917. [Google Scholar]
- He, Y.; Huang, Y.; Zhou, L.; Lu, L.M.; Zhu, Y.C.; Yao, T. All-trans retinoic acid inhibited angiotensin II-induced increase in cell growth and collagen secretion of neonatal cardiac fibroblasts. Acta Pharmacol. Sin. 2006, 27, 423–429. [Google Scholar]
- Oliveira, L.C.; Azevedo, P.S.; Minicucci, M.E.; Rafacho, B.P.; Duarte, D.R.; Matsubara, L.S.; Matsubara, B.B.; Paiva, S.A.; Zornoff, L.A. Retinoic acid prevents ventricular remodelling induced by tobacco smoke exposure in rats. Acta Cardiol. 2011, 66, 3–7. [Google Scholar]
- Lu, L.; Yao, T.; Zhu, Y.Z.; Huang, G.Y.; Cao, Y.X.; Zhu, Y.C. Chronic all-trans retinoic acid treatment prevents medial thickening of intramyocardial and intrarenal arteries in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H1370–H1377. [Google Scholar]
- Paiva, S.A.; Matsubara, L.S.; Matsubara, B.B.; Minicucci, M.F.; Azevedo, P.S.; Campana, A.O.; Zornoff, L.A. Retinoic acid supplementation attenuates ventricular remodeling after myocardial infarction in rats. J. Nutr. 2005, 135, 2326–2328. [Google Scholar]
- Choudhary, R.; Palm-Leis, A.; Scott, R.C., III.; Guleria, R.S.; Rachut, E.; Baker, K.M.; Pan, J. All-trans retinoic acid prevents development of cardiac remodeling in aortic banded rats by inhibiting the renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H633–H644. [Google Scholar] [CrossRef]
- Mortuza, R.; Chakrabarti, S. Glucose-induced cell signaling in the pathogenesis of diabetic cardiomyopathy. Heart Fail. Rev. 2014, 19, 75–86. [Google Scholar] [CrossRef]
- Camps, M.; Castello, A.; Munoz, P.; Monfar, M.; Testar, X.; Palacin, M.; Zorzano, A. Effect of diabetes and fasting on GLUT-4 (muscle/fat) glucose-transporter expression in insulin-sensitive tissues. Heterogeneous response in heart, red and white muscle. Biochem. J. 1992, 282, 765–772. [Google Scholar]
- Montessuit, C.; Lerch, R. Regulation and dysregulation of glucose transport in cardiomyocytes. Biochim. Biophys. Acta 2013, 1833, 848–856. [Google Scholar] [CrossRef]
- Coort, S.L.; Bonen, A.; van der Vusse, G.J.; Glatz, J.F.; Luiken, J.J. Cardiac substrate uptake and metabolism in obesity and type-2 diabetes: Role of sarcolemmal substrate transporters. Mol. Cell. Biochem. 2007, 299, 5–18. [Google Scholar] [CrossRef]
- Young, M.E.; Guthrie, P.H.; Razeghi, P.; Leighton, B.; Abbasi, S.; Patil, S.; Youker, K.A.; Taegtmeyer, H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes 2002, 51, 2587–2595. [Google Scholar] [CrossRef]
- Van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef]
- Basu, R.; Oudit, G.Y.; Wang, X.; Zhang, L.; Ussher, J.R.; Lopaschuk, G.D.; Kassiri, Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2096–H2108. [Google Scholar] [CrossRef]
- Pulinilkunnil, T.; Kienesberger, P.C.; Nagendran, J.; Waller, T.J.; Young, M.E.; Kershaw, E.E.; Korbutt, G.; Haemmerle, G.; Zechner, R.; Dyck, J.R. Myocardial adipose triglyceride lipase overexpression protects diabetic mice from the development of lipotoxic cardiomyopathy. Diabetes 2013, 62, 1464–1477. [Google Scholar] [CrossRef]
- Ouwens, D.M.; Diamant, M.; Fodor, M.; Habets, D.D.; Pelsers, M.M.; El Hasnaoui, M.; Dang, Z.C.; van den Brom, C.E.; Vlasblom, R.; Rietdijk, A.; et al. Cardiac contractile dysfunction in insulin-resistant rats fed a high-fat diet is associated with elevated CD36-mediated fatty acid uptake and esterification. Diabetologia 2007, 50, 1938–1948. [Google Scholar]
- Chiu, H.C.; Kovacs, A.; Blanton, R.M.; Han, X.; Courtois, M.; Weinheimer, C.J.; Yamada, K.A.; Brunet, S.; Xu, H.; Nerbonne, J.M.; et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 2005, 96, 225–233. [Google Scholar] [CrossRef]
- Johansen, J.S.; Harris, A.K.; Rychly, D.J.; Ergul, A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovasc. Diabetol. 2005, 4. [Google Scholar] [CrossRef] [Green Version]
- Ungvari, Z.; Gupte, S.A.; Recchia, F.A.; Batkai, S.; Pacher, P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr. Vasc. Pharmacol. 2005, 3, 221–229. [Google Scholar] [CrossRef]
- Cai, L.; Kang, Y.J. Oxidative stress and diabetic cardiomyopathy: A brief review. Cardiovasc. Toxicol. 2001, 1, 181–193. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar]
- Dirkx, E.; Schwenk, R.W.; Glatz, J.F.; Luiken, J.J.; van Eys, G.J. High fat diet induced diabetic cardiomyopathy. Prostaglandins Leukot. Essent. Fatty Acids 2011, 85, 219–225. [Google Scholar] [CrossRef]
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef]
- Mukherjee, R.; Strasser, J.; Jow, L.; Hoener, P.; Paterniti, J.R., Jr.; Heyman, R.A. RXR agonists activate PPARα-inducible genes, lower triglycerides, and raise HDL levels in vivo. Arterioscler Thromb. Vasc. Biol. 1998, 18, 272–276. [Google Scholar] [CrossRef]
- Aranda, A.; Pascual, A. Nuclear hormone receptors and gene expression. Physiol. Rev. 2001, 81, 1269–1304. [Google Scholar]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef]
- Poulsen, L.; Siersbaek, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef]
- Diamant, M.; Lamb, H.J.; Smit, J.W.; de Roos, A.; Heine, R.J. Diabetic cardiomyopathy in uncomplicated type 2 diabetes is associated with the metabolic syndrome and systemic inflammation. Diabetologia 2005, 48, 1669–1670. [Google Scholar] [CrossRef]
- Westermann, D.; Rutschow, S.; Jager, S.; Linderer, A.; Anker, S.; Riad, A.; Unger, T.; Schultheiss, H.P.; Pauschinger, M.; Tschope, C. Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: The role of angiotensin type 1 receptor antagonism. Diabetes 2007, 56, 641–646. [Google Scholar] [CrossRef]
- Palomer, X.; Salvado, L.; Barroso, E.; Vazquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef]
- Gullestad, L.; Ueland, T.; Vinge, L.E.; Finsen, A.; Yndestad, A.; Aukrust, P. Inflammatory cytokines in heart failure: Mediators and markers. Cardiology 2012, 122, 23–35. [Google Scholar] [CrossRef]
- Paulus, W.J. Cytokines and heart failure. Heart Fail. Monit. 2000, 1, 50–56. [Google Scholar]
- Wan, F.; Lenardo, M.J. The nuclear signaling of NF-κB: Current knowledge, new insights, and future perspectives. Cell Res. 2010, 20, 24–33. [Google Scholar] [CrossRef]
- Valen, G.; Yan, Z.Q.; Hansson, G.K. Nuclear factor κ-B and the heart. J. Am. Coll. Cardiol. 2001, 38, 307–314. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Schiekofer, S.; Isermann, B.; Kanitz, M.; Henkels, M.; Joswig, M.; Treusch, A.; Morcos, M.; Weiss, T.; Borcea, V.; et al. Peripheral blood mononuclear cells isolated from patients with diabetic nephropathy show increased activation of the oxidative-stress sensitive transcription factor NF-κB. Diabetologia 1999, 42, 222–232. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-κB. Diabetes 2001, 50, 2792–2808. [Google Scholar]
- Granic, I.; Dolga, A.M.; Nijholt, I.M.; van Dijk, G.; Eisel, U.L. Inflammation and NF-κB in Alzheimer’s disease and diabetes. J. Alzheimers Dis. 2009, 16, 809–821. [Google Scholar]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef]
- Chen, S.; Khan, Z.A.; Cukiernik, M.; Chakrabarti, S. Differential activation of NF-κB and AP-1 in increased fibronectin synthesis in target organs of diabetic complications. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E1089–E1097. [Google Scholar]
- Lorenzo, O.; Picatoste, B.; Ares-Carrasco, S.; Ramirez, E.; Egido, J.; Tunon, J. Potential role of nuclear factor κB in diabetic cardiomyopathy. Mediat. Inflamm. 2011, 2011. [Google Scholar] [CrossRef]
- Xu, J.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Peroxisome proliferator-activated receptor-α and retinoid, X receptor agonists inhibit inflammatory responses of astrocytes. J. Neuroimmunol. 2006, 176, 95–105. [Google Scholar] [CrossRef]
- Lalloyer, F.; Fievet, C.; Lestavel, S.; Torpier, G.; van der Veen, J.; Touche, V.; Bultel, S.; Yous, S.; Kuipers, F.; Paumelle, R.; et al. The RXR agonist bexarotene improves cholesterol homeostasis and inhibits atherosclerosis progression in a mouse model of mixed dyslipidemia. Arterioscler Thromb. Vasc. Biol. 2006, 26, 2731–2737. [Google Scholar] [CrossRef]
- Villamor, E.; Fawzi, W.W. Effects of vitamin a supplementation on immune responses and correlation with clinical outcomes. Clin. Microbiol. Rev. 2005, 18, 446–464. [Google Scholar]
- Long, K.Z.; Garcia, C.; Ko, G.; Santos, J.I.; Al Mamun, A.; Rosado, J.L.; DuPont, H.L.; Nathakumar, N. Vitamin A modifies the intestinal chemokine and cytokine responses to norovirus infection in Mexican children. J. Nutr. 2011, 141, 957–963. [Google Scholar] [CrossRef]
- Reifen, R.; Nur, T.; Ghebermeskel, K.; Zaiger, G.; Urizky, R.; Pines, M. Vitamin A deficiency exacerbates inflammation in a rat model of colitis through activation of nuclear factor-κB and collagen formation. J. Nutr. 2002, 132, 2743–2747. [Google Scholar]
- Austenaa, L.M.; Carlsen, H.; Ertesvag, A.; Alexander, G.; Blomhoff, H.K.; Blomhoff, R. Vitamin A status significantly alters nuclear factor-κB activity assessed by in vivo imaging. FASEB J. 2004, 18, 1255–1257. [Google Scholar]
- Gatica, L.; Alvarez, S.; Gomez, N.; Zago, M.P.; Oteiza, P.; Oliveros, L.; Gimenez, M.S. Vitamin A deficiency induces prooxidant environment and inflammation in rat aorta. Free Radic Res. 2005, 39, 621–628. [Google Scholar] [CrossRef]
- Ning, R.B.; Zhu, J.; Chai, D.J.; Xu, C.S.; Xie, H.; Lin, X.Y.; Zeng, J.Z.; Lin, J.X. RXR agonists inhibit high glucose-induced upregulation of inflammation by suppressing activation of the NADPH oxidase-nuclear factor-κB pathway in human endothelial cells. Genet Mol. Res. 2013, 12, 6692–6707. [Google Scholar] [CrossRef]
- Beitelshees, A.L.; Zineh, I. Renin-angiotensin-aldosterone system (RAAS) pharmacogenomics: Implications in heart failure management. Heart Fail. Rev. 2010, 15, 209–217. [Google Scholar] [CrossRef]
- Lang, C.C.; Struthers, A.D. Targeting the renin-angiotensin-aldosterone system in heart failure. Nat. Rev. Cardiol. 2013, 10, 125–134. [Google Scholar] [CrossRef]
- Hershon, K.S. Mechanistic and clinical aspects of renin-angiotensin-aldosterone system blockade in the prevention of diabetes mellitus and cardiovascular disease. Endocr. Pract. 2011, 17, 430–440. [Google Scholar] [CrossRef]
- Frustaci, A.; Kajstura, J.; Chimenti, C.; Jakoniuk, I.; Leri, A.; Maseri, A.; Nadal-Ginard, B.; Anversa, P. Myocardial cell death in human diabetes. Circ. Res. 2000, 87, 1123–1132. [Google Scholar] [CrossRef]
- Fiordaliso, F.; Li, B.; Latini, R.; Sonnenblick, E.H.; Anversa, P.; Leri, A.; Kajstura, J. Myocyte death in streptozotocin-induced diabetes in rats in angiotensin, II-dependent. Lab. Invest. 2000, 80, 513–527. [Google Scholar] [CrossRef]
- Brown, L.; Wall, D.; Marchant, C.; Sernia, C. Tissue-specific changes in angiotensin II receptors in streptozotocin-diabetic rats. J. Endocrinol. 1997, 154, 355–362. [Google Scholar] [CrossRef]
- Fiordaliso, F.; Leri, A.; Cesselli, D.; Limana, F.; Safai, B.; Nadal-Ginard, B.; Anversa, P.; Kajstura, J. Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes 2001, 50, 2363–2375. [Google Scholar] [CrossRef]
- Sechi, L.A.; Griffin, C.A.; Schambelan, M. The cardiac renin-angiotensin system in STZ-induced diabetes. Diabetes 1994, 43, 1180–1184. [Google Scholar] [CrossRef]
- Singh, V.P.; Le, B.; Khode, R.; Baker, K.M.; Kumar, R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 2008, 57, 3297–3306. [Google Scholar] [CrossRef]
- Dechow, C.; Morath, C.; Peters, J.; Lehrke, I.; Waldherr, R.; Haxsen, V.; Ritz, E.; Wagner, J. Effects of all-trans retinoic acid on renin-angiotensin system in rats with experimental nephritis. Am. J. Physiol. Renal Physiol. 2001, 281, F909–F919. [Google Scholar]
- Takeda, K.; Ichiki, T.; Funakoshi, Y.; Ito, K.; Takeshita, A. Downregulation of angiotensin II type 1 receptor by all-trans retinoic acid in vascular smooth muscle cells. Hypertension 2000, 35, 297–302. [Google Scholar] [CrossRef]
- Zhong, J.C.; Huang, D.Y.; Yang, Y.M.; Li, Y.F.; Liu, G.F.; Song, X.H.; Du, K. Upregulation of angiotensin-converting enzyme 2 by all-trans retinoic acid in spontaneously hypertensive rats. Hypertension 2004, 44, 907–912. [Google Scholar] [CrossRef]
- Haxsen, V.; Adam-Stitah, S.; Ritz, E.; Wagner, J. Retinoids inhibit the actions of angiotensin II on vascular smooth muscle cells. Circ. Res. 2001, 88, 637–644. [Google Scholar] [CrossRef]
- Zhong, J.C.; Huang, D.Y.; Liu, G.F.; Jin, H.Y.; Yang, Y.M.; Li, Y.F.; Song, X.H.; Du, K. Effects of all-trans retinoic acid on orphan receptor APJ signaling in spontaneously hypertensive rats. Cardiovasc. Res. 2005, 65, 743–750. [Google Scholar] [CrossRef]
- Leid, M.; Kastner, P.; Chambon, P. Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem. Sci. 1992, 17, 427–433. [Google Scholar] [CrossRef]
- Lufkin, T.; Lohnes, D.; Mark, M.; Dierich, A.; Gorry, P.; Gaub, M.P.; LeMeur, M.; Chambon, P. High postnatal lethality and testis degeneration in retinoic acid receptor α mutant mice. Proc. Natl. Acad. Sci. USA 1993, 90, 7225–7229. [Google Scholar] [CrossRef]
- Sucov, H.M.; Dyson, E.; Gumeringer, C.L.; Price, J.; Chien, K.R.; Evans, R.M. RXRα mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis. Genes Dev. 1994, 8, 1007–1018. [Google Scholar] [CrossRef]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-α in pacing-induced heart failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef]
- Feingold, K.; Kim, M.S.; Shigenaga, J.; Moser, A.; Grunfeld, C. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E201–E207. [Google Scholar]
- Singh, A.B.; Guleria, R.S.; Nizamutdinova, I.T.; Baker, K.M.; Pan, J. High Glucose-induced repression of RAR/RXR in cardiomyocytes is mediated through oxidative stress/JNK signaling. J. Cell. Physiol. 2012, 227, 2632–2644. [Google Scholar] [CrossRef]
- Zhu, S.; Guleria, G.S.; Thomas, C.M.; Kumar, R.; Roth, A.; Baker, K.M.; Pan, J. Cardiomyocyte-specific deletion of RARα and RXRα induces diastolic heart failure. Criculation Res. in preparation. 2014. [Google Scholar]
- Bastien, J.; Rochette-Egly, C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004, 328, 1–16. [Google Scholar] [CrossRef]
- Zassadowski, F.; Rochette-Egly, C.; Chomienne, C.; Cassinat, B. Regulation of the transcriptional activity of nuclear receptors by the, MEK/ERK1/2 pathway. Cell. Signal. 2012, 24, 2369–2377. [Google Scholar] [CrossRef]
- Macoritto, M.; Nguyen-Yamamoto, L.; Huang, D.C.; Samuel, S.; Yang, X.F.; Wang, T.T.; White, J.H.; Kremer, R. Phosphorylation of the human retinoid X receptor α at serine 260 impairs coactivator(s) recruitment and induces hormone resistance to multiple ligands. J. Biol. Chem. 2008, 283, 4943–4956. [Google Scholar] [CrossRef]
- Rochette-Egly, C. Nuclear receptors: Integration of multiple signalling pathways through phosphorylation. Cell. Signal. 2003, 15, 355–366. [Google Scholar] [CrossRef]
- Solomon, C.; White, J.H.; Kremer, R. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin, D3-dependent signal transduction by phosphorylating human retinoid X receptor α. J. Clin. Invest. 1999, 103, 1729–1735. [Google Scholar] [CrossRef]
- Adam-Stitah, S.; Penna, L.; Chambon, P.; Rochette-Egly, C. Hyperphosphorylation of the retinoid X receptor α by activated c-Jun, NH2-terminal kinases. J. Biol. Chem. 1999, 274, 18932–18941. [Google Scholar]
- Hoshikawa, Y.; Kanki, K.; Ashla, A.A.; Arakaki, Y.; Azumi, J.; Yasui, T.; Tezuka, Y.; Matsumi, Y.; Tsuchiya, H.; Kurimasa, A.; et al. c-Jun N-terminal kinase activation by oxidative stress suppresses retinoid signaling through proteasomal degradation of retinoic acid receptor α protein in hepatic cells. Cancer Sci. 2011, 102, 934–941. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Pan, J.; Guleria, R.S.; Zhu, S.; Baker, K.M. Molecular Mechanisms of Retinoid Receptors in Diabetes-Induced Cardiac Remodeling. J. Clin. Med. 2014, 3, 566-594. https://doi.org/10.3390/jcm3020566
Pan J, Guleria RS, Zhu S, Baker KM. Molecular Mechanisms of Retinoid Receptors in Diabetes-Induced Cardiac Remodeling. Journal of Clinical Medicine. 2014; 3(2):566-594. https://doi.org/10.3390/jcm3020566
Chicago/Turabian StylePan, Jing, Rakeshwar S. Guleria, Sen Zhu, and Kenneth M. Baker. 2014. "Molecular Mechanisms of Retinoid Receptors in Diabetes-Induced Cardiac Remodeling" Journal of Clinical Medicine 3, no. 2: 566-594. https://doi.org/10.3390/jcm3020566