Chromosomal Mosaicism in Human Feto-Placental Development: Implications for Prenatal Diagnosis

Abstract
:
1. Introduction
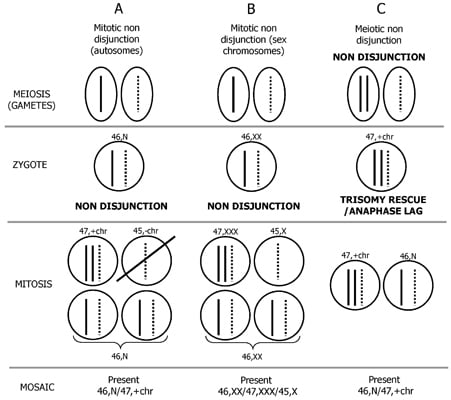
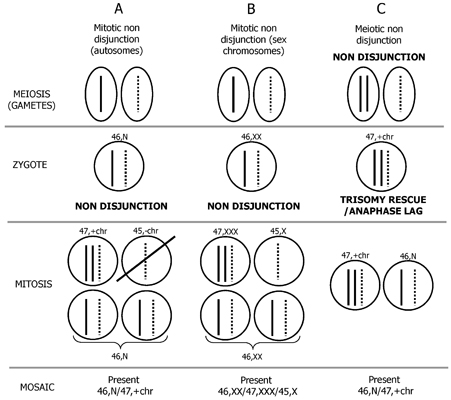
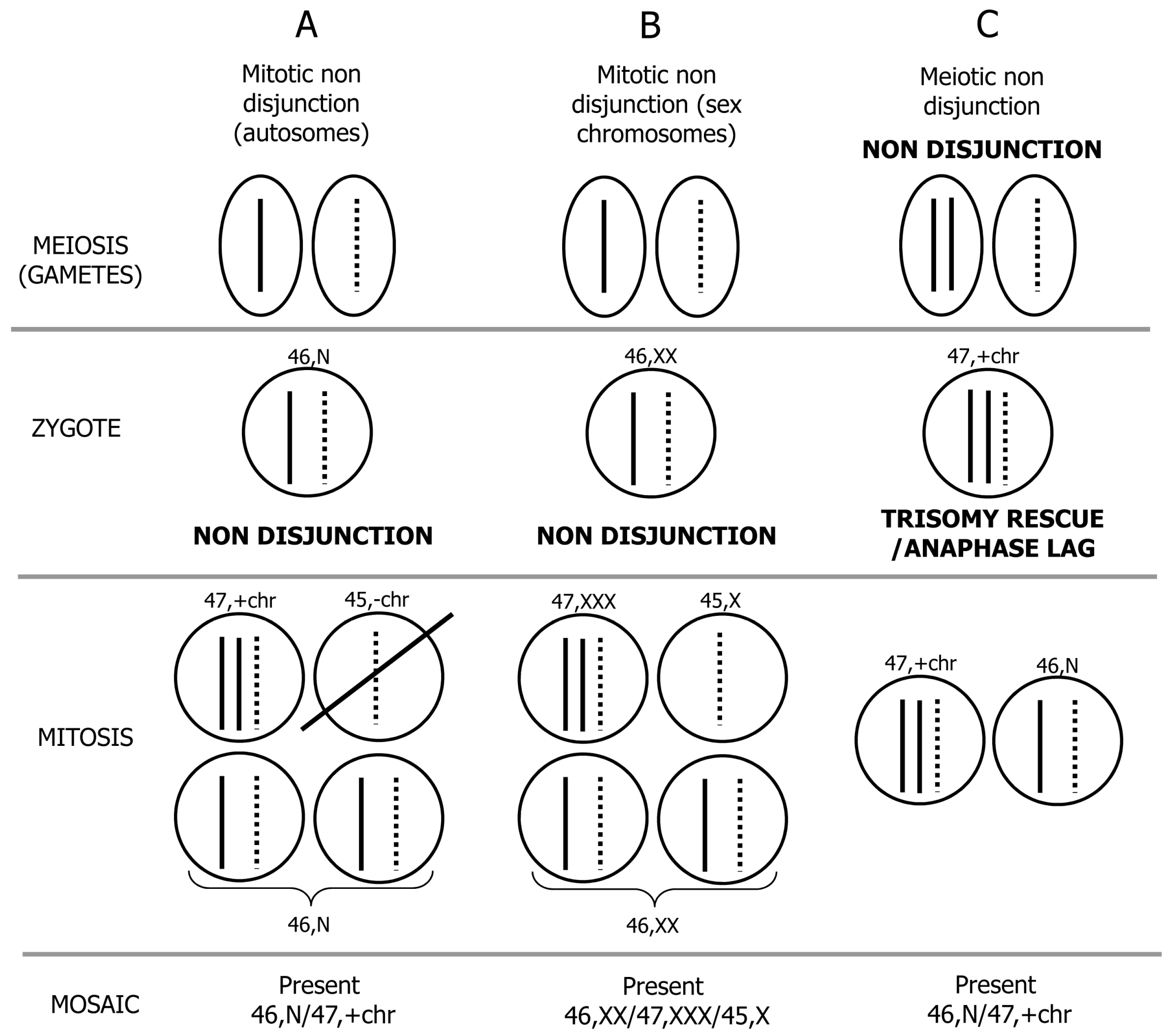
2. Mechanism of Formation of Chromosome Mosaicism
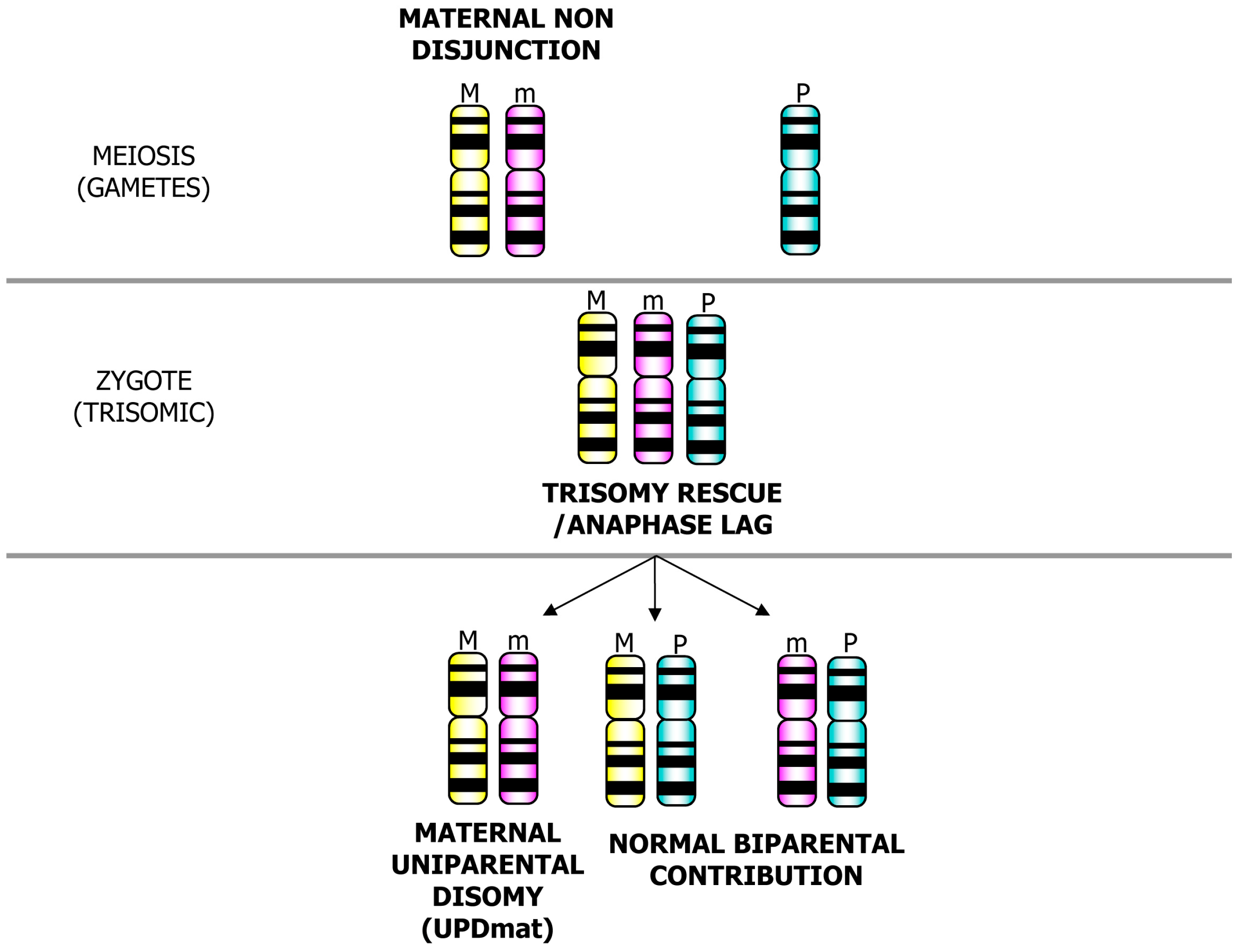
3. Postzygotic Correction of Aneuploidy and Uniparental Disomy (UPD)



{kind=link}
{kind=link}
{kind=link}
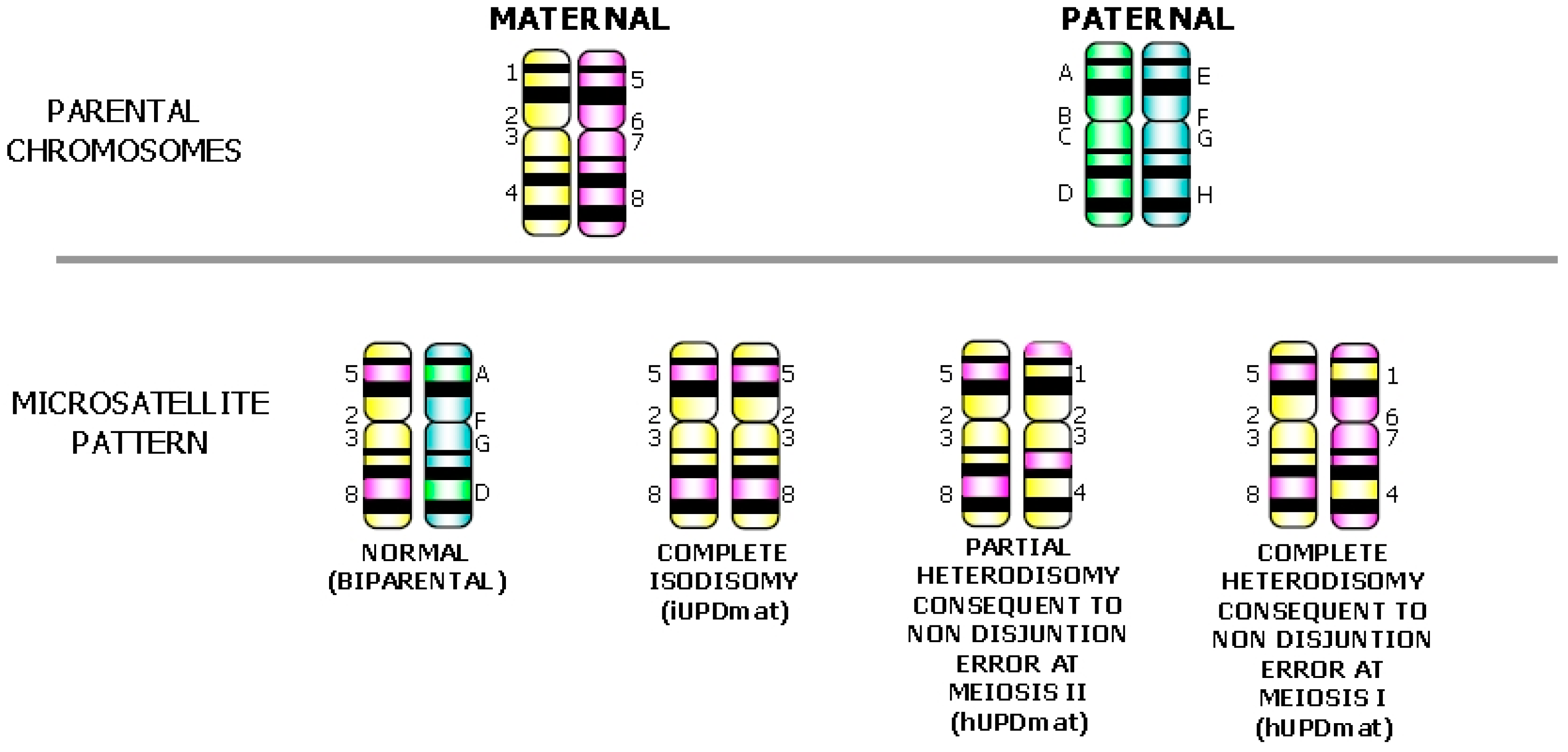{kind=link}
{kind=link}
| UPD type | Syndrome/Disease | OMIM reference ID | Phenotype |
|---|---|---|---|
| paternal UPD6 | Transient neonatal diabete mellitus (TNDM) | #601410 | IUGR, neonatal diabetes |
| maternal UPD7 | Silver-Russell | #180860 | IUGR/PNGR, dysmorfisms |
| maternal UPD11 | Silver-Russell | #180860 | IUGR/PNGR, dysmorfisms |
| paternal UPD11 | Beckwith-Wiedemann | #130650 | Overgrowth, dysmorfisms, tumors (or isolated hemihyperplasia) |
| maternal UPD14 | Temple syndrome | *605636 and #176270 | IUGR, dysmorfisms |
| paternal UPD14 | Bell-shaped thorax, developmental retardation | #608149 | Dwarfisms, dysmorfisms |
| maternal UPD15 | Prader-Willi | #176270 | Obesity, dymorfisms, MR |
| paternal UPD15 | Angelman | #105830 | MR, dysmorfisms |
| maternal UPD20 | Growth failure, hyperactivity | *139320 | IUGR/PNGR |
| paternal UPD20 | Pseudohypoparathyroidism | *139320 | Pseudohypoparathyroidism |
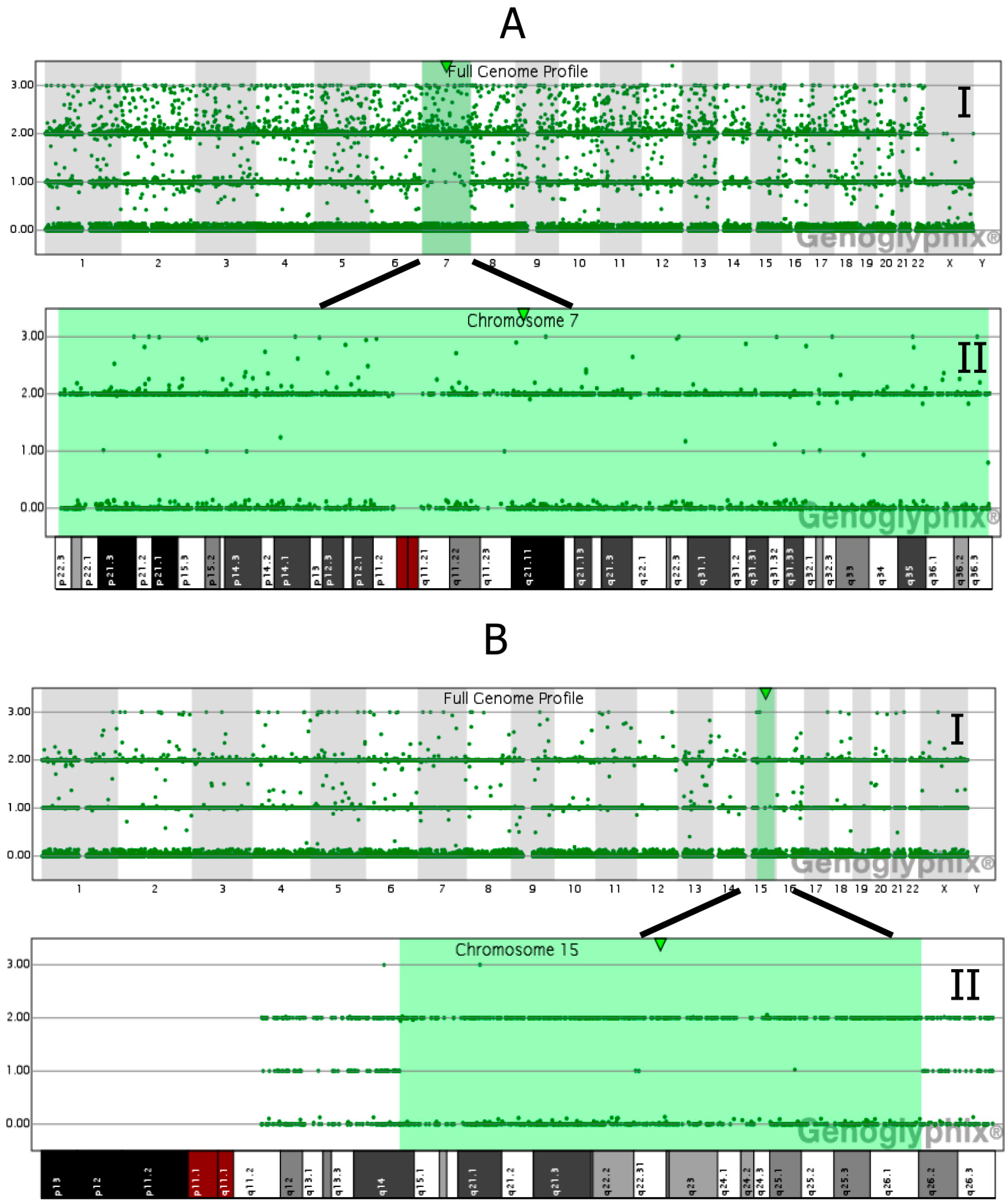
4. Detection of UPD and Discrimination of the Mechanism Generating the Uniparental Disomy Condition

5. Confined Placental and True Fetal Mosaicisms
5.1. Mosaicism in Amniotic Fluid

5.2. Mosaicism in Chorionic Villi
| Type | Nature | Trophoblast | Mesenchyme | Amniocytes | Relative frequencies |
|---|---|---|---|---|---|
| (direct) | (culture) | ||||
| I | CPM | Abnormal | Normal | Normal | 34.76% (308/886) |
| II | CPM | Normal | Abnormal | Normal | 42.32% (375/886) |
| III | CPM | Abnormal | Abnormal | Normal | 10.16% (90/886) |
| IV | TFM | Abnormal | Normal | Abnormal | 1.58% (14/886) |
| V | TFM | Normal | Abnormal | Abnormal | 5.76% (51/886) |
| VI | TFM | Abnormal | Abnormal | Abnormal | 5.42% (48/886) |
5.3. Risk of Fetal Confirmation by Amniocentesis of a Mosaic Abnormality: An Examination of 52,673 Chorionic Villi Samples
| Trophoblast | Mesenchyme | Confirmation |
|---|---|---|
| (direct) | (culture) | |
| A | N | TFM IV/(CPM I + TFM IV) = 14/322 = 4.4% |
| MA | N | 7/(236 + 7) = 2.9% |
| NMA | N | 7/(72 + 7) = 8.9% |
| N | A | TFM V/(CPM II + TFM V) = 51/426 = 12.0% |
| N | MA | 35/(335 + 35) = 9.5% |
| N | NMA | 16/(40 + 16) = 28.6 % |
| A | A | TFM VI/(CPM III + TFM VI) = 48/138 = 34.8% |
| MA | MA | 20/(52 + 20) = 27.8% |
| NMA | MA | 13/(28 + 13) = 31.7 % |
| MA | NMA | 10/(3 + 10) = 76.9 % |
| NMA | NMA | 5/(7 + 5) = 41.7% |
| Chromosome abnormality | Risk of confirmation stratified by type of mosaicism in chorionic villi [Tfm/(Tfm + Cpm)] | Risk of confirmation of each chromosome abnormality | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Placental tissue involvement | |||||||||
| Only cytotrophoblast (CPMI/TFMIV) | Only mesenchyme (CPMII/TFMV) | Both placental tissues (CPMIII/TFMVI) | |||||||
| NMA | MA | NMA | MA | NMA-C/MA-M | MA-C/NMA-M | MA-CM | NMA-CM | ||
| Trisomy 1 | 0/1 = 0 | 0/1 = 0 | |||||||
| Trisomy 2 | 0/1 = 0 | 0/4 = 0 | 0/9 = 0 | 0/48 = 0 | 0/1 = 0 | 0/1 = 0 | 0/64 = 0 | ||
| Trisomy 3 | 0/3 = 0 | 0/20 = 0 | 0/2 = 0 | 0/25 = 0 | |||||
| Trisomy 4 | 0/1 = 0 | 0/1 = 0 | 0/1 = 0 | 1/1 = 100% | 1/4 = 25% | ||||
| Trisomy 5 | 0/2 = 0 | 0/2 = 0 | |||||||
| Trisomy 6 | 0/1 = 0 | 0/1 = 0 | 0/2 = 0 | ||||||
| Trisomy 7 | 0/4 = 0 | 0/32 = 0 | 0/2 = 0 | 0/22 = 0 | 0/3 = 0 | 0/1 = 0 | 0/64 = 0 | ||
| Trisomy 8 | 0/3 = 0 | 0/6 = 0 | 0/2 = 0 | 2/14 = 14.3% | 0/1 = 0 | 2/26 = 7.7% | |||
| Trisomy 9 | 0/1 = 0 | 0/2 = 0 | 0/11 = 0 | 0/1 = 0 | 0/2 = 0 | 0/17 = 0 | |||
| Trisomy 10 | 0/2 = 0 | 0/2 = 0 | 0/1 = 0 | 0/5 = 0 | 0/10 = 0 | ||||
| Trisomy 11 | 0/1 = 0 | 0/2 = 0 | 0/3 = 0 | ||||||
| Trisomy 12 | 0/1 = 0 | 0/1 = 0 | 1/8 = 12.5% | 1/10 = 10% | |||||
| Trisomy 13 | 0/2 = 0 | 1/20 = 5% | 0/10 = 0 | 0/4 = 0 | 0/3 = 0 | 1/39 = 2.6% | |||
| Trisomy 14 | 0/4 * = 0 | 0/3 = 0 | 0/3 = 0 | 0/10 = 0 | |||||
| Trisomy 15 | 0/2 = 0 | 0/12 = 0 | 0/2 * = 0 | 0/5 = 0 | 0/1 = 0 | 0/2 = 0 | 0/24 = 0 | ||
| Trisomy 16 | 0/1 = 0 | 0/2 = 0 | 0/1 = 0 | 0/6 = 0 | 0/3 = 0 | 1/2 = 50% | 1 */2 = 50% | 2/17 = 11.8% | |
| Trisomy 17 | 0/2 = 0 | 0/2 = 0 | |||||||
| Trisomy 18 | 0/10 = 0 | 5/6 = 83.3% | 1/25 = 4% | 0/2 = 0 | 1/2 = 50% | 1/1 = 100% | 8/46 = 17.4% | ||
| Trisomy 19 | 0/1 = 0 | 0/1 = 0 | |||||||
| Trisomy 20 | 0/10 = 0 | 1/1 = 100% | 1/9 = 11.1% | 0/3 = 0 | 0/2 = 0 | 2/25 = 8% | |||
| Trisomy 21 | 1/3 = 33.3% | 0/6 = 0 | 3/5 = 60% | 4/26 = 15.4% | 5/7 = 71.4% | 3/3 = 100% | 0/1 = 0 | 16/51 = 31.4% | |
| Trisomy 22 | 0/3 = 0 | 0/1 = 0 | 0/2 = 0 | 0/1 = 0 | 0/2 = 0 | 0/9 = 0 | |||
| Multiple trisomies | 0/2 = 0 | 0/5 = 0 | 0/2 = 0 | 0/11 = 0 | 0/2 = 0 | 0/1 = 0 | 0/23 = 0 | ||
| All autosomal trisomies | 1/29 = 3.5% | 1/142 = 0.7% | 9/33 = 27.3% | 9/210 = 4.3% | 5/25 = 20% | 4/6 = 66.7% | 2/24 = 8.3% | 2/6 = 33.3% | 33/475 = 6.9% |
| 47,XYY | 0/2 = 0 | 0/2 = 0 | |||||||
| 45,X | 4/15 = 26.7% | 3/30 = 10% | 3/4 = 75% | 8/33 = 24.2% | 0/2 = 0 | 1/1 = 100% | 7/16 = 43.8% | 26/101 = 25.7% | |
| 47,XXY | 0/5 = 0 | 1/1 = 100% | 3/7 = 42.9% | 1/1 = 100% | 2/2 = 100% | 0/1 = 0 | 7/17 = 42.2% | ||
| 47,XXX | 1/2 = 50% | 0/4 = 0 | 0/1 = 0 | 2/2 = 100% | 1/1 = 100% | 4/10 = 40% | |||
| 45,X/46,XX/47,XXX | 1/1 = 100% | 1/1 = 100% | 4/4 = 100% | 6/6 = 100% | |||||
| All sex chromosome aneuploidies | 5/17 = 29.4% | 3/41 = 7.3% | 4/5 = 80% | 12/42 = 28.6% | 2/4 = 50% | 5/5 = 100% | 12/22 = 37.5% | 43/136 = 31.6% | |
| 45,−22 | 0/1 = 0 | 0/1 = 0 | |||||||
| 47,+mar | 1/1 = 100% | 3/12 = 25% | 1/2 = 50% | 6/26 = 23.1% | 2/2 = 100% | 1/2 = 50% | 5/8 = 62.5% | 19/50 = 35.8% | |
| 47,+der | 0/2 = 0 | 0/4 = 0 | 0/1 = 0 | 0/2 = 0 | 0/9 = 0 | ||||
| 46,der | 0/10 = 0 | 0/26 = 0 | 0/5 = 0 | 3/54 = 5.6% | 1/1 = 100% | 1/1 = 100% | 5/97 = 5.2% | ||
| Triploid | 1/1 = 100% | 1/1 = 100% | 2/2 = 100% | ||||||
| Tetraploidy | 0/16 = 0 | 0/9 = 0 | 0/6 = 0 | 0/4 = 0 | 0/6 = 0 | 0/14 = 0 | 0/1 = 0 | 0/53 = 0 | |
| 47,+i(13q) | 0/4 = 0 | 0/4 = 0 | 0/4 = 0 | ||||||
| 47,+i(7p) | 0/3 = 0 | 0/3 = 0 | |||||||
| Other § | 1/4 = 25% | 2/2 = 100% | 3/5 = 60% | 6/11 = 54.5% | |||||
| 46,t or 45,rob | 0/4 = 0 | 0/5 = 0 | 2/4 = 50% | 3/21 = 14.3% | 0/1 = 0 | 5/35 = 14.3% | |||
| All remaining abnormalities | 1/33 = 3% | 3/60 = 5% | 3/18 = 16.7% | 14/118 = 11.9% | 6/12 = 50% | 1/2 = 50% | 6/26 = 23.1% | 3/6 = 50% | 37/275 = 13.5% |
| Type of mosaicism | Trophoblast | Mesenchyme | Amniocytes |
|---|---|---|---|
| CPM II | 46,XX[15] | 47,XX,+9[12]/47,XX,+9,der(22)[2] | 46,XX |
| 46,XX[11] | 47,XX,+12,t(12;12)[4]/46,XX[26] | 46,XX | |
| 46,XX[10] | 46,XX,fra(10)(q12) * | 46,XX | |
| CPM III | 46,XX,add(8)[14] | 46,XX,add(2)[13] | 46,XX |
| 46,X,+mar[20] | 45,X[9] | 46,XY | |
| TFM V | 46,XY[14] | 47,XY,+7[3]/46,XY[9] | mos 45,X[9]/46,XY[41] |
| TFM VI | 46,X,der(Y)[15] | 46,X,+mar[3]/45,X[2]/47,XX,+mar[3]/46,XX[24]/46,XY[3] | mos 45,X[40]/46,XY[11] |
| 46,XX,add(13)[16] | 46,XX,add(13)[3]/46,XX[7] | 46,XX,inv(13)(q11.1q32.1)dn.ish inv(13)(q11.1q32.1)dn(D13Z1/D21Z1+,D13S319+,LAMP1+,D13S1160+).arr(1-22,X)x2 | |
| 46,XY,r(22)[19] | 45,XY,der(21;22)[5] | 46,XY,r(22)(p11;q13) | |
| 46,XY,add(7)[15] | 46,XY,del(7)[15] | 46,XY,del(7)(q32).ish del(7)(q32)(ELN+,LIMK1+,D7S613+,D7S486+,D7S522+,D7S427−) | |
| 46,XX,add(6)[12] | 47,XX,6ps,+mar[11] | 46,XX,del(6)(p25.3) |
6. Risk of Fetal Uniparental Disomy (UPD) after the Detection of a Mosaic Abnormality Involving an Imprinted Chromosome
| Type of abnormality | No. investigated cases | No. UPD | Type of CPM or TFM | UPD incidence (%) |
|---|---|---|---|---|
| trisomy 2 | 62 | - | - | - |
| trisomy 7 | 60 | - | - | - |
| trisomy 6 | 2 | - | - | - |
| trisomy 11 | 3 | - | - | - |
| trisomy 14 | 10 | 2 | 2 (CPM type I) | 20 |
| trisomy 15 | 24 | 1 | 1 (CPM type II) | 4.2 |
| trisomy 16 | 17 | 3 | 2 (CPM type III) + 1 (TFM type VI) | 17.6 |
| trisomy 20 | 25 | - | - | - |
| sSMC, others | 40 | - | - | - |
| Total | 243 | 6 | 5 CPM and 1 TFM | 2.5 |
| Study | Total No. of CVS sample | No. of CVS samples with trisomy 2 | Prevalence of trisomy 2 in CV | No. of TFM with trisomy 2 after a mosaic in CV | No. of cases with UPD investigation | No. of UPD2 retrieved | Incidence of UPD in mosaic trisomy 2 in CV (%) | |
|---|---|---|---|---|---|---|---|---|
| % | 1/x | |||||||
| Wolstenholme, 1996 | 66,129 | 41 | 0.06 | 1613 | na | na | na | na |
| Hahnemann and Vejerslev, 1997 | 92,246 | 11 | 0.01 | 8386 | 0 | na | na | na |
| Sago et al., 1997 | 10,500 | 11 | 0.10 | 955 | na | 11 | 0 | 0 |
| Sifakis et al., 2010 | 37,474 | 45 | 0.12 | 833 | 0 | 43 | 0 | 0 |
| Present Study * | 52,673 | 74 | 0.14 | 712 | 0 | 62 | 0 | 0 |
| Total | 259,022 | 182 | 0.07 | 1423 | 0 | 116 | 0 | 0 |
| Study | Total No. of CVS sample | No. of CVS samples with trisomy 7 | Prevalence of trisomy 7 in CV | No. of TFM with trisomy 7 after a mosaic in CV | No. of cases with UPD investigation | No. of UPD7 retrieved | Incidence of UPD in mosaic trisomy 7 in CV (%) | |
|---|---|---|---|---|---|---|---|---|
| % | 1/x | |||||||
| Wolstenholme, 1996 | 66,129 | 60 | 0.09 | 1102 | na | na | na | na |
| Hahnemann and Vejerslev, 1997 | 92,246 | 32 | 0.03 | 2883 | 0 | na | na | na |
| Sachs et al., 1990 | 3000 | 5 | 0.17 | 600 | 0 | na | na | na |
| Kalousek et al., 1996 | na | na | na | na | na | 14 | 1 | 7.1 |
| Present Study * | 52,673 | 73 | 0.14 | 722 | 0 | 62 | 0 | 0 |
| Total | 214,048 | 170 | 0.08 | 1259 | 0 | 76 | 1 | 1.3 |
7. Molecular Techniques for Detection of Chromosomal Mosaicism: Fluorescence in Situ Hybridization Analysis (FISH), Quantitative Fluorescent Polymerase Chain Reaction (QF-PCR), and Chromosomal Microarrays (Array Comparative Genomic Hybridization, aCGH; Single Nucleotide Polymorphism Array, SNP Array)
8. Potential False Positive and False Negative Results Using only (Semi-)Direct Preparation (STC) or Long-Term Culture (LTC)
9. Potential False Positive and False Negative Results with Non-invasive Prenatal Screening (NIPS) for Screening (NIPS) for Common Aneuploidies Due to Feto-Placental Mosaicism
10. Conclusions
Supplementary Files
Supplementary File 1Abbreviations
| aCGH | array comparative genomic hybridization |
| AF | amniotic fluid |
| cffDNA | cell free fetal DNA |
| cfpDNA | cell free placental DNA |
| CNV | copy number variation |
| CPM | confined placental mosaicism |
| CVS | chorionic villous sample |
| FISH | fluorescence in situ hybridization |
| FN | false negative |
| FP | false positive |
| h/UPDmat/pat | maternal/paternal heterodisomy/isodisomy in uniparental disomy condition |
| IUGR | intrauterine growth restriction |
| LTC | long term culture |
| MA | mosaic abnormality |
| MCC | maternal cell contamination |
| NDJ | non disjunction |
| NIPS | non-invasive prenatal screening |
| NGS | next generation sequencing |
| NMA | non mosaic abnormality |
| PNGR | postnatal growth retardation |
| QF-PCR | quantitative fluorescence polymerase chain reaction |
| STC | short term culture |
| SNP | single nucleotide polymorphism |
| STR | short tandem repeat |
| TFM | true fetal mosaicism |
| UPD | uniparental disomy |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Brambati, B.; Simoni, G. Diagnosis of fetal trisomy 21 in first trimester. Lancet 1983, 1, 586. [Google Scholar]
- Simoni, G.; Fraccaro, M.; Gimelli, G.; Maggi, F.; Dagna Bricarelli, F. False-positive and false-negative findings on chorionic villus sampling. Prenat. Diagn. 1987, 7, 671–672. [Google Scholar] [CrossRef]
- Miny, P.; Basaran, S.; Holzgreve, W.; Horst, J.; Pawlowitzki, I.H.; Ngo, T.K. False negative cytogenetic result in direct preparations after CVS. Prenat. Diagn. 1988, 8, 633. [Google Scholar] [CrossRef]
- Tomkins, D.J.; Vekemans, M.J. False-positive and false-negative cytogenetic findings on chorionic villus sampling. Prenat. Diagn. 1989, 9, 139–140. [Google Scholar] [CrossRef]
- Simoni, G.; Sirchia, S.M. Confined placental mosaicism. Prenat. Diagn. 1994, 14, 1185–1189. [Google Scholar] [CrossRef]
- Kalousek, D.K.; Barrett, I. Confined placental mosaicism and stillbirth. Pediatr. Pathol. 1994, 14, 151–159. [Google Scholar] [CrossRef]
- Ledbetter, D.H.; Zachary, J.M.; Simpson, J.L.; Golbus, M.S.; Pergament, E.; Jackson, L.; Mahoney, M.J.; Desnick, R.J.; Schulman, J.; Copeland, K.L.; et al. Cytogenetic results from the U.S. Collaborative Study on CVS. Prenat. Diagn. 1992, 12, 317–345. [Google Scholar] [CrossRef]
- Cytogenetic analysis of chorionic villi for prenatal diagnosis: An ACC collaborative study of UK data. Association of Clinical Cytogeneticists Working Party on Chorionic Villi in Prenatal Diagnosis. Prenat. Diagn. 1994, 14, 363–379. [CrossRef]
- Strachan, T.; Read, A.P. Human Molecular Genetics; Garland Science: New York, NY, USA, 2011. [Google Scholar]
- Hook, E.B. Exclusion of chromosomal mosaicism: Tables of 90%, 95% and 99% confidence limits and comments on use. Am. J. Hum. Genet. 1977, 29, 94–97. [Google Scholar]
- Iourov, I.Y.; Vorsanova, S.G.; Yurov, Y.B. Chromosomal mosaicism goes global. Mol. Cytogenet. 2008, 1. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef]
- Porter, S.; Wilson, E.; Tyler, X.; Warren, R.; ffrench-Constant, C.; Pearson, J. A case of discordant related abnormal karyotypes from chorionic villi and amniocytes. Prenat. Diagn. 1999, 19, 887–890. [Google Scholar] [CrossRef]
- Soler, A.; Sánchez, A.; Carrió, A.; Badenas, C.; Milà, M.; Borrell, A. Fetoplacental discrepancy involving structural abnormalities of chromosome 8 detected by prenatal diagnosis. Prenat. Diagn. 2003, 23, 319–322. [Google Scholar] [CrossRef]
- Brisset, S.; Aboura, A.; Audibert, F.; Costa, J.M.; L’Herminé, A.C.; Gautier, V.; Frydman, R.; Tachdjian, G. Discordant prenatal diagnosis of trisomy 21 due to mosaic structural rearrangements of chromosome 21. Prenat. Diagn. 2003, 23, 461–469. [Google Scholar] [CrossRef]
- Vejerslev, L.O.; Mikkelsen, M. The European collaborative study on mosaicism in chorionic villus sampling: Data from 1986 to 1987. Prenat. Diagn. 1989, 9, 575–588. [Google Scholar] [CrossRef]
- Medical Research Council working party on the evaluation of chorionic villus sampling: Medical Research Council European trial of chorionic villus sampling. Lancet 1991, 337, 1491–1499. [CrossRef]
- Teshima, I.E.; Kalousek, D.K.; Vekemans, M.J.; Markovic, V.; Cox, D.M.; Dallaire, L.; Gagne, R.; Lin, J.C.; Ray, M.; Sergovich, F.R.; et al. Canadian multicenter randomized clinical trial of chorion villus sampling and amniocentesis. Chromosome mosaicism in CVS and amniocentesis samples. Prenat. Diagn. 1992, 12, 443–466. [Google Scholar] [CrossRef]
- Smidt-Jensen, S.; Lind, A.M.; Permin, M.; Zachary, J.M.; Lundsteen, C.; Philip, J. Cytogenetic analysis of 2928 CVS samples and 1075 amniocenteses from randomized studies. Prenat. Diagn. 1993, 13, 723–740. [Google Scholar] [CrossRef]
- Wang, B.B.; Rubin, C.H.; Williams, J., III. Mosaicism in chorionic villus sampling: An analysis of incidence and chromosomes involved in 2612 consecutive cases. Prenat. Diagn. 1993, 13, 179–190. [Google Scholar] [CrossRef]
- Wolstenholme, J.; Rooney, D.E.; Davison, E.V. Confined placental mosaicism; IUGR; and adverse pregnancy outcome: A controlled retrospective UK collaborative survey. Prenat. Diagn. 1994, 14, 345–361. [Google Scholar] [CrossRef]
- Stetten, G.; Escallon, C.S.; South, S.T.; McMichael, J.L.; Saul, D.O.; Blakemore, K.J. Reevaluating confined placental mosaicism. Am. J. Med. Genet. 2004, 131, 232–239. [Google Scholar]
- Grati, F.R.; Grimi, B.; Frascoli, G.; di Meco, A.M.; Liuti, R.; Milani, S.; Trotta, A.; Dulcetti, F.; Grosso, E.; Miozzo, M.; et al. Confirmation of mosaicism and uniparental disomy in amniocytes; after detection of mosaic chromosome abnormalities in chorionic villi. Eur. J. Hum. Genet. 2006, 14, 282–288. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Avramopoulos, D.; Blouin, J.L.; Talbot, C.C., Jr.; Schinzel, A.A. Mitotic errors in somatic cells cause trisomy 21 in about 4.5% of cases and are not associated with advanced maternal age. Nat. Genet. 1993, 3, 146–150. [Google Scholar] [CrossRef]
- Kalousek, D.K.; Langlois, S.; Robinson, W.P.; Telenius, A.; Bernard, L.; Barrett, I.J.; Howard-Peebles, P.N.; Wilson, R.D. Trisomy 7 CVS mosaicism: Pregnancy outcome; placental and DNA analysis in 14 cases. Am. J. Med. Genet. 1996, 65, 348–352. [Google Scholar] [CrossRef]
- Sirchia, S.M.; Garagiola, I.; Colucci, G.; Guerneri, S.; Lalatta, F.; Grimoldi, M.G.; Simoni, G. Trisomic zygote rescue revealed by DNA polymorphism analysis in confined placental mosaicism. Prenat. Diagn. 1998, 18, 201–206. [Google Scholar] [CrossRef]
- Engel, E. A new genetic concept: Uniparental disomy and its potential effect; isodisomy. Am. J. Med. Genet. 1980, 6, 137–143. [Google Scholar] [CrossRef]
- Ledbetter, D.H.; Engel, E. Uniparental disomy in humans: Development of an imprinting map and its implications for prenatal diagnosis. Hum. Mol. Genet. 1995, 4, 1757–1764. [Google Scholar]
- Engel, E.; DeLozier-Blanchet, C.D. Uniparental disomy; isodisomy; and imprinting: Probable effects in man and strategies for their detection. Am. J. Med. Genet. 1991, 40, 432–439. [Google Scholar] [CrossRef]
- Engel, E. Uniparental disomy revisited: The first twelve years. Am. J. Med. Genet. 1993, 46, 670–674. [Google Scholar] [CrossRef]
- Miozzo, M.; Simoni, G. The role of imprinted genes in fetal growth. Biol. Neonate 2002, 81, 217–228. [Google Scholar] [CrossRef]
- Yamazawa, K.; Ogata, T.; Ferguson-Smith, A.C. Uniparental disomy and human disease: An overview. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154C, 329–334. [Google Scholar] [CrossRef]
- Liehr, T. Cytogenetic contribution to uniparental disomy (UPD). Mol. Cytogenet. 2010, 3. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Agan, N.; Goldberg, J.D.; Ledbetter, D.H.; Longshore, J.W.; Cassidy, S.B. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet. Med. 2001, 3, 206–211. [Google Scholar] [CrossRef]
- Engel, E.; Antonarakis, S.E. Genomic Imprinting and Uniparental Disomy in Medicine: Clinical and Molecular Aspects; Wiley-Liss: New York, NY, USA, 2001. [Google Scholar]
- Kotzot, D. Prenatal testing for uniparental disomy: Indications and clinical relevance. Ultrasound Obstet. Gynecol. 2008, 31, 100–105. [Google Scholar] [CrossRef]
- Dawson, A.J.; Chernos, J.; McGowan-Jordan, J.; Lavoie, J.; Shetty, S.; Steinraths, M.; Wang, J.-C.; Xu, J. CCMG guidelines: Prenatal and postnatal diagnostic testing for uniparental disomy. Clin. Genet. 2011, 79, 118–124. [Google Scholar] [CrossRef]
- Fernández-Rebollo, E.; Lecumberri, B.; Garin, I.; Arroyo, J.; Bernal-Chico, A.; Goñi, F.; Orduña, R.; Spanish PHP Group; Castaño, L.; de Nanclares, G.P. New mechanisms involved in paternal 20q disomy associated with pseudohypoparathyroidism. Eur. J. Endocrinol. 2010, 163, 953–962. [Google Scholar] [CrossRef]
- Miozzo, M.; Grati, F.R.; Bulfamante, G.; Rossella, F.; Cribiù, M.; Radaelli, T.; Cassani, B.; Persico, T.; Cetin, I.; Pardi, G.; et al. Post-zygotic origin of complete maternal chromosome 7 isodisomy and consequent loss of placental PEG1/MEST expression. Placenta 2001, 22, 813–821. [Google Scholar] [CrossRef]
- Robinson, W.P.; Barrett, I.J.; Bernard, L.; Telenius, A.; Bernasconi, F.; Wilson, R.D.; Best, R.G.; Howard-Peebles, P.N.; Langlois, S.; Kalousek, D.K. Meiotic origin of trisomy in confined placental mosaicism is correlated with presence of fetal uniparental disomy; high levels of trisomy in trophoblast; and increased risk of fetal intrauterine growth restriction. Am. J. Hum. Genet. 1997, 60, 917–927. [Google Scholar]
- Tucker, T.; Schlade-Bartusiak, K.; Eydoux, P.; Nelson, T.N.; Brown, L. Uniparental disomy: Can SNP array data be used for diagnosis? Genet. Med. 2012, 14, 753–756. [Google Scholar] [CrossRef]
- Hubbard, V.S.; Davis, P.B.; di Sant’Agnese, P.A.; Gorden, P.; Schwartz, R.H. Isolated growth hormone deficiency and cystic fibrosis: A report of two cases. Am. J. Dis. Child. 1980, 134, 317–319. [Google Scholar]
- Spotila, L.D.; Sereda, L.; Prockop, D.J. Partial isodisomy for maternal chromosome 7 and short stature in an individual with a mutation at the COL1A2 locus. Am. J. Hum. Genet. 1992, 51, 1396–1405. [Google Scholar]
- Höglund, P.; Holmberg, C.; de la Chapelle, A.; Kere, J. Paternal isodisomy for chromosome 7 is compatible with normal growth and development in a patient with congenital chloride diarrhea. Am. J. Hum. Genet. 1994, 55, 747–752. [Google Scholar]
- Brzustowicz, L.M.; Allitto, B.A.; Matseoane, D.; Theve, R.; Michaud, L.; Chatkupt, S.; Sugarman, E.; Penchaszadeh, G.K.; Suslak, L.; Koenigsberger, M.R.; et al. Paternal isodisomy for chromosome 5 in a child with spinal muscular atrophy. Am. J. Hum. Genet. 1994, 54, 482–488. [Google Scholar]
- Woodage, T.; Prasad, M.; Dixon, J.W.; Selby, R.E.; Romain, D.R.; Columbano-Green, L.M.; Graham, D.; Rogan, P.K.; Seip, J.R.; Smith, A.; et al. Bloom syndrome and maternal uniparental disomy for chromosome 15. Am. J. Hum. Genet. 1994, 55, 74–80. [Google Scholar]
- Worton, R.G.; Stern, R. A Canadian collaborative study of mosaicism in amniotic fluid cell cultures. Prenat. Diagn. 1984, 4, 131–144. [Google Scholar] [CrossRef]
- Hsu, L.Y.; Kaffe, S.; Jenkins, E.C.; Alonso, L.; Benn, P.A.; David, K.; Hirschhorn, K.; Lieber, E.; Shanske, A.; Shapiro, L.R.; et al. Proposed guidelines for diagnosis of chromosome mosaicism in amniocytes based on data derived from chromosome mosaicism and pseudomosaicism studies. Prenat. Diagn. 1992, 12, 555–573. [Google Scholar] [CrossRef]
- Hsu, L.Y.; Benn, P.A. Revised guidelines for the diagnosis of mosaicism in amniocytes. Prenat. Diagn. 1999, 19, 1081–1082. [Google Scholar] [CrossRef]
- Gardner, R.J.; Sutherland, G.R.; Shaffer, L.G. Chromosome Abnormalities and Genetic Counseling, 4th ed.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Bianchi, D.W.; Wilkins-Haug, L.E.; Enders, A.C.; Hay, E.D. Origin of extraembryonic mesoderm in experimental animals: Relevance to chorionic mosaicism in humans. Am. J. Med. Genet. 1993, 46, 542–550. [Google Scholar] [CrossRef]
- Crane, J.P.; Cheung, S.W. An embryogenic model to explain cytogenetic inconsistencies observed in chorionic villus vs. fetal tissue. Prenat. Diagn. 1988, 8, 119–129. [Google Scholar] [CrossRef]
- Shalev, E.; Zalel, Y.; Weiner, E.; Cohen, H.; Shneur, Y. The role of cordocentesis in assessment of mosaicism found in amniotic fluid cell culture. Acta Obstet. Gynecol. Scand. 1994, 73, 119–122. [Google Scholar] [CrossRef]
- Chiesa, J.; Hoffet, M.; Rousseau, O.; Bourgeois, J.M.; Sarda, P.; Mares, P.; Bureau, J.P. Pallister-Killian syndrome [i(12p)]: First pre-natal diagnosis using cordocentesis in the second trimester confirmed by in situ hybridization. Clin. Genet. 1998, 54, 294–302. [Google Scholar]
- Moradkhani, K.; Puechberty, J.; Blanchet, P.; Coubes, C.; Lallaoui, H.; Lewin, P.; Lefort, G.; Sarda, P. Mosaic trisomy 16 in a fetus: The complex relationship between phenotype and genetic mechanisms. Prenat. Diagn. 2006, 26, 1179–1182. [Google Scholar] [CrossRef]
- Hartmann, A.; Hofmann, U.B.; Hoehn, H.; Broecker, E.B.; Hamm, H. Postnatal confirmation of prenatally diagnosed trisomy 20 mosaicism in a patient with linear and whorled nevoid hypermelanosis. Pediatr. Dermatol. 2004, 21, 636–641. [Google Scholar] [CrossRef]
- Chen, C.P.; Su, Y.N.; Chern, S.R.; Chen, Y.T.; Wu, P.S.; Su, J.W.; Pan, C.W.; Wang, W. Mosaic trisomy 2 at amniocentesis: Prenatal diagnosis and molecular genetic analysis. Taiwan J. Obstet. Gynecol. 2012, 51, 603–611. [Google Scholar] [CrossRef]
- Simoni, G.; Fraccaro, M. Does confined placental mosaicism affect the fetus? Hum. Reprod. 1992, 7, 139–140. [Google Scholar]
- Kalousek, D.K. Pathogenesis of chromosomal mosaicism and its effect on early human development. Am. J. Med. Genet. 2000, 91, 39–45. [Google Scholar] [CrossRef]
- Robinson, W.P.; McFadden, D.E.; Barrett, I.J.; Kuchinka, B.; Peñaherrera, M.S.; Bruyère, H.; Best, R.G.; Pedreira, D.A.; Langlois, S.; Kalousek, D.K. Origin of amnion and implications for evaluation of the fetal genotype in cases of mosaicism. Prenat. Diagn. 2002, 22, 1076–1085. [Google Scholar] [CrossRef]
- Kalousek, D.K.; Barrett, I.J.; Gartner, A.B. Spontaneous abortion and confined chromosomal mosaicism. Hum. Genet. 1992, 88, 642–646. [Google Scholar] [CrossRef]
- Hahnemann, J.M.; Vejerslev, L.O. European collaborative research on mosaicism in CVS (EUCROMIC)—Fetal and extrafetal cell lineages in 192 gestations with CVS mosaicism involving single autosomal trisomy. Am. J. Med. Genet. 1997, 70, 179–187. [Google Scholar] [CrossRef]
- Daniel, A.; Wu, Z.; Darmanian, A.; Malafiej, P.; Tembe, V.; Peters, G.; Kennedy, C.; Adès, L. Issues arising from the prenatal diagnosis of some rare trisomy mosaics—The importance of cryptic fetal mosaicism. Prenat. Diagn. 2004, 24, 524–536. [Google Scholar] [CrossRef]
- Robinson, W.P.; Peñaherrera, M.S.; Jiang, R.; Avila, L.; Sloan, J.; McFadden, D.E.; Langlois, S.; von Dadelszen, P. Assessing the role of placental trisomy in preeclampsia and intrauterine growth restriction. Prenat. Diagn. 2010, 30, 1–8. [Google Scholar] [CrossRef]
- Kalousek, D.K. Current topic: Confined placental mosaicism and intrauterine fetal development. Placenta 1994, 15, 219–230. [Google Scholar] [CrossRef]
- Amor, D.J.; Neo, W.T.; Waters, E.; Heussler, H.; Pertile, M.; Halliday, J. Health and developmental outcome of children following prenatal diagnosis of confined placental mosaicism. Prenat. Diagn. 2006, 26, 443–448. [Google Scholar] [CrossRef]
- Simoni, G.; Brambati, B.; Danesino, C.; Rossella, F.; Terzoli, G.L.; Ferrari, M.; Fraccaro, M. Efficient direct chromosome analyses and enzyme determinations from chorionic villi samples in the first trimester of pregnancy. Hum. Genet. 1983, 63, 349–357. [Google Scholar] [CrossRef]
- Verma, R.S.; Babu, A. Human Chromosomes Principles and Techniques; McGraw-Hill Inc.: Milano, Italy, 1995; Chapter 2.16; pp. 24–26. [Google Scholar]
- Grati, F.R.; Malvestiti, F.; Ferreira, J.C.P.B.; Bajaj, K.; Gaetani, E.; Agrati, C.; Grimi, B.; Dulcetti, F.; Ruggeri, A.M.; de Toffol, S.; et al. Feto-placental mosacism: Potential implications for false positive and false negative non-invasive prenatal screening results. Genet. Med. 2014. [Google Scholar] [CrossRef]
- Gentile, M.; Volpe, P.; Cariola, F.; di Carlo, A.; Marotta, V.; Buonadonna, A.L.; Boscia, F.M. Prenatal diagnosis of chromosome 4 mosaicism: Prognostic role of cytogenetic; molecular; and ultrasound/MRI characterization. Am. J. Med. Genet. A 2005, 136, 66–70. [Google Scholar]
- Brady, A.N.; May, K.M.; Fernhoff, P.M. Mosaic trisomy 4: Long-term outcome on the first reported liveborn. Am. J. Med. Genet. A 2005, 132, 411–413. [Google Scholar] [CrossRef]
- Chen, C.P.; Chern, S.R.; Lee, C.C.; Chang, T.Y.; Wang, W.; Tzen, C.Y. Clinical; cytogenetic; and molecular findings of prenatally diagnosed mosaic trisomy 4. Prenat. Diagn. 2004, 24, 38–44. [Google Scholar] [CrossRef]
- Wolstenholme, J. Confined placental mosaicism for trisomies 2; 3; 7; 8; 9; 16; and 22: Their incidence; likely origins; and mechanisms for cell lineage compartmentalization. Prenat. Diagn. 1996, 16, 511–524. [Google Scholar] [CrossRef]
- Malvestiti, F.; de Toffol, S.; Grimi, B.; Chinetti, S.; Marcato, L.; Agrati, C.; di Meco, A.M.; Frascoli, G.; Trotta, A.; Malvestiti, B.; et al. De novo small supernumerary marker chromosomes detected on 143,000 consecutive prenatal diagnoses: Chromosomal distribution; frequencies and characterization combining molecular-cytogenetics approaches. Prenat. Diagn. 2014. [Google Scholar] [CrossRef]
- Hsu, L.Y.; Yu, M.T.; Richkind, K.E.; van Dyke, D.L.; Crandall, B.F.; Saxe, D.F.; Khodr, G.S.; Mennuti, M.; Stetten, G.; Miller, W.A.; et al. Incidence and significance of chromosome mosaicism involving an autosomal structural abnormality diagnosed prenatally through amniocentesis: A collaborative study. Prenat. Diagn. 1996, 16, 1–28. [Google Scholar] [CrossRef]
- Grati, F.R.; Malvestiti, F.; Grimi, B.; Gaetani, E.; di Meco, A.M.; Trotta, A.; Liuti, R.; Chinetti, S.; Dulcetti, F.; Ruggeri, A.M.; et al. QF-PCR as a substitute for karyotyping of cytotrophoblast for the analysis of chorionic villi: Advantages and limitations from a cytogenetic retrospective audit of 44,727 first-trimester prenatal diagnoses. Prenat. Diagn. 2013, 33, 502–508. [Google Scholar] [CrossRef]
- Sago, H.; Chen, E.; Conte, W.J.; Cox, V.A.; Goldberg, J.D.; Lebo, R.V.; Golabi, M. True trisomy 2 mosaicism in amniocytes and newborn liver associated with multiple system abnormalities. Am. J. Med. Genet. 1997, 72, 343–346. [Google Scholar] [CrossRef]
- Sifakis, S.; Staboulidou, I.; Maiz, N.; Velissariou, V.; Nicolaides, K.H. Outcome of pregnancies with trisomy 2 cells in chorionic villi. Prenat. Diagn. 2010, 30, 329–332. [Google Scholar]
- Magenis, E.; Webb, M.J.; Spears, B.; Opitz, J.M. Blaschkolinear malformation syndrome in complex trisomy-7 mosaicism. Am. J. Med. Genet. 1999, 87, 375–383. [Google Scholar] [CrossRef]
- Hsu, L.Y. United States survey on chromosome mosaicism and pseudomosaicism in prenatal diagnosis. Prenat. Diagn. 1984, 4, 97–130. [Google Scholar] [CrossRef]
- Hahnemann, J.M.; Vejerslev, L.O. Accuracy of cytogenetic findings on chorionic villus sampling (CVS)—Diagnostic consequences of CVS mosaicism and non-mosaic discrepancy in centres contributing to EUCROMIC 1986–1992. Prenat. Diagn. 1997, 17, 801–820. [Google Scholar] [CrossRef]
- Sachs, E.S.; Jahoda, M.G.; Los, F.J.; Pijpers, L.; Reuss, A.; Wladimiroff, J.W. Interpretation of chromosome mosaicism and discrepancies in chorionic villi studies. Am. J. Med. Genet. 1990, 37, 268–271. [Google Scholar] [CrossRef]
- Klinger, K.; Landes, G.; Shook, D.; Harvey, R.; Lopez, L.; Locke, P.; Lerner, T.; Osathanondh, R.; Leverone, B.; Houseal, T.; et al. Rapid detection of chromosome aneuploidies in uncultured amniocytes by using fluorescence in situ hybridization (FISH). Am. J. Hum. Genet. 1992, 51, 55–65. [Google Scholar]
- Morris, A.; Boyd, E.; Dhanjal, S.; Lowther, G.W.; Aitken, D.A.; Young, J.; Menzies, A.L.; Imrie, S.J.; Connor, J.M. Two years’ prospective experience using fluorescence in situ hybridization on uncultured amniotic fluid cells for rapid prenatal diagnosis of common chromosomal aneuploidies. Prenat. Diagn. 1999, 19, 546–551. [Google Scholar] [CrossRef]
- Weremowicz, S.; Sandstrom, D.J.; Morton, C.C.; Niedzwiecki, C.A.; Sandstrom, M.M.; Bieber, F.R. Fluorescence in situ hybridization (FISH) for rapid detection of aneuploidy: Experience in 911 prenatal cases. Prenat. Diagn. 2001, 21, 262–269. [Google Scholar] [CrossRef]
- Baart, E.B.; Martini, E.; van Opstal, D. Screening for aneuploidies of ten different chromosomes in two rounds of FISH: A short and reliable protocol. Prenat. Diagn. 2004, 24, 955–961. [Google Scholar] [CrossRef]
- Feldman, B.; Ebrahim, S.A.; Gyi, K.; Flore, L.A.; Evans, M.I. Rapid confirmation of previously detected prenatal mosaicism by fluorescence in situ hybridization in interphase uncultured amniocytes. Genet. Test. 2000, 4, 61–63. [Google Scholar] [CrossRef]
- Mann, K.; Ogilvie, C.M. QF-PCR: Application, overview and review of the literature. Prenat. Diagn. 2012, 32, 309–314. [Google Scholar] [CrossRef]
- Grati, F.R.; Barlocco, A.; Grimi, B.; Milani, S.; Frascoli, G.; di Meco, A.M.; Liuti, R.; Trotta, A.; Chinetti, S.; Dulcetti, F.; et al. Chromosome abnormalities investigated by non-invasive prenatal testing account for approximately 50% of fetal unbalances associated with relevant clinical phenotypes. Am. J. Med. Genet. A 2010, 152A, 1434–1442. [Google Scholar]
- Speevak, M.D.; McGowan-Jordan, J.; Chun, K. The detection of chromosome anomalies by QF-PCR and residual risks as compared to G-banded analysis. Prenat. Diagn. 2011, 31, 454–458. [Google Scholar] [CrossRef]
- Shaffer, L.G.; Dabell, M.P.; Rosenfeld, J.A.; Neill, N.J.; Ballif, B.C.; Coppinger, J.; Diwan, N.R.; Chong, K.; Shohat, M.; Chitayat, D. Referral patterns for microarray testing in prenatal diagnosis. Prenat. Diagn. 2012, 32, 344–350. [Google Scholar] [CrossRef]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray vs. karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef]
- Vetro, A.; Bouman, K.; Hastings, R.; McMullan, D.J.; Vermeesch, J.R.; Miller, K.; Sikkema-Raddatz, B.; Ledbetter, D.H.; Zuffardi, O.; van Ravenswaaij-Arts, C.M. The introduction of arrays in prenatal diagnosis: A special challenge. Hum. Mutat. 2012, 33, 923–929. [Google Scholar] [CrossRef]
- Dondorp, W.; Sikkema-Raddatz, B.; de Die-Smulders, C.; de Wert, G. Arrays in postnatal and prenatal diagnosis: An exploration of the ethics of consent. Hum. Mutat. 2012, 33, 916–922. [Google Scholar] [CrossRef]
- Ballif, B.C.; Rorem, E.A.; Sundin, K.; Lincicum, M.; Gaskin, S.; Coppinger, J.; Kashork, C.D.; Shaffer, L.G.; Bejjani, B.A. Detection of low-level mosaicism by array CGH in routine diagnostic specimens. Am. J. Med. Genet. A 2006, 140, 2757–2767. [Google Scholar]
- Conlin, L.K.; Kaur, M.; Izumi, K.; Campbell, L.; Wilkens, A.; Clark, D.; Deardorff, M.A.; Zackai, E.H.; Pallister, P.; Hakonarson, H.; et al. Utility of SNP arrays in detecting, quantifying, and determining meiotic origin of tetrasomy 12p in blood from individuals with Pallister-Killian syndrome. Am. J. Med. Genet. A 2012, 158A, 3046–3053. [Google Scholar] [CrossRef]
- Filges, I.; Kang, A.; Klug, V.; Wenzel, F.; Heinimann, K.; Tercanli, S.; Miny, P. aCGH on chorionic villi mirrors the complexity of fetoplacental mosaicism in prenatal diagnosis. Prenat. Diagn. 2011, 31, 473–478. [Google Scholar] [CrossRef]
- Karampetsou, E.; Morrogh, D.; Ballard, T.; Waters, J.J.; Lench, N.; Chitty, L.S. Confined placental mosaicism: Implications for fetal chromosomal analysis using microarray comparative genomic hybridization. Prenat. Diagn. 2014, 34, 98–101. [Google Scholar] [CrossRef]
- Saura, R.; Roux, D.; Maugey-Laulon, B.; Taine, L.; Wen, Z.Q.; Vergnaud, A.; Horovitz, J. False-negative results of trisomy 21 on direct analysis on chorionic villus sampling. Prenat. Diagn. 1998, 18, 866–867. [Google Scholar] [CrossRef]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Platt, L.D.; Goldberg, J.D.; Abuhamad, A.Z.; Sehnert, A.J.; Rava, R.P.; MatErnal BLood, I.S.; Source to Accurately Diagnose Fetal Aneuploidy (MELISSA) Study Group. Genome-wide fetal aneuploidy detection by maternal plasma DNA sequencing. Obstet. Gynecol. 2012, 119, 890–901. [Google Scholar] [CrossRef]
- Chiu, R.W.; Akolekar, R.; Zheng, Y.W.; Leung, T.Y.; Sun, H.; Chan, K.C.; Lun, F.M.; Go, A.T.; Lau, E.T.; To, W.W.; et al. Non-invasive prenatal assessment of trisomy 21 by multiplexed maternal plasma DNA sequencing: Large scale validity study. BMJ 2011, 342. [Google Scholar] [CrossRef] [Green Version]
- Tjoa, M.L.; Cindrova-Davies, T.; Spasic-Boskovic, O.; Bianchi, D.W.; Burton, G.J. Trophoblastic oxidative stress and the release of cell-free feto-placental DNA. Am. J. Pathol. 2006, 169, 400–404. [Google Scholar] [CrossRef]
- Flori, E.; Doray, B.; Gautier, E.; Kohler, M.; Ernault, P.; Flori, J.; Costa, J.M. Circulating cell-free fetal DNA in maternal serum appears to originate from cyto- and syncytio-trophoblastic cells. Hum. Reprod. 2004, 19, 723–724. [Google Scholar] [CrossRef]
- Faas, B.H.; de Ligt, J.; Janssen, I.; Eggink, A.J.; Wijnberger, L.D.; van Vugt, J.M.; Vissers, L.; Geurts van Kessel, A. Non-invasive prenatal diagnosis of fetal aneuploidies using massively parallel sequencing-by-ligation and evidence that cell-free fetal DNA in the maternal plasma originates from cytotrophoblastic cells. Expert Opin. Biol. Ther. 2012, 12, S19–S26. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Wilkins-Haug, L. Integration of noninvasive DNA testing for aneuploidy into prenatal care: What has happened since the rubber met the road? Clin. Chem. 2014, 60, 78–87. [Google Scholar] [CrossRef]
- Futch, T.; Spinosa, J.; Bhatt, S.; de Feo, E.; Rava, R.P.; Sehnert, A.J. Initial clinical laboratory experience in noninvasive prenatal testing for fetal aneuploidy from maternal plasma DNA samples. Prenat. Diagn. 2013, 33, 569–574. [Google Scholar] [CrossRef]
- Yao, H.; Zhang, L.; Zhang, H.; Jiang, F.; Hu, H.; Chen, F.; Jiang, H.; Mu, F.; Zhao, L.; Liang, Z.; Wang, W. Noninvasive prenatal genetic testing for fetal aneuploidy detects maternal trisomy X. Prenat. Diagn. 2012, 32, 1114–1116. [Google Scholar] [CrossRef]
- Osborne, C.M.; Hardisty, E.; Devers, P.; Kaiser-Rogers, K.; Hayden, M.A.; Goodnight, W.; Vora, N.L. Discordant noninvasive prenatal testing results in a patient subsequently diagnosed with metastatic disease. Prenat. Diagn. 2013, 33, 609–611. [Google Scholar] [CrossRef]
- Allen, R.; Kezmarsky, P.; Lescale, K. False Negative NIPT and Potential Implications for Genetic Counseling. Available online: http://ww2.aievolution.com/acm1301/index.cfm?do=abs.viewAbs&abs=1427 (accessed on 24 January 2014).
- Gao, Y.; Stejskal, D.; Jiang, F.; Wang, W. A T18 false negative result by NIPT in a XXX; T18 case due to placental mosaicism. Ultrasound Obstet. Gynecol. 2013. [Google Scholar] [CrossRef]
- Pan, M.; Li, F.T.; Li, Y.; Jiang, F.M.; Li, D.Z.; Lau, T.K.; Liao, C. Discordant results between fetal karyotyping and non-invasive prenatal testing by maternal plasma sequencing in a case of uniparental disomy 21 due to trisomic rescue. Prenat. Diagn. 2013, 33, 598–601. [Google Scholar] [CrossRef]
- Henderson, K.G.; Shaw, T.E.; Barrett, I.J.; Telenius, A.H.; Wilson, R.D.; Kalousek, D.K. Distribution of mosaicism in human placentae. Hum. Genet. 1996, 97, 650–654. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Van Mieghem, T.; Shaffer, L.G.; Faas, B.H.; Chitty, L.S.; Ghidini, A.; Deprest, J. In case you missed it: The Prenatal Diagnosis section editors bring you the most significant advances of 2013. Prenat. Diagn 2014, 34, 1–5. [Google Scholar]
- Shaffer, L.G.; Dabell, M.P.; Fisher, A.J.; Coppinger, J.; Bandholz, A.M.; Ellison, J.W.; Ravnan, J.B.; Torchia, B.S.; Ballif, B.C.; Rosenfeld, J.A. Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat. Diagn. 2012, 32, 976–985. [Google Scholar] [CrossRef]
- Crolla, J.A.; Wapner, R.; van Lith, J.M. Controversies in prenatal diagnosis 3: Should everyone undergoing invasive testing have a microarray? Prenat. Diagn. 2014, 34, 18–22. [Google Scholar] [CrossRef]
- De Jong, A.; Dondorp, W.J.; Macville, M.V.; de Die-Smulders, C.E.; van Lith, J.M.; de Wert, G.M. Microarrays as a diagnostic tool in prenatal screening strategies: Ethical reflection. Hum. Genet. 2014, 133, 163–172. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Grati, F.R. Chromosomal Mosaicism in Human Feto-Placental Development: Implications for Prenatal Diagnosis. J. Clin. Med. 2014, 3, 809-837. https://doi.org/10.3390/jcm3030809
Grati FR. Chromosomal Mosaicism in Human Feto-Placental Development: Implications for Prenatal Diagnosis. Journal of Clinical Medicine. 2014; 3(3):809-837. https://doi.org/10.3390/jcm3030809
Chicago/Turabian StyleGrati, Francesca Romana. 2014. "Chromosomal Mosaicism in Human Feto-Placental Development: Implications for Prenatal Diagnosis" Journal of Clinical Medicine 3, no. 3: 809-837. https://doi.org/10.3390/jcm3030809