Microarray Technology for the Diagnosis of Fetal Chromosomal Aberrations: Which Platform Should We Use?

Abstract
:
1. Introduction


2. Types of Array Platforms
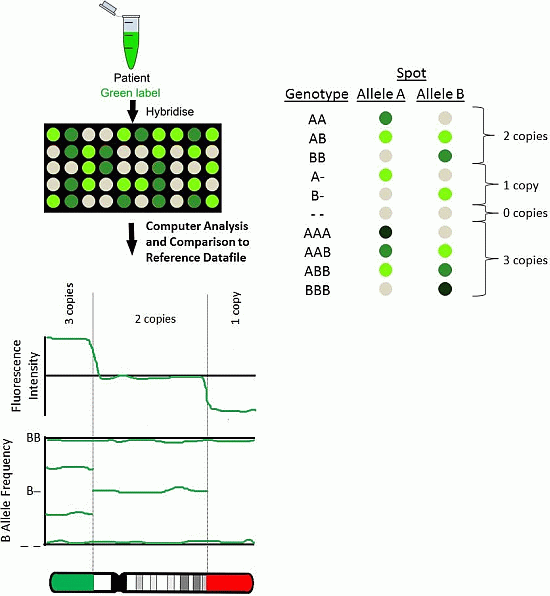
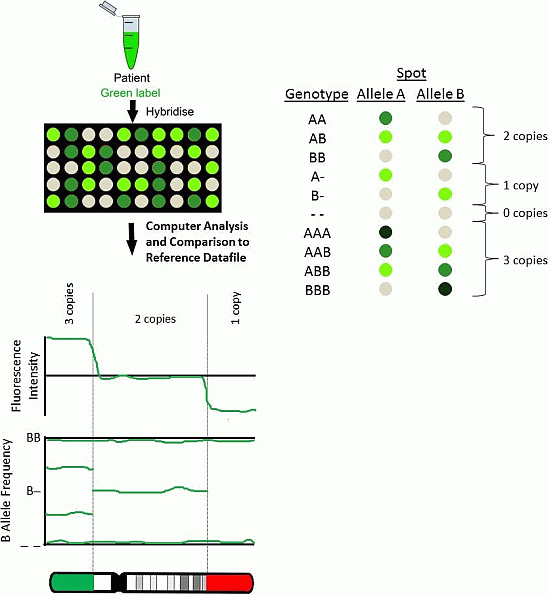
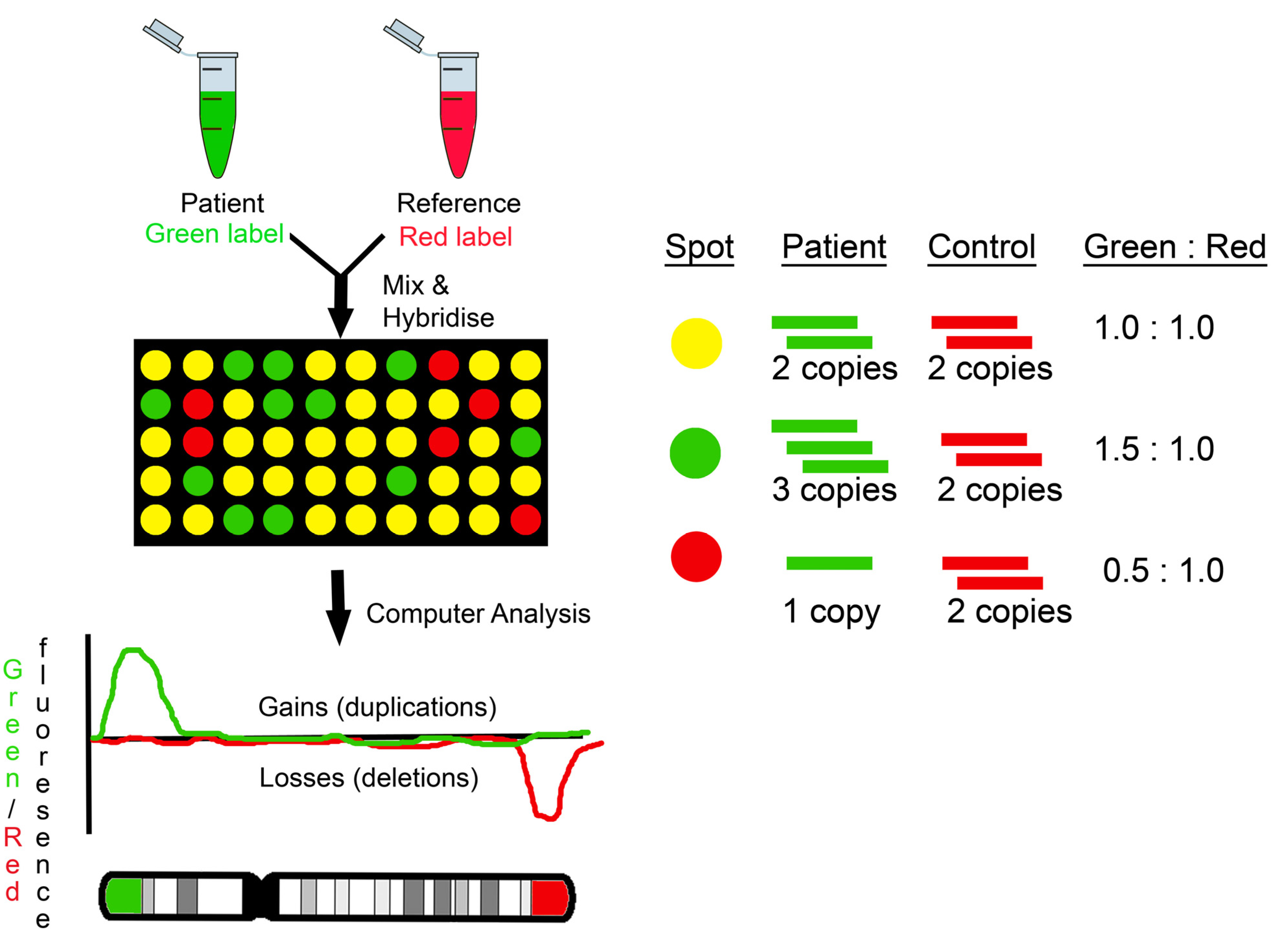
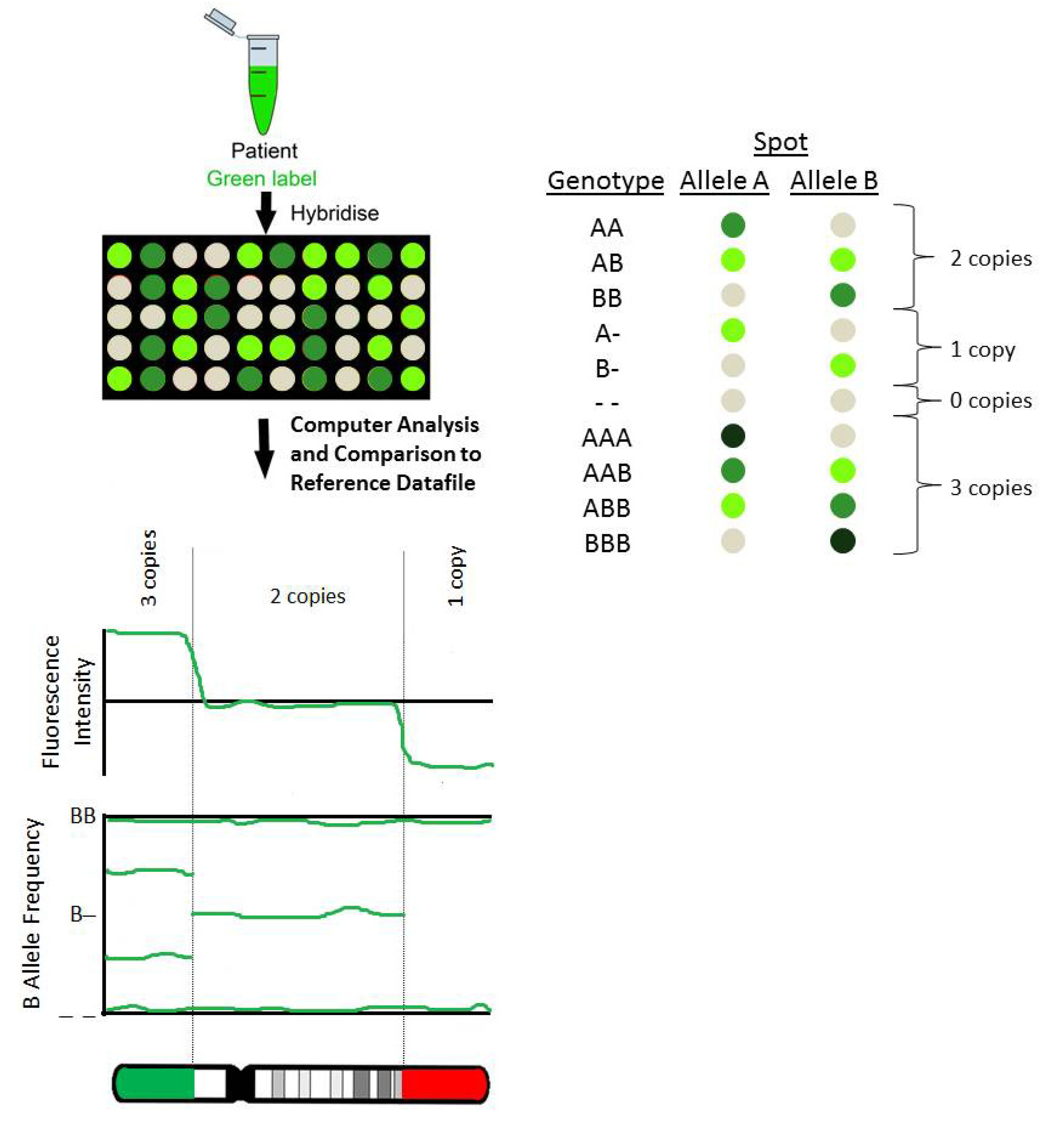
2.1. BAC Arrays

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Array Platform | ||||
|---|---|---|---|---|
| Karyotype | BAC | Oligo CGH | SNP | |
| Resolution | 5–10 Mb | 0.5–1 Mb | 0.05–0.4 Mb | 0.05–0.4 Mb |
| depending on specific platform, design and calling settings | ||||
| Diagnostic yield (excluding common aneuploidies) | around 5% | higher than karyotype | higher than BAC arrays | higher than BAC arrays |
| Diagnostic yield almost double compared to karyotype | ||||
| Detection of CNVs of unknown significance | + | + | ++ | ++ |
| Detection of CNVs of reduced penetrance and variable expressivity | − | + | + | + |
| Starting material (ng) | 50 | 1000 (200–2000) | 200–250 | |
| Turnaround time (working days) (+ 1 day if a rapid result is needed in advance) | 6–10 | 3 | 4 | 4–7 |
| Multiplexing/Throughput | − | 2 samples per slide | 1–8 samples per slide multiplexing in 94-well plates possible | 1 sample per chip or 8 samples per slide multiplexing in 94-well plates possible |
| Detection of MCC | possible only if the fetus is male | − | − | + |
| Detection of triploidy | + | − | − | + |
| Detection of LOH/UPIC | − | − | − | + |
| Detection of mosaicism | + | Depends on size of the locus, type of aberration, platform, normalisation and calling algorithms. Possibly easier detection using SNP arrays | ||
| Cost | comparable to microarray | Depends on throughput, specific platform and overhead costs | ||
2.2. OligoCGH Arrays
2.3. SNP Arrays
3. Array Design
3.1. Targeted
3.2. Whole-Genome
3.3. Whole-Genome and Targeted
3.4. Custom Designs
4. Other Factors to Consider When Implementing a Prenatal Array Service
4.1. Experience
4.2. Costs
4.3. Analysis and Analysis Software
5. Conclusions
| 1 | Current platform used by the laboratory; incorporation in current workflow |
| 2 | Existing experience with and results in-house database from current/previous platforms |
| 3 | Platform resolution; diagnostic yield and potential detection of CNVs of unknown significance |
| 4 | Platform design; whole-genome/targeted, off-the-self/custom-made |
| 5 | Cost for setting-up a prenatal microarray service |
| 6 | Reagents cost per patient; potential for multiplexing |
| 7 | Starting material; DNA extraction method and DNA yield |
| 8 | Report turnaround time |
| 9 | SNP information; detection of triploidy/MCC/LOH |
| 10 | Analysis software |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Dabell, M.P.; Fisher, A.J.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Rosenfeld, J.A. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat. Diagn. 2012, 32, 976–985. [Google Scholar] [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef]
- Fiorentino, F.; Napoletano, S.; Caiazzo, F.; Sessa, M.; Bono, S.; Spizzichino, L.; Gordon, A.; Nuccitelli, A.; Rizzo, G.; Baldi, M. Chromosomal microarray analysis as a first-line test in pregnancies with a priori low risk for the detection of submicroscopic chromosomal abnormalities. Eur. J. Hum. Genet. 2013, 21, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Callaway, J.L.; Shaffer, L.G.; Chitty, L.S.; Rosenfeld, J.A.; Crolla, J.A. The clinical utility of microarray technologies applied to prenatal cytogenetics in the presence of a normal conventional karyotype: A review of the literature. Prenat. Diagn. 2013, 33, 1119–1123. [Google Scholar] [CrossRef]
- Hillman, S.C.; McMullan, D.J.; Hall, G.; Togneri, F.S.; James, N.; Maher, E.J.; Meller, C.H.; Williams, D.; Wapner, R.J.; Maher, E.R.; et al. Use of prenatal chromosomal microarray: Prospective cohort study and systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2013, 41, 610–620. [Google Scholar] [CrossRef]
- American College of Obstetricians and Gynecologists’ Committee on Genetics. Committee Opinion No. 581: The use of chromosomal microarray analysis in prenatal diagnosis. Obstet. Gynecol. 2013, 122, 1374–1377. [Google Scholar] [CrossRef]
- Carter, N.P. Methods and strategies for analyzing copy number variation using DNA microarrays. Nat. Genet. 2007, 39, S16–S21. [Google Scholar] [CrossRef]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef]
- Hochstenbach, R.; Buizer-Voskamp, J.E.; Vorstman, J.A.; Ophoff, R.A. Genome arrays for the detection of copy number variations in idiopathic mental retardation, idiopathic generalized epilepsy and neuropsychiatric disorders: Lessons for diagnostic workflow and research. Cytogenet. Genome Res. 2011, 135, 174–202. [Google Scholar]
- Shaffer, L.G.; Dabell, M.P.; Rosenfeld, J.A.; Neill, N.J.; Ballif, B.C.; Coppinger, J.; Diwan, N.R.; Chong, K.; Shohat, M.; Chitayat, D. Referral patterns for microarray testing in prenatal diagnosis. Prenat. Diagn. 2012, 32, 344–350. [Google Scholar] [CrossRef]
- Hillman, S.C.; McMullan, D.J.; Silcock, L.; Maher, E.R.; Kilby, M.D. How does altering the resolution of chromosomal microarray analysis in the prenatal setting affect the rates of pathological and uncertain findings? J. Matern. Fetal Neonatal Med. 2014, 27, 649–657. [Google Scholar] [CrossRef]
- South, S.T.; Lee, C.; Lamb, A.N.; Higgins, A.W.; Kearney, H.M.; Working Group for the American College of Medical Genetics and Genomics Laboratory Quality Assurance Committee. ACMG Standards and Guidelines for constitutional cytogenomic microarray analysis, including postnatal and prenatal applications: Revision 2013. Genet. Med. 2013, 15, 901–909. [Google Scholar]
- Neill, N.J.; Torchia, B.S.; Bejjani, B.A.; Shaffer, L.G.; Ballif, B.C. Comparative analysis of copy number detection by whole-genome BAC and oligonucleotide array CGH. Mol. Cytogenet. 2010, 3. [Google Scholar] [CrossRef]
- Macdonald, J.R.; Ziman, R.; Yuen, R.K.; Feuk, L.; Scherer, S.W. The Database of Genomic Variants: A curated collection of structural variation in the human genome. Nucleic Acids Res. 2014, 42, D986–D992. [Google Scholar] [CrossRef]
- Bragin, E.; Chatzimichali, E.A.; Wright, C.F.; Hurles, M.E.; Firth, H.V.; Bevan, A.P.; Swaminathan, G.J. DECIPHER: Database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res. 2014, 42, D993–D1000. [Google Scholar] [CrossRef]
- Vulto-Van Silfhout, A.T.; van Ravenswaaij, C.M.; Hehir-Kwa, J.Y.; Verwiel, E.T.; Dirks, R.; van Vooren, S.; Schinzel, A.; de Vries, B.B.; de Leeuw, N. An update on ECARUCA, the European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations. Eur. J. Med. Genet. 2013, 56, 471–474. [Google Scholar] [CrossRef]
- Riggs, E.R.; Church, D.M.; Hanson, K.; Horner, V.L.; Kaminsky, E.B.; Kuhn, R.M.; Wain, K.E.; Williams, E.S.; Aradhya, S.; Kearney, H.M.; et al. Towards an evidence-based process for the clinical interpretation of copy number variation. Clin. Genet. 2012, 81, 403–412. [Google Scholar] [CrossRef]
- Rosenfeld, J.A.; Coe, B.P.; Eichler, E.E.; Cuckle, H.; Shaffer, L.G. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet. Med. 2013, 15, 478–481. [Google Scholar] [CrossRef]
- Cooper, G.M.; Coe, B.P.; Girirajan, S.; Rosenfeld, J.A.; Vu, T.H.; Baker, C.; Williams, C.; Stalker, H.; Hamid, R.; Hannig, V.; et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011, 43, 838–846. [Google Scholar] [CrossRef]
- Mann, K.; Hills, A.; Donaghue, C.; Thomas, H.; Ogilvie, C.M. Quantitative fluorescence PCR analysis of >40,000 prenatal samples for the rapid diagnosis of trisomies 13, 18 and 21 and monosomy X. Prenat. Diagn. 2012, 32, 1197–1204. [Google Scholar] [CrossRef]
- Robinson, W.P. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 2000, 22, 452–459. [Google Scholar] [CrossRef]
- Liao, C.; Fu, F.; Li, R.; Xie, G.E.; Zhang, Y.L.; Li, J.; Li, D.Z. Implementation of high-resolution SNP arrays in the investigation of fetuses with ultrasound malformations: Five years of clinical experience. Clin. Genet. 2013. [Google Scholar] [CrossRef]
- Coppinger, J.; Alliman, S.; Lamb, A.N.; Torchia, B.S.; Bejjani, B.A.; Shaffer, L.G. Whole-genomemicroarray analysis in prenatal specimens identifies clinically significant chromosome alterations without increase in results of unclear significance compared to targeted microarray. Prenat. Diagn. 2009, 29, 1156–1166. [Google Scholar] [CrossRef]
- Santen, G.W.; Aten, E.; Sun, Y.; Almomani, R.; Gilissen, C.; Nielsen, M.; Kant, S.G.; Snoeck, I.N.; Peeters, E.A.; Hilhorst-Hofstee, Y.; et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 2012, 44, 379–380. [Google Scholar] [CrossRef]
- Hoyer, J.; Ekici, A.B.; Endele, S.; Popp, B.; Zweier, C.; Wiesener, A.; Wohlleber, E.; Dufke, A.; Rossier, E.; Petsch, C.; et al. Haplo-insufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 2012, 90, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Vetro, A.; Bouman, K.; Hastings, R.; McMullan, D.J.; Vermeesch, J.R.; Miller, K.; Sikkema-Raddatz, B.; Ledbetter, D.H.; Zuffardi, O.; van Ravenswaaij-Arts, C.M. The introduction of arrays in prenatal diagnosis: A special challenge. Hum. Mutat. 2012, 33, 923–929. [Google Scholar] [CrossRef]
- Lockwood, W.W.; Chari, R.; Chi, B.; Lam, W.L. Recent advances in array comparative genomic hybridization technologies and their applications in human genetics. Eur. J. Hum. Genet. 2006, 14, 139–148. [Google Scholar]
- Lamy, P.; Grove, J.; Wiuf, C. A review of software for microarray genotyping. Hum. Genomics 2011, 5, 304–309. [Google Scholar]
- Boon, E.M.; Faas, B.H. Benefits and limitations of whole genome versus targeted approaches for non-invasive prenatal testing for fetal aneuploidies. Prenat. Diagn. 2013, 33, 563–568. [Google Scholar] [CrossRef]
- Chitty, L.S.; Hill, M.; White, H.; Wright, D.; Morris, S. Non-invasive prenatal testing for aneuploidy—Ready for prime time? Am. J. Obstet. Gynecol. 2012, 206, 269–275. [Google Scholar] [CrossRef]
- Srinivasan, A.; Bianchi, D.W.; Huang, H.; Sehnert, A.J.; Rava, R.P. Non-invasive detection of fetal sub-chromosome abnormalities via deep sequencing of maternal plasma. Am. J. Hum. Genet. 2013, 92, 167–176. [Google Scholar] [CrossRef]
- Snyder, M.W.; Simmons, L.E.; Kitzman, J.O.; Santillan, D.A.; Santillan, M.K.; Gammill, H.S.; Shendure, J. Non-invasive fetal genome sequencing: A primer. Prenat. Diagn. 2013, 33, 547–554. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Karampetsou, E.; Morrogh, D.; Chitty, L. Microarray Technology for the Diagnosis of Fetal Chromosomal Aberrations: Which Platform Should We Use? J. Clin. Med. 2014, 3, 663-678. https://doi.org/10.3390/jcm3020663
Karampetsou E, Morrogh D, Chitty L. Microarray Technology for the Diagnosis of Fetal Chromosomal Aberrations: Which Platform Should We Use? Journal of Clinical Medicine. 2014; 3(2):663-678. https://doi.org/10.3390/jcm3020663
Chicago/Turabian StyleKarampetsou, Evangelia, Deborah Morrogh, and Lyn Chitty. 2014. "Microarray Technology for the Diagnosis of Fetal Chromosomal Aberrations: Which Platform Should We Use?" Journal of Clinical Medicine 3, no. 2: 663-678. https://doi.org/10.3390/jcm3020663