An Evaluation of Ischaemic Preconditioning as a Method of Reducing Ischaemia Reperfusion Injury in Liver Surgery and Transplantation


Abstract
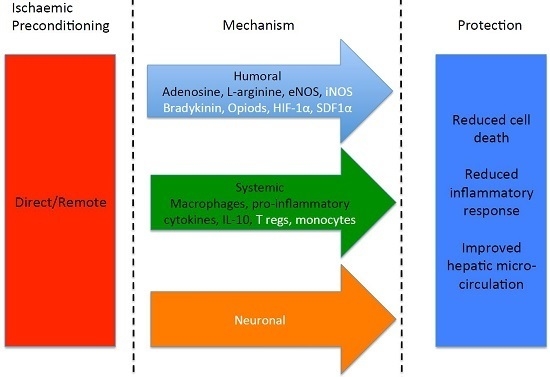
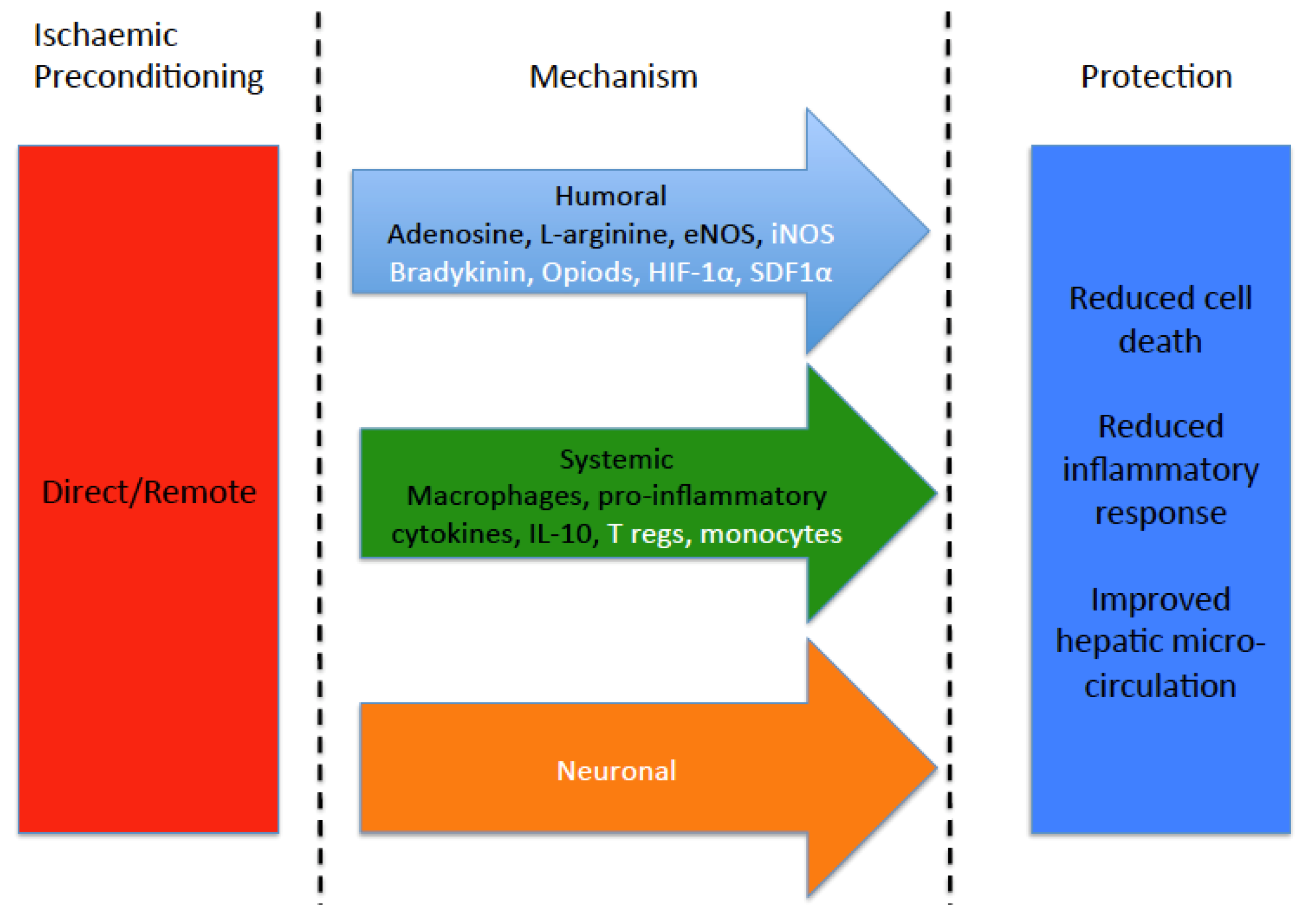
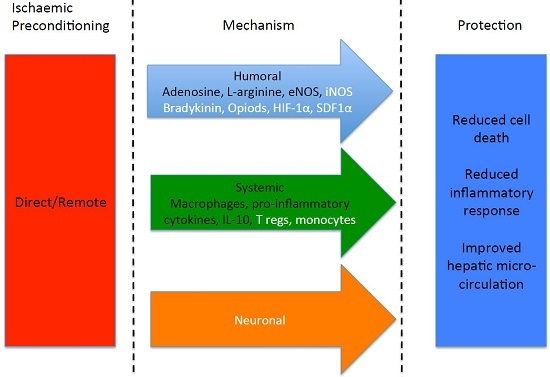
:
1. Introduction
2. Methods
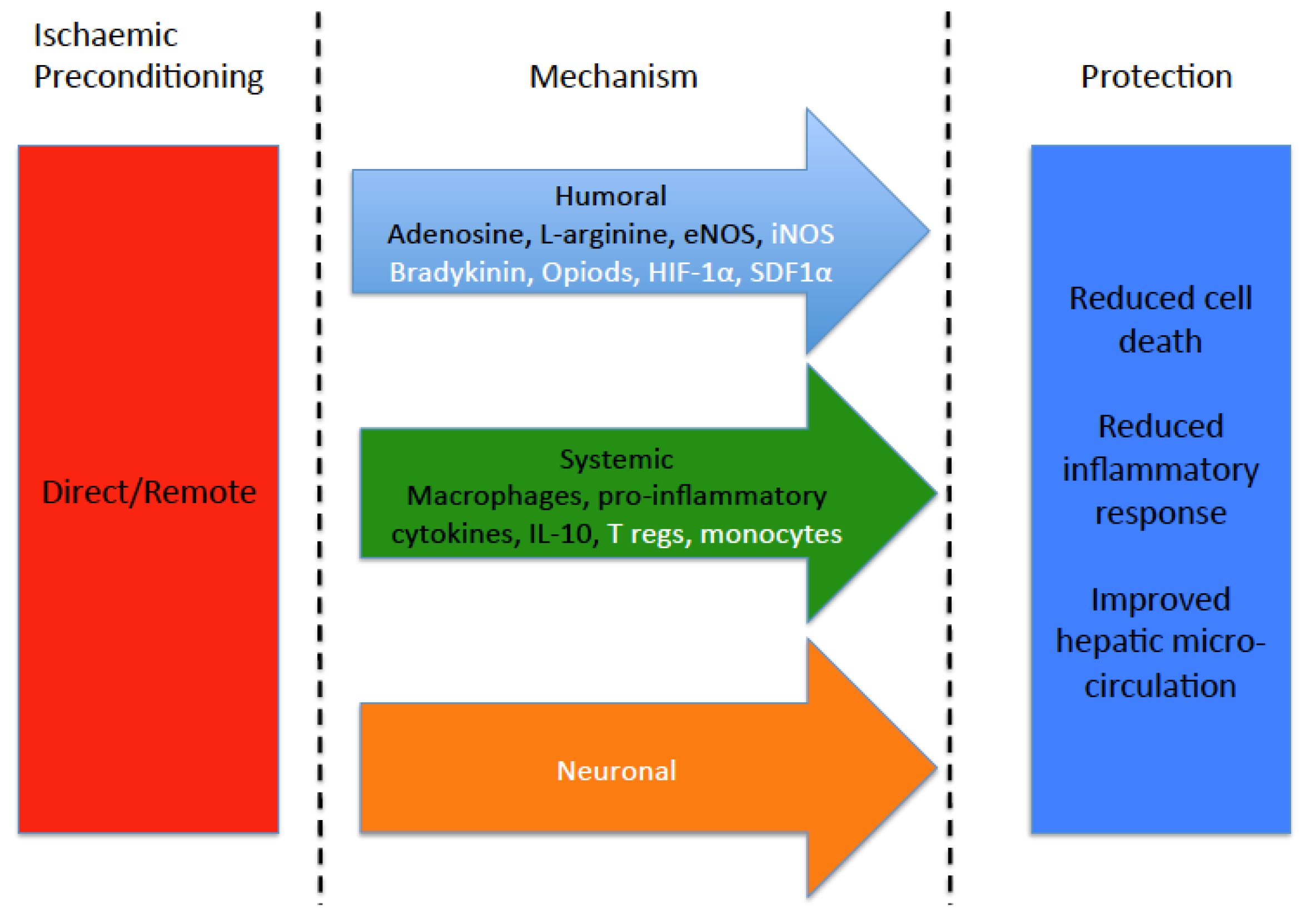
3. Protective Effects of Preconditioning
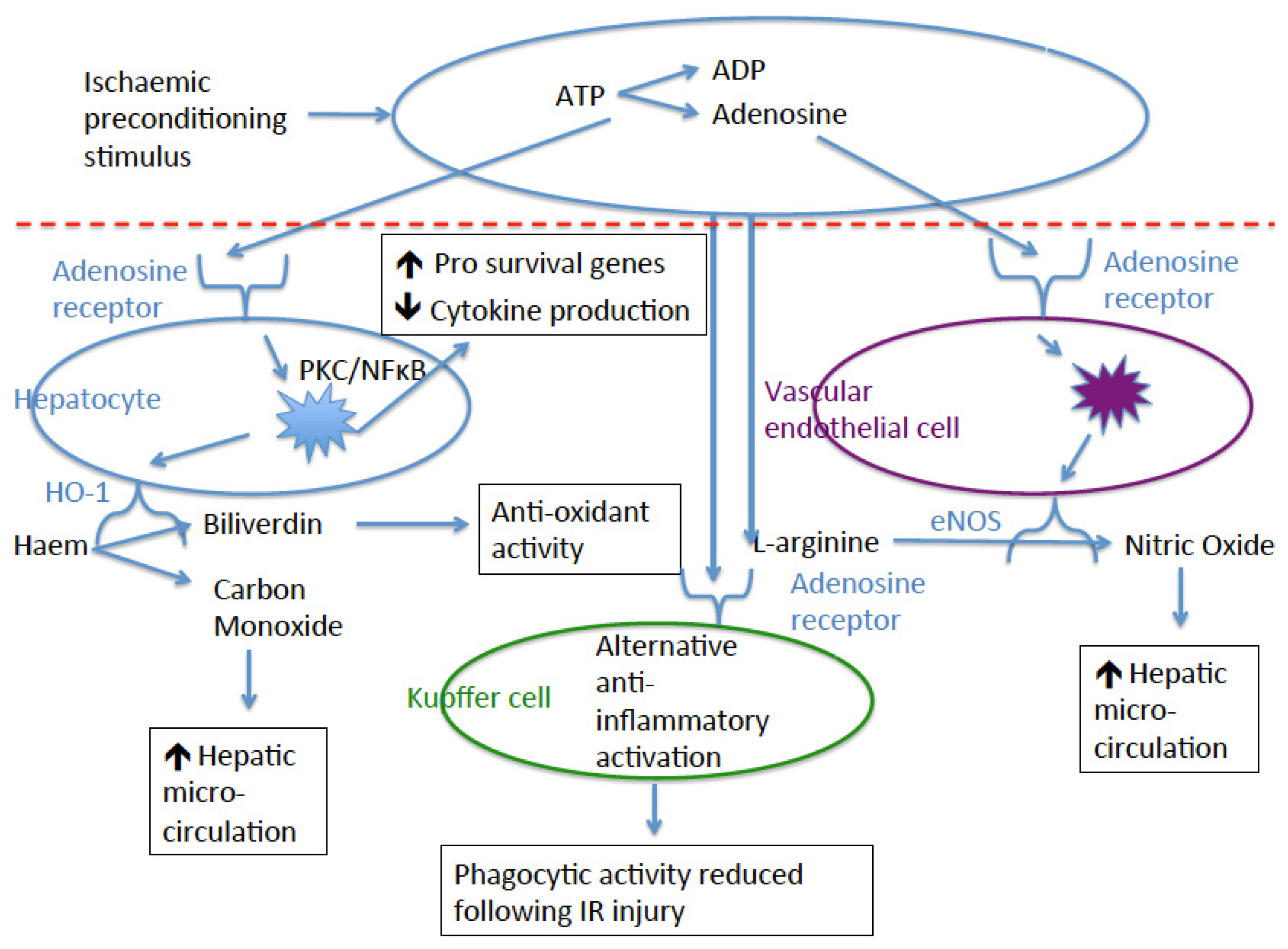
4. Adenosine
5. The A1 Receptor
6. The A2A Receptor
7. The A2B Receptor
8. The A3 Receptor
9. Adenosine and Its Receptors
10. Nitric Oxide and Nitric Oxide Synthase
11. Protein Kinase C
12. Nuclear Factor Kappa-Light-Chain-Enhancer of B Cells (NF-κB)
13. Haem-Oxygenase-1 (HO-1)
14. The Immune System
15. CD4+ T Cells
16. Macrophages
17. Monocytes
18. Cytokines
19. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Veteläinen, R.; van Vliet, A.; Gouma, D.J.; van Gulik, T.M. Steatosis as a risk factor in liver surgery. Ann. Surg. 2007, 245, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Selzner, M.; Clavien, P.-A. Fatty liver in liver transplantation and surgery. Semin. Liver Dis. 2001, 21, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, C.J.; Charman, S.C.; Muiesan, P.; Powell, J.J.; Gimson, A.E.; van der Meulen, J.H.P. Outcomes of transplantation of livers from donation after circulatory death donors in the UK: A cohort study. BMJ Open 2013, 3, e003287. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Przyklenk, K.; Bauer, B.; Ovize, M.; Kloner, R.A.; Whittaker, P. Regional ischemic “preconditioning” protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993, 87, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Clavien, P.A.; Yadav, S.; Sindram, D.; Bentley, R.C. Protective effects of ischemic preconditioning for liver resection performed under inflow occlusion in humans. Ann. Surg. 2000, 232, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Clavien, P.-A.; Selzner, M.; Rüdiger, H.A.; Graf, R.; Kadry, Z.; Rousson, V.; Jochum, W. A prospective randomized study in 100 consecutive patients undergoing major liver resection with versus without ischemic preconditioning. Ann. Surg. 2003, 238, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Kanoria, S.; Robertson, F.P.; Mehta, N.N.; Fusai, G.; Sharma, D.; Davidson, B.R. Effect of Remote Ischaemic Preconditioning on Liver Injury in Patients Undergoing Major Hepatectomy for Colorectal Liver Metastasis: A Pilot Randomised Controlled Feasibility Trial. World J. Surg. 2016. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; Leuschner, S.; McNally, S.J.; Garden, O.J.; Wigmore, S.J.; Harrison, E.M. Meta-analysis of ischaemic preconditioning for liver resections. Br. J. Surg. 2013, 100, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.P.; Magill, L.J.; Wright, G.P.; Fuller, B.; Davidson, B.R. A systematic review and meta-analysis of donor ischaemic preconditioning in liver transplantation. Transpl. Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, K.; Forbes, S.; Thiemermann, C.; Yaqoob, M.M. The challenge of translating ischemic conditioning from animal models to humans: The role of comorbidities. Dis. Model. Mech. 2014, 7, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N. Engl. J. Med. 2015, 373, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Meybohm, P.; Bein, B.; Brosteanu, O.; Cremer, J.; Gruenewald, M.; Stoppe, C.; Coburn, M.; Schaelte, G.; Böning, A.; Niemann, B.; et al. A Multicenter Trial of Remote Ischemic Preconditioning for Heart Surgery. N. Engl. J. Med. 2015, 373, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Robertson, F.P.; Goswami, R.; Wright, G.P.; Imber, C.; Sharma, D.; Malago, M.; Fuller, B.J.; Davidson, B.R. Remote ischaemic preconditioning in orthotopic liver transplantation (RIPCOLT trial): A pilot randomized controlled feasibility study. HPB 2017. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Hotter, G.; Closa, D.; Gelpí, E.; Bulbena, O.; Rosello-Catafau, J. Protective effect of preconditioning on the injury associated to hepatic ischemia-reperfusion in the rat: Role of nitric oxide and adenosine. Hepatology 1997, 25, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Closa, D.; Xaus, C.; Gelpí, E.; Roselló-catafau, J.; Hotter, G. Hepatic preconditioning in rats is defined by a balance of adenosine and xanthine. Hepatology 1998, 28, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Yamamoto, Y.; Kume, M.; Yamagami, K.; Yamamoto, H.; Kimoto, S.; Ishikawa, Y.; Ozaki, N.; Shimahara, Y.; Yamaoka, Y. Pharmacologic stimulation of adenosine A2 receptor supplants ischemic preconditioning in providing ischemic tolerance in rat livers. Surgery 1999, 126, 945–954. [Google Scholar] [CrossRef]
- Peralta, C.; Hotter, G.; Closa, D.; Prats, N.; Xaus, C.; Gelpí, E.; Roselló-Catafau, J. The protective role of adenosine in inducing nitric oxide synthesis in rat liver ischemia preconditioning is mediated by activation of adenosine A2 receptors. Hepatology 1999, 29, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Ajamieh, H.H.; Candelario-Jalil, E.; Fernández, O.S.L.; Gerbes, A.L. Ischaemic and pharmacological preconditionings protect liver via adenosine and redox status following hepatic ischaemia/reperfusion in rats. Clin. Sci. 2008, 115, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Schauer, R.; Gerbes, A.L.; Vonier, D.; op den Winkel, M.; Fraunberger, P.; Bilzer, M. Induction of cellular resistance against Kupffer cell–derived oxidant stress: A novel concept of hepatoprotection by ischemic preconditioning. Hepatology 2003, 37, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Chouker, A.; Ohta, A.; Martignoni, A.; Lukashev, D.; Zacharia, L.C.; Jackson, E.K.; Schnermann, J.; Ward, J.M.; Kaufmann, I.; Klaunberg, B.; et al. In Vivo Hypoxic Preconditioning Protects From Warm Liver Ischemia-Reperfusion Injury Through the Adenosine A2B Receptor. Transplantation 2012, 94, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Koti, R.S.; Tsui, J.; Lobos, E.; Yang, W.; Seifalian, A.M.; Davidson, B.R. Nitric oxide synthase distribution and expression with ischemic preconditioning of the rat liver. FASEB J. 2005, 19, 1155–1157. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amara, M.; Yang, S.Y.; Quaglia, A.; Rowley, P.; Fuller, B.; Seifalian, A.; Davidson, B. Role of endothelial nitric oxide synthase in remote ischemic preconditioning of the mouse liver. Liver Transpl. 2011, 17, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Carini, R.; De Cesaris, M.G.; Splendore, R.; Bagnati, M.; Albano, E. Ischemic preconditioning reduces Na+ accumulation and cell killing in isolated rat hepatocytes exposed to hypoxia. Hepatology 2000, 31, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Carini, R.; De Cesaris, M.G.; Splendore, R.; Vay, D.; Domenicotti, C.; Nitti, M.P.; Paola, D.; Pronzato, M.A.; Albano, E. Signal pathway involved in the development of hypoxic preconditioning in rat hepatocytes. Hepatology 2001, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, R.; Meyers, W.C.; Schaffer, B.K.; Kim, R.D.; Shah, S.A.; Wheeler, S.M.; Donohue, S.E.; Sheth, K.R.; Callery, M.P.; Chari, R.S. Protein kinase C inhibition abrogates hepatic ischemic preconditioning responses. J. Surg. Res. 2001, 97, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, R.; Shah, S.A.; Wheeler, S.M.; Quarfordt, S.H.; Callery, M.P.; Meyers, W.C.; Chari, R.S. Regulation of NFκB in hepatic ischemic preconditioning. J. Am. Coll. Surg. 2002, 195, 319–326. [Google Scholar] [CrossRef]
- Lai, I.-R.; Chang, K.-J.; Chen, C.-F.; Tsai, H.-W. Transient limb ischemia induces remote preconditioning in liver among rats: The protective role of heme oxygenase-1. Transplantation 2006, 81, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Datta, G.; Luong, T.V.; Fuller, B.J.; Davidson, B.R. Endothelial nitric oxide synthase and heme oxygenase-1 act independently in liver ischemic preconditioning. J. Surg. Res. 2014, 186, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, J.; Xiong, X.; Xu, Y.; Zhang, H.; Huang, C.; Tian, Y.; Jiao, C.; Wang, X.; Li, X. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. PLoS ONE 2014, 9, e98834. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, G.R.; Huang, L.; Vergis, A.L.; Li, L.; Okusa, M.D. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int. 2010, 77, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.Y.; Choi, H.M.; Lee, S.Y.; Kim, M.G.; Kim, H.-K.; Jo, S.-K. The role of Tregs and CD11c(+) macrophages/dendritic cells in ischemic preconditioning of the kidney. Kidney Int. 2010, 78, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Devey, L.R.; Richards, J.A.; O’Connor, R.A.; Borthwick, G.; Clay, S.; Howie, A.F.; Wigmore, S.J.; Anderton, S.M.; Howie, S.E.M. Ischemic preconditioning in the liver is independent of regulatory T cell activity. PLoS ONE 2012, 7, e49647. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Prats, N.; Xaus, C.; Gelpí, E.; Roselló-Catafau, J. Protective effect of liver ischemic preconditioning on liver and lung injury induced by hepatic ischemia-reperfusion in the rat. Hepatology 1999, 30, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Fernández, L.; Panés, J.; Prats, N.; Sans, M.; Piqué, J.M.; Gelpí, E.; Roselló-Catafau, J.; Roselló-Catafau, J. Preconditioning protects against systemic disorders associated with hepatic ischemia-reperfusion through blockade of tumor necrosis factor–induced P-selectin up-regulation in the rat. Hepatology 2001, 33, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Glanemann, M.; Vollmar, B.; Nussler, A.K.; Schaefer, T.; Neuhaus, P.; Menger, M.D. Ischemic preconditioning protects from hepatic ischemia/reperfusion-injury by preservation of microcirculation and mitochondrial redox-state. J. Hepatol. 2003, 38, 59–66. [Google Scholar] [CrossRef]
- Arai, M.; Ikeda, H.; Tomiya, T.; Yanase, M.; Inoue, Y.; Nagashima, K.; Nishikawa, T.; Watanabe, N.; Omata, M.; Fujiwara, K.; et al. Ischemic preconditioning protects hepatocytes via reactive oxygen species derived from Kupffer cells in rats. Gastroenterology 2004, 127, 1488–1496. [Google Scholar] [CrossRef]
- Funaki, H.; Shimizu, K.; Harada, S.-I.; Tsuyama, H.; Fushida, S.; Tani, T.; Miwa, K. Essential role for nuclear factor kappaB in ischemic preconditioning for ischemia-reperfusion injury of the mouse liver. Transplantation 2002, 74, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Zhu, C.-J.; Zhao, Y.-F.; Li, J.; Guo, W.-Z. Different ischemic preconditioning for rat liver graft: Protection and mechanism. Hepatobiliary Pancreat. Dis. Int. 2003, 2, 509–512. [Google Scholar] [PubMed]
- Yao, A.; Li, X.; Pu, L.; Zhong, J.; Liu, X.; Yu, Y.; Zhang, F.; Kong, L.; Sun, B.; Wang, X.; et al. Impaired hepatic regeneration by ischemic preconditioning in a rat model of small-for-size liver transplantation. Transpl. Immunol. 2007, 18, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Koneru, B.; Shareef, A.; Dikdan, G.; Desai, K.; Klein, K.M.; Peng, B.; Wachsberg, R.H.; de la Torre, A.N.; Debroy, M.; Fisher, A.; et al. The ischemic preconditioning paradox in deceased donor liver transplantation—Evidence from a prospective randomized single blind clinical trial. Am. J. Transplant. 2007, 7, 2788–2796. [Google Scholar] [CrossRef] [PubMed]
- Guimarães Filho, M.A.C.; Cortez, E.; Garcia-Souza, É.P.; Soares, V.M.; Moura, A.S.; Carvalho, L.; Maya, M.C.A.; Pitombo, M.B. Effect of remote ischemic preconditioning in the expression of IL-6 and IL-10 in a rat model of liver ischemia-reperfusion injury. Acta Cir. Bras. 2015, 30, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-Y.; Shi, X.-J.; Li, W.; Sun, X.-D.; Wang, G.-Y. Ischemic preconditioning and remote ischemic preconditioning provide combined protective effect against ischemia/reperfusion injury. Life Sci. 2016, 150, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.; Yellon, D.M. New directions for protecting the heart against ischaemia–reperfusion injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc. Res. 2004, 61, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Marber, M.S.; Latchman, D.S.; Walker, J.M.; Yellon, D.M. Cardiac stress protein elevation 24 hours after brief ischemia or heat stress is associated with resistance to myocardial infarction. Circulation 1993, 88, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, T.; Hoshida, S.; Yamashita, N.; Fuji, H.; Oe, H.; Hori, M.; Kamada, T.; Tada, M. Delayed effects of sublethal ischemia on the acquisition of tolerance to ischemia. Circ. Res. 1993, 72, 1293–1299. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. The second window of preconditioning (SWOP) where are we now? Cardiovasc. Drugs Ther. 2010, 24, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Hausenloy, D.J. Remote ischemic conditioning: From bench to bedside. Front. Physiol. 2012, 3, 27. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007, 14, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [Google Scholar] [PubMed]
- Laubach, V.E.; French, B.A.; Okusa, M.D. Targeting of adenosine receptors in ischemia-reperfusion injury. Expert Opin. Ther. Targets 2011, 15, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Todo, S.; Zhu, Y.; Zhang, S.; Jin, M.B.; Ishizaki, N.; Tanaka, H.; Subbotin, V.; Starzl, T.E. Attenuation of ischemic liver injury by augmentation of endogenous adenosine. Transplantation 1997, 63, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Fenton, R.A.; Cutler, B.S.; Dobson, J.G. Adenosine enhances nitric oxide production by vascular endothelial cells. Am. J. Physiol. 1995, 269, C519–C523. [Google Scholar] [PubMed]
- Koneru, B.; Fisher, A.; He, Y.; Klein, K.M.; Skurnick, J.; Wilson, D.J.; de la Torre, D.N.; Merchant, A.; Arora, R.; Samanta, A.K.; et al. Ischemic preconditioning in deceased donor liver transplantation: A prospective randomized clinical trial of safety and efficacy. Liver Transplant. 2005, 11, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Jassem, W.; Fuggle, S.V.; Cerundolo, L.; Heaton, N.D.; Rela, M. Ischemic preconditioning of cadaver donor livers protects allografts following transplantation. Transplantation 2006, 81, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, M.; Song, J.H.; Lee, H.T. Endogenous A1 adenosine receptors protect against hepatic ischemia reperfusion injury in mice. Liver Transplant. 2008, 14, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Magata, S.; Taniguchi, M.; Suzuki, T.; Shimamura, T.; Fukai, M.; Furukawa, H.; Fujita, M.; Todo, S. The Effect of Antagonism of Adenosine A1 Receptor Against Ischemia and Reperfusion Injury of the Liver. J. Surg. Res. 2007, 139, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Ben-Ari, Z.; Pappo, O.; Sulkes, J.; Cheporko, Y.; Vidne, B.A.; Hochhauser, E. Effect of adenosine A2A receptor agonist (CGS) on ischemia/reperfusion injury in isolated rat liver. Apoptosis 2005, 10, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.G.; Lemasters, J.J.; Arai, M.; Thurman, R.G.; Lemasters, J.J. Contribution of adenosine A(2) receptors and cyclic adenosine monophosphate to protective ischemic preconditioning of sinusoidal endothelial cells against Storage/Reperfusion injury in rat livers. Hepatology 2000, 32, 297–302. [Google Scholar] [CrossRef]
- Day, Y.-J.; Marshall, M.A.; Huang, L.; McDuffie, M.J.; Okusa, M.D.; Linden, J. Protection from ischemic liver injury by activation of A2A adenosine receptors during reperfusion: inhibition of chemokine induction. AJP Gastrointest. Liver Physiol. 2004, 286, G285–G293. [Google Scholar] [CrossRef] [PubMed]
- Lappas, C.M.; Day, Y.-J.; Marshall, M.A.; Engelhard, V.H.; Linden, J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J. Exp. Med. 2006, 203, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.A.; Grenz, A.; Tak, E.; Kaplan, M.; Ridyard, D.; Brodsky, K.S.; Mandell, M.S.; Kam, I.; Eltzschig, H.K. Signaling through hepatocellular A2B adenosine receptors dampens ischemia and reperfusion injury of the liver. Proc. Natl. Acad. Sci. USA 2013, 110, 12012–12017. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amara, M.; Yang, S.Y.; Seifalian, A.; Davidson, B.; Fuller, B. The nitric oxide pathway—Evidence and mechanisms for protection against liver ischaemia reperfusion injury. Liver Int. 2012, 32, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amara, M.; Yang, S.Y.; Quaglia, A.; Rowley, P.; Tapuria, N.; Seifalian, A.M.; Fuller, B.J.; Davidson, B.R. Effect of remote ischemic preconditioning on liver ischemia/reperfusion injury using a new mouse model. Liver Transpl. 2011, 17, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Peralta, C.; Rull, R.; Rimola, A.; Deulofeu, R.; Roselló-Catafau, J.; Gelpí, E.; Rodés, J. Endogenous nitric oxide and exogenous nitric oxide supplementation in hepatic ischemia-reperfusion injury in the rat. Transplantation 2001, 71, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Hines, I.N.; Kawachi, S.; Harada, H.; Pavlick, K.P.; Hoffman, J.M.; Bharwani, S.; Wolf, R.E.; Grisham, M.B. Role of nitric oxide in liver ischemia and reperfusion injury. Mol. Cell. Biochem. 2002, 234, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Hines, I.N.; Harada, H.; Flores, S.; Gao, B.; McCord, J.M.; Grisham, M.B. Endothelial nitric oxide synthase protects the post-ischemic liver: potential interactions with superoxide. Biomed. Pharmacother. 2005, 59, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Kawachi, S.; Hines, I.N.; Laroux, F.S.; Hoffman, J.; Bharwani, S.; Gray, L.; Leffer, D.; Grisham, M.B. Nitric Oxide Synthase and Postischemic Liver Injury. Biochem. Biophys. Res. Commun. 2000, 276, 851–854. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.G.; Johnson, M.L.; Baust, J.; Laubach, V.E.; Watkins, S.C.; Billiar, T.R. The roles of iNOS in liver ischemia-reperfusion injury. Shock 2001, 16, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Theruvath, T.P.; Zhong, Z.; Currin, R.T.; Ramshesh, V.K.; Lemasters, J.J. Endothelial nitric oxide synthase protects transplanted mouse livers against storage/reperfusion injury: Role of vasodilatory and innate immunity pathways. Transplant. Proc. 2006, 38, 3351–3357. [Google Scholar] [CrossRef] [PubMed]
- Duranski, M.R.; Elrod, J.W.; Calvert, J.W.; Bryan, N.S.; Feelisch, M.; Lefer, D.J. Genetic overexpression of eNOS attenuates hepatic ischemia-reperfusion injury. AJP Hear. Circ. Physiol. 2006, 291, H2980–H2986. [Google Scholar] [CrossRef] [PubMed]
- Serracino-Inglott, F.; Virlos, I.T.; Habib, N.A.; Williamson, R.C.N.; Mathie, R.T. Adenosine preconditioning attenuates hepatic reperfusion injury in the rat by preventing the down-regulation of endothelial nitric oxide synthase. BMC Gastroenterol. 2002, 2, 22. [Google Scholar] [CrossRef]
- Wang, L.-M.; Tian, X.-F.; Song, Q.-Y.; Gao, Z.-M.; Luo, F.-W.; Yang, C.-M. Expression and role of inducible nitric oxide synthase in ischemia-reperfusion liver in rats. Hepatobiliary Pancreat. Dis. Int. 2003, 2, 252–258. [Google Scholar] [PubMed]
- Zwacka, R.M.; Zhang, Y.; Halldorson, J.; Schlossberg, H.; Dudus, L.; Engelhardt, J.F. CD4(+) T-lymphocytes mediate ischemia/reperfusion-induced inflammatory responses in mouse liver. J. Clin. Investig. 1997, 100, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Bradham, C.A.; Stachlewitz, R.F.; Gao, W.; Qian, T.; Jayadev, S.; Jenkins, G.; Hannun, Y.; Lemasters, J.J.; Thurman, R.G.; Brenner, D.A. Reperfusion after liver transplantation in rats differentially activates the mitogen-activated protein kinases. Hepatology 1997, 25, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Cohen, M.V.; Downey, J.M. Ischemic preconditioning: from basic mechanisms to clinical applications. Pharmacol. Ther. 2000, 86, 263–275. [Google Scholar] [CrossRef]
- Carini, R.; Grazia De Cesaris, M.; Splendore, R.; Domenicotti, C.; Nitti, M.P.; Pronzato, M.A.; Albano, E. Signal pathway responsible for hepatocyte preconditioning by nitric oxide. Free Radic. Biol. Med. 2003, 34, 1047–1055. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Kawahara, T.; Kumakura, S.; Hua, J.; Kugimiya, T.; Nagaoka, I.; Inada, E. Effect of olprinone, a phosphodiesterase iii inhibitor, on hepatic ischemia-reperfusion injury in rats. Shock 2010, 33, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.-X.; Yuan, Q.; Wei, L.-P.; Meng, H.; Wang, Y.-J. Protein kinase C-β inhibitor treatment attenuates hepatic ischemia and reperfusion injury in diabetic rats. Exp. Ther. Med. 2016, 11, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, R.; Schaffer, B.K.; Kim, R.D.; Shah, S.A.; Donohue, S.E.; Wheeler, S.M.; Quarfordt, S.H.; Callery, M.P.; Meyers, W.C.; Chari, R.S.; et al. Protective effects of ischemic preconditioning on the cold-preserved liver are tyrosine kinase dependent. Transplantation 2001, 72, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Hur, G.M.; Ryu, Y.S.; Yun, H.Y.; Jeon, B.H.; Kim, Y.M.; Seok, J.H.; Lee, J.H. Hepatic Ischemia/Reperfusion in Rats Induces iNOS Gene Transcription by Activation of NF-κB. Biochem. Biophys. Res. Commun. 1999, 261, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.K.; Alam, J. Heme Oxygenase-1: Function, Regulation, and Implication of a Novel Stress-inducible Protein in Oxidant-induced Lung Injury. Am. J. Respir. Cell Mol. Biol. 1996, 15, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Origassa, C.S.T.; Câmara, N.O.S. Cytoprotective role of heme oxygenase-1 and heme degradation derived end products in liver injury. World J. Hepatol. 2013, 5, 541–549. [Google Scholar] [PubMed]
- Devey, L.; Ferenbach, D.; Mohr, E.; Sangster, K.; Bellamy, C.O.; Hughes, J.; Wigmore, S.J. Tissue-resident macrophages protect the liver from ischemia reperfusion injury via a heme oxygenase-1-dependent mechanism. Mol. Ther. 2009, 17, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.M.; Ham, S.A.; Kim, Y.M.; Lee, Y.S.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Park, M.K.; Chang, K.C. β1-Adrenergic receptor-mediated HO-1 induction, via PI3K and p38 MAPK, by isoproterenol in RAW 264.7 cells leads to inhibition of HMGB1 release in LPS-activated RAW 264.7 cells and increases in survival rate of CLP-induced septic mice. Biochem. Pharmacol. 2011, 82, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, X.; Fu, W.; Xie, J.; Zhou, X.; Jiang, H. Isoproterenol-mediated heme oxygenase-1 induction inhibits high mobility group box 1 protein release and protects against rat myocardial ischemia/reperfusion injury in vivo. Mol. Med. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Shi, T.; Qiao, H.; Jiang, X.; Jiang, H.; Krissansen, G.W.; Sun, X. Hepatic Overexpression of Heme Oxygenase-1 Improves Liver Allograft Survival by Expanding T Regulatory Cells. J. Surg. Res. 2011, 166, e187–e194. [Google Scholar] [CrossRef] [PubMed]
- Burne, M.J.; Daniels, F.; El Ghandour, A.; Mauiyyedi, S.; Colvin, R.B.; O’Donnell, M.P.; Rabb, H. Identification of the CD4(+) T cell as a major pathogenic factor in ischemic acute renal failure. J. Clin. Investig. 2001, 108, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Sharma, A.K.; Linden, J.; Kron, I.L.; Laubach, V.E. CD4+ T lymphocytes mediate acute pulmonary ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2009, 137, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Dienz, O.; Rincon, M. The effects of IL-6 on CD4 T cell responses. Clin. Immunol. 2009, 130, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Nish, S.A.; Schenten, D.; Wunderlich, F.T.; Pope, S.D.; Gao, Y.; Hoshi, N.; Yu, S.; Yan, X.; Lee, H.K.; Pasman, L.; et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife 2014, 3, e01949. [Google Scholar] [CrossRef] [PubMed]
- Caldwell-Kenkel, J.C.; Currin, R.T.; Tanaka, Y.; Thurman, R.G.; Lemasters, J.J. Kupffer cell activation and endothelial cell damage after storage of rat livers: effects of reperfusion. Hepatology 1991, 13, 83–95. [Google Scholar] [PubMed]
- Shiratori, Y.; Kiriyama, H.; Fukushi, Y.; Nagura, T.; Takada, H.; Hai, K.; Kamii, K. Modulation of ischemia-reperfusion-induced hepatic injury by Kupffer cells. Dig. Dis. Sci. 1994, 39, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Reinke, L.A. Antioxidants and gadolinium chloride attenuate hepatic parenchymal and endothelial cell injury induced by low flow ischemia and reperfusion in perfused rat livers. Free Radic. Res. 2000, 32, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Yamaguchi, Y.; Matsumura, F.; Goto, M.; Akizuki, E.; Matsuda, T.; Okabe, K.; Ohshiro, H.; Ishihara, K.; Yamada, S.; et al. Calcium-Channel Blocker Attenuates Kupffer Cell Production of Cytokine-Induced Neutrophil Chemoattractant Following Ischemia–Reperfusion in Rat Liver. Dig. Dis. Sci. 2000, 45, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Hirano, K.; Yamamoto, T.; Hasegawa, G.; Hatakeyama, K.; Suematsu, M.; Naito, M. The protective role of Kupffer cells in the ischemia-reperfused rat liver. Arch. Histol. Cytol. 2002, 65, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. New Mechanisms and Pathways for Monocyte Recruitment. J. Exp. Med. 2001, 194, F47–F52. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Mossanen, J.C.; Tacke, F. Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg. Nutr. 2014, 3, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Bamboat, Z.Z.M.; Ocuin, L.M.; Balachandran, V.P.; Obaid, H.; Plitas, G.; DeMatteo, R.R.P.; Lotze, M.; Clavien, P.; Harvey, P.; Strasberg, S.; et al. Conventional DCs reduce liver ischemia/reperfusion injury in mice via IL-10 secretion. J. Clin. Investig. 2010, 120, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Colletti, L.M.; Remick, D.G.; Burtch, G.D.; Kunkel, S.L.; Strieter, R.M.; Campbell, D.A., Jr. Role of tumor necrosis factor-alpha in the pathophysiologic alterations after hepatic ischemia/reperfusion injury in the rat. J. Clin. Investig. 1990, 85, 1936–1943. [Google Scholar] [CrossRef] [PubMed]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A.; et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 2005, 201, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, C.C.; Okaya, T.; Martignoni, A.; Husted, T.; Schuster, R.; Lentsch, A.B. Divergent functions of CD4+ T lymphocytes in acute liver inflammation and injury after ischemia-reperfusion. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G969–G976. [Google Scholar] [CrossRef] [PubMed]
- Camargo, C.A.; Madden, J.F.; Gao, W.; Selvan, R.S.; Clavien, P. Interleukin-6 protects liver against warm ischemia/reperfusion injury and promotes hepatocyte proliferation in the rodent. Hepatology 1997, 26, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Kirillova, I.; Peschon, J.J.; Fausto, N. Initiation of liver growth by tumor necrosis factor: Deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Dal-Secco, D.; Wang, J.; Zeng, Z.; Kolaczkowska, E.; Wong, C.H.Y.; Petri, B.; Ransohoff, R.M.; Charo, I.F.; Jenne, C.N.; Kubes, P. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 2015, 212, 447–456. [Google Scholar] [CrossRef] [PubMed]
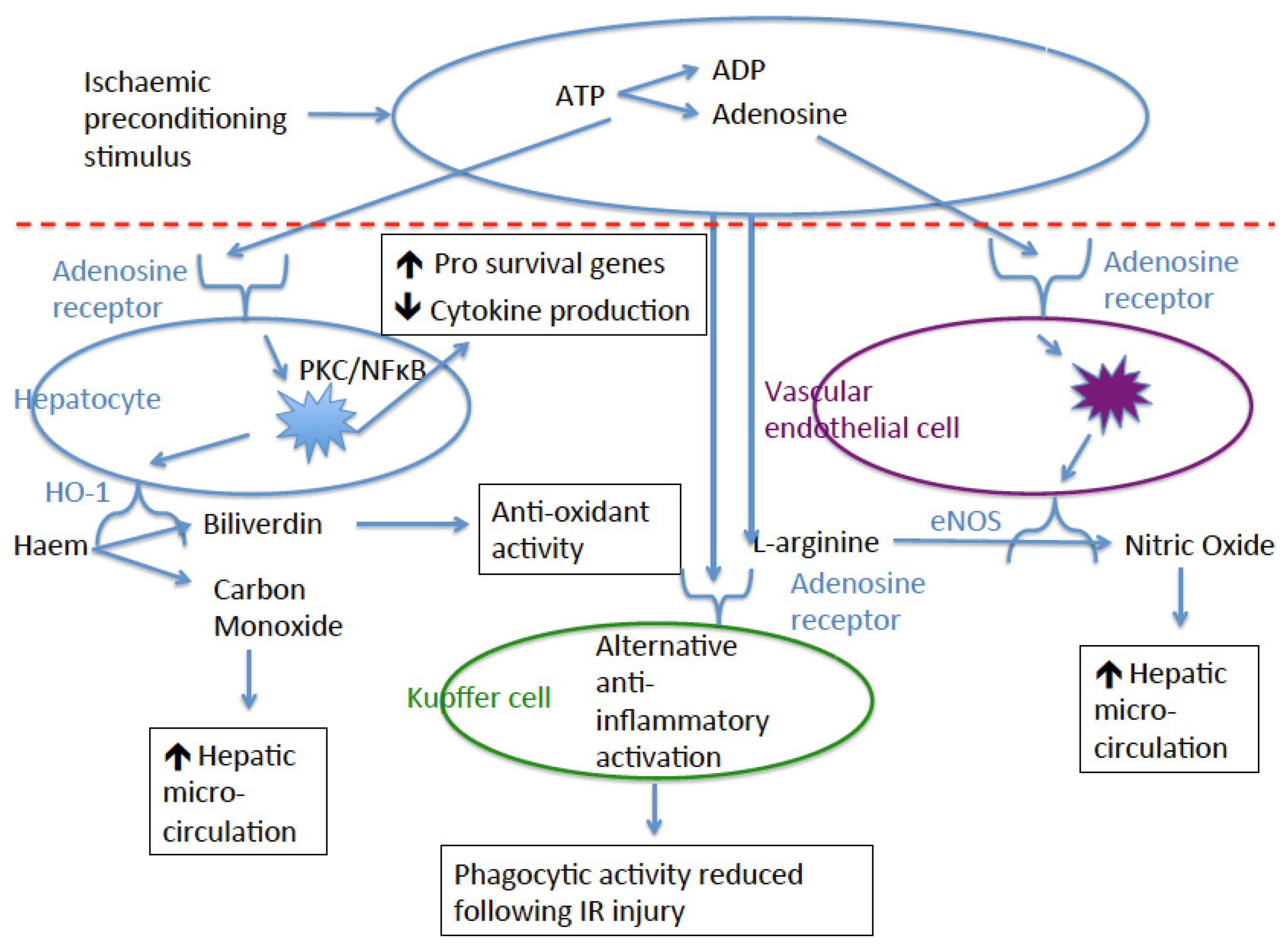

{kind=link}
{kind=link}
{kind=link}
| Study Group | Year | Species | IPC Time (min) | Ischaemic Time (min) | Reperfusion Time (min) | Hepatic Ischaemia | Pharmacological Manipulations | Parameters Assessed | Outcome of IPC | Proposed Mechanism |
|---|---|---|---|---|---|---|---|---|---|---|
| Adenosine | ||||||||||
| Peralta [15] | 1997 | rat | 10 | 90 | 90 | partial | Adenosine and NO | LFTs | ↓ LFTs | Adenosine/NO |
| Hepatic blood flow | ↑ blood flow | |||||||||
| Peralta [16] | 1998 | rat | Variable | 90 | 90 | partial | Adenosine and NO | LFTs Adenosine Inosine Xanthine | ↓ LFTs ↑ Adenosine | Adenosine |
| Nakayama [17] | 1999 | rat | 10 | 45 | Up to 7 days | unclear | A1 and A2 receptors | LFTs | ↑ 7 day survival | Adenosine via A2 receptor |
| 7 day survival | ↓ LFTs | |||||||||
| Adenosine | ↑ Adenosine | |||||||||
| A1 receptor | ||||||||||
| Peralta [18] | 1999 | rat | 10 | 90 | 90 | partial | A1, A2 receptors and NO | LFTs | ↓ LFTs | NO production through action of adenosine on A2R |
| Hepatic blood flow | ↑ blood flow | |||||||||
| NO production | ↑ NO production | |||||||||
| Ajamieh [19] | 2008 | rat | 10 | 90 | 24 h | partial | A1 receptor | LFTs | ↓ LFTs | A1 receptor |
| TNFα levels | ↓ TNFα levels | |||||||||
| MPO activity | ↓ oxidative stress | |||||||||
| A2A receptor | ||||||||||
| Perlata [18] | 1999 | rat | 10 | 90 | 90 | partial | A1, A2 receptors and NO | LFTs | ↓ LFTs | NO production through action of adenosine on A2R |
| Hepatic blood flow | ↑ blood flow | |||||||||
| NO production | ↑ NO production | |||||||||
| Schaeur [20] | 2003 | rat | 10 | 90 | 120 | partial | A2A receptor and p38 MAPK | LFTs | ↓ LFTs | p38 MAPK stimulation not A2A receptor |
| Hepatic perfusion | ↓ KC induce liver damage | |||||||||
| A2B receptor | ||||||||||
| Chouker [21] | 2012 | mouse | 10 | 45 | 240 | partial | A2A, A2B receptors | LFTs | ↓ LFTs | A2B receptor but not A2A receptor |
| TNFα levels | ↓ TNFα levels | |||||||||
| IL-6 levels | ↓ IL-6 levels | |||||||||
| A3 receptor | ||||||||||
| None | ||||||||||
| eNOS | ||||||||||
| Koti [22] | 2005 | rat | 5 | 45 | 120 | partial | l-arginine and NO | LFTs | ↓ LFTs | NO formed from eNOS is hepatoprotective |
| NO | ↑ NO levels | |||||||||
| eNOS | ↑ eNOS | |||||||||
| iNOS | no change in iNOS | |||||||||
| Abu-Amara [23] | 2011 | mouse | 4 | 40 | 120 | partial | eNOS genetic knockout | LFTs Hepatic blood flow Pathological injurye NOS expression | ↓ LFTs ↓ injury eNOS expression not upregulated in wild type mice. | RIPC provided no protection in eNOS−/− mice |
| RIPC did not upregulate eNOS expression in wild type mice | ||||||||||
| iNOS | ||||||||||
| Koti [22] | 2005 | rat | 5 | 45 | 120 | partial | l-arginine and NO | LFTs | ↓ LFTs | NO formed from eNOS is hepatoprotective |
| NO | ↑ NO levels | |||||||||
| eNOS | ↑ eNOS | |||||||||
| iNOS | no change in iNOS | |||||||||
| PKC | ||||||||||
| Carini [24] | 2000 | rat | 10 | 90 | 0 | hepatocytes | PKC | Intracellular pH | ↑ cell survival | PKC necessary to allow IPC |
| Intracellular Na | ↓ pH | |||||||||
| Cell viability | ↓ Na accumulation | |||||||||
| Carini [25] | 2001 | rat | 10 | 90 | 90 | hepatocytes | A2A receptor and PKC | Cell viability | ↑ cell survival | PKC activation following A2A receptor stimulation |
| PK levels | ↑ p38 MAPK phosphorylation | |||||||||
| Ricciardi [26] | 2001 | pig | 15 | 120 | 240 | total | PKC | Graft function | ↑ Graft function | PKC translocation to nucleus is necessary for IPC |
| Hepatic perfusion | ↑ Hepatic perfusion | |||||||||
| Graft injury | ↓ Graft injury | |||||||||
| NF-κB | ||||||||||
| Ricciardi [27] | 2002 | pig | 15 | 120 | 240 | total | PKC | NF-κB | ↑NF-κB | IPC increases translocation of NF-κB |
| HO-1 | ||||||||||
| Lai [28] | 2006 | rat | 10 | 45 | 240 | partial | HO-1 | LFTs | ↓ LFTs | RIPC increases HO-1 expression and activity |
| HO-1 expression | ↑ HO-1 expression | |||||||||
| HO activity | ↑ HO activity | |||||||||
| Datta [29] | 2014 | mouse | 5 | 45 | 120 | partial | eNOS genetic knockout | LFTs Hepatic perfusion HO-1 expression | ↓ LFTs ↑ Hepatic perfusion | eNOS−/− mice had reduced effect from IPC. HO-1 mRNA no significantly increased by IPC |
| Wang [30] | 2014 | mouse | 4 | 45 | 24 h | partial | HO-1 | LFTs | ↓ LFTs | RIPC lead to increased autophagy in a HO-1 dependant manner |
| HO-1 expression | ↑ HO-1 expression | |||||||||
| Autophagy | ↑ Autophagy | |||||||||
| Tregs | ||||||||||
| Kinsey [31] | 2010 | mouse | 24 (bilateral) | 28 (7 days post IPC) | unclear | Renal (1 kidney) | Treg depletion and adoptive transfer | Serum creatinine Renal Treg number and IL-10 production | ↓ Creatinine ↑ Treg accumulation ↑ Treg IL-10 production | Treg accumulation took 7 days. Treg depletion ablated effect of IPC |
| Cho [32] | 2010 | mouse | 24 (bilateral) | 28 (7 days post IPC) | 24 h | Renal (1 kidney) | Treg depletion and adoptive transfer | Serum creatinine | ↓ Creatinine | Treg depletion ablated effect of IPC. Stimulated lymphocytes from mice undergoing IPC were less pro-inflammatory. |
| Treg number | ↑ Treg accumulation | |||||||||
| Splenocytes cytokine and proliferation | ↓ Splenocyte proliferation and cytokine production | |||||||||
| Devey [33] | 2012 | mouse | 15 | 50 | 24 h | partial | Treg depletion and adoptive transfer | LFTs Treg numbers Circulating cytokines | ↓ LFTs Treg recruitment | IPC mechanism not related to Tregs |
| Macrophages | ||||||||||
| Peralta [34] | 1999 | rat | 10 | 90 | 90 | partial | TNFα treatment and macrophage depletion with Gadolinium Chloride. | LFTs | ↓ LFTs | TNFα production by macrophages drives hepatic IR injury |
| Hepatic oedema | ↓ TNFα release | |||||||||
| TNFα release | ↓ hepatic oedema | |||||||||
| Peralta [35] | 2001 | rat | 19 | 90 | 90 | partial | Antibody inhibition of I-CAM and macrophage depletion with Gadolinium Chloride | LFTs Neutrophil accumulation and activity in distant organs | ↓ neutrophil accumulation and activity in distant end organs | IPC reduce neutrophil infiltration into distant organs but not the liver itself. Likely secondary to macrophage TNFα production |
| Glanemann [36] | 2003 | rat | 5 | 45 | 90 | global | Nil | LFTs | ↓ LFTs | IPC reduction macrophage activation in early staged of IR injury |
| Kupffer cell phagocytosis | ↓ Kupffer cell activation | |||||||||
| Hepatic perfusion and oxygenation | ↑ hepatic perfusion and oxygenation | |||||||||
| Tejima [37] | 2004 | rat | 10 | 40 | 60 | partial | Macrophage depletion with Gadolinium Chloride and treatment with anti-oxidants | LFTs | ↓ LFTs | Macrophages were essential for the preconditioning stimulus to be effective |
| Sinusoidal epithelial cell injury | no change in sinusoidal epithelial cell injury | |||||||||
| Cytokines | ||||||||||
| Funaki [38] | 2002 | mouse | 15 | 70 | 240 | global | NF-κB an tyrosine kinase inhibition | Hepatic TNFα | ↓ TNFα | IPC reduced hepatic TNFα levels |
| Zhu [39] | 2003 | rat | 10 | 240 (cold) | 24 h | global | nil | LFTs | ↓ LFTs | IPC lead to reduced apoptosis and TNFα release |
| Serum TNFα | ↓ TNFα | |||||||||
| Apoptosis | ↓ Apoptosis | |||||||||
| Yao [40] | 2007 | rat | 10 | 55 | 7 days | global | nil | Survival | No change in survival | IPC increased IR injury in small for size grafts |
| LFTs | ↑ LFTs | |||||||||
| Hepatic IL-6 | No change in TNFα | |||||||||
| Hepatic TNFα | ↓ IL-6 | |||||||||
| Koneru [41] | 2007 | human | 10 | 329-505 | n/a | global | nil | Survival LFTs Post-op complications Serum TNFα Serum IL-6 Serum IL-10 | No change in survival ↑ LFTs in the first 2 days ↓ episodes of acute rejection ↑ IL-10 levels post reperfusion No change in TNFα or IL-6 levels | |
| Ajamieh [19] | 2008 | rat | 10 | 90 | 24 h | partial | A1 receptor | LFTs | ↓ LFTs | A1 receptor |
| TNFα levels | ↓ TNFα levels | |||||||||
| MPO activity | ↓ oxidative stress | |||||||||
| Guimaraes [42] | 2015 | rat | 4 | 45 | 180 | partial | nil | LFTs Serum IL-6 Serum IL-10 | ↓ LFTs ↑ IL-6 at 1 h ↓ IL-6 at 3 h | IL-6 levels were raised 1 h post IPC but were significantly less at 3 h |
| Li [43] | 2016 | mouse | 10 | 120 (cold) | 3 days | global | nil | Survival | No change in survival | IPC reduced liver injury but did not improve survival |
| LFTs | ↓ LFTs | |||||||||
| Serum TNFα | ↓ TNFα | |||||||||
| Innate immune response | ↓ Apoptosis | |||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robertson, F.P.; Fuller, B.J.; Davidson, B.R. An Evaluation of Ischaemic Preconditioning as a Method of Reducing Ischaemia Reperfusion Injury in Liver Surgery and Transplantation. J. Clin. Med. 2017, 6, 69. https://doi.org/10.3390/jcm6070069
Robertson FP, Fuller BJ, Davidson BR. An Evaluation of Ischaemic Preconditioning as a Method of Reducing Ischaemia Reperfusion Injury in Liver Surgery and Transplantation. Journal of Clinical Medicine. 2017; 6(7):69. https://doi.org/10.3390/jcm6070069
Chicago/Turabian StyleRobertson, Francis P., Barry J. Fuller, and Brian R. Davidson. 2017. "An Evaluation of Ischaemic Preconditioning as a Method of Reducing Ischaemia Reperfusion Injury in Liver Surgery and Transplantation" Journal of Clinical Medicine 6, no. 7: 69. https://doi.org/10.3390/jcm6070069