
Emerging Metabolic Therapies in Pulmonary Arterial Hypertension


1
Medical Scientist Training Program, University of Pittsburgh School of Medicine and University of Pittsburgh Medical Center, Pittsburgh, PA 15213, USA
2
Division of Cardiology, Center for Pulmonary Vascular Biology and Medicine, Pittsburgh Heart, Lung, Blood, and Vascular Medicine Institute, University of Pittsburgh School of Medicine and University of Pittsburgh Medical Center, Pittsburgh, PA 15213, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2017, 6(4), 43; https://doi.org/10.3390/jcm6040043
Submission received: 15 February 2017
/
Revised: 28 March 2017
/
Accepted: 29 March 2017
/
Published: 4 April 2017
(This article belongs to the Special Issue Novel Therapeutic Approaches for Pulmonary Arterial Hypertension)
Abstract
:Pulmonary hypertension (PH) is an enigmatic vascular disorder characterized by pulmonary vascular remodeling and increased pulmonary vascular resistance, ultimately resulting in pressure overload, dysfunction, and failure of the right ventricle. Current medications for PH do not reverse or prevent disease progression, and current diagnostic strategies are suboptimal for detecting early-stage disease. Thus, there is a substantial need to develop new diagnostics and therapies that target the molecular origins of PH. Emerging investigations have defined metabolic aberrations as fundamental and early components of disease manifestation in both pulmonary vasculature and the right ventricle. As such, the elucidation of metabolic dysregulation in pulmonary hypertension allows for greater therapeutic insight into preventing, halting, or even reversing disease progression. This review will aim to discuss (1) the reprogramming and dysregulation of metabolic pathways in pulmonary hypertension; (2) the emerging therapeutic interventions targeting these metabolic pathways; and (3) further innovation needed to overcome barriers in the treatment of this devastating disease.
1. Introduction
Pulmonary hypertension (PH) is an enigmatic vascular disorder characterized by complex pulmonary vascular remodeling and subsequent increased pulmonary vascular resistance. The World Health Organization (WHO) has classified PH into five major groups based on clinical associations and histologic appearance [1,2,3]. Group I comprises a severe form of this disease that has idiopathic, heritable, and comorbid etiologies (e.g., connective tissue disorders, HIV infection, schistosomiasis, etc.), termed pulmonary arterial hypertension (PAH) [1]. PAH results from the obliteration of pulmonary arterioles, thereby causing an increase in the pulmonary vascular resistance, subsequent right ventricular hypertrophy, and culminating as right heart failure [4]. Histologically, this panvasculopathy demonstrates intimal hyperplasia, medial hypertrophy, adventitial proliferation, and pathognomonic plexiform lesions in pulmonary arterioles [5]. Groups II, III, IV, and V represent a wide variety of conditions that can cause PH, such as left heart disease, lung diseases and/or hypoxia, thromboembolic diseases, and unclear multifactorial mechanisms, respectively [1,2,3].
PH and particularly PAH are highly morbid conditions. Unfortunately, the manifestations of symptoms including shortness of breath and right heart failure often come late in the course of the disease, portending a worsened clinical prognosis. Survival rates of PAH without treatment are grim: with 68%, 48%, and 34% alive at 1, 3, and 5 years, respectively [6]. Current clinical therapies primarily improve pulmonary vasomotor tone and provide symptomatic relief and lengthening of the time to clinical worsening. However, they do not reverse or prevent the disease process. As such, there is an obvious and critical need for novel therapies capable of affecting more than vasodilation.
Tracing the historical arc of PH research reveals a foundational basis of understanding rooted in pathological manifestations within the pulmonary arterioles [7]. Early investigations focused on hypoxia and the vasoconstrictive effects on pulmonary vasculature. Under normal conditions, hypoxic pulmonary vasoconstriction (HPV) is a physiologically protective response that optimizes ventilation-perfusion matching by diverting blood flow away from alveoli with poor ventilation [8]. HPV is mediated through pulmonary arterial smooth muscle cell (PASMC) mitochondria that act as oxygen sensors capable of altering reactive oxygen species (ROS) dynamics under acute hypoxic conditions [9]. Chronic hypoxia sustains vasoconstriction and induces activation of hypoxia-inducible factor (HIF), resulting in pulmonary vascular remodeling and the development of hypoxia-induced PH [10]. Induction of HIF can also occur under normoxic conditions when aberrations of this oxygen-sensing mechanism result in its inappropriate activation and the subsequent pathogenesis of PAH. Under conditions of chronic hypoxia, HIF regulates adaptive cellular responses, namely the metabolic reprogramming of mitochondria to meet cellular bioenergetic demands [11]. Thus, as a primary adaptive response to hypoxia, it now makes sense to have expected mitochondrial metabolic reprogramming as involved in the pathogenesis of hypoxia-induced PH and PAH [12,13].
The early idea of PH being mediated through hypoxia-mediated vasoconstriction served as an impetus for therapeutic intervention with the development and use of selective vasodilatory agents. Accordingly, the mainstay of current PH pharmacotherapy relies on endothelin-1 receptor antagonists, enhancers of nitric oxide (NO) signaling, and prostacyclin analogues [14]. The paradigm has shifted, however, over time toward the notion that the origin of PH is founded instead in myriad molecular aberrations that manifest as a remodeling of the pulmonary vasculature characterized by pathological hyperproliferation and a resistance to apoptosis [4]. As such, a metabolic theory of PAH has been put forth to unify several molecular signatures of the disease around an etiologic origin within mitochondrial dysregulation and metabolic reprogramming [15].
2. A Metabolic Theory of PAH
The basis of this metabolic theory is founded in several molecular and cellular processes observed in both PH and cancer that rely upon the mitochondrion as the epicenter of metabolic control and regulation. Mammalian cells under hypoxic conditions demonstrate a metabolic shift away from oxidative phosphorylation toward glycolysis and lactic acid fermentation to sustain ATP production and facilitate acute survival—known as the Pasteur effect. This metabolic shift toward glycolysis—first described by Otto Warburg in cancer cells under normal oxygen tension—serves to sustain uncontrolled, neoplastic growth [16]. Similarly, the hyperproliferative, antiapoptotic, and glycolytic phenotype of the vasculature in PH has drawn comparisons to the cellular phenotype observed in cancer, collectively contributing to proliferation and resistance to apoptosis [4,17].
As our understanding of metabolic dysregulation in PH has increased, the metabolic theory of PH has expanded and evolved beyond its original premise constrained by the Warburg effect. Recent expansions of the metabolic theory have included dysregulation of a multitude of pathways, which is further complicated by evidence of genetic polymorphisms altering these responses. In addition, the anatomic epicenter of metabolic dysregulation has widened. Early investigations and theories assumed metabolic dysregulation would logically manifest at the site of disease pathology—the pulmonary vasculature. However, increasing evidence reveals aberration of metabolism within the right ventricle (RV) and perhaps in skeletal muscle, possibly suggestive of paracrine and even systemic effects in disease pathogenesis [18,19]. The significance of metabolic dysfunction beyond the pulmonary vasculature has implications in reframing our understanding of metabolism in PH. For example, cells of the pulmonary vascular microenvironment and cardiomyocytes have inherently different metabolic demands and mechanisms of energy production, namely the high reliance of cardiomyocytes on fatty acid oxidation. These metabolic principles will be important in creating a more unified and comprehensive metabolic theory of PH.
Beyond a hyperproliferative and antiapoptotic phenotype, our expanding view of metabolic dysregulation has led to the discovery of other phenotypes influenced by metabolic reprogramming in PH, such as angiogenesis, vessel stiffening and fibrosis, inflammation, cell migration, and cell identity. Beyond endothelial cells, smooth muscle cells, and cardiomyocytes—where much of the initial observations of metabolic dysfunction in PH have centered—the role of metabolic dysregulation is expanding into other cell types within the vasculature, such as infiltrating inflammatory cells of the immune system and resident tissue stem cells [20]. The temporal relationship of metabolic dysregulation and its reprogramming is another critical question to be answered. Whether metabolic aberrations occur early or late in disease pathogenesis are important in understanding the overarching breadth of metabolic dysfunction in PH.
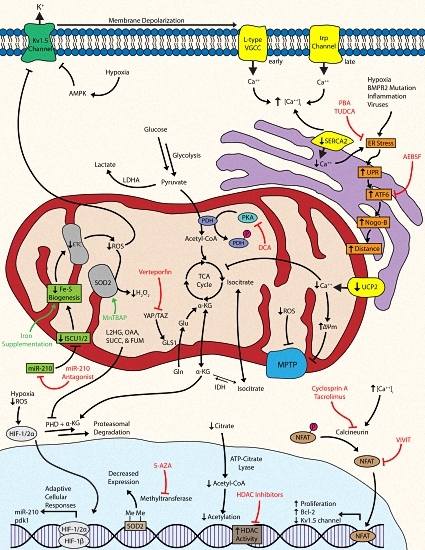
Ultimately, there are a series of coordinated levels of mitochondrial dysregulation that give rise to the cellular phenotypes of PH. The proliferation of pulmonary vascular cells, such as fibroblasts, endothelial cells (ECs), and smooth muscle cells (SMCs) are hallmarks of PH pathology [4]. The observed metabolic shift toward glycolysis has been associated with numerous molecular events that confer the ability to adapt to short bouts of cellular stress and resist apoptosis: the activation of master regulatory factors like hypoxia-inducible factor and nuclear factor of activated T cells, dysregulation of bioactive metal homeostasis, compensatory anaplerosis, a reduction in mitochondrial reactive oxygen species, the inhibition and internalization of oxygen-sensitive voltage-gated potassium channels, mitochondrial membrane hyperpolarization, dysregulation of calcium dynamics, endoplasmic reticulum stress, and the suppression of histone acetylation (Figure 1) [4,11,21,22,23]. Despite these short-term adaptations, the cell must continue to produce energy and mass at a level sufficient for metabolic demands, which typically cannot be sustained solely via enhanced glycolysis and thus requires multiple levels of molecular reprogramming.
In the context of such complexity, the therapeutic targeting of metabolism in PH confers both advantages and disadvantages. Metabolism is an exquisitely complex series of molecular events, which encompasses control of many downstream features of PH; however, metabolism is also tightly regulated in time and space, meaning that harnessing therapeutic control of metabolic pathways can be difficult and can lead to unintended side-effects. Conserved metabolic processes may allow for interventions capable of widespread targeting in various cell types and tissues, yet the efficacy of targeting a conserved pathway may be a challenge when considering systemic versus local administration of an intervention. Moreover, the intricacy inherent to metabolism poses a challenge when selecting an effector to target, which ideally has a master regulatory role in reprogramming events. Metabolic variation further serves as an obstacle in treatment with potential genetic, epigenetic, and PH-subtype differences that limit novel therapies to specific patient populations. Despite such barriers, therapeutic targeting of metabolism has great potential to treat PH early in its course, allowing for the prevention or reversal of pathologic cellular phenotypes. The targeting of novel metabolic effectors, in addition to current therapies, may provide synergistic effects in disease treatment or even reversal. A difficulty of studying efficacies of new therapies is that novel agents would be tested in patients with the current standard of care—combination therapy—further complicating clinical trials. Furthermore, new therapies could be tested in other subtypes of PH that may not lend themselves directly with the metabolic theory of PAH. In these contexts, we will discuss our understanding of the metabolic pathways involved in the development of PAH and the possibility of targeting such effectors to develop more robust diagnostics and therapeutics for this disease.
3. Novel Targets of Emerging Metabolic Therapies
3.1. Targeting Hypoxia-Inducible Factor and Downstream Metabolic Effectors
A master regulatory, oxygen sensitive transcription factor expressed in all nucleated metazoan cells—hypoxia-inducible factor (HIF)—is central to the reprogramming of metabolic activity in response to hypoxic stimulus [11]. The role of HIF in the pathogenesis of PAH is well-documented with its inappropriate induction resulting from a failure of prolyl hydroxylases (PHDs) in facilitating proteasomal degradation of the HIF-1α/HIF-2α subunit [24]. Specifically, HIF activation under normoxic or hypoxic conditions is important in the development of WHO Group I PAH and Group III hypoxia-induced PH, respectively, with possible contributions to other PH subtypes as well. The role of HIF under conditions of hypoxia and in the pathogenesis of PH has been reviewed (see [11,25,26]). In metazoan cells, hypoxic stimulus represses proteasomal degradation of the HIF-1α/HIF-2α subunit, thereby allowing for its nuclear translocation and binding with the HIF-1β subunit [26]. The resulting heterodimeric transcription factor binds to a hypoxic response element (HRE), resulting in the activation of more than a hundred genes involved in adaptive cellular responses, such as the acute metabolic shift from oxidative phosphorylation to glycolysis [26,27,28]. Studies have demonstrated the pathogenic potential of HIF-1α/HIF-2α by demonstrating protection from chronic hypoxia and resistance to the development of hypoxia-induced PH in mice heterozygous for either subunit [29,30]. Other animal models such as the fawn-hooded rat (FHR) demonstrated inappropriate HIF induction under normoxia and a predisposition to develop PAH [31,32]. Additionally, normoxic HIF activation was observed in pulmonary artery smooth muscle cells (PASMCs) from patients with PAH [31,33]. Taken together, the induction of HIF under normoxia or hypoxia lays the foundation through which subsequent molecular, cellular, and metabolic events occur in idiopathic PAH and hypoxia-induced PH, respectively.
Once activated, HIF induces transcription and expression of glycolytic genes and suppresses oxidative metabolism via increased transcription of pyruvate dehydrogenase kinase (PDK) [4]. The mitochondrial enzyme pyruvate dehydrogenase (PDH) catalyzes irreversible oxidation of pyruvate into acetyl-CoA for entry into the tricarboxylic acid (TCA) cycle and subsequent glucose oxidation (GO); however, phosphorylation of PDH by PDK results in its inhibition and a shunting of pyruvate via lactate dehydrogenase A (LDHA) into lactate for anaerobic respiration. Essentially, PDH serves as the gatekeeper that controls the rate of oxidative metabolism [34]. HIF upregulation of PDK is essential in the suppression of GO and the upregulation of glycolysis under hypoxic conditions. Under acute circumstances, this adaptive mechanism is cytoprotective, conferring short-term resistance to cellular stress; however, chronicity of environmental stressors and the dysregulation of these protective mechanisms can serve as drivers in the pathogenesis and manifestation of PH.
A mitochondrial-targeted strategy can partially correct for the resulting metabolic abnormalities observed after PDK inhibition of PDH. Dichloroacetate (DCA), an inhibitor of PDK, can attenuate the shift from oxidative phosphorylation to glycolysis by maintaining production of the acetyl-CoA necessary for the TCA cycle [35]. The use of DCA has shown promising preclinical results in the treatment of animal models of PH. One study demonstrated that DCA prevented and even reversed monocrotaline (MCT)-induced PAH in rat models via restoration of PDH activity and GO through stimulation of mitochondria-dependent apoptosis [36]. In FHRs, DCA treatment reversed normoxic HIF activation and increased voltage-gated potassium (Kv1.5) channel expression, thereby reducing PAH and improving survival [31]. In addition, this same study also inhibited HIF-1α using a selective dominant-negative construct in FHR PASMCs to show attenuation of HIF translocation and increased Kv1.5 expression [31]. Another study showed that DCA therapy in a chronic hypoxia (CH)-induced rat model enhanced activity and expression of Kv2.1 channels, improved hemodynamic parameters, and prevented and reversed CH-induced PH [21]. The culmination of such promising preclinical work was a phase I clinical trial assessing the effects of DCA treatment in patients with advanced PAH that was completed in 2013; however, the results of this study have yet to be published (NCT01083524).
3.2. Targeting Bioactive Metals: Iron-Sulfur, Iron, and Zinc
Bioactive metals are essential components of enzymes, functioning as cofactors necessary for metabolic reactions to occur and their dysregulation has been linked to PH. The emerging field of microRNA biology has recently shown that regulation of the metabolism is not limited to protein-coding genes, implicating a role for these small non-coding RNAs in the glycolytic shift. MicroRNA-210 (miR-210) is a known transcriptional target of activated HIF that has shown contributory regulation and facilitation of the metabolic shift toward glycolysis under hypoxic conditions [37]. Specifically, miR-210 was found to downregulate expression of the iron-sulfur cluster assembly proteins 1 and 2 (ISCU1/2) [38]. ISCU1/2 are responsible for the assembly of iron-sulfur (Fe-S) clusters—namely [4Fe-4S] and [2Fe-2S]—that are essential prosthetic groups in proper operation of the TCA cycle and mitochondrial complexes I, II, and III in the electron transport chain (ETC) [39,40]. Downregulation of ISCU1/2 by hypoxic miR-210 was found to attenuate Fe-S-dependent mitochondrial respiration in favor of glycolysis in pulmonary arterial endothelial cells (PAECs). Although this metabolic shift was protective under acute circumstances, its chronic repression drove dysfunction and reprogramming of mitochondrial metabolism, thereby promoting PH in rodent models [41]. Notably, a woman with a genetic deficiency in ISCU1/2 was found to suffer from exercise-induced pulmonary hypertension, providing important proof of the relevance of this mechanism in human disease. This principle of linking Fe-S deficiency to PH has also been supported by epidemiologic data showing the histologic manifestations of PAH in infants with genetic deficiency in NFU1, another Fe-S cluster assembly protein [42,43]. Although no small molecules have been discovered to directly regulate Fe-S cluster expression, pharmacological inhibition of upstream miR-210 with pulmonary vascular specific anti-miR delivery may serve as a promising therapeutic option as proven in rodent models of PH [41].
Iron deficiency has been observed in PAH populations, raising the notion that iron deficiency may serve as a mechanism of repressed Fe-S clusters by substrate deprivation [44,45,46,47]. Additionally, the enzymes responsible for HIF hydroxylation—like PHD—require a ferrous iron ion at their active site for electron transfer, implying that hydroxylation of HIF may be dependent on the availability of intracellular iron [48,49]. Iron-regulatory protein 1 (Irp1) binds to iron-responsive element (IRE) machinery as an apoprotein under iron-deficient conditions to regulate iron metabolism proteins. Moreover, Irp1 can also become a cytosolic aconitase in iron-replete states upon the acquisition of a [4Fe-S] cluster [50,51,52]. Thus, when hypoxia, anemia, or iron-deficient conditions are present, stabilization of HIF-2α induces erythropoietin (EPO) expression and red blood cell (RBC) development, thereby consuming iron via hemoglobin synthesis. After a certain threshold, iron deficiency halts Fe-S cluster production and Irp1 converts to the apoprotein form that binds to IREs to repress HIF-2α translation and decrease EPO production, thereby halting further iron consumption via hematopoiesis. Thus, a failure to repress HIF activation would result in its inappropriate and sustained expression as a driver in the pathogenesis of PH. Recently, it was reported that mice deficient of Irp1 (Irp1−/−) failed to repress HIF-2α translation, causing its increased expression and thus stimulated EPO synthesis to drive RBC production, iron consumption, and the development of polycythemia and PH. In vitro, PAECs from these mice showed increased expression of HIF-2α and the potent vasoconstrictor endothelin-1 [53]. Endothelin-1 binds to its receptor on SMCs to induce vasoconstriction and on cardiomyocytes and fibroblasts to induce their proliferation, which subsequently leads to PH, cardiac hypertrophy, and fibrosis [54,55]. Thus, pharmacologic activation of Irp1 binding to IREs with agents like the nitroxide Tempol may serve as therapeutic strategy in the treatment of PH [56].
Additionally, rats fed an iron-deficient diet for one month exhibited profound pulmonary vascular remodeling with upregulation of important regulators like HIF-1α and HIF-2α, decreased mitochondrial complex I activity, and upregulation of glycolytic genes like pdk1. Notably, the consequences of iron deficiency were reversed with iron replacement therapy via intravenous ferric carboxymaltose [57]. As such, iron replacement therapy in PAH patients with iron deficiency has been trialed with intravenous ferric carboxymaltose. The study, while it demonstrated improved exercise endurance, did not significantly alter the six-minute walk distance at 12 weeks (NCT01288651) [58]. In a recently published clinical study, iron deficiency caused exaggerated hypoxia-induced hypertension when compared to controls that could be reversed with ferric carboxymaltose administration, suggesting a role of iron deficiency in hypoxia-sensing mechanisms (NCT01847352) [59]. Other clinical trials focusing on iron homeostasis are currently underway, examining if single dose iron supplementation influences cardiopulmonary hemodynamics in PAH (NCT01447628) and if low Fe-S clusters predispose patients to the development of PH (NCT02594917). Of note, however, this biology may be context-specific, as seemingly disparate data are also emerging regarding the predisposition to PH in sickle cell patients with iron overload [60].
The dysregulation of the bioactive ion zinc has also been established in the pathogenesis of PH. Whole-genome sequencing from rats identified Slc39a12 as a candidate gene, which encodes the solute carrier 39 zinc transporter family member 12 (ZIP12), involved in the predisposition to hypoxia-induced PH [61]. ZIP12 promotes the extracellular uptake and intracellular release of zinc in various cell types, serving as an important regulator in zinc homeostasis [62]. In hypoxia-induced PH rats, markedly increased ZIP12 mRNA levels and protein expression were observed in lung and remodeled pulmonary arterial tissues when compared to normoxic controls. Downstream of the ZIP12 transcription start site was a HRE containing HIF-1/2α binding motifs. Chromatin immunoprecipitation following hypoxic exposure in human PASMCs revealed enrichment of HIF-1/2α bound to the ZIP12 HRE, indicating a relationship with hypoxia as a driver of aberrant pathology [61]. Additionally, increased ZIP12 expression was observed in cattle with naturally occurring PH (i.e., Brisket disease) and in human populations residing above 2500 m within the remodeled pulmonary vasculature, suggesting that ZIP12 upregulation is broadly observed in response to a variety of conditions seen with chronic hypoxia. Human PASMCs exposed to hypoxia demonstrated increased ZIP12 expression, intracellular zinc, and proliferation [61]. Inhibition of ZIP12 with siRNA reversed proliferation of hypoxia-exposed human PASMCs, indicating ZIP12 involvement in cellular proliferation in response to hypoxia. Lastly, genetically ablated (ZIP12−/−) rats exposed to chronic hypoxia for two weeks had lower pulmonary arterial pressures, reduced RV hypertrophy, and less pulmonary remodeling than wild-type controls [61]. Pathologic aberrations of zinc dynamics, an ion involved in the structural components of enzymes and transcription factors, further implicated its regulation within the cell. These data cumulatively support a role for ZIP12 as a critical regulator of zinc homeostasis in the pathogenesis of PH in response to chronic hypoxic stimulus, thus suggesting its utility as a potential drug target. Taken together, targeting the dynamics of bioactive metal homeostasis, a critical regulator of metabolic events, may serve as a modulatory treatment strategy.
3.3. Targeting Other Tricarboxylic Acid Cycle Intermediates: Connections to Hypoxia
Aberrations of the TCA cycle and its intermediates serve to stabilize HIF and facilitate its activation. The TCA cycle-derived α-ketoglutarate (α-KG) is a required cofactor for PHD, the enzyme that under normal conditions causes the proteasomal degradation of HIF [24,63]. Other intermediates such as succinate, fumarate, and oxaloacetate inhibit the activity of PHD at high concentrations to allow translocation and activation of HIF [64]. The influence of TCA cycle intermediates also has nuclear implications with acetylation and methylation of nuclear histones both regulated by citrate and α-KG, respectively [65,66]. Citrate that leaks out of mitochondria enters the nucleus where ATP-citrate lyase creates the acetyl-CoA necessary for histone acetylation [65]. Coupled with the knowledge of a shutdown of the TCA cycle during the glycolytic shift, the notion of decreased citrate and thus decreased histone acetylation via substrate deprivation becomes an area of therapeutic interest. In addition, histone deacetylase (HDAC) activity is elevated in human PAH, indicating a potential aberration from proper epigenetic regulation via decreased histone acetylation. Accordingly, the use of the HDAC inhibitors valproic acid (VPA) and suberoylanilide hydroxamic acid (SAHA; vorinostat) were found to be effective in reversing PH in a CH-induced rat model [67]. Of note, this compensatory mechanism in the cytoplasm can synthesize citrate via the glutamine pathway, which has been established in cancer but not yet in PH [68,69].
Analyses of metabolic flux indicate that transformed cell lines may add carboxyl residues to α-KG via isocitrate dehydrogenase (IDH) to compensate for decreased levels of citrate normally derived from GO [70,71,72]. Hypoxia causes an increase in the α-KG-reduced metabolite 2-hydroxyglutarate (2HG) with both L and D enantiomers (L2HG and D2HG) that can inhibit PHDs [66]. A recent study showed that hypoxia in human PAECs and PASMCs specifically and dose-dependently increased L2HG in response to mitochondrial reductive stress via TCA cycle dysfunction, where it served to inhibit glycolysis and oxidative phosphorylation to regulate the cellular redox state [73]. The complexity of metabolic dysregulation clearly extends beyond the overly simplified Warburg model, requiring many more studies in understanding the elaborate metabolic network occurring within the vascular microenvironment.
As would be expected, enzymatic mutations of the TCA cycle have been associated with PAH. Mutations in the bone morphogenetic protein receptor 2 (BMPR2) are linked to the pathogenesis of PH, and expression arrays from Bmpr2 mutant mice revealed that nearly half of the significantly altered genes fell into metabolic gene ontology groups [74]. Metabolomic analysis downstream of BMPR2 mutations revealed a profound failure of anaplerosis and depletion of TCA cycle intermediates, indicating that metabolic defects in PAH are not solely due to the Warburg effect and instead are present in multiple coexisting and interdependent pathways. This study focused on the TCA cycle as the major point of metabolic integration and specifically observed that the activity of IDH is elevated in human pulmonary microvascular endothelial cells with mutant BMPR2 and in the serum of PAH patients [75]. IDH can convert α-KG into isocitrate, catalyzing against the normal flow of the TCA cycle. As such, increased reversal of IDH activity deprives PHD of its α-KG cofactor to hydroxylate HIF for proteasomal degradation and disrupts further processing of glucose for oxidation, serving as a mechanism to sustain HIF activation and translocation. In fact, increased activity of IDH has been correlated with disease severity in PAH patients [75]. The therapeutic implications of metabolomic analyses suggest that most or perhaps even all errant metabolic pathways may need to be addressed therapeutically to drive a more robust and significant improvement in clinical outcome.
3.4. Targeting Anaplerosis and Glutaminolysis
The metabolic shift away from oxidative phosphorylation to glycolysis alone would not be expected to serve as a favorable condition for excessive proliferation of pulmonary vascular cells. While ATP production is less efficient for glycolysis per molecule of glucose, prior studies have suggested that there is enough glucose available in the pulmonary vasculature to allow for sufficient ATP production for proliferation [22]. Independent of ATP production, however, is the concept that a decrease of carbon intermediates also results if glucose is shunted away from the TCA cycle, thus negatively affecting the carbon substrates used to produce cell mass (i.e., nucleotides and proteins). Anaplerosis, or the replenishing of TCA carbon intermediates, is therefore critically important in maintaining cell mass, particularly if glycolysis predominates. One pathway for replenishment of carbon intermediates is through the deamidation of glutamine via glutaminase (GLS1). GLS1 activity in glutaminolysis provides energy for rapidly proliferating cells, and it has been established as necessary for the formation of aspartate for malignant cell proliferation [76,77,78]. Moreover, the dysregulation of cardiac glutaminolysis has been documented in maladaptive RV hypertrophy in the MCT-induced PAH model [79]. A recent study demonstrated that two transcriptional coactivators activated by mechanotransduction—Yes-associated protein 1 (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ)—are necessary for GLS1 upregulation and subsequent glutaminolysis to sustain proliferation and migration within stiff extracellular matrix. Thus, in addition to hypoxia, this study defined a mechanical stimulus, vascular stiffness, as a metabolic mechanism by which cellular proliferation is induced in PH [80]. Importantly, the same study revealed a marked improvement in a MCT-induced PH model using a YAP inhibitor verteporfin, which is already a FDA-approved intravenous medication for age-related macular degeneration [81]. Moreover, the same effects were seen with two separate glutaminase inhibitors, one of which (CB-839) is currently under clinical investigation in human cancer trials [82] (NCT02071862). Taken together, these findings suggest that the repurposing of currently available therapies affecting vascular stiffness and glutaminolysis may have additive and perhaps synergistic effects on ameliorating or preventing PH.
3.5. Targeting Mitochondria-Dependent Reactive Oxygen Species and Oxidative Stress
The inhibition of pyruvate processing for aerobic respiration results in an attenuation of the TCA cycle and mitochondrial ROS generation within the ETC. Although well-known for their damaging and detrimental effects, ROS have been implicated as important signaling molecules [83], ultimately contributing to a state of pulmonary vasodilation or vasoconstriction [84]. The major sites of ROS generation are classically regarded at Complexes I and III; however, more recent studies have demonstrated other mitochondrial enzymes involved in ROS generation, such as complex II [85]. And, in vascular SMCs, the ETC has been put forth as an oxygen sensor that generates ROS in proportion to alveolar PaO2, suggestive of a regulatory role [86]. Accordingly, the downstream effects of decreased ROS generation cause HIF activation and the inhibition of Kv1.5 channels, leading to cellular depolarization, calcium influx via l-type voltage-gated calcium channels (VGCCs), and pulmonary vasoconstriction.
Directly related to the disruption of ROS dynamics is the corollary that manganese superoxide dismutase (Mn-SOD or SOD2), an enzyme found only in the mitochondria, is decreased in PH [87]. SOD2 converts the ROS superoxide produced in the ETC into the diffusible second messenger hydrogen peroxide (H2O2), which inhibits the activity of HIF and increases the expression and activation of Kv1.5 channels [88,89,90]. As such, decreased ROS generation coupled with low-yield transformation into H2O2 by reduced SOD2 further contributes to ionic dysregulation and downstream pulmonary vasoconstriction. One avenue of targeting SOD expression includes miR biology. The siRNA knockdown of miR-23a in human small vascular endothelial cells demonstrated significantly increased mRNA transcripts of SOD, suggesting that miR-23a antagonism might serve to sustain H2O2 production and its inhibition of HIF [91]. A study of patients with severe PAH noted decreased SOD2 expression and activity, further supporting a role in disease pathology [87]. Methylation of SOD2 has been proposed as a potential epigenetic mechanism in the development in both FHRs and heritable PAH. Downregulation of SOD2 is reversible with the use of the DNA methyltransferase inhibitor 5-aza-2’-deoxycytidine (5-AZA), causing a restoration of mitochondrial function, inhibition of proliferation, and apoptosis in PAH PASMCs. The in vivo administration of the SOD-mimetic metalloporphyrin Mn(III)tetrakis (4-benzoic acid) porphyrin (MnTBAP) caused partial reversal of PAH in the FHR model [92]. In consolidating these findings, the ROS-SOD2-HIF-PDK nexus has emerged as a relevant pathogenic axis in PH, revealing multiple potential opportunities for therapy.
A recent study has implicated a role for the third isoform of SOD, which exists in the extracellular environment (EC-SOD or SOD3), in the pathogenesis of PH. SOD3 is the most abundant and active isoform within the vasculature [93]. Numerous studies and various models of lung injury or vascular injury have shown that loss of SOD3 and various polymorphisms contribute to disease severity, indicating a possible epigenetic regulatory mechanism of SOD3 [94]. In human PASMCs, SOD3 mRNA expression was decreased in lung tissue from patients with idiopathic PAH at transplantation versus failed donors and SOD2 protein expression was unchanged, contrary to the results of a previous report [94]. In contrast to the previous work demonstrating 5-AZA effects in increasing SOD2 expression, treatment with 5-AZA did not increase PASMC SOD3 mRNA, implying that its expression is not under the regulatory control of methylation [92,94]. Class I HDAC inhibitors, however, demonstrated an increase in SOD3 and a reduction in proliferation in idiopathic PAH PASMCs, which reflects a functional connection between SOD3 regulation and class I HDACs [94].
Thus, despite some conflicting data among separate reports, the role of ROS generation and downstream metabolic signaling are clearly important components of PH pathogenesis. Whether pharmacologic targeting of SOD isoforms can be effective in treatment of PH remains to be elucidated, and to do so with predictable and limited side-effects would require several complexities in redox biology to be better defined.
3.6. Targeting Kv1.5 Channels and AMP Kinase in Mitochondria
A direct consequence of reduced mitochondrial ROS production is the inhibition of oxygen-sensitive, voltage-gated potassium (Kv) channels, namely Kv1.5 [95,96]. As such, the metabolic shift toward anaerobic respiration, which reduces ROS generation in the ETC, further mediates downstream effects on ion homeostasis that ultimately contributes to a self-sustaining cycle of aberrant biology. The Kv1.5 channel serves as an additional oxygen sensor for the cell via intracellular ROS detection, meaning that its inhibition can keep the cell in a (pseudo)hypoxia-induced metabolic state even if normal oxygen levels have returned. The downregulation of the Kv1.5 channel is observed in human PASMCs with PAH and rodent models of PAH [4,96,97]. The downregulation and inhibition of these channels results in a buildup of cytosolic potassium, causing depolarization of the cellular membrane and the activation of L-type VGCCs. Thus, depolarization of the membrane causes an inappropriate influx of calcium and the subsequent initiation of signaling cascades that promote pulmonary vasoconstriction and resistance to apoptosis [86]. Using a CH-induced rat model, a study demonstrated that in vivo gene transfer of Kv1.5 channels protected from the development of PH [98].
Kv1.5 channels are functionally associated with upstream metabolic effectors. Inhibition of mitochondrial oxidative phosphorylation with either phenformin or hypoxia was found to result in AMP-activated protein kinase (AMPK)-mediated inhibition of Kv1.5 channels in rat PASMCs. The same study showed that the use of various activators of AMPK also produced a marked reduction in Kv1.5 channel currents [99]. The anti-diabetic drug metformin, a known stimulator of AMPK, protected against PAH in both CH- and MCT-induced models, while also demonstrating anti-remodeling properties [100,101]. A recently published study revealed downregulation of AMPK in PAECs in both hypoxia-induced mice and PAH patients, and it showed that knockdown of endothelial AMPK promoted the development of PH in the hypoxia-induced model. Additionally, the study demonstrated that endothelial AMPK-knockdown promoted proliferation in PASMCs, suggestive of a complex relationship within the vascular microenvironment, and that the application of PAH patient serum increased proliferation of PASMCs and reduced proliferation of PAECs [102].
The pathogenic importance of AMPK signaling extends beyond PAH. Left ventricular diastolic dysfunction, or heart failure with preserved ejection fraction (HFpEF), is recognized as a clinical complication of metabolic syndrome [103,104]. As a cause of Group II PH, the relationship between HFpEF secondary to metabolic syndrome and the pathogenesis of PH is of increasing interest. The development of both metabolic syndrome and PH has been linked to the reduced production and bioavailability of nitric oxide (NO)—a known activator of AMPK [105,106,107,108,109,110]. Accordingly, treatment with nitrite or metformin at time of SU5416 injection—a VEGF receptor blocker and model of PH—in rats reduced pulmonary arterial pressures and reversed vascular remodeling [111]. Clinically, a phase 2 trial evaluating the effects of metformin in PAH at Nantes University Hospital was withdrawn prior to enrollment due difficulty in attracting patients and other universities (NCT01352026); however, a current clinical trial is actively recruiting patients to examine the effects of metformin in improving pulmonary vascular function in patients with PAH (NCT01884051).
Taken together, these recent studies suggest that either AMPK or its downstream effector Kv1.5 may serve as attractive drug targets. AMPK has great potential as a metabolic drug target in the context of the sheer quantity of molecules known to stimulate its activation, such as salicylate and methotrexate [112,113,114].
3.7. Targeting Calcium Ion Homeostasis and Mitochondrial Electrical Dynamics
Central to the proper metabolic functioning of mitochondria is the careful control and regulation of intramitochondrial calcium dynamics. Many of the reactions inherent to mitochondrial metabolism are highly dependent on finely-tuned calcium ion homeostasis, meaning that even slight deviations from calcium homeostasis can have profound influence on metabolic events. As is expected, intracellular and mitochondrial calcium dynamics are of great interest in the metabolic events underlying PH pathology, especially since downstream sequelae following the downregulation of Kv channels and thus retention of cytoplasmic potassium include increased intracellular calcium-mediated pulmonary vasoconstriction and cellular proliferation [36].
The intracellular calcium overload, initially driven by pathologic depolarization of the L-type VGCCs, is later maintained and reinforced by upregulation and activation of transient receptor potential (trp) channels [115,116]. Increased vascular cell proliferation—a hallmark of PAH histopathology—is in part due to activation of the calcium-calcineurin-dependent proliferative transcription factor nuclear factor of activated T cells (NFAT) [117]. The nuclear translocation of NFAT promotes proliferation and reduces expression of Kv1.5 channels in PAH PASMCs, promoting a positive feedback loop via increased intracellular potassium and subsequent depolarization-mediated calcium influx [117]. Furthermore, activation of NFAT contributes to the upregulation of the antiapoptotic protein bcl-2 (and mitochondrial hyperpolarization), which is also upregulated in human idiopathic PAH [117]. Increased expression of bcl-2 prevents mitochondrial depolarization by proapoptotic mediators via enhanced hydrogen efflux, thereby contributing to the observed hyperpolarized state [118].
As a tightly regulated ion, there exist many cellular transporters involved in maintaining calcium homeostasis. Mitochondrial calcium dynamics are regulated by a calcium-uniporter called uncoupling protein 2 (UCP2) that, unlike its name implies, moves calcium from the ER into mitochondria [119,120]. Abnormalities of UCP2 would presumably cause a deficiency in mitochondrial calcium influx from the ER, resulting in suppression of GO and induce a pathophysiological state characterized by proliferation. Furthermore, a description of polymorphisms in UCP2 causing decreased gene expression suggests this may be a clinically relevant target [121]. Genetic ablation of UCP2 in PASMCs demonstrated mitochondrial hyperpolarization, decreased activity of calcium-sensitive mitochondrial enzymes, resistance to apoptosis, and established a proliferative phenotype. The same study showed that in vivo knockout of Ucp2 in mice models activated HIF-1α and NFAT, increased remodeling of the pulmonary vasculature, and developed PH [122,123]. In ECs, the loss of UCP2 resulted in exaggerated PTEN-induced putative kinase 1 (Pink1)-induced mitophagy, decreased mitochondrial biosynthesis, and increased apoptosis, indicating that a UCP2-Pink1 axis might serve as a therapeutic target [124]. Additionally, impaired function of another calcium uniporter—the mitochondrial calcium uniporter complex (MCUC)—through its downregulation decreases mitochondrial calcium levels that inhibits PDH and GO, causing the PAH phenotype in PASMCs. The in vivo administration of anti-miR-25 and anti-miR-138 restored MCUC expression and reversed established PAH in a MCT-induced rat model [125].
Intracellular calcium dynamics are also dysregulated at the level of the sarco-/endoplasmic reticulum calcium-ATPase (SERCA), a transporter that sequesters calcium into the sarcoplasmic reticulum after its depletion during excitation-contraction coupling [126]. In rat vascular injury models, the SERCA2 transporter is downregulated in the vascular media, causing increased intracellular calcium and subsequent NFAT-mediated vascular SMC proliferation and neointima formation [127]. SERCA2 expression is also decreased in human PAH PASMCs in comparison to controls, which indicates it as a target for disease treatment. Gene transfer of human SERCA2a using an adeno-associated virus serotype 1 (AAV1.SERCA2a) via aerosolized inhalation in both a MCT-induced rat PAH model and a pulmonary vein banding porcine model demonstrated increased expression of SERCA2 in pulmonary arteries, decreased pulmonary artery pressure, and improved RV function [128,129].
A further consequence of metabolically dysregulated ion homeostasis alters the electrical dynamics of the cell and ultimately mitochondria. The resistance to apoptosis observed in PAH results in part from a failure of proapoptotic mediators to efflux from mitochondria through the mitochondrial permeability transition pore (MPTP) [130]. The MPTP is a voltage- and redox-sensitive channel that closes under hyperpolarized mitochondrial membrane potential, thereby limiting the efflux of proapoptotic mediators and establishing a state of apoptotic resistance [131,132]. Inhibition of PDH prevents the movement of pyruvate into the mitochondria as acetyl-CoA for GO, thereby triggering glycolytic compensatory mechanisms. Increased activity of hexokinase-II (HK-II) during the glycolytic shift inhibits a portion of the MPTP—the voltage-dependent anion channel (VDAC) [130]. Inhibition of the VDAC causes retention of anions and hyperpolarization of the mitochondrial membrane, further limiting the function of the MPTP and inducing a relative state of apoptotic resistance [133]. Inhibition of Akt causes the activation glycogen synthase kinase 3β (GSK3β) and its translocation to the outer mitochondrial membrane, where it disrupts HK-II interaction with the VDAC preventing its inhibition [130]. As such, GSK3β prevents nuclear translocation of NFAT that upregulates anti-apoptotic mediators and downregulates Kv1.5 channel expression, further propagating and sustaining a reactive cycle. Accordingly, prevention of NFAT translocation, either directly or through other mechanisms such as the stabilization of calcium dynamics, may be a promising target for therapeutic intervention. Maintenance of proper calcium dynamics and/or prevention of NFAT translocation would prevent the upregulation of antiapoptotic molecules, the hyperpolarization of the mitochondrial membrane and thus MPTP closure, and the downregulation of Kv1.5 channels, thereby halting the metabolically-induced state of pro-proliferation and resistance to apoptosis.
The use of nonspecific calcineurin inhibitors that prevent the dephosphorylation and thus translocation of NFAT, such as cyclosporin A and tacrolimus, are current therapeutic candidates. A selective NFAT peptide inhibitor, VIVIT, specifically inhibits the docking of calcineurin onto NFAT instead of broad calcineurin inhibition, thereby limiting the adverse effects of nonspecific inhibitors [134]. One study demonstrated inhibition of NFAT using cyclosporin A or VIVIT in vitro increased Kv1.5 expression, reduced intracellular potassium and calcium levels, decreased mitochondrial membrane potential, and attenuated expression of bcl-2 [117]. The use of cyclosporin A in MCT-induced rat model produced a decrease in pulmonary vascular resistance and pulmonary arterial pressure, while increasing cardiac output [117].
Additionally, administration of low-dose tacrolimus restored signaling through the BMPR2, and reversed severe PAH in the MCT-induced model and in the VEGF receptor blockade/chronic hypoxia model, suggesting potential clinical benefit with low-dose therapy [135]. Loss of function mutations in BMPR2 are well-documented in sporadic and familial PAH [3]. Mutations in BMPR2 signaling have various roles in the pathogenesis of PH, exerting modulatory influence in certain aspects of metabolism. For example, in mice with a smooth muscle cell-specific overexpression of BMPR2, there is an observed downregulation of Kv1.5 channel expression resulting in PASMC depolarization and vasoconstriction, indicating Kv1.5 channel regulation as downstream of BMPR2 signaling [136]. Accordingly, the safety and efficacy of low-dose tacrolimus was trialed in a phase II study (NCT01647945) that has yet to be published; however, the authors recently reported a case of three patients with end-stage PAH who, ineligible for enrollment, instead received compassionate low-dose tacrolimus. Although the data are preliminary, the authors note a marked stabilization in cardiac function and freedom from hospitalization due to RV failure [137].
3.8. Targeting Mitochondrial Fission, Fusion, and Function
Mitochondrial networks within the cell represent a dynamic balance between fusion and fission. During cell division, mitochondrial fission evenly partitions mitochondria into daughter cells, whereas mitochondrial fusion is the joining of mitochondria to create an interconnected network for the distribution of mitochondrial proteins and DNA [138,139]. In hyperproliferative states, mitochondrial fission is necessary to sustain daughter cell production, which identifies this process as a possible intervention. Accordingly, the activity of dynamin-related protein-1 (DRP1)—a GTPase that regulates mitochondrial fission and fragmentation via the formation of oligomeric complexes and mechanical constriction of mitochondria—is of interest [140,141]. DRP1 is activated via phosphorylation by cyclin B1/cyclin-dependent kinase 1 (CDK1) at serine 616, causing its translocation to the cytoplasm for mitochondrial fragmentation [142]. Conversely, phosphorylation by protein kinase A at serine 637 results in its inhibition [143,144].
It has been reported that HIF-1α activation in human PAH PASMCs resulted in mitochondrial fission via cyclin B1/CDK1 phosphorylation of DRP1, in comparison to human control PASMCs that could be stimulated to undergo DRP1-mediated fission through chemical activation of HIF-1α [142]. The same study demonstrated that the inhibition of DRP1 with the small molecule Mdivi-1 reduced its activation, mitochondrial fission, and PASMC proliferation. Furthermore, in vivo administration of Mdivi-1 inhibited proliferation, while improving exercise capacity and RV function. Thus, the hyperproliferative phenotype characteristic in PAH, which is sustained through metabolic reprogramming, can also be targeted by locking mitochondria into a homogenous network, thereby causing cell-cycle arrest and preventing proliferation.
Decreased activation of the peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α), a transcription factor involved in mitochondrial biogenesis, is associated with increased mitochondrial fission [145]. In both EC and SMCs, BMPR2 regulates the peroxisome proliferator-activated receptor-γ (PPARγ)-mediated gene transcription [146,147]. Thus, a dysfunction in PGC1α may serve a key role in the biogenesis of mitochondria. Accordingly, loss of BMPR2 in human PAECs under normoxia increased PGC1α transcriptional activity, mitochondrial ATP production, glycolysis, and mitochondrial fission [148]. However, during hypoxia-reoxygenation, a reduction in BMPR2 demonstrated decreased mitochondrial biogenesis and energy metabolism that promoted EC apoptosis, which has been linked to SMC proliferation due to the loss of EC repressive growth factors [148,149,150]. The loss of PAECs via BMPR2 mutations contributes to a pro-inflammatory state that would allow for immune cell infiltration, PASMC hyperproliferation, and occlusion of the vessel lumen, which is observed in PAH histology [20]. Activation of nuclear factor erythroid 2-related factor 2 (Nrf2) has been shown to improve mitochondrial function and reduce the generation of ROS and inflammation, while also reducing arterial and RV remodeling [151,152]. Currently, bardoxolone methyl, an inducer of Nrf2, is underway in a phase II clinical study in PAH patients (NCT02036970) [152]. Taken together, the relationship among mitochondrial biogenesis, fusion, fission, and function are important in the observed hyperproliferation observed in PAH.
The upstream mechanisms underlying alterations in mitochondrial structure and metabolic reprogramming are unclear. One regulator, the deacetylase Sirtuin 3 (SIRT3), organizes multiple levels of mitochondrial function through mitochondrial protein deacetylation and thus activation of enzymes and ETC complexes [153]. A single nucleotide polymorphism (SNP) in the SIRT3 gene led to a decrease in enzymatic activity and was associated with the development of metabolic syndrome in humans [154]. Thus, decreased expression or function of SIRT3 could serve as an explanation for the metabolic abnormalities in PAH. Mouse PASMCs deficient in SIRT3 revealed hyper-acetylation, which presumably caused the decreased PDH activity and depletion of TCA cycle intermediates. Interestingly, genetic ablation of SIRT3 also caused the activation of NFAT, the previously discussed transcription factor that promotes PH pathogenesis [155]. The traditional MCT-induced rat model of PAH demonstrated attenuation of PGC1α and SORT3 mRNA levels, which when coupled with the finding that SIRT3 KO mice developed spontaneous PAH, further implicates SIR3 as having a broad regulatory role in the events underpinning PAH metabolic reprogramming [155]. Of note, PGC-1α can activate Sirt3 gene expression, which was previously discussed as being dysregulated downstream of BMPR2 mutations [148,156].
3.9. Targeting Endoplasmic Reticulum Stress
Dysregulation of calcium ion dynamics within the cell has the potential to manifest as endoplasmic reticulum (ER) stress. Intracellular calcium is one of the most important intracellular signaling molecules, requiring tight regulation in time, space, and concentration for proper homeostatic functioning of the cell [157]. The ER serves a critical homeostatic function in the storage of intracellular calcium and the maintenance of its homeostasis [158]. Furthermore, the mitochondrion, as an organelle with tightly controlled calcium dynamics, is dependent on the ER for maintenance of calcium homeostasis and proper functioning of calcium-dependent mitochondrial enzymes such as PDH, α-ketoglutarate dehydrogenase, and isocitrate dehydrogenase [15,159]. As previously discussed, the MPTP opens in response to depolarization and ROS to mediate the translocation of proapoptotic mediators [130]. An inability of calcium transfer between the ER and mitochondria would predispose mitochondria to remain in a relatively hyperpolarized state, conferring resistance to apoptosis and contributing to the observed phenotype seen in PAH [131].
The transfer of calcium from the ER to mitochondria is dependent on the tubular structure of the ER in forming a functional ER-mitochondrial unit, which is critically regulated by the protein Nogo [23]. Specifically, the isoform Nogo-B, the only isoform found in pulmonary vasculature, is implicated as having a role in the disruption of the ER-mitochondria unit [160,161]. The upregulation of Nogo-B in mice SMCs is seen under conditions of ER stress induced by hypoxia via activation of the unfolded protein response (UPR) through activating transcription factor 6 (ATF6)—one of three prongs of the UPR [160,162]. The dysregulation of calcium dynamics predisposes the cell to increased unfolded or misfolded proteins, resulting in ER stress and activation of the UPR—a fundamentally adaptive cytoprotective mechanism meant to overcome cellular stress via the upregulation of relevant chaperones and the global shutdown of protein synthesis [163]. Increased Nogo-B widened the distance between the ER and mitochondria and decreased mitochondria-dependent apoptosis; however, genetic ablation in Nogo-A/B−/− mice prevented the observed hypoxia-induced changes, conferring resistance to PAH development [160]. Inhibition of ATF6 using the protease inhibitor 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) to prevent its cleavage and activation or using an ATF6 small interfering RNA prevented Nogo-B induction in PASMCs under ER stress and normalized Nogo-B expression in PAH PASMCs [160,164].
Chemical chaperones like 4-phenylbutyrate (PBA) and tauroursodeoxycholic acid (TUDCA) are other potential therapeutic agents capable of reducing ER stress. Administration of PBA prevented and reversed PAH in CH-induced mice and in MCT-induced rats. Moreover, PBA and TUDCA inhibited the ATF6 prong of the UPR in PASMCs under hypoxia, causing a subsequent decrease in Nogo expression, mitochondrial calcium levels, and mitochondrial membrane potential. Both in vitro and in vivo, these agents reduced proliferation and induced apoptosis in PASMCs, which reversed and prevented pulmonary vascular remodeling [165]. Interestingly, BMPR2 mutations have been implicated in protein misfolding, aggregation, and subsequent ER stress, and PBA treatment has been shown to restore BMPR2 signaling in HeLa cells transfected with the human disease-causing BMPR2 mutant [166]. Notably, PBA and TUDCA already have FDA approval for urea cycle and cholestatic liver disorders, meaning there is a potential for rapid clinical translation [167]. Another modulator of the UPR prong mediated through PKR-like ER kinase (PERK), salubrinal, prevented and attenuated MCT-induced PAH in rats [168].
The significance of the therapeutic potential underlying ER stress is that its stimuli are broad and diverse, meaning that therapeutic treatment may be broadly relevant for multiple subtypes of PH. For example, ER stress can be triggered via hypoxia, mutations in BMPR2, HIV or HSV infected PASMCs, and overexpression of Notch3—all of which have been linked to the pathogenesis of PAH [166,169,170,171,172]. Such capacity to integrate many disparate signals to mediate adaptive responses via mitochondria or other cytoprotective mechanisms implicates the ER as a convergence point in metabolic dysfunction.
3.10. Targeting Metabolic Dysfunction beyond the Pulmonary Vasculature: Fatty Acid Oxidation in the Right Ventricle
The natural course of PAH is described as a pathological remodeling of pulmonary vessels that results in increased pulmonary vasculature resistance, causing increased afterload and strain, hypertrophy, dysfunction, and ultimately failure of the RV. Traditionally, the obvious therapeutic approaches for PAH sought to target the upstream events of this pathological cascade: pulmonary vascular remodeling and resistance. However, experimental evidence has demonstrated that therapeutic intervention targeting the RV may also be worthwhile, especially since RV function has shown to be a powerful prognostic factor over other measures of pulmonary vascular obstruction [173]. Positron emission tomography (PET) studies have shown an increase in glucose uptake by the RV in both animals and patients with RVH, which implies an upregulation of glycolysis and dysregulation of mitochondrial metabolism [174]. Under normal conditions, fatty acid oxidation (FAO) accounts for the vast majority (60%–90%) of energy production in cardiomyocytes, while the rest (10%–40%) is driven by GO [175]. Notably, there exists a mutually competitive relationship between FAO and GO, known as the Randle cycle, that can be exploited in the treatment of RV hypertrophy [176]. The Randle cycle acts through an increased production of citrate during FAO, which inhibits phosphofructokinase (PFK) and causing a subsequent accumulation of glucose-6-phosphate (G6P). The increase in G6P inhibits hexokinase, resulting in a decrease in the production of pyruvate. Pyruvate processing is further inhibited by the production of acetyl-CoA during FAO [177]. The cumulative effect of these metabolic events facilitates a preference toward FAO over GO in cardiomyocytes (Figure 2). In comparison to the LV, the RV has lower oxygen requirements in part due to lower mass and lower energy expenditure that is concordantly matched with lower coronary blood flow. Accordingly, RV hypertrophy and an increased metabolic demand coupled with less reservoir capacity of coronary blood flow implies a deficiency of oxygen despite a greater oxygen demand.
A loss of adequate oxygen supply causes activation of HIF-1α in cardiomyocytes and the upregulation of relevant glycolytic genes, which may account for the observed increased glucose uptake previously discussed [178]. In fact, the upregulation of HIF-1α in hypertrophied RV has been demonstrated in both CH-induced and MCT-induced models [179,180]. In both MCT- and pulmonary arterial banding (PAB)-induced RV hypertrophy, the administration of DCA reduced PDH phosphorylation, improved GO, partially restored Kv1.5 channel expression, and increased cardiac output and function [18]. These same beneficial effects were seen in the pulmonary vasculature, as previously mentioned [31,36]. The Randle cycle can also be exploited using FAO inhibitors to improve GO [181]. The FAO inhibitors trimetazidine and ranolazine can reverse the glycolytic shift back toward GO [182,183]. In a PAB-induced model, the use of trimetazidine and ranolazine reversed the metabolic changes in RV hypertrophy, enhanced GO, and improved RV function [184]. Accordingly, multiple clinical trials are currently underway or have been completed using FAO inhibitors.
A completed phase III study examined if three months of ranolazine treatment improved blood flow the heart, exercise capacity, and quality of life in symptomatic PAH patients with echocardiographic evidence of RV dysfunction (NCT01174173). At the end of the three months, there was an improvement in functional class, reduction and improvement of the RV, and an increasing trend toward better exercise capacity; however, there was no observed improvement in hemodynamic parameters [185]. A recently finished phase IV study examined the effects of six months of ranolazine in PH associated with LV diastolic dysfunction and has yet to be published (NCT02133352). Two ongoing studies sponsored by the University of Pennsylvania are examining the effects of ranolazine in subjects on stable PH therapies with RV dysfunction (RVEF < 45%) and comparing baseline metabolic profiles of subjects with and without RV dysfunction (NCT01839110 and NCT02829034). Additionally, a study is examining the differences in metabolism and functional imaging in PH patients with normal and persistent RV dysfunction. Metabolic and structural changes in response to treatment with ranolazine are also being measured with 11C-acetate and 18-FDG PET/CT imaging and cardiac MRI (NCT01917136). Lastly, a phase II trial has begun enrollment, studying the effects of trimetazidine on RV function, ventricular remodeling, and miRNA expression (NCT02102672). Overall, the relatively rapid time from the first proposal of FAO inhibitors as a treatment for PAH to their current use in multiple clinical trials shows the power behind the repurposing of currently approved therapies. Taken together, targeting dysfunction at the RV separately from dysregulation of pulmonary vascular remodeling provides another avenue, if used in combination to classical therapeutic approaches, in the treatment of PAH.
4. Future Directions
Since its original conception, the metabolic theory of PH has expanded in molecular scope and complexity outside the umbrella of the Warburg model. As a result, it serves as a powerful conceptual foundation for the elucidation of molecular dysregulation, disease manifestation, and future therapeutic intervention. As has been evidenced by the numerous multicenter, randomized, placebo-controlled clinical trials discussed in this review, strides are being made within the realm of metabolic intervention for PH; however, despite these successes, there are still barriers and challenges that must be overcome.
The current mainstay of PH treatment involves repurposed drugs that do more to support and preserve RV function than to halt or reverse disease pathology [14]. The lack of therapeutic agents capable of preventing and/or reversing structural alterations within the pulmonary vasculature and RV warrants the discovery of novel drug targets, such as the preclinical studies of novel drugs summarized in Table 1. Unfortunately, challenges in the identification of novel drug targets are inherent to the disease process of PH. As a relatively rare disease with multifactorial mechanisms of pathogenesis, identification of potential targets is variable based on the underlying disease-inciting stimulus. The elucidation of novel metabolic targets—when coupled with our recent understanding of genetic polymorphisms and their role in PH—becomes highly specific to subpopulations of patients, which further reduces an already limited population for clinical testing of new interventions.
Much of the rapid translation of preclinical findings in animal models to clinical trials in PH patients, as summarized in Table 2, can be attributed to the repurposing of already FDA-approved medications. Since the metabolic theory of PH share parallels in metabolic dysfunction with cancer, there is great potential for overlap and use of current therapies. In addition, the further development of new small molecules will be of critical importance, especially if targeting metabolic aberrations based on genetic polymorphisms, such as those downstream of BMPR2 mutations. The development of new small molecules will require detailed analyses of genomic and metabolomic screenings to identify polymorphisms and metabolites involved in disease manifestation and progression. The multitude of metabolic anomalies suggests that perhaps multiple errant pathways may need to be targeted for an efficacious clinical response. This rationale of combination therapy has been demonstrated in many chronic diseases, and long-term outcome trials are increasingly indicating that monotherapy is less effective than immediate combination therapy in delaying PH progression over time [186]. With many emerging metabolic therapies in clinical trials, there is an obvious need for studies that examine the combined effects of these new therapeutics.
Beyond therapeutics, the utility of metabolomic screening may also serve in the identification of novel clinical biomarkers for early diagnosis and prognosis. Mitochondria produce diffusible metabolites, which can serve as markers of disease progression, severity, and perhaps even signature when considering tailored therapies. As such, the use of precision medicine is central to the future of PH treatment, for the selective targeting of patient-specific pathways has the power of producing highly efficacious clinical responses and deepen our understanding of disease heterogeneity.
Noninvasive molecular imaging is an emerging application of technology in the study, staging, and potentially treatment of PH. The use of imaging can visualize metabolic shifts occurring in PH, such as enhanced glucose uptake and glycolysis following the inhibition of mitochondrial oxidative phosphorylation. The PET marker 18F-fluorodeoxyglucose (18FDG) is analogous to glucose and its uptake is driven by glucose transporters, meaning that its accumulation within cells can be visualized as a measure of cellular energy utilization. Diseased pulmonary vasculature in most patients with PAH has demonstrated via PET a chronic induction of the Warburg phenotype, as evidenced by increased glucose uptake; however, there is an observed variability among patients that is indicative of metabolic heterogeneity in patient populations [187,188]. Moreover, PET imaging of MCT-induced PAH rat model treated with DCA showed reductions in 18FDG uptake, revealing PET utility in investigating PH molecular pathology and its response to treatment [188]. The pulmonary vascular remodeling underlying PH involves ECs, SMCs, fibroblasts, and the infiltration of inflammatory cells, which limits the resolution of 18FDG PET imaging in being able to differentiate among cell types. Thus, increased 18FDG uptake is assumed to be due to a combination of a hyperproliferative state and an invasive inflammatory component [189,190,191]. Similar to other findings implicating the RV in the metabolic disease process, 18FDG uptake is markedly increased in the RV of both patients and rat models [18,174,192,193]. Future studies using PET can examine metabolism between two distinct anatomic compartments—the pulmonary vasculature and the RV—perhaps revealing previously undiscovered spatiotemporal relationships. Additional uses of PET can investigate drug distribution, drug target-binding, and drug-induced biochemical responses; however, its ease of use in preclinical and clinical application is unfortunately limited by cost and availability.
The complexity surrounding metabolite heterogeneity in PH patient populations can be addressed with novel applications of current technology, such as nuclear magnetic resonance, mass spectrometry, and chromatography. A recently published study utilized high-performance liquid and gas chromatography in combination with mass spectrometry in PAH and control populations to examine metabolic fingerprints. The study found a significant shift in metabolites related to the glycolytic shift and lipid-related energy imbalance that could be considered pathological hallmarks of PAH. In addition, novel metabolites not previously associated with PAH were discovered, suggestive of novel biochemical insight into metabolic dysfunction and potential use as clinical biomarkers [194].
5. Conclusions
The advancement of a metabolic theory in PH has been critical in the proposal of novel therapeutic strategies targeting metabolism and mitochondria. However, this theory has yet to comprehensively connect the seemingly discordant metabolic aberrations underlying PH pathophysiology. Understanding those fundamental molecular links will be essential in realizing the application of novel and potentially combinatorial therapies in the human population. For example, genomic and metabolomic profiles of PH patient populations will illuminate molecular explanations of individualized disease manifestations, allowing for the development of future treatment strategies based on a stratification of patient populations. Accordingly, due to the exponentially increasing possibilities of metabolic targets in PH, future work in the field will require not only further insight into underpinnings of the metabolic theory as a framework for molecular organization but also a better application of each person’s metabolic profile in tailoring and prioritizing specific therapeutic interventions.
Acknowledgments
This work was supported by the NIH Medical Scientist Training Program (L.D.H.), as well as NIH grants HL096834, HL124021, and the American Heart Association grant 14GRNT19600012 (S.Y.C.).
Author Contributions
Both authors contributed to the composition of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Simonneau, G.; Robbins, I.M.; Beghetti, M.; Channick, R.N.; Delcroix, M.; Denton, C.P.; Elliott, C.G.; Gaine, S.P.; Gladwin, M.T.; Jing, Z.C.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2009, 54, S43–S54. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Simonneau, G. The fifth world symposium on pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D1–D3. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Archer, S.L.; Badesch, D.B.; Barst, R.J.; Farber, H.W.; Lindner, J.R.; Mathier, M.A.; McGoon, M.D.; Park, M.H.; Rosenson, R.S.; et al. Accf/aha 2009 expert consensus document on pulmonary hypertension a report of the american college of cardiology foundation task force on expert consensus documents and the american heart association developed in collaboration with the american college of chest physicians; american thoracic society, inc.; and the pulmonary hypertension association. J. Am. Coll. Cardiol. 2009, 53, 1573–1619. [Google Scholar] [PubMed]
- Archer, S.L.; Weir, E.K.; Wilkins, M.R. Basic science of pulmonary arterial hypertension for clinicians: New concepts and experimental therapies. Circulation 2010, 121, 2045–2066. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Marecki, J.C.; Richter, A.; Fijalkowska, I.; Flores, S. Pathology of pulmonary hypertension. Clin. Chest Med. 2007, 28, 23–42. [Google Scholar] [CrossRef] [PubMed]
- D’Alonzo, G.E.; Barst, R.J.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Kernis, J.T.; et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann. Intern. Med. 1991, 115, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Dresdale, D.T.; Schultz, M.; Michtom, R.J. Primary pulmonary hypertension. I. Clinical and hemodynamic study. Am. J. Med. 1951, 11, 686–705. [Google Scholar] [CrossRef]
- Ward, J.P.; McMurtry, I.F. Mechanisms of hypoxic pulmonary vasoconstriction and their roles in pulmonary hypertension: New findings for an old problem. Curr. Opin. Pharmacol. 2009, 9, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. Hypoxic pulmonary vasoconstriction. J. Appl. Physiol. 2005, 98, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Dunham-Snary, K.J.; Wu, D.; Sykes, E.A.; Thakrar, A.; Parlow, L.R.; Mewburn, J.D.; Parlow, J.L.; Archer, S.L. Hypoxic pulmonary vasoconstriction: From molecular mechanisms to medicine. Chest 2017, 151, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Cottrill, K.A.; Chan, S.Y. Metabolic dysfunction in pulmonary hypertension: The expanding relevance of the warburg effect. Eur. J. Clin. Invest. 2013, 43, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Thebaud, B.; Weir, E.K.; Archer, S.L. Hypoxic pulmonary vasoconstriction: Redox regulation of o2-sensitive k+ channels by a mitochondrial o2-sensor in resistance artery smooth muscle cells. J. Mol. Cell Cardiol. 2004, 37, 1119–1136. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Sitbon, O.; Simonneau, G. Treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1425–1436. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Davis, L.A.; Graham, B.B. Targeting energetic metabolism: A new frontier in the pathogenesis and treatment of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Fang, Y.H.; Cadete, V.J.; Wietholt, C.; Urboniene, D.; Toth, P.T.; Marsboom, G.; Zhang, H.J.; Haber, I.; Rehman, J.; et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: Resuscitating the hibernating right ventricle. J. Mol. Med. 2010, 88, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Malenfant, S.; Potus, F.; Fournier, F.; Breuils-Bonnet, S.; Pflieger, A.; Bourassa, S.; Tremblay, E.; Nehme, B.; Droit, A.; Bonnet, S.; et al. Skeletal muscle proteomic signature and metabolic impairment in pulmonary hypertension. J. Mol. Med. 2015, 93, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Sawada, H.; Saito, T.; Nickel, N.P.; Alastalo, T.P.; Glotzbach, J.P.; Chan, R.; Haghighat, L.; Fuchs, G.; Januszyk, M.; Cao, A.; et al. Reduced bmpr2 expression induces gm-csf translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J. Exp. Med. 2014, 211, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; McMurtry, M.S.; Wu, X.C.; Dyck, J.R.; Moudgil, R.; Hopkins, T.A.; Lopaschuk, G.D.; Puttagunta, L.; Waite, R.; Archer, S.L. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 2002, 105, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Nozik-Grayck, E.; Gerasimovskaya, E.; Anwar, A.; Li, M.; Riddle, S.; Frid, M. The adventitia: Essential role in pulmonary vascular remodeling. Compr. Physiol. 2011, 1, 141–161. [Google Scholar] [PubMed]
- Voeltz, G.K.; Prinz, W.A.; Shibata, Y.; Rist, J.M.; Rapoport, T.A. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 2006, 124, 573–586. [Google Scholar] [CrossRef] [PubMed]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, L.A.; Semenza, G.L. Hif and the lung: Role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 2011, 183, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 284, 16767–16775. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.Y.; Shimoda, L.A.; Iyer, N.V.; Huso, D.L.; Sun, X.; McWilliams, R.; Beaty, T.; Sham, J.S.; Wiener, C.M.; Sylvester, J.T.; et al. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J. Clin. Invest. 1999, 103, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Compernolle, V.; Tjwa, M.; Wiesener, M.S.; Maxwell, P.H.; Collen, D.; Carmeliet, P. Heterozygous deficiency of hypoxia-inducible factor-2alpha protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J. Clin. Invest. 2003, 111, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Michelakis, E.D.; Porter, C.J.; Andrade-Navarro, M.A.; Thebaud, B.; Bonnet, S.; Haromy, A.; Harry, G.; Moudgil, R.; McMurtry, M.S.; et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: Similarities to human pulmonary arterial hypertension. Circulation 2006, 113, 2630–2641. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Webb, S.; Tucker, A.; Rabinovitch, M.; O'Brien, R.F.; McMurtry, I.F.; Stelzner, T.J. Factors influencing the idiopathic development of pulmonary hypertension in the fawn hooded rat. Am. Rev. Respir. Dis. 1992, 145, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Dang, C.V. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 2005, 30, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Bersin, R.M.; Stacpoole, P.W. Dichloroacetate as metabolic therapy for myocardial ischemia and failure. Am. Heart J. 1997, 134, 841–855. [Google Scholar] [CrossRef]
- McMurtry, M.S.; Bonnet, S.; Wu, X.; Dyck, J.R.; Haromy, A.; Hashimoto, K.; Michelakis, E.D. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 2004, 95, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Loscalzo, J. Microrna-210: A unique and pleiotropic hypoxamir. Cell Cycle 2010, 9, 1072–1083. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. Microrna-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins iscu1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H.; Holm, R.H.; Munck, E. Iron-sulfur clusters: Nature's modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A.; Tong, W.H. Iron-sulfur cluster biogenesis and human disease. Trends Genet. 2008, 24, 398–407. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Lu, Y.; Annis, S.; Hale, A.E.; Chau, B.N.; Dahlman, J.E.; Hemann, C.; Opotowsky, A.R.; Vargas, S.O.; Rosas, I.; et al. Genetic and hypoxic alterations of the microrna-210-iscu1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol. Med. 2015, 7, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sastre, A.; Tort, F.; Stehling, O.; Uzarska, M.A.; Arranz, J.A.; Del Toro, M.; Labayru, M.T.; Landa, J.; Font, A.; Garcia-Villoria, J.; et al. A fatal mitochondrial disease is associated with defective nfu1 function in the maturation of a subset of mitochondrial fe-s proteins. Am. J. Hum. Genet. 2011, 89, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Ahting, U.; Mayr, J.A.; Vanlander, A.V.; Hardy, S.A.; Santra, S.; Makowski, C.; Alston, C.L.; Zimmermann, F.A.; Abela, L.; Plecko, B.; et al. Clinical, biochemical, and genetic spectrum of seven patients with nfu1 deficiency. Front. Genet. 2015, 6, 123. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Howard, L.S.; Busbridge, M.; Ashby, D.; Kondili, E.; Gibbs, J.S.; Wharton, J.; Wilkins, M.R. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: Clinical prevalence, outcomes, and mechanistic insights. J. Am. Coll. Cardiol. 2011, 58, 300–309. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.J.; Wharton, J.; Howard, L.; Gibbs, J.S.; Vonk-Noordegraaf, A.; Wilkins, M.R. Iron deficiency in pulmonary arterial hypertension: A potential therapeutic target. Eur. Respir. J. 2011, 38, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Lankhorst, S.; Boonstra, A.; Postmus, P.E.; Zweegman, S.; Westerhof, N.; van der Laarse, W.J.; Vonk-Noordegraaf, A. Iron deficiency is common in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2011, 37, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Treacy, C.M.; Toshner, M.R.; MacKenzie-Ross, R.; Manglam, V.; Busbridge, M.; Sinclair-McGarvie, M.; Arnold, J.; Sheares, K.K.; Morrell, N.W.; et al. Unexplained iron deficiency in idiopathic and heritable pulmonary arterial hypertension. Thorax 2011, 66, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Clifton, I.J.; McDonough, M.A.; Ehrismann, D.; Kershaw, N.J.; Granatino, N.; Schofield, C.J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded beta-helix fold proteins. J. Inorg. Biochem. 2006, 100, 644–669. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R. Iron homeostasis and its interaction with prolyl hydroxylases. Antioxid. Redox. Signal 2010, 12, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Walden, W.E.; Selezneva, A.I.; Dupuy, J.; Volbeda, A.; Fontecilla-Camps, J.C.; Theil, E.C.; Volz, K. Structure of dual function iron regulatory protein 1 complexed with ferritin ire-rna. Science 2006, 314, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.C.; Zhang, D.L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of hif2alpha. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Park, J.E.; Wort, S.J. The role of endothelin-1 in the pathogenesis of pulmonary arterial hypertension. Pharmacol. Res. 2011, 63, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Thorin, E.; Clozel, M. The cardiovascular physiology and pharmacology of endothelin-1. Adv. Pharmacol. 2010, 60, 1–26. [Google Scholar] [PubMed]
- Ghosh, M.C.; Tong, W.H.; Zhang, D.; Ollivierre-Wilson, H.; Singh, A.; Krishna, M.C.; Mitchell, J.B.; Rouault, T.A. Tempol-mediated activation of latent iron regulatory protein activity prevents symptoms of neurodegenerative disease in irp2 knockout mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12028–12033. [Google Scholar] [CrossRef] [PubMed]
- Cotroneo, E.; Ashek, A.; Wang, L.; Wharton, J.; Dubois, O.; Bozorgi, S.; Busbridge, M.; Alavian, K.N.; Wilkins, M.R.; Zhao, L. Iron homeostasis and pulmonary hypertension: Iron deficiency leads to pulmonary vascular remodeling in the rat. Circ. Res. 2015, 116, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, G.; Manders, E.; Happe, C.M.; Schalij, I.; Groepenhoff, H.; Howard, L.S.; Wilkins, M.R.; Bogaard, H.J.; Westerhof, N.; van der Laarse, W.J.; et al. Intravenous iron therapy in patients with idiopathic pulmonary arterial hypertension and iron deficiency. Pulm Circ. 2015, 5, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Frise, M.C.; Cheng, H.Y.; Nickol, A.H.; Curtis, M.K.; Pollard, K.A.; Roberts, D.J.; Ratcliffe, P.J.; Dorrington, K.L.; Robbins, P.A. Clinical iron deficiency disturbs normal human responses to hypoxia. J. Clin. Invest. 2016, 126, 2139–2150. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mendelsohn, L.; Rogers, H.; Leitman, S.; Raghavachari, N.; Yang, Y.; Yau, Y.Y.; Tallack, M.; Perkins, A.; Taylor, J.G.; et al. Heme-bound iron activates placenta growth factor in erythroid cells via erythroid kruppel-like factor. Blood 2014, 124, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Oliver, E.; Maratou, K.; Atanur, S.S.; Dubois, O.D.; Cotroneo, E.; Chen, C.N.; Wang, L.; Arce, C.; Chabosseau, P.L.; et al. The zinc transporter zip12 regulates the pulmonary vascular response to chronic hypoxia. Nature 2015, 524, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Eide, D.J. The slc39 family of zinc transporters. Mol. Aspects Med. 2013, 34, 612–619. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, E.D.; Selak, M.A.; Tennant, D.A.; Payne, L.J.; Crosby, S.; Frederiksen, C.M.; Watson, D.G.; Gottlieb, E. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell Biol. 2007, 27, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, N.; Baysal, B.E.; Shadel, G.S. Revisiting the tca cycle: Signaling to tumor formation. Trends Mol. Med. 2011, 17, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. Atp-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, C.N.; Hajji, N.; Oliver, E.; Cotroneo, E.; Wharton, J.; Wang, D.; Li, M.; McKinsey, T.A.; Stenmark, K.R.; et al. Histone deacetylation inhibition in pulmonary hypertension: Therapeutic potential of valproic acid and suberoylanilide hydroxamic acid. Circulation 2012, 126, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Michelakis, E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Perez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; Lopez-Larrubia, P.; et al. In vivo hif-mediated reductive carboxylation is regulated by citrate levels and sensitizes vhl-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-mediated increases in l-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Majka, S.; Hagen, M.; Blackwell, T.; Harral, J.; Johnson, J.A.; Gendron, R.; Paradis, H.; Crona, D.; Loyd, J.E.; Nozik-Grayck, E.; et al. Physiologic and molecular consequences of endothelial bmpr2 mutation. Respir. Res. 2011, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Fessel, J.P.; Hamid, R.; Wittmann, B.M.; Robinson, L.J.; Blackwell, T.; Tada, Y.; Tanabe, N.; Tatsumi, K.; Hemnes, A.R.; West, J.D. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ. 2012, 2, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Fang, Y.H.; Parikh, K.; Ryan, J.J.; Toth, P.T.; Archer, S.L. Cardiac glutaminolysis: A maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J. Mol. Med. 2013, 91, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates yap/taz-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Invest. 2016, 126, 3313–3335. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.L. Age-related macular degeneration: Beyond anti-angiogenesis. Mol. Vis. 2014, 20, 46–55. [Google Scholar] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Will, J.A.; Weir, E.K. Redox status in the control of pulmonary vascular tone. Herz 1986, 11, 127–141. [Google Scholar] [PubMed]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox. Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Hampl, V.; Nsair, A.; Wu, X.; Harry, G.; Haromy, A.; Gurtu, R.; Archer, S.L. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ. Res. 2002, 90, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.; Cool, C.; Murphy, R.C.; Tuder, R.M.; Hopken, M.W.; Flores, S.C.; Voelkel, N.F. Oxidative stress in severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2004, 169, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Semenza, G.L. Effect of altered redox states on expression and DNA-binding activity of hypoxia-inducible factor 1. Biochem. Biophys. Res. Commun. 1995, 212, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Weir, E.K.; Lopez-Barneo, J.; Buckler, K.J.; Archer, S.L. Acute oxygen-sensing mechanisms. N. Engl. J. Med. 2005, 353, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Bunn, H.F. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef] [PubMed]
- Sarrion, I.; Milian, L.; Juan, G.; Ramon, M.; Furest, I.; Carda, C.; Cortijo Gimeno, J.; Mata Roig, M. Role of circulating mirnas as biomarkers in idiopathic pulmonary arterial hypertension: Possible relevance of mir-23a. Oxid. Med. Cell Longev. 2015, 2015, 792846. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thebaud, B.; Husain, A.N.; et al. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular superoxide dismutase. Int. J. Biochem. Cell Biol. 2005, 37, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Woods, C.; Stearman, R.S.; Venkataraman, S.; Ferguson, B.S.; Swain, K.; Bowler, R.P.; Geraci, M.W.; Ihida-Stansbury, K.; Stenmark, K.R.; et al. Histone deacetylation contributes to low extracellular superoxide dismutase expression in human idiopathic pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L124–L134. [Google Scholar] [CrossRef] [PubMed]
- Krick, S.; Platoshyn, O.; McDaniel, S.S.; Rubin, L.J.; Yuan, J.X. Augmented K(+) currents and mitochondrial membrane depolarization in pulmonary artery myocyte apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L887–L894. [Google Scholar] [PubMed]
- Remillard, C.V.; Tigno, D.D.; Platoshyn, O.; Burg, E.D.; Brevnova, E.E.; Conger, D.; Nicholson, A.; Rana, B.K.; Channick, R.N.; Rubin, L.J.; et al. Function of kv1.5 channels and genetic variations of kcna5 in patients with idiopathic pulmonary arterial hypertension. Am. J. Physiol. Cell Physiol. 2007, 292, C1837–C1853. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.X.; Aldinger, A.M.; Juhaszova, M.; Wang, J.; Conte, J.V., Jr.; Gaine, S.P.; Orens, J.B.; Rubin, L.J. Dysfunctional voltage-gated K+ channels in pulmonary artery smooth muscle cells of patients with primary pulmonary hypertension. Circulation 1998, 98, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thebaud, B.; Wu, X.C.; Dyck, J.R.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In Vivo gene transfer of the o2-sensitive potassium channel kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Moral-Sanz, J.; Mahmoud, A.D.; Ross, F.A.; Eldstrom, J.; Fedida, D.; Hardie, D.G.; Evans, A.M. Amp-activated protein kinase inhibits kv 1.5 channel currents of pulmonary arterial myocytes in response to hypoxia and inhibition of mitochondrial oxidative phosphorylation. J. Physiol. 2016, 594, 4901–4915. [Google Scholar] [CrossRef] [PubMed]
- Agard, C.; Rolli-Derkinderen, M.; Dumas-de-La-Roque, E.; Rio, M.; Sagan, C.; Savineau, J.P.; Loirand, G.; Pacaud, P. Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br. J. Pharmacol. 2009, 158, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of amp-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Omura, J.; Satoh, K.; Kikuchi, N.; Satoh, T.; Kurosawa, R.; Nogi, M.; Otsuki, T.; Kozu, K.; Numano, K.; Suzuki, K.; et al. Protective roles of endothelial amp-activated protein kinase against hypoxia-induced pulmonary hypertension in mice. Circ. Res. 2016, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M. Pulmonary hypertension in heart failure preserved ejection fraction: Prevalence, pathophysiology, and clinical perspectives. Circ. Heart Fail 2014, 7, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, T.; Shah, S.J.; Gomberg-Maitland, M.; Collander, B.; Vallakati, A.; Shroff, P.; Rich, S. Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circ. Heart Fail 2011, 4, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Duplain, H.; Burcelin, R.; Sartori, C.; Cook, S.; Egli, M.; Lepori, M.; Vollenweider, P.; Pedrazzini, T.; Nicod, P.; Thorens, B.; et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001, 104, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.L.; Ruiz, R.; Gonzalez, M.A.; Ramirez-Lorca, R.; Couto, C.; Ramos, A.; Gutierrez-Tous, R.; Rivera, J.M.; Ruiz, A.; Real, L.M.; et al. Association of nos3 gene with metabolic syndrome in hypertensive patients. Thromb. Haemost 2004, 92, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Giaid, A.; Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995, 333, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Higaki, Y.; Hirshman, M.F.; Fujii, N.; Goodyear, L.J. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes 2001, 50, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Kamga Pride, C.; Mo, L.; Quesnelle, K.; Dagda, R.K.; Murillo, D.; Geary, L.; Corey, C.; Portella, R.; Zharikov, S.; St Croix, C.; et al. Nitrite activates protein kinase a in normoxia to mediate mitochondrial fusion and tolerance to ischaemia/reperfusion. Cardiovasc. Res. 2014, 101, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Lira, V.A.; Soltow, Q.A.; Long, J.H.; Betters, J.L.; Sellman, J.E.; Criswell, D.S. Nitric oxide increases glut4 expression and regulates AMPK signaling in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1062–E1068. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Tabima, D.M.; Dube, J.J.; Hughan, K.S.; Vanderpool, R.R.; Goncharov, D.A.; St Croix, C.M.; Garcia-Ocana, A.; Goncharova, E.A.; Tofovic, S.P.; et al. Sirt3-amp-activated protein kinase activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction. Circulation 2016, 133, 717–731. [Google Scholar] [PubMed]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Pirkmajer, S.; Kulkarni, S.S.; Tom, R.Z.; Ross, F.A.; Hawley, S.A.; Hardie, D.G.; Zierath, J.R.; Chibalin, A.V. Methotrexate promotes glucose uptake and lipid oxidation in skeletal muscle via ampk activation. Diabetes 2015, 64, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Grahame Hardie, D. Regulation of amp-activated protein kinase by natural and synthetic activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Landsberg, J.W.; Yuan, J.X. Calcium and trp channels in pulmonary vascular smooth muscle cell proliferation. News Physiol. Sci. 2004, 19, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Fantozzi, I.; Remillard, C.V.; Landsberg, J.W.; Kunichika, N.; Platoshyn, O.; Tigno, D.D.; Thistlethwaite, P.A.; Rubin, L.J.; Yuan, J.X. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 13861–13866. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Rochefort, G.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Bonnet, S.N.; Michelakis, E.D. The nuclear factor of activated T cells in pulmonary arterial hypertension can be therapeutically targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Eguchi, Y.; Kamiike, W.; Funahashi, Y.; Mignon, A.; Lacronique, V.; Matsuda, H.; Tsujimoto, Y. Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc. Natl. Acad. Sci. USA 1998, 95, 1455–1459. [Google Scholar] [CrossRef] [PubMed]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Malli, R.; Naghdi, S.; Trenker, M.; Kahn, M.J.; Graier, W.F. The contribution of UCP2 and UCP3 to mitochondrial Ca(2+) uptake is differentially determined by the source of supplied Ca(2+). Cell Calcium 2010, 47, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.J.; Zhang, X.; Ge, C.R.; Jois, M. The polymorphisms of UCP2 and UCP3 genes associated with fat metabolism, obesity and diabetes. Obes. Rev. 2009, 10, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Pak, O.; Sommer, N.; Hoeres, T.; Bakr, A.; Waisbrod, S.; Sydykov, A.; Haag, D.; Esfandiary, A.; Kojonazarov, B.; Veit, F.; et al. Mitochondrial hyperpolarization in pulmonary vascular remodeling. Mitochondrial uncoupling protein deficiency as disease model. Am. J. Respir. Cell Mol. Biol. 2013, 49, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Dromparis, P.; Paulin, R.; Sutendra, G.; Qi, A.C.; Bonnet, S.; Michelakis, E.D. Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ. Res. 2013, 113, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Haslip, M.; Dostanic, I.; Huang, Y.; Zhang, Y.; Russell, K.S.; Jurczak, M.J.; Mannam, P.; Giordano, F.; Erzurum, S.C.; Lee, P.J. Endothelial uncoupling protein 2 regulates mitophagy and pulmonary hypertension during intermittent hypoxia. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Chen, K.H.; Dasgupta, A.; Potus, F.; Dunham-Snary, K.; Bonnet, S.; Tian, L.; Fu, J.; Breuils-Bonnet, S.; Provencher, S.; et al. Mir-138 and mir-25 downregulate mcu, causing pulmonary arterial hypertension's cancer phenotype. Am. J. Respir. Crit. Care Med. 2016, 195, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, G.; Ducret, T.; Marthan, R.; Savineau, J.P.; Quignard, J.F. Stretch-induced Ca2+ signalling in vascular smooth muscle cells depends on Ca2+ store segregation. Cardiovasc. Res. 2014, 103, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Lipskaia, L.; del Monte, F.; Capiod, T.; Yacoubi, S.; Hadri, L.; Hours, M.; Hajjar, R.J.; Lompre, A.M. Sarco/endoplasmic reticulum Ca2+-atpase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ. Res. 2005, 97, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Aguero, J.; Ishikawa, K.; Hadri, L.; Santos-Gallego, C.G.; Fish, K.M.; Kohlbrenner, E.; Hammoudi, N.; Kho, C.; Lee, A.; Ibanez, B.; et al. Intratracheal gene delivery of serca2a ameliorates chronic post-capillary pulmonary hypertension: A large animal model. J. Am. Coll. Cardiol. 2016, 67, 2032–2046. [Google Scholar] [CrossRef] [PubMed]
- Hadri, L.; Kratlian, R.G.; Benard, L.; Maron, B.A.; Dorfmuller, P.; Ladage, D.; Guignabert, C.; Ishikawa, K.; Aguero, J.; Ibanez, B.; et al. Therapeutic efficacy of aav1.Serca2a in monocrotaline-induced pulmonary arterial hypertension. Circulation 2013, 128, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B.; Shulga, N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005, 65, 10545–10554. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How pandora's box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Marchetti, P.; Castedo, M.; Hirsch, T.; Susin, S.A.; Masse, B.; Kroemer, G. Inhibitors of permeability transition interfere with the disruption of the mitochondrial transmembrane potential during apoptosis. FEBS Lett. 1996, 384, 53–57. [Google Scholar] [CrossRef]
- Camara, A.K.; Lesnefsky, E.J.; Stowe, D.F. Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox. Signal 2010, 13, 279–347. [Google Scholar] [CrossRef] [PubMed]
- Aramburu, J.; Yaffe, M.B.; Lopez-Rodriguez, C.; Cantley, L.C.; Hogan, P.G.; Rao, A. Affinity-driven peptide selection of an nfat inhibitor more selective than cyclosporin a. Science 1999, 285, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Tian, X.; Cai, J.; Hopper, R.K.; Sudheendra, D.; Li, C.G.; El-Bizri, N.; Sawada, H.; Haghighat, R.; Chan, R.; et al. FK506 activates Bmpr2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Invest. 2013, 123, 3600–3613. [Google Scholar] [CrossRef] [PubMed]
- Young, K.A.; Ivester, C.; West, J.; Carr, M.; Rodman, D.M. Bmp signaling controls pasmc Kv channel expression In Vitro and In Vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L841–L848. [Google Scholar] [CrossRef] [PubMed]
- Spiekerkoetter, E.; Sung, Y.K.; Sudheendra, D.; Bill, M.; Aldred, M.A.; van de Veerdonk, M.C.; Vonk Noordegraaf, A.; Long-Boyle, J.; Dash, R.; Yang, P.C.; et al. Low-dose Fk506 (tacrolimus) in end-stage pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chomyn, A.; Chan, D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005, 280, 26185–26192. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase DRP1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondrial dynamics in disease. N. Engl. J. Med. 2007, 356, 1707–1709. [Google Scholar] [CrossRef] [PubMed]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Marsboom, G.; Toth, P.T.; Ryan, J.J.; Hong, Z.; Wu, X.; Fang, Y.H.; Thenappan, T.; Piao, L.; Zhang, H.J.; Pogoriler, J.; et al. Dynamin-related protein 1-mediated mitochondrial mitotic fission permits hyperproliferation of vascular smooth muscle cells and offers a novel therapeutic target in pulmonary hypertension. Circ. Res. 2012, 110, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.R.; Blackstone, C. Cyclic amp-dependent protein kinase phosphorylation of DRP1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 2007, 282, 21583–21587. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, J.T.; Strack, S. Reversible phosphorylation of drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007, 8, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.J.; Marsboom, G.; Fang, Y.H.; Toth, P.T.; Morrow, E.; Luo, N.; Piao, L.; Hong, Z.; Ericson, K.; Zhang, H.J.; et al. Pgc1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Alvira, C.M.; Alastalo, T.P.; Sawada, H.; Hansmann, G.; Zhao, M.; Wang, L.; El-Bizri, N.; Rabinovitch, M. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes pdgf receptor-beta-dependent pulmonary arterial muscularization. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L1082–L1090. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, G.; de Jesus Perez, V.A.; Alastalo, T.P.; Alvira, C.M.; Guignabert, C.; Bekker, J.M.; Schellong, S.; Urashima, T.; Wang, L.; Morrell, N.W.; et al. An antiproliferative bmp-2/ppargamma/apoe axis in human and murine smcs and its role in pulmonary hypertension. J. Clin. Invest. 2008, 118, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Hennigs, J.K.; Miyagawa, K.; Li, C.G.; Nickel, N.P.; Kaschwich, M.; Cao, A.; Wang, L.; Reddy, S.; Chen, P.I.; et al. Bmpr2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 2015, 21, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Alastalo, T.P.; Li, M.; Perez Vde, J.; Pham, D.; Sawada, H.; Wang, J.K.; Koskenvuo, M.; Wang, L.; Freeman, B.A.; Chang, H.Y.; et al. Disruption of ppargamma/beta-catenin-mediated regulation of apelin impairs bmp-induced mouse and human pulmonary arterial ec survival. J. Clin. Invest. 2011, 121, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.; Ali, Z.A.; Kojima, Y.; Kundu, R.K.; Sheikh, A.Y.; Agrawal, R.; Zheng, L.; Leeper, N.J.; Pearl, N.E.; Patterson, A.J.; et al. Apelin signaling antagonizes ang ii effects in mouse models of atherosclerosis. J. Clin. Invest. 2008, 118, 3343–3354. [Google Scholar] [CrossRef] [PubMed]
- Eba, S.; Hoshikawa, Y.; Moriguchi, T.; Mitsuishi, Y.; Satoh, H.; Ishida, K.; Watanabe, T.; Shimizu, T.; Shimokawa, H.; Okada, Y.; et al. The nuclear factor erythroid 2-related factor 2 activator oltipraz attenuates chronic hypoxia-induced cardiopulmonary alterations in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Xu, Q.; McTiernan, C.; Lai, Y.C.; Osei-Hwedieh, D.; Gladwin, M. Novel targets of drug treatment for pulmonary hypertension. Am. J. Cardiovasc. Drugs 2015, 15, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.A.; Hirschey, M.D. Mitochondrial protein acetylation regulates metabolism. Essays Biochem. 2012, 52, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Huang, J.Y.; Schwer, B.; Verdin, E. Sirt3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Dromparis, P.; Sutendra, G.; Gurtu, V.; Zervopoulos, S.; Bowers, L.; Haromy, A.; Webster, L.; Provencher, S.; Bonnet, S.; et al. Sirtuin 3 deficiency is associated with inhibited mitochondrial function and pulmonary arterial hypertension in rodents and humans. Cell Metab. 2014, 20, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Giralt, A.; Hondares, E.; Villena, J.A.; Ribas, F.; Diaz-Delfin, J.; Giralt, M.; Iglesias, R.; Villarroya, F. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J. Biol. Chem. 2011, 286, 16958–16966. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Duchen, M.R.; Pozzan, T. Flirting in little space: The er/mitochondria Ca2+ liaison. Sci. STKE 2004, 2004, re1. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Dromparis, P.; Wright, P.; Bonnet, S.; Haromy, A.; Hao, Z.; McMurtry, M.S.; Michalak, M.; Vance, J.E.; Sessa, W.C.; et al. The role of nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci. Transl. Med. 2011, 3, 88ra55. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.Y.; Tang, B.L. Cell autonomous function of nogo and reticulons: The emerging story at the endoplasmic reticulum. J. Cell Physiol. 2008, 216, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.J.; Tadimalla, A.; Chen, W.J.; Martindale, J.J.; Thuerauf, D.J.; Marcinko, M.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Coordination of growth and endoplasmic reticulum stress signaling by regulator of calcineurin 1 (rcan1), a novel atf6-inducible gene. J. Biol. Chem. 2008, 283, 14012–14021. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Haze, K.; Nadanaka, S.; Yoshida, H.; Seidah, N.G.; Hirano, Y.; Sato, R.; Negishi, M.; Mori, K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor atf6. J. Biol. Chem. 2003, 278, 31024–31032. [Google Scholar] [CrossRef] [PubMed]
- Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Sutendra, G.; Michelakis, E.D. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation 2013, 127, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Sobolewski, A.; Rudarakanchana, N.; Upton, P.D.; Yang, J.; Crilley, T.K.; Trembath, R.C.; Morrell, N.W. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: Potential for rescue. Hum. Mol. Genet. 2008, 17, 3180–3190. [Google Scholar] [CrossRef] [PubMed]
- Engin, F.; Hotamisligil, G.S. Restoring endoplasmic reticulum function by chemical chaperones: An emerging therapeutic approach for metabolic diseases. Diabetes Obes. Metab. 2010, 12, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.E.; Reddy, M.B.; Nguyen, C.M.; Colvin, K.L.; Ivy, D.D.; Stenmark, K.R. Activation of the unfolded protein response is associated with pulmonary hypertension. Pulm Circ. 2012, 2, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Voelkel, N.F.; Cool, C.D.; Flores, S. From viral infection to pulmonary arterial hypertension: A role for viral proteins? AIDS 2008, 22, S49–S53. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B.; Mukhopadhyay, S.; Patel, K.; Xu, F.; Almodovar, S.; Tuder, R.M.; Flores, S.C. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in shiv-nef-infected macaques. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L729–L737. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Adachi, K.; Yoshizaki, K.; Kunimoto, S.; Kalaria, R.N.; Watanabe, A. Mutations in notch3 cause the formation and retention of aggregates in the endoplasmic reticulum, leading to impaired cell proliferation. Hum. Mol. Genet. 2010, 19, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Leathers, R.; Makino, A.; Huang, C.; Parsa, P.; Macias, J.; Yuan, J.X.; Jamieson, S.W.; Thistlethwaite, P.A. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 2009, 15, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Van Wolferen, S.A.; Marcus, J.T.; Boonstra, A.; Marques, K.M.; Bronzwaer, J.G.; Spreeuwenberg, M.D.; Postmus, P.E.; Vonk-Noordegraaf, A. Prognostic value of right ventricular mass, volume, and function in idiopathic pulmonary arterial hypertension. Eur. Heart J. 2007, 28, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, M.; Kagaya, Y.; Otani, H.; Sakuma, M.; Demachi, J.; Suzuki, J.; Takahashi, T.; Nawata, J.; Ido, T.; Watanabe, J.; et al. Increased [18f]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J. Am. Coll. Cardiol. 2005, 45, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Lopaschuk, G.D.; Hall, J.L.; McCormack, J.G. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc. Res. 1997, 33, 243–257. [Google Scholar] [CrossRef]
- Archer, S.L.; Fang, Y.H.; Ryan, J.J.; Piao, L. Metabolism and bioenergetics in the right ventricle and pulmonary vasculature in pulmonary hypertension. Pulm Circ. 2013, 3, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.M.; Frazier, D.P.; Thompson, J.W.; Haliko, S.; Li, H.; Wasserlauf, B.J.; Spiga, M.G.; Bishopric, N.H.; Webster, K.A. A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J. Exp. Biol. 2004, 207, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Bogaard, H.J.; Natarajan, R.; Henderson, S.C.; Long, C.S.; Kraskauskas, D.; Smithson, L.; Ockaili, R.; McCord, J.M.; Voelkel, N.F. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 2009, 120, 1951–1960. [Google Scholar] [CrossRef] [PubMed]
- Redout, E.M.; Wagner, M.J.; Zuidwijk, M.J.; Boer, C.; Musters, R.J.; van Hardeveld, C.; Paulus, W.J.; Simonides, W.S. Right-ventricular failure is associated with increased mitochondrial complex II activity and production of reactive oxygen species. Cardiovasc. Res. 2007, 75, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.; Marsboom, G.; Archer, S.L. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J. Mol. Med. 2010, 88, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Kantor, P.F.; Lucien, A.; Kozak, R.; Lopaschuk, G.D. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme a thiolase. Circ. Res. 2000, 86, 580–588. [Google Scholar] [CrossRef] [PubMed]
- McCormack, J.G.; Barr, R.L.; Wolff, A.A.; Lopaschuk, G.D. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation 1996, 93, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting randle’s cycle. J. Mol. Med. 2012, 90, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.S.; Cuttica, M.J.; Beussink-Nelson, L.; Kozyleva, A.; Sanchez, C.; Mkrdichian, H.; Selvaraj, S.; Dematte, J.E.; Lee, D.C.; Shah, S.J. Effects of ranolazine on exercise capacity, right ventricular indices, and hemodynamic characteristics in pulmonary arterial hypertension: A pilot study. Pulm Circ. 2015, 5, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Gaine, S. Beyond a single pathway: Combination therapy in pulmonary arterial hypertension. Eur. Respir. Rev. 2016, 25, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ashek, A.; Wang, L.; Fang, W.; Dabral, S.; Dubois, O.; Cupitt, J.; Pullamsetti, S.S.; Cotroneo, E.; Jones, H.; et al. Heterogeneity in lung (18)FDG uptake in pulmonary arterial hypertension: Potential of dynamic (18)FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation 2013, 128, 1214–1224. [Google Scholar] [PubMed]
- Price, L.C.; Wort, S.J.; Perros, F.; Dorfmuller, P.; Huertas, A.; Montani, D.; Cohen-Kaminsky, S.; Humbert, M. Inflammation in pulmonary arterial hypertension. Chest 2012, 141, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Stacher, E.; Graham, B.B.; Hunt, J.M.; Gandjeva, A.; Groshong, S.D.; McLaughlin, V.V.; Jessup, M.; Grizzle, W.E.; Aldred, M.A.; Cool, C.D.; et al. Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M. Pathology of pulmonary arterial hypertension. Semin. Respir. Crit. Care Med. 2009, 30, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Can, M.M.; Kaymaz, C.; Tanboga, I.H.; Tokgoz, H.C.; Canpolat, N.; Turkyilmaz, E.; Sonmez, K.; Ozdemir, N. Increased right ventricular glucose metabolism in patients with pulmonary arterial hypertension. Clin. Nucl. Med. 2011, 36, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Zhao, L.; Xiong, C.M.; Ni, X.H.; He, Z.X.; He, J.G.; Wilkins, M.R. Comparison of 18f-FDG uptake by right ventricular myocardium in idiopathic pulmonary arterial hypertension and pulmonary arterial hypertension associated with congenital heart disease. Pulm Circ. 2012, 2, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Bujak, R.; Mateo, J.; Blanco, I.; Izquierdo-Garcia, J.L.; Dudzik, D.; Markuszewski, M.J.; Peinado, V.I.; Laclaustra, M.; Barbera, J.A.; Barbas, C.; et al. New biochemical insights into the mechanisms of pulmonary arterial hypertension in humans. PLoS ONE 2016, 11, e0160505. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A Metabolic Theory of Pulmonary Hypertension. The activation of hypoxia-inducible factor-1/2α (HIF-1/2α) results in its nuclear translocation and binding with HIF-1β. The heterodimeric transcription factor binds to a hypoxic response element, where genes involved in adaptive cellular responses are upregulated (i.e., pdk1 and micro-RNA 210 [miR-210]). Upregulation of pyruvate dehydrogenase kinase (PDK) results in the phosphorylation and inhibition of pyruvate dehydrogenase (PDH), limiting the flow of acetyl-CoA into the tricarboxylic acid (TCA) cycle for glucose oxidation. Instead, pyruvate is shunted via lactose dehydrogenase A (LDHA) into lactate for anaerobic respiration. HIF-mediated upregulation of miR-210 negatively regulations expression of iron-sulfur cluster assembly proteins 1 and 2 (ISCU1/2), causing a decrease in iron-sulfur biogenesis. The decrease in iron-sulfur cluster biogenesis attenuates mitochondrial respiration in the electron transport chain (ETC) and reactive oxygen species (ROS) generation. A decrease in ROS coupled with downregulation of SOD2 via methylation by methyltransferase reduces the concentration of hydrogen peroxide (H2O2), which normally inhibits HIF-1/2α activation. Decreases in ROS cause the activation of HIF-1/2α and the inhibition of Kv1.5 channels, resulting in depolarization and the activation of early current L-type voltage-gated calcium channels (VGCC) and late current transient receptor potential (trp) channels. The inhibition of Kv1.5 channels is further mediated through hypoxia-induced activation of AMP-activated protein kinase (AMPK). Increases in intracellular calcium ([Ca++]) inappropriately activate calcineurin, which dephosphorylates nuclear factor of activated T cells (NFAT). NFAT translocates to the nucleus where it increases proliferation, expression of bcl-2, and inhibits Kv1.5 channel expression—contributing to a self-propagating cycle. Calcium dynamics are further dysregulated with reduced activity of uncoupling protein 2 (UCP2), contributing to the dysfunction of mitochondrial enzymes and hyperpolarization of the mitochondrial membrane (ΔΨm). An increase in ΔΨm causes inhibition of the mitochondrial permeability transition pore (MPTP) and facilitates a phenotype resistant to apoptosis. A failure of the sarco-/endoplasmic reticulum calcium-ATPase 2 (SERCA2) further contributes to increased cytosolic calcium concentration. Disruption of endoplasmic reticulum (ER) calcium dynamics causes ER stress and, if prolonged, the activation of the unfolded protein response (UPR). Activating transcription factor 6 (ATF6)—a prong of the UPR—mediates the upregulation of Nogo-B, which widens the distance between the ER and mitochondrion and prevents mitochondrion-dependent apoptosis. TCA cycle intermediates such as oxaloacetate (OAA), succinate (SUCC), and fumarate (FUM) inhibit prolyl dehydrogenase (PDH)-mediated proteasomal degradation of HIF-1/2α. Decreases in the concentration of citrate reduce its nuclear conversion into acetyl-CoA by ATP-citrate lyase, thereby reducing histone acetylation. Coupled with increased histone deacetylase activity, there is a disrupted balance favoring the deacetylation of histones. Reduced levels of citrate trigger isocitrate dehydrogenase (IDH) to convert α-ketoglutarate (α-KG) into isocitrate to replenish citrate, which then deprives PHD of its cofactor necessary for hydroxylation of HIF-1/2α. Moreover, increases in the α-KG metabolite 2-hydroxyglutrate (L2HG) result in the inhibition of PHD. Activation of Yes-associated protein 1 (YAP) and transcription coactivator with a PDZ-binding motif (TAZ) upregulate glutaminase (GLS1), which converts glutamine into glutamate to ultimately replenish α-KG.
Figure 1.
A Metabolic Theory of Pulmonary Hypertension. The activation of hypoxia-inducible factor-1/2α (HIF-1/2α) results in its nuclear translocation and binding with HIF-1β. The heterodimeric transcription factor binds to a hypoxic response element, where genes involved in adaptive cellular responses are upregulated (i.e., pdk1 and micro-RNA 210 [miR-210]). Upregulation of pyruvate dehydrogenase kinase (PDK) results in the phosphorylation and inhibition of pyruvate dehydrogenase (PDH), limiting the flow of acetyl-CoA into the tricarboxylic acid (TCA) cycle for glucose oxidation. Instead, pyruvate is shunted via lactose dehydrogenase A (LDHA) into lactate for anaerobic respiration. HIF-mediated upregulation of miR-210 negatively regulations expression of iron-sulfur cluster assembly proteins 1 and 2 (ISCU1/2), causing a decrease in iron-sulfur biogenesis. The decrease in iron-sulfur cluster biogenesis attenuates mitochondrial respiration in the electron transport chain (ETC) and reactive oxygen species (ROS) generation. A decrease in ROS coupled with downregulation of SOD2 via methylation by methyltransferase reduces the concentration of hydrogen peroxide (H2O2), which normally inhibits HIF-1/2α activation. Decreases in ROS cause the activation of HIF-1/2α and the inhibition of Kv1.5 channels, resulting in depolarization and the activation of early current L-type voltage-gated calcium channels (VGCC) and late current transient receptor potential (trp) channels. The inhibition of Kv1.5 channels is further mediated through hypoxia-induced activation of AMP-activated protein kinase (AMPK). Increases in intracellular calcium ([Ca++]) inappropriately activate calcineurin, which dephosphorylates nuclear factor of activated T cells (NFAT). NFAT translocates to the nucleus where it increases proliferation, expression of bcl-2, and inhibits Kv1.5 channel expression—contributing to a self-propagating cycle. Calcium dynamics are further dysregulated with reduced activity of uncoupling protein 2 (UCP2), contributing to the dysfunction of mitochondrial enzymes and hyperpolarization of the mitochondrial membrane (ΔΨm). An increase in ΔΨm causes inhibition of the mitochondrial permeability transition pore (MPTP) and facilitates a phenotype resistant to apoptosis. A failure of the sarco-/endoplasmic reticulum calcium-ATPase 2 (SERCA2) further contributes to increased cytosolic calcium concentration. Disruption of endoplasmic reticulum (ER) calcium dynamics causes ER stress and, if prolonged, the activation of the unfolded protein response (UPR). Activating transcription factor 6 (ATF6)—a prong of the UPR—mediates the upregulation of Nogo-B, which widens the distance between the ER and mitochondrion and prevents mitochondrion-dependent apoptosis. TCA cycle intermediates such as oxaloacetate (OAA), succinate (SUCC), and fumarate (FUM) inhibit prolyl dehydrogenase (PDH)-mediated proteasomal degradation of HIF-1/2α. Decreases in the concentration of citrate reduce its nuclear conversion into acetyl-CoA by ATP-citrate lyase, thereby reducing histone acetylation. Coupled with increased histone deacetylase activity, there is a disrupted balance favoring the deacetylation of histones. Reduced levels of citrate trigger isocitrate dehydrogenase (IDH) to convert α-ketoglutarate (α-KG) into isocitrate to replenish citrate, which then deprives PHD of its cofactor necessary for hydroxylation of HIF-1/2α. Moreover, increases in the α-KG metabolite 2-hydroxyglutrate (L2HG) result in the inhibition of PHD. Activation of Yes-associated protein 1 (YAP) and transcription coactivator with a PDZ-binding motif (TAZ) upregulate glutaminase (GLS1), which converts glutamine into glutamate to ultimately replenish α-KG.
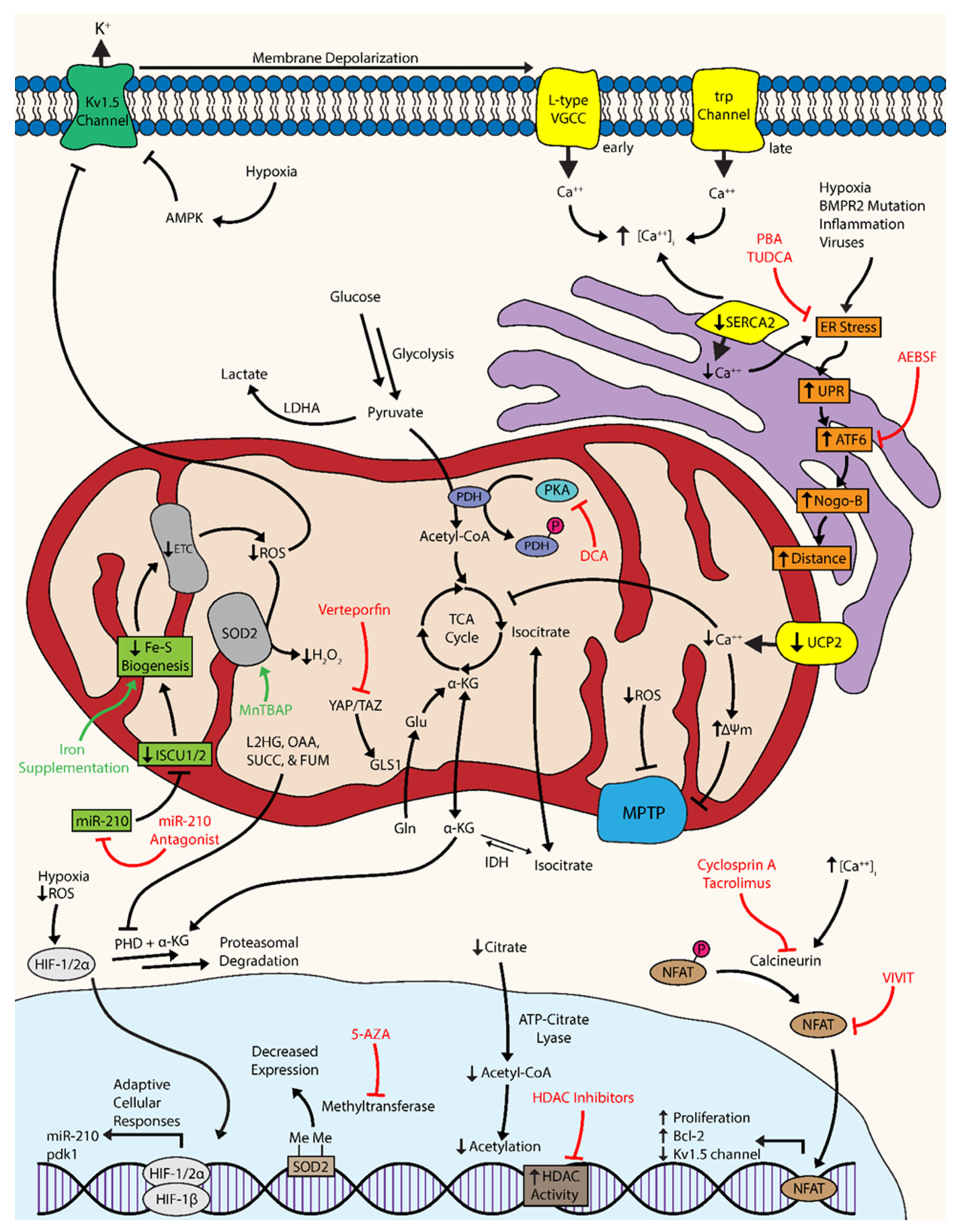
Figure 2.
The Randle Cycle in Right Ventricular Dysfunction. The Randle cycle depicts a reciprocal, mutually-inhibitory relationship between glucose oxidation and fatty acid oxidation (FAO). The accumulation of acetyl-CoA and citrate from fatty acid β-oxidation inhibits pyruvate dehydrogenase (PDH) and phosphofructokinase, respectively. Moreover, the accumulation of glucose-6-phosphate inhibits hexokinase, further attenuating glycolysis. Inhibition of PDH, phosphofructokinase and hexokinase results in the inhibition of glucose oxidation; thus, FAO inhibits glucose oxidation in cardiomyocytes. Intervention with dichloroacetate (DCA) inhibits pyruvate dehydrogenase kinase (PDK)-mediated inhibition of PDH. The FAO inhibitors trimetazidine and ranolazine prevent acetyl-CoA and citrate accumulation, thereby preventing FAO-mediated shutdown of glucose oxidation.
Figure 2.
The Randle Cycle in Right Ventricular Dysfunction. The Randle cycle depicts a reciprocal, mutually-inhibitory relationship between glucose oxidation and fatty acid oxidation (FAO). The accumulation of acetyl-CoA and citrate from fatty acid β-oxidation inhibits pyruvate dehydrogenase (PDH) and phosphofructokinase, respectively. Moreover, the accumulation of glucose-6-phosphate inhibits hexokinase, further attenuating glycolysis. Inhibition of PDH, phosphofructokinase and hexokinase results in the inhibition of glucose oxidation; thus, FAO inhibits glucose oxidation in cardiomyocytes. Intervention with dichloroacetate (DCA) inhibits pyruvate dehydrogenase kinase (PDK)-mediated inhibition of PDH. The FAO inhibitors trimetazidine and ranolazine prevent acetyl-CoA and citrate accumulation, thereby preventing FAO-mediated shutdown of glucose oxidation.
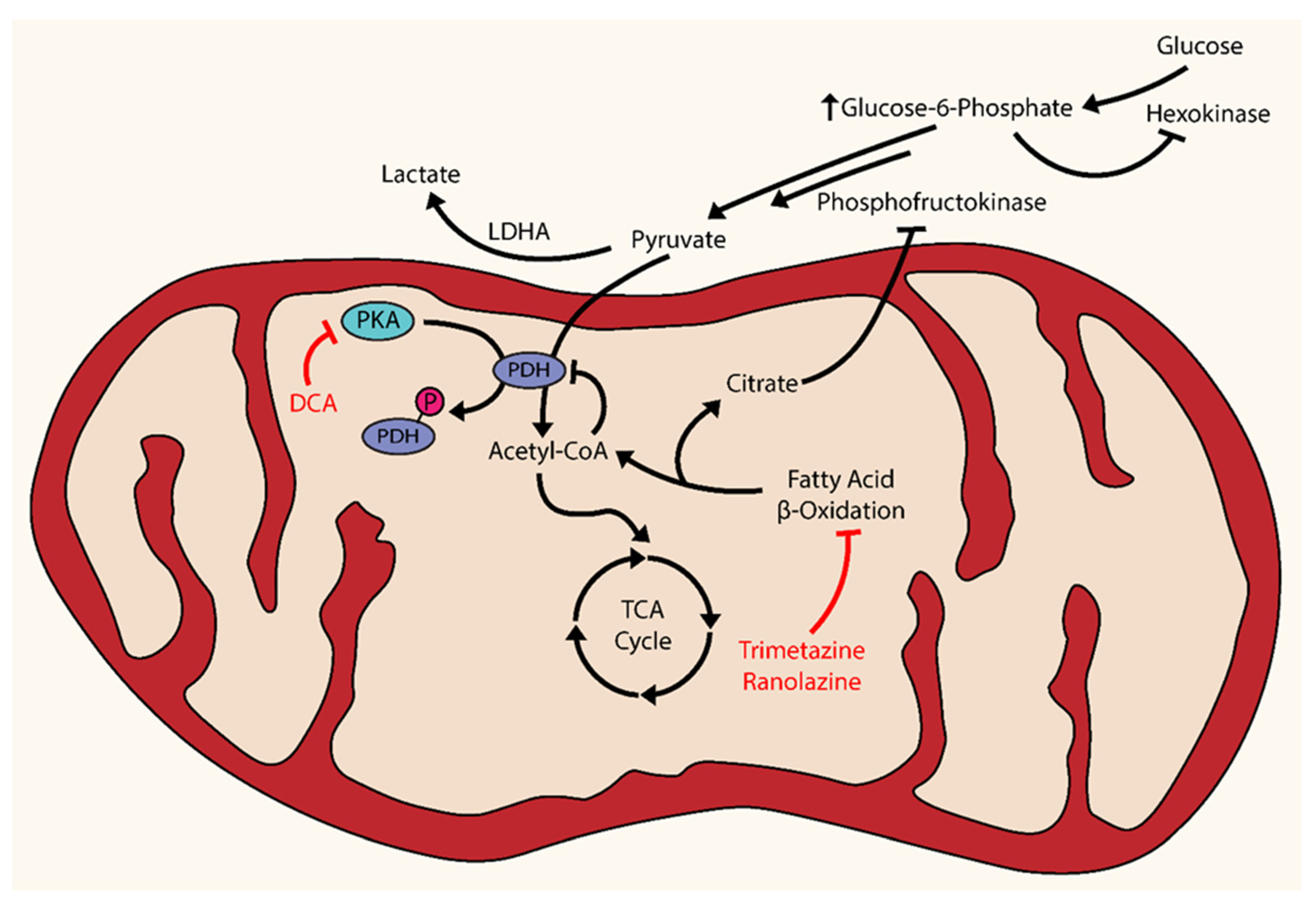

{kind=link}
{kind=link}
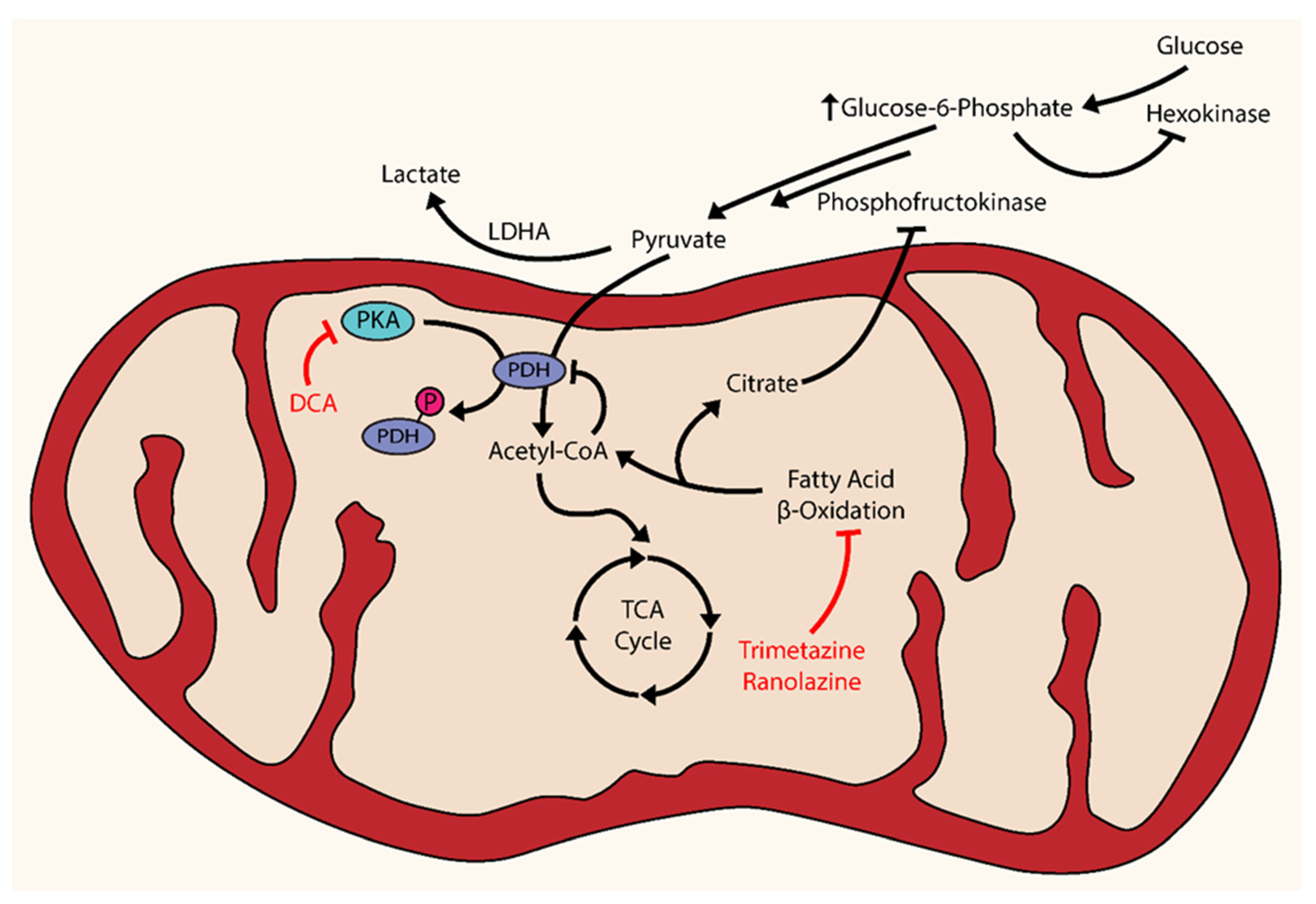{kind=link}
Table 1.
Preclinical Interventions in Models of Pulmonary Hypertension. ATF6: activating transcription factor 6. BMPR2: bone morphogenetic protein 2. CH: chronic hypoxia. CO: cardiac output. DRP1: Dynamin-related protein 1. EC: endothelial cell. ER: endoplasmic reticulum. FAO: fatty acid oxidation. FHR: Fawn-Hooded rat. GLS1: glutaminase. HIF: hypoxia-inducible factor. HK1: hexokinase 1. KO: knockout. LDHA: lactose dehydrogenase A. MCT: monocrotaline. NFAT: nuclear factor of activated T cells. PAB: pulmonary arterial banding. PAP: pulmonary artery pressure. PASMC: pulmonary arterial smooth muscle cell. PDH: Pyruvate dehydrogenase. PVR: pulmonary vascular resistance. RV: right ventricle. RVH: right ventricular hypertrophy. RVSP: right ventricular systolic pressure. SOD: superoxide dismutase. VEGFR: vascular endothelial growth factor receptor.
Table 1.
Preclinical Interventions in Models of Pulmonary Hypertension. ATF6: activating transcription factor 6. BMPR2: bone morphogenetic protein 2. CH: chronic hypoxia. CO: cardiac output. DRP1: Dynamin-related protein 1. EC: endothelial cell. ER: endoplasmic reticulum. FAO: fatty acid oxidation. FHR: Fawn-Hooded rat. GLS1: glutaminase. HIF: hypoxia-inducible factor. HK1: hexokinase 1. KO: knockout. LDHA: lactose dehydrogenase A. MCT: monocrotaline. NFAT: nuclear factor of activated T cells. PAB: pulmonary arterial banding. PAP: pulmonary artery pressure. PASMC: pulmonary arterial smooth muscle cell. PDH: Pyruvate dehydrogenase. PVR: pulmonary vascular resistance. RV: right ventricle. RVH: right ventricular hypertrophy. RVSP: right ventricular systolic pressure. SOD: superoxide dismutase. VEGFR: vascular endothelial growth factor receptor.
| Therapy | Finding | Citation |
|---|---|---|
| Dichloroacetate (DCA) | Restores PDH activity glucose oxidation; reverses MCT-induced PAH in rat model | [36] |
| Reverses HIF activation, increases Kv1.5 channel expression; reduces PAH in FHRs | [31] | |
| Enhances activity and expression of Kv2.1 channels, improves hemodynamic parameters; prevents and reverses CH-induced PH in rat model | [21] | |
| Reduces PDH phosphorylation, increases glucose oxidation, restores Kv1.5 channel expression, and increases cardiac output and function in MCT- and PAB-induced RV hypertrophy | [18] | |
| Ferric Carboxymaltose | Reverses consequences of iron deficiency, such as HIF upregulation, upregulation of pdk1, and decreases mitochondrial complex I activity | [57] |
| HDAC Inhibitors (Valproic Acid & Suberoylanilide Hydroxamic Acid) | Inhibit proliferative phenotype, exert anti-inflammatory effects, and reverse CH-induced PH in rats | [67] |
| Verteporfin | Decreases GLS1 expression and activity, decreases pulmonary arteriolar stiffness, reduces vascular remodeling, RVSP, and RV remodeling in MCT-induced PH | [80] |
| CB-839 | Decreases GLS activity, proliferation, and pulmonary arteriolar remodeling in MCT-induced | [80] |
| 5-aza-2’-deoxycytidine (5-AZA) | Restores SOD2 expression and mitochondrial function, inhibits PASMC proliferation, and increases cell apoptosis in vitro | [92] |
| Metalloporphyrin Mn(III)tetrakis (4-benzoic acid) porphyrin (MnTBAP) | Induces partial regression of PH, decreases vascular remodeling, and reverses the hyperproliferative phenotype in FHRs | [92] |
| Class I HDAC Inhibitors | Increases SOD3 expression and reduces proliferation of human idiopathic PAH PASMCs | [94] |
| Metformin | Inhibits PASMC proliferation in vitro; normalizes PAP and RVH in hypoxia- and MCT-induced PH in rats | [100] |
| Reverses hypoxia-induced PH in mice | [102] | |
| Reduces PAP and reverses vascular remodeling in VEGFR blockade-induced PH rat model | [111] | |
| VIVIT | Inhibits docking of calcineurin onto NFAT and thereby prevents its translocation | [134] |
| Increases Kv1.5 channel expression, reduces intracellular potassium and calcium, mitochondrial membrane potential, and expression of bcl-2 in vitro | [117] | |
| Cyclosporin A | Increases Kv1.5 channel expression, reduces intracellular potassium and calcium, mitochondrial membrane potential, and expression of bcl-2 in vitro; reduces PVR and PAP in MCT-induced rat model | [117] |
| Tacrolimus | Restores BMPR2 signaling in human PASMCs; reverses PH in a Bmpr2 EC KO mouse model and in MCT-induced or VEGFR blockage/CH-induced rat models | [135] |
| Mdivi-1 | Reduces DRP1 activation, mitochondrial fission, and PASMC proliferation; in vivo administration inhibits proliferation and improves exercise capacity and RV function | [142] |
| 4-(2-aminoethyl)benzenesulfonyl fluouride hydrochloride (AEBSF) | Prevents nuclear translocation of ER stress-induced ATF6 | [164] |
| 4-phenylbutyrate (PBA) | Inhibits ATF6 in hypoxic PASMCs in vitro, decreasing expression of Nogo and restoration of mitochondrial calcium dynamics and function; reverses PH in CH-induced mice and MCT-induced rats | [165] |
| Restores BMPR2 signaling in HeLa cells transfected with the human BMPR2 mutant | [166] | |
| Tauroursodeoyxcholic Acid (TUDCA) | Inhibits ATF6 in hypoxic PASMCs in vitro, decreasing expression of Nogo and restoration of mitochondrial calcium dynamics and function | [165] |
| Salubrinal | Decreases lung macrophages, pro-inflammatory cytokines, PAP, and vascular remodeling in a MCT-induced PH rat model | [168] |
| Ranolazine | Stimulates glucose oxidation via PDH activation and reduces FAO in isolated rat hearts | [183] |
| Increases CO and exercise capacity in PAB-induced PH rat model; increases oxygen consumption and ATP production; reduces expression of glycolytic mediators HK1 and LDHA | [184] | |
| Trimetazidine | Enhances glucose oxidation via increased PDH activity and reduces FAO in rat hearts | [182] |
| Increases CO and exercise capacity in PAB-induced PH rat model; increases oxygen consumption and ATP production; reduces expression of glycolytic mediators HK1 and LDHA | [184] |
Table 2.
Clinical Trials in Pulmonary Hypertension. 6MWD: 6-minute walk distance. DCA: dichloroacetate. 18FDG: 18F-fluorodeoxyglucose. PAP: pulmonary artery pressure. PAOP: pulmonary artery wedge pressure. PASP: pulmonary artery systolic pressure. PVR: pulmonary vascular resistance. MRI: magnetic resonance imaging. RVEF: right ventricular ejection fraction.
Table 2.
Clinical Trials in Pulmonary Hypertension. 6MWD: 6-minute walk distance. DCA: dichloroacetate. 18FDG: 18F-fluorodeoxyglucose. PAP: pulmonary artery pressure. PAOP: pulmonary artery wedge pressure. PASP: pulmonary artery systolic pressure. PVR: pulmonary vascular resistance. MRI: magnetic resonance imaging. RVEF: right ventricular ejection fraction.
| Therapy | Clinical Trial Identification | Design | Primary Endpoints | Treatment Duration | Status as of Publication |
|---|---|---|---|---|---|
| Dichloroacetate Sodium | NCT01083524 | Phase I, interventional, open-label, non-randomized in idiopathic, familial, or anorexigen-associated PAH patients | Safety and tolerability of DCA | 16 weeks | Completed September 2013 [unpublished] |
| Ferric carboxymaltose | NCT01288651 | Phase IV, interventional, open-label, single group assignment in iron deficient patients with idiopathic PAH | Change in 6MWD | 12 weeks | Completed [58] |
| Ferric carboxymaltose | NCT01847352 | Single-blind, interventional, non-randomized in iron-deficient and iron-replete healthy volunteers | Change in PASP under subacute hypoxia with and without prior intravenous iron infusion | 6 hours | Completed [59] |
| Ferric carboxymaltose (Europe) or Iron Dextran (China) | NCT01447628 | Phase II, interventional, randomized, double-blind in patients with idiopathic, heritable, or anorexigen-associated PAH | Change in PVR and exercise capacity | 12 weeks | Recruiting |
| Observing Low Fe-S Clusters | NCT02594917 | Observational, cohort, prospective in patients with low Fe-S clusters | Change in 6MWD and PAP | — | Recruiting |
| Metformin | NCT01352026 | Phase II, interventional, single group assignment, open-label in patients with PAH | — | — | Withdrawn due to lack of recruiting |
| Metformin | NCT01884051 | Observational, cohort, prospective and Phase I, interventional in patients with idiopathic, heritable, scleroderma-, or anorexigen-associated PAH | Safety and tolerability of metformin (& secondary efficacy outcome measures) | — | Recruiting |
| Tacrolimus | NCT01647945 | Phase II, interventional, randomized, double-blind in Group I PAH patients | Safety of low-dose tacrolimus (& 6MWD as secondary outcome) | 18 weeks | Completed [unpublished] |
| Bardoxolone methyl | NCT02036970 | Phase II, interventional, randomized, parallel assignment in PAH | Change in 6MWD | 16 weeks | Ongoing |
| Ranolazine | NCT01174173 | Phase III, interventional, single group assignment, open-label in patients with angina and PAH | Change in angina symptoms, 6MWD, and quality of life | 3 months | Completed [185] |
| Ranolazine | NCT02133352 | Phase IV, interventional, single group assignment, open-label in Group II PH patients | Change in mean PAP, PAOP, and PVR | 6 months | Completed [unpublished] |
| Ranolazine | NCT01839110 | Interventional, randomized, double-blind in subjects on stable PH therapies with RV dysfunction (RVEF <45%) | Number and percentage of subjects with high risk profile | 26 weeks | Ongoing |
| Ranolazine | NCT02829034 | Interventional, randomized, double-blind in subjects on stable PH therapies with RV dysfunction (RVEF <45%) | Percent change in RVEF as measured by MRI | 26 weeks | Recruiting |
| Ranolazine 11C-Acetate 18FDG | NCT01917136 | Phase II, interventional, single group assignment, open-label in PH patients with and without RV dysfunction | Change in myocardial oxygen consumption, 18FDG uptake, and myocardial perfusion | 26 weeks | Ongoing |
| Trimetazidine | NCT02102672 | Phase II, interventional, randomized, double-blind in Group I PAH patients | Changes in RV function assessed by echo 3d | 3 months | Recruiting |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Harvey, L.D.; Chan, S.Y. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. J. Clin. Med. 2017, 6, 43. https://doi.org/10.3390/jcm6040043
AMA Style
Harvey LD, Chan SY. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. Journal of Clinical Medicine. 2017; 6(4):43. https://doi.org/10.3390/jcm6040043
Chicago/Turabian StyleHarvey, Lloyd D., and Stephen Y. Chan. 2017. "Emerging Metabolic Therapies in Pulmonary Arterial Hypertension" Journal of Clinical Medicine 6, no. 4: 43. https://doi.org/10.3390/jcm6040043
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.