Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
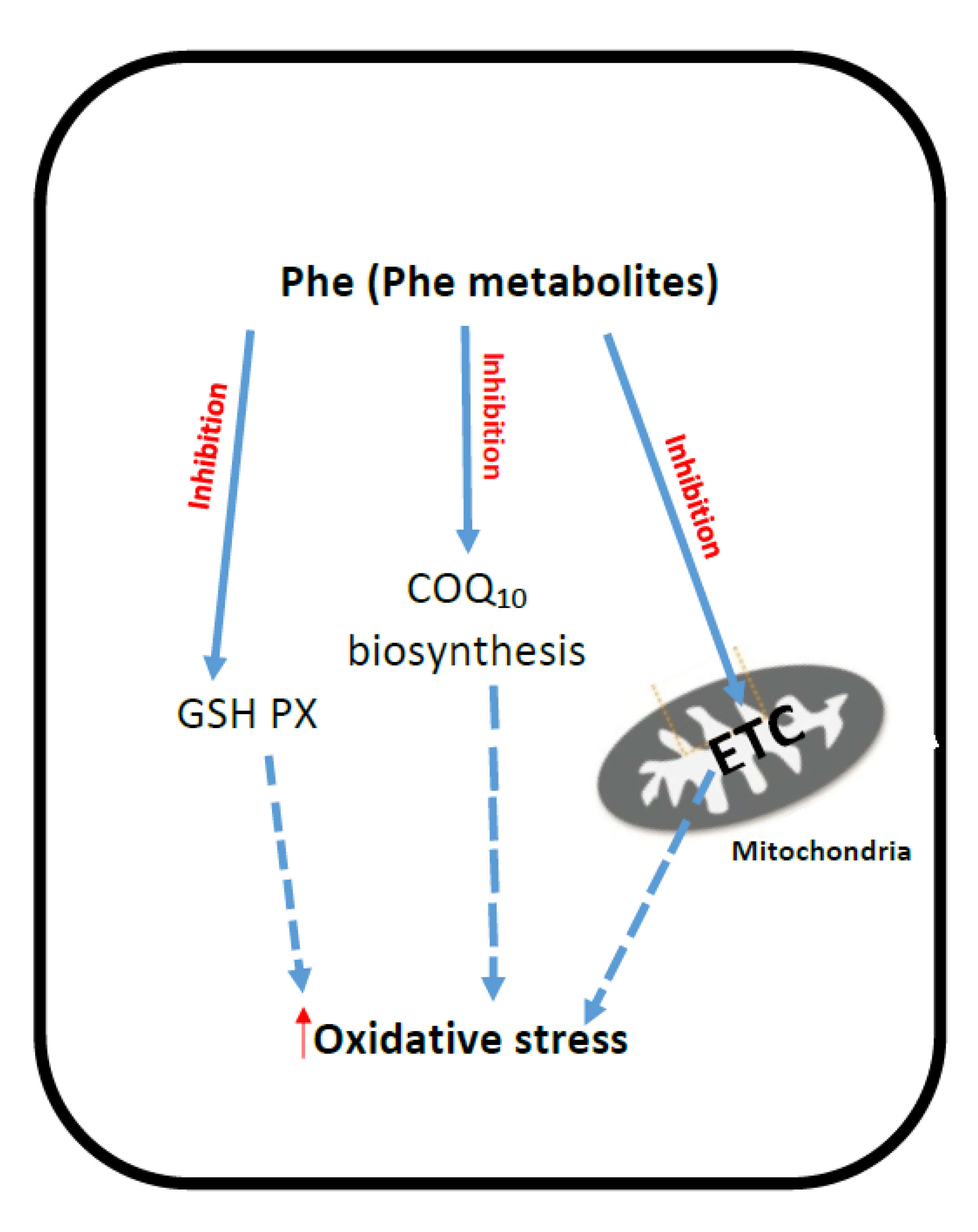
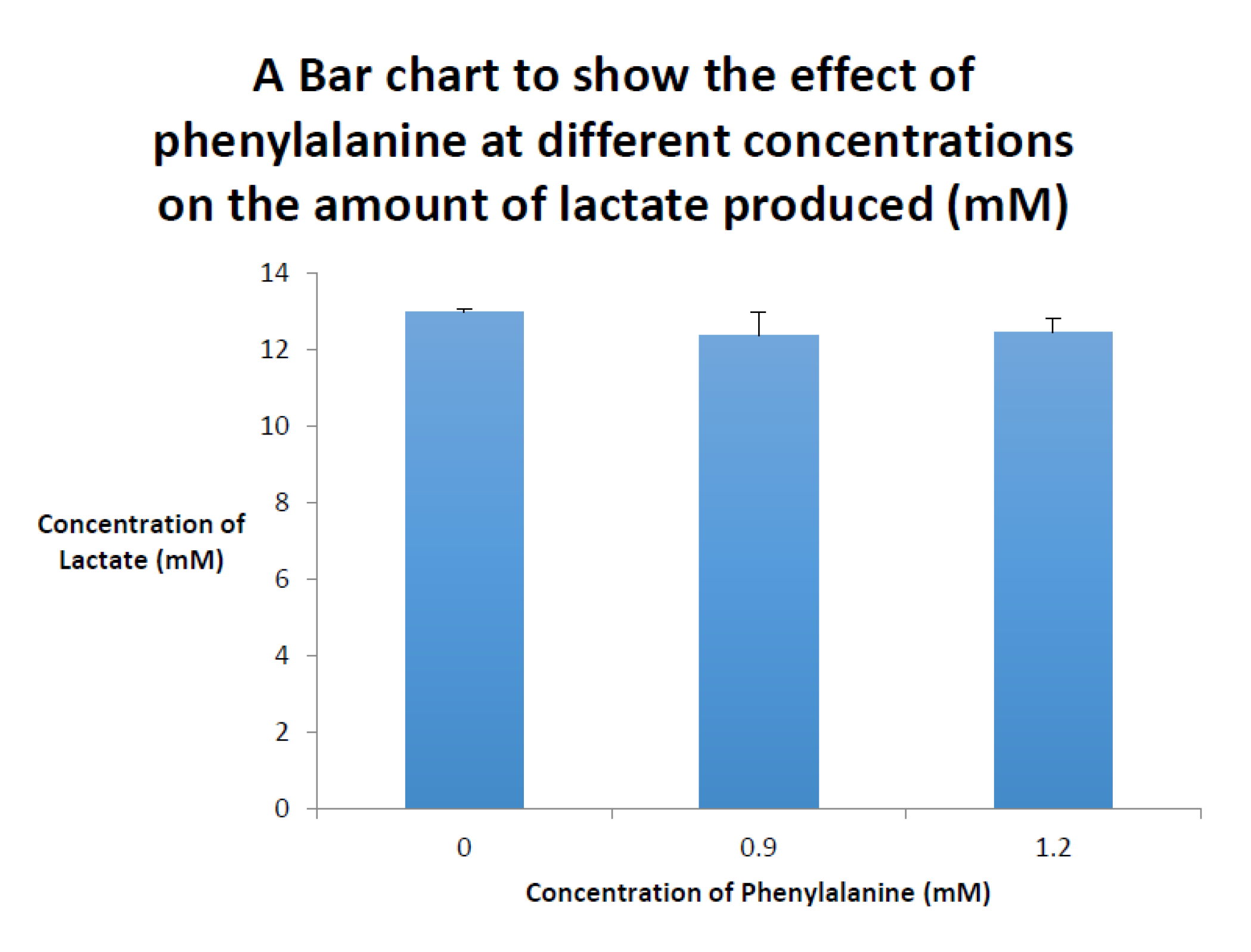
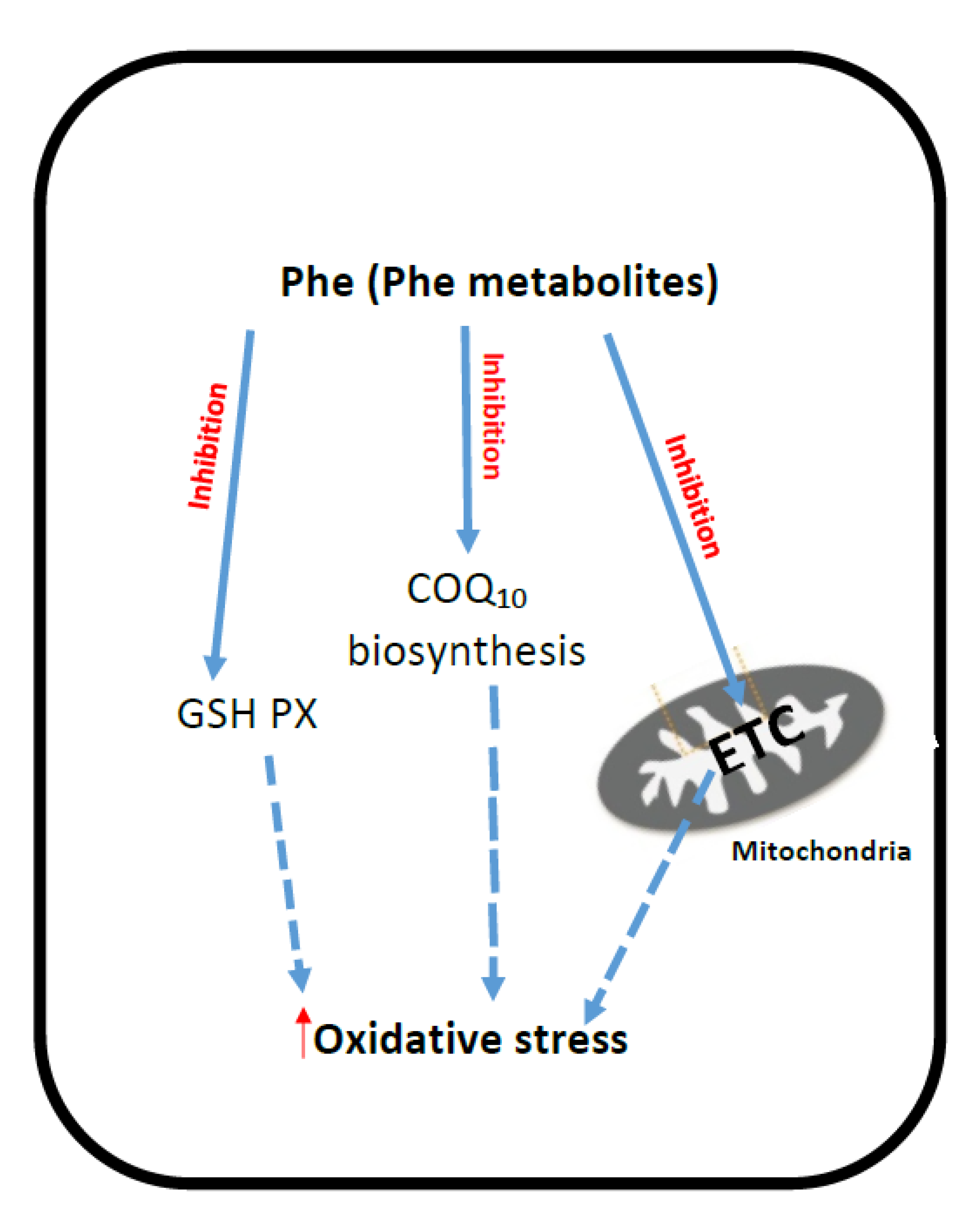
2. Phenyloketonuria (PKU)
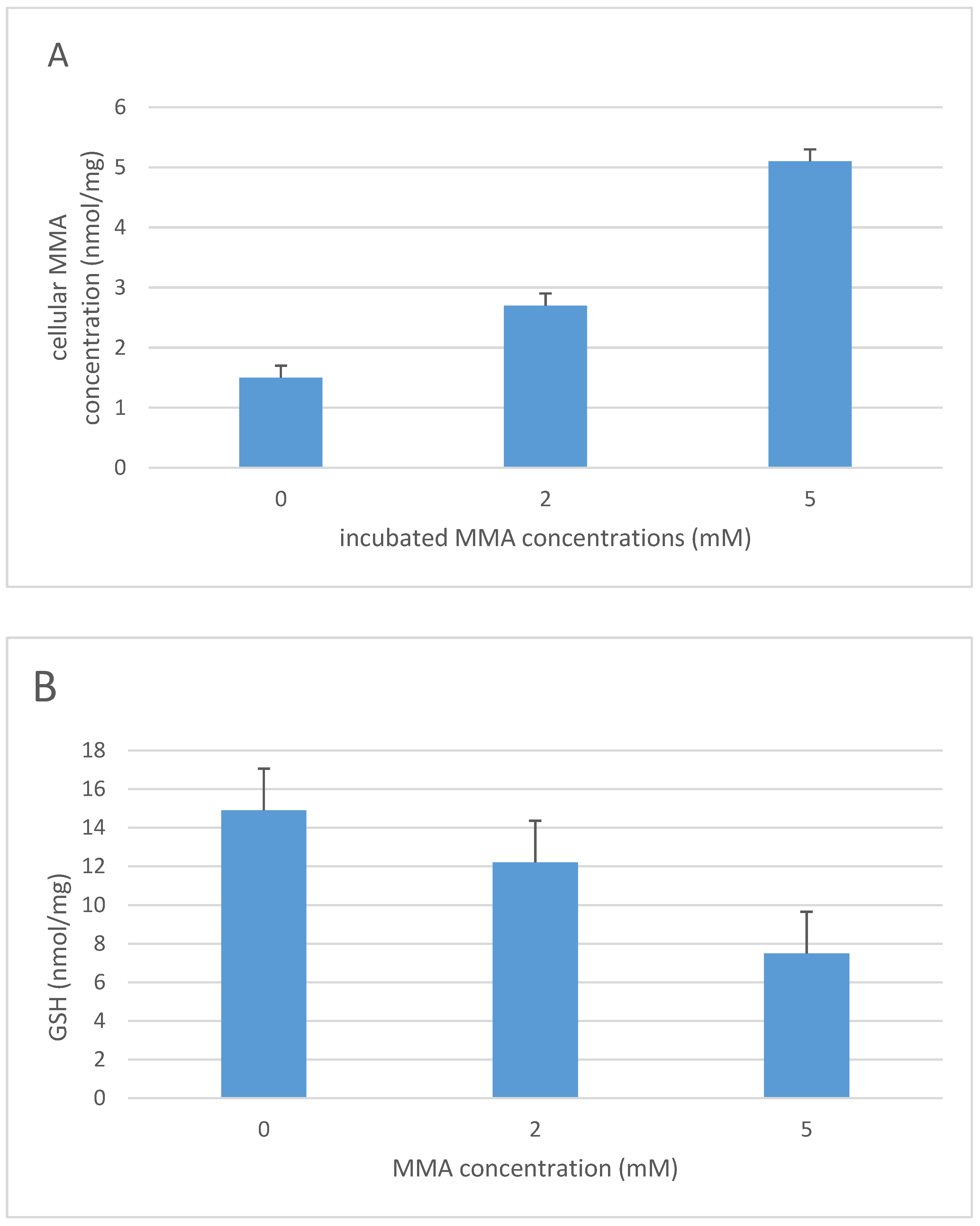
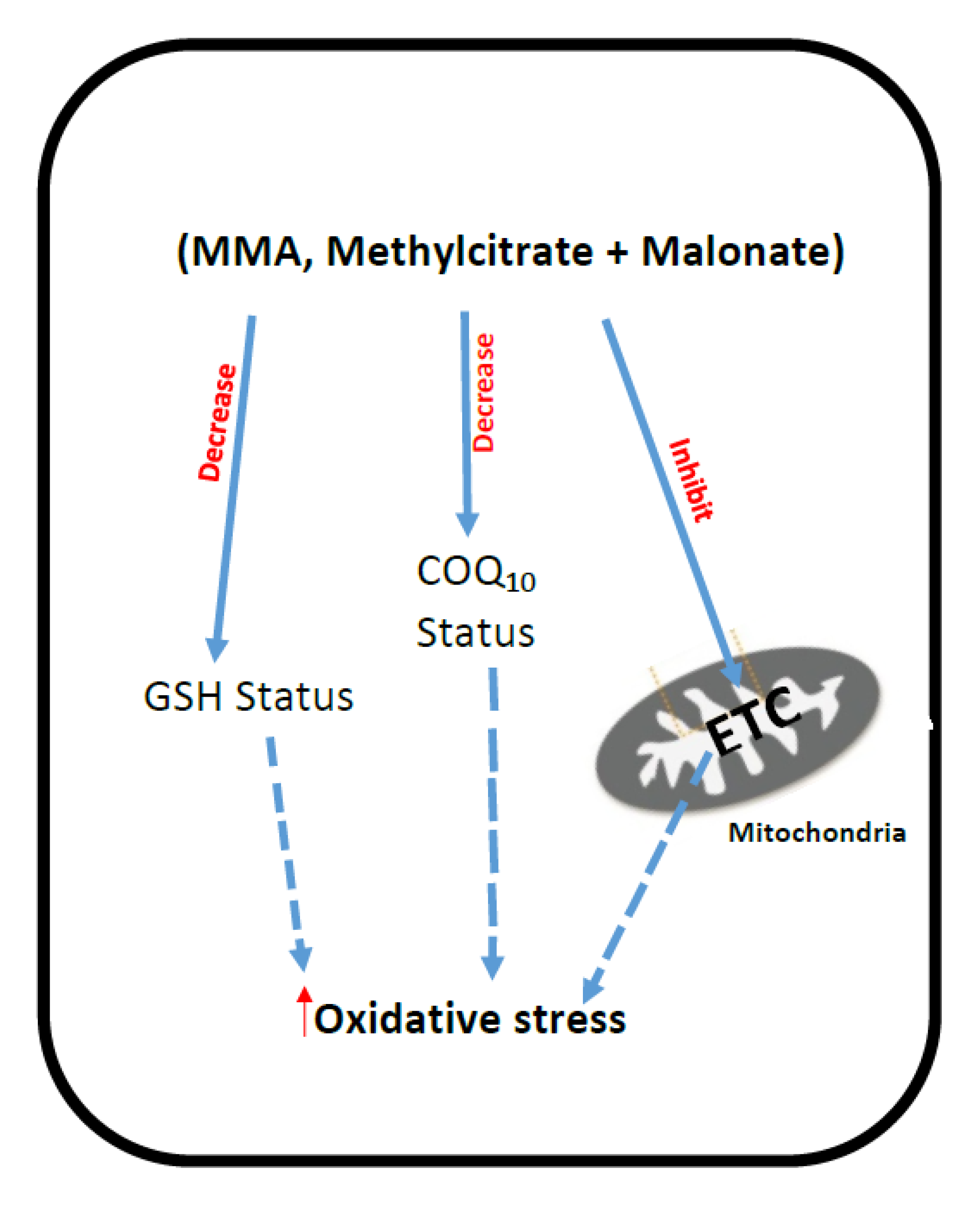
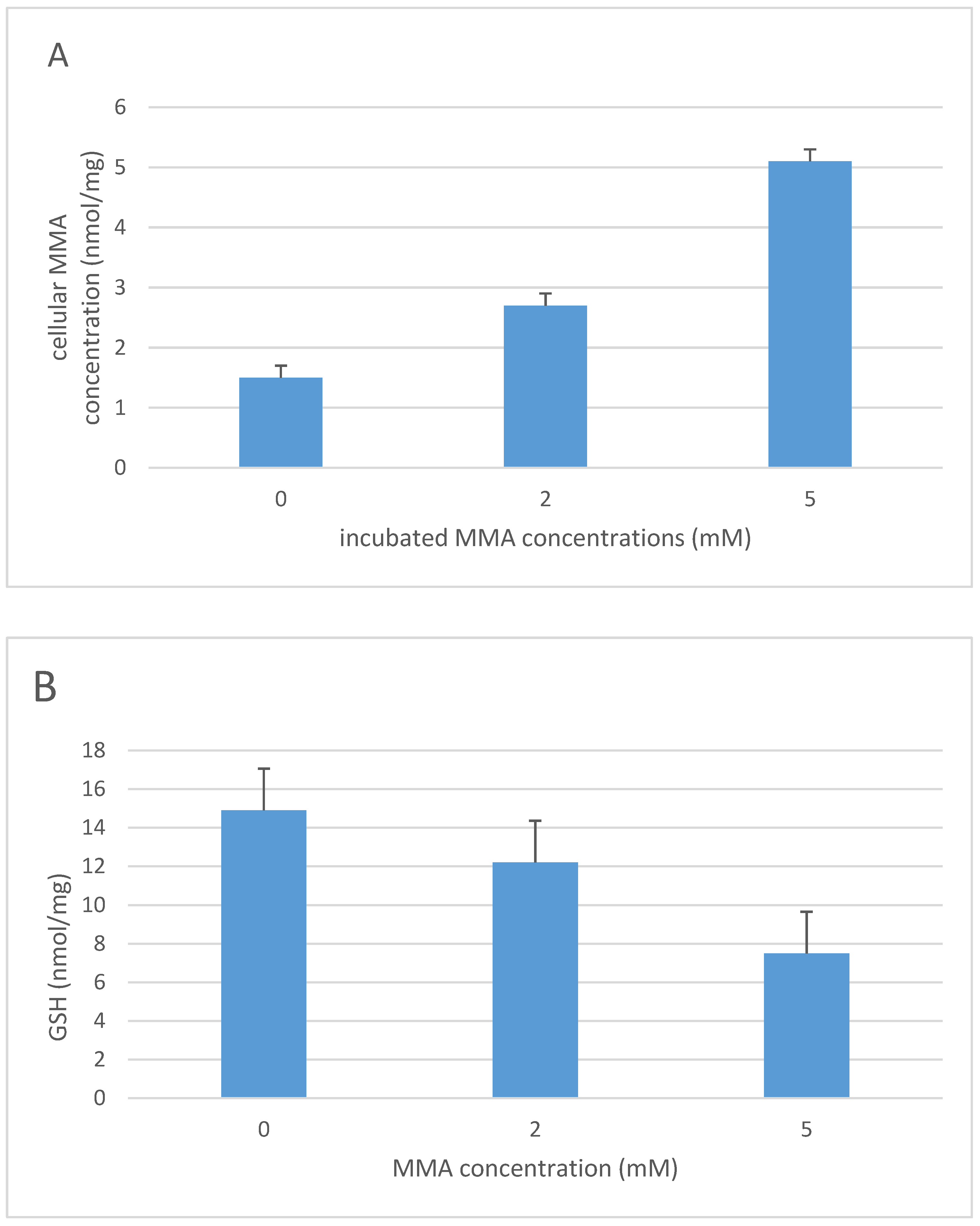
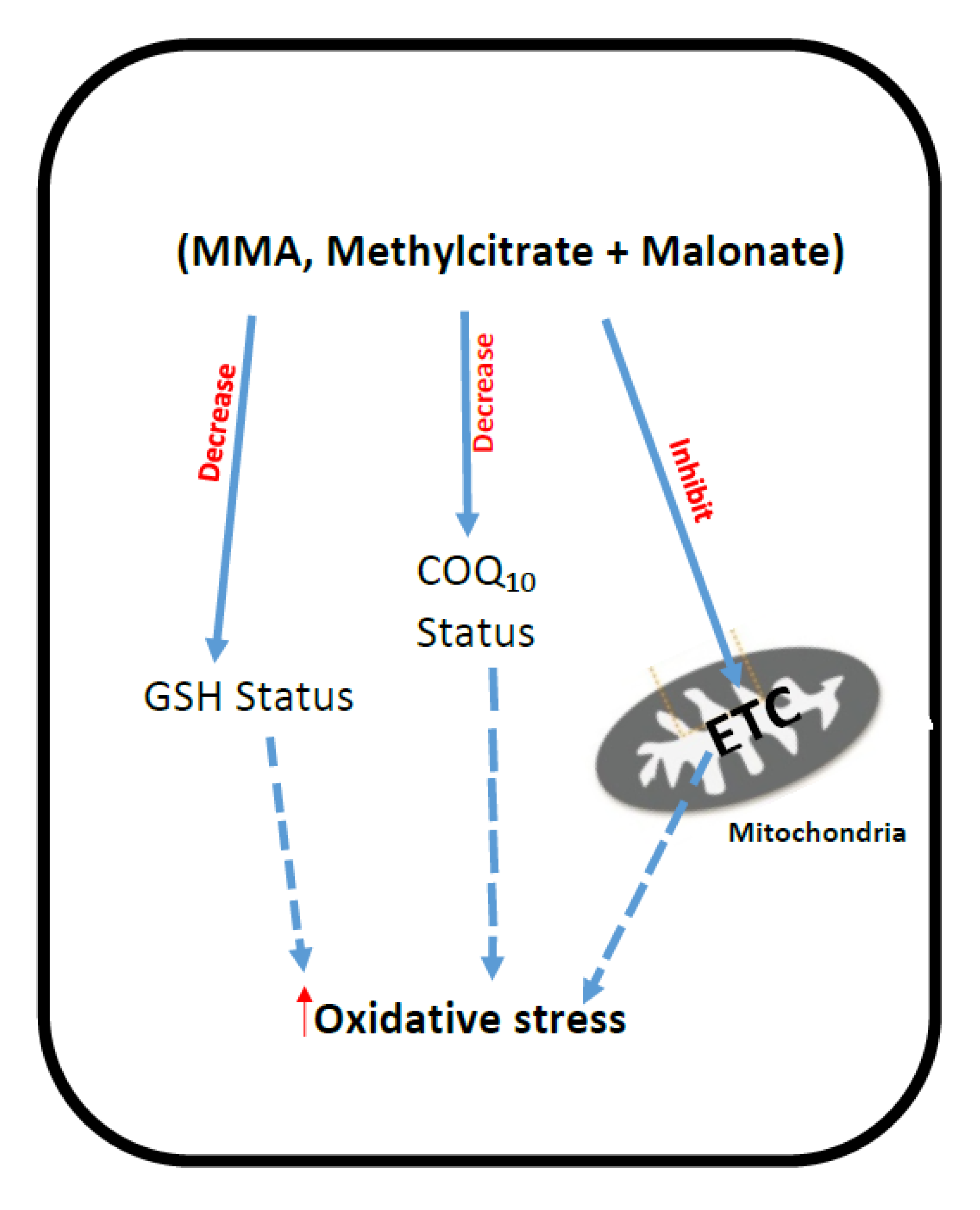
3. Methylmalonic Acidemia
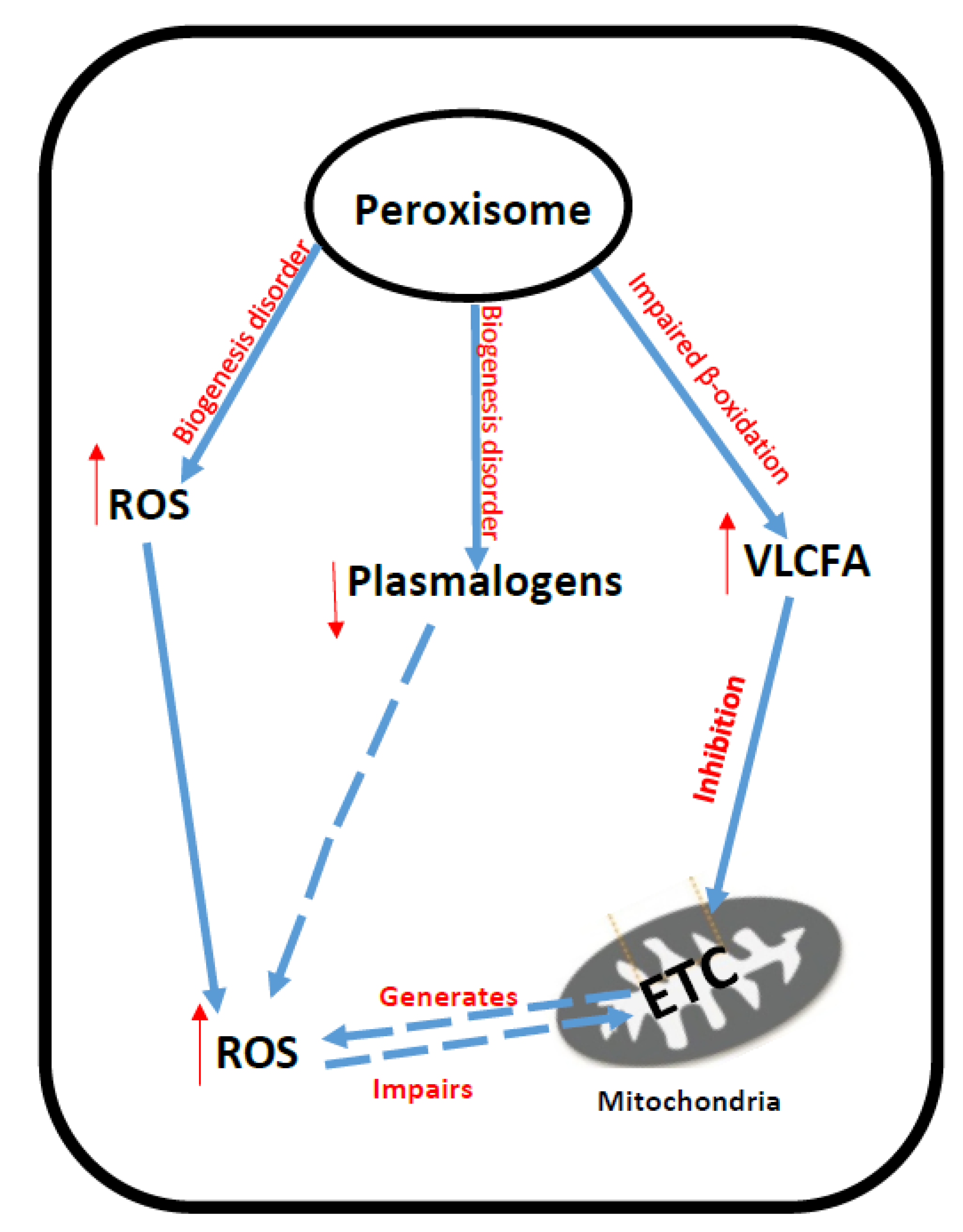
4. Peroxisomal Disorders
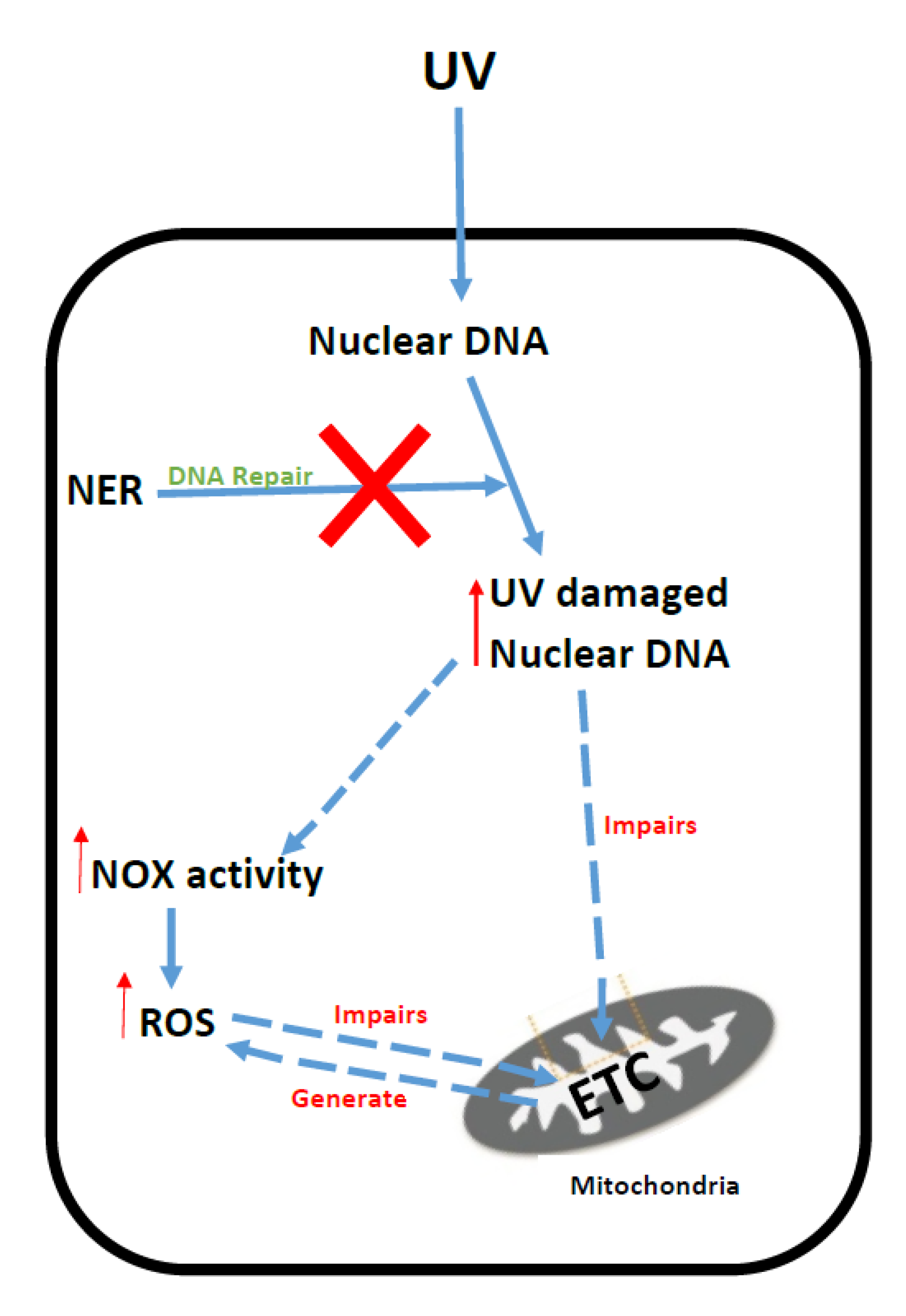
5. Xeroderma Pigmentosum
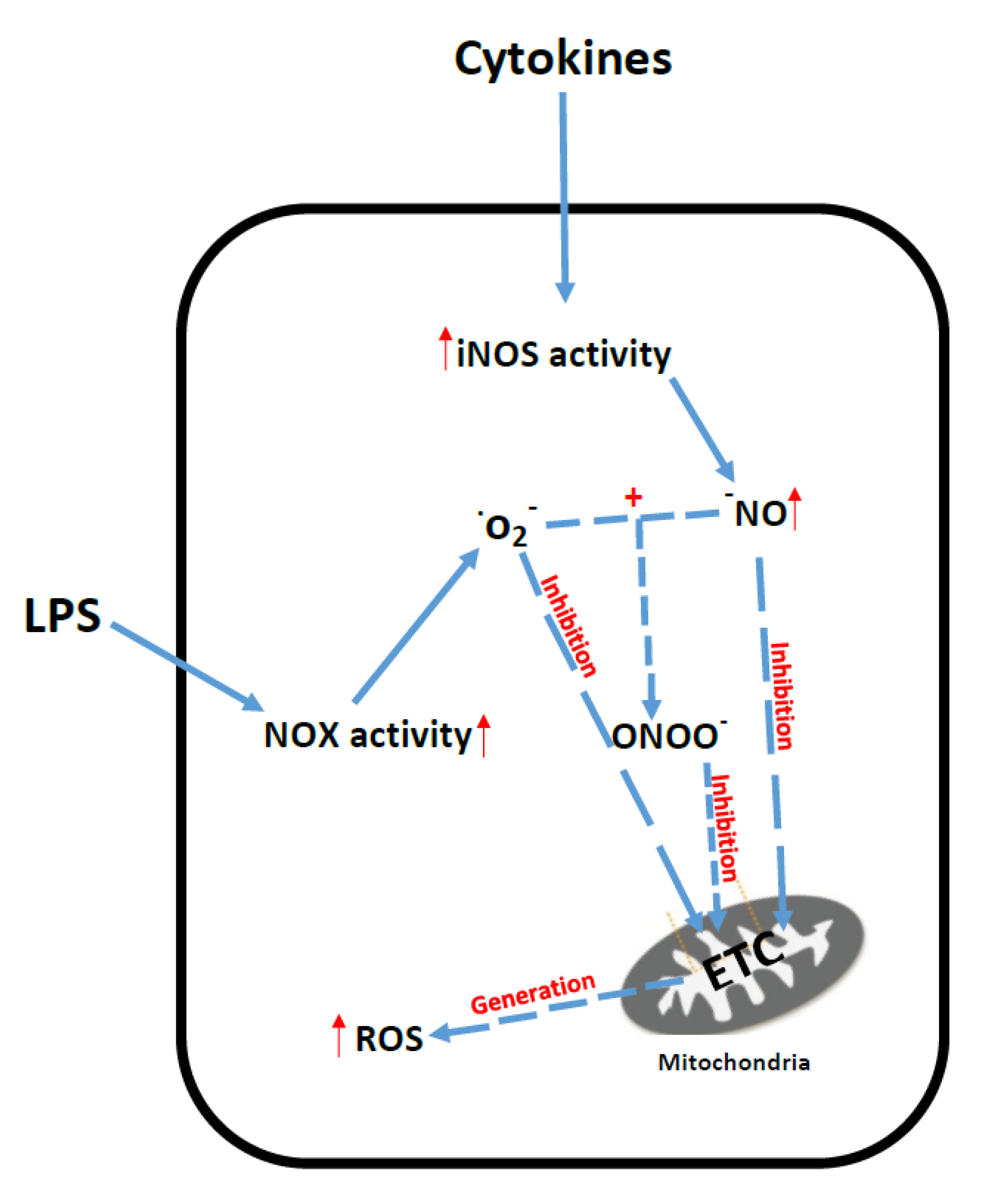
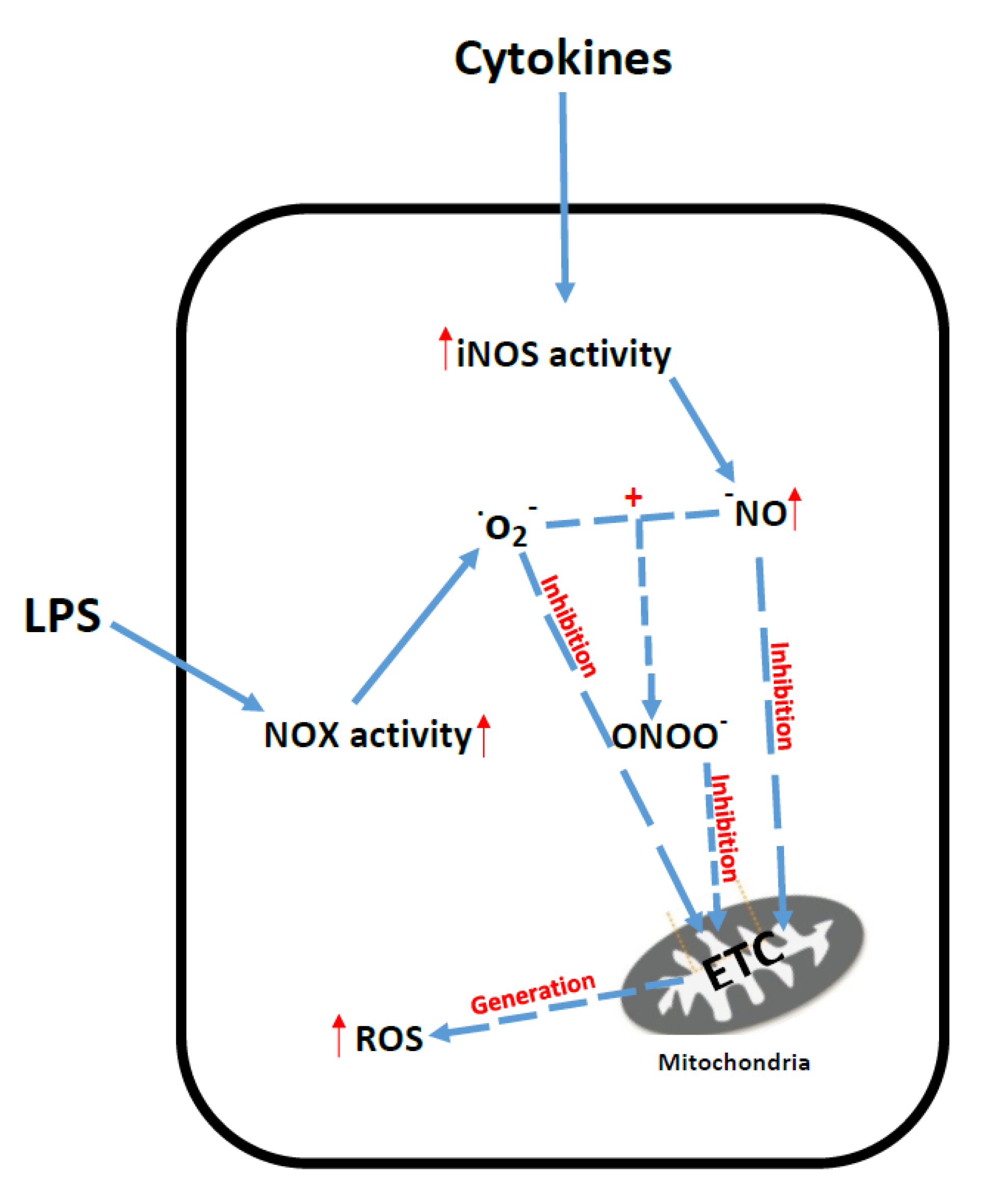
6. Sepsis
7. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of interest
Abbreviations
| ALD | adrenoleuklodystrophy |
| CSF | cerebrospinal fluid |
| ETC | electron transport chain |
| CoQ10 | coenzyme C10 |
| Fe | iron |
| GSH | glutathione |
| GSH-PX | glutathione peroxidase |
| GSSG | the oxidised form of GSH |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-CoA |
| H2O2 | hydrogen peroxide |
| LPS | lipopolysaccharides |
| Inos | induction of nitric oxide synthase |
| MCM | l-methylmalonyl-CoA mutase |
| MMA | methylmalonic acid |
| mtDNA | mitochondrial DNA |
| NADPH | nicotinamide adenine dinucleotide phosphate-oxidase |
| NAC | N-acetyl-cysteine cysteine |
| NER | nucleotide excision repair system |
| NOx | NADPH oxidase |
| Phe | phenylalanine |
| PKU | Phenyloketonuria |
| RCDP | rhizomeric chondrodysplasia punctate |
| SOD | superoxide dismutase |
| SIRS | systemic inflammatory response syndrome |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
| Se | selenium |
| VLCFA | very-long-chain fatty acids |
| VLCAC | very-long-chain acyl-CoAs |
| TBAR | thiobarbituric acid-reactive species |
| XP | Xeroderma pigmentosum |
References
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Leonard, L. Oxidative stress and cancer: An overview. Aging Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davis, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef]
- Heales, S.J.; Bolanos, J.P.; Brand, M.P.; Clark, J.B.; Land, J.M. Mitochondrial damage: An important feature in a number of inborn errors of metabolism? J. Inherit. Metab. Dis. 1996, 19, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Moyano, D.; Vilaseca, M.A.; Pineda, M.; Campistol, J.; Vernet, A.; Poo, P.; Artuch, R.; Sierra, C. Tocopherol in inborn errors of intermediary metabolism. Clin. Chim. Acta 1997, 263, 147–155. [Google Scholar] [CrossRef]
- Sierra, C.; Vilaseca, M.A.; Moyano, D.; Brandi, N.; Campistol, J.; Lambruschini, N.; Cambra, F.J.; Deulofeu, R.; Mira, A. Antioxidant status in hyperphenylalaninemia. Clin. Chim. Acta 1998, 276, 1–9. [Google Scholar] [CrossRef]
- Artuch, R.; Vilaseca, M.A.; Moreno, J.; Lambruschini, N.; Cambra, F.J.; Campistol, J. Decreased serum ubiquinone-10 concentrations in phenylketonuria. Am. J. Clin. Nutr. 1999, 70, 892–895. [Google Scholar] [PubMed]
- Fisberg, R.M.; Silva-Femandes, M.E.; Fisberg, M.; Schmidt, B.J. Plasma zinc, copper, and erythrocyte superoxide dismutase in children with phenylketonuria. Nutrition 1999, 15, 449–452. [Google Scholar] [CrossRef]
- Van Bakel, M.M.E.; Printzen, G.; Wermuth, B.; Wiesmann, U.N. Antioxidant and thyroid hormone status in selenium-deficient phenylketonuric and hyperphenylalaninemic patients. Am. J. Clin. Nutr. 2000, 72, 976–981. [Google Scholar] [PubMed]
- Rani, V.; Deep, G.; Singh, R.K. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Kolker, S.; Schwab, M.; Horster, F.; Sauer, S.; Hinz, A.; Wolf, N.I.; Mayatepek, E.; Hoffmann, G.F.; Smeitink, J.A.M.; Okun, J.G. Methylmalonic acid, a biochemical hallmark of methylmalonic acidurias but no inhibitor of mitochondrial respiratory chain. J. Biol. Chem. 2003, 278, 47388–47393. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Reiser, G. Rotenone-like action of the branch chain phytanic acid induces oxidative stress in mitochondria. J. Biol. Chem. 2006, 281, 7136–7142. [Google Scholar] [CrossRef] [PubMed]
- Zapelini, P.H.; Retin, G.T.; Cardoso, M.R.; Ritter, C.; Ritter, C.; Klamt, F.; Moreira, J.C.; Streck, E.L.; Dal-Pizzol, F. Antioxidant treatment reverses mitochondrial dysfunction in a sepsis animal model. Mitochondrion 2008, 8, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Paradies, G.; Ruggiero, F.M.; Petrosillo, G.; Quagliariello, E. Peroxidative damage to cardiac mitochondria: Cytochrome c oxidase and cardiolipin alterations. FEBS Lett. 1998, 424, 155–158. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.; Martins, M.J. Oxidative stress in phenylketonuria: Future directions. J. Inherit. Metab. Dis. 2012, 35, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Prauchner, C.A. Oxidative stress in sepsis: Pathophysiological implications justifying antioxidant co-therapy. Burns 2016, 43, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Scriver, C.R.; Kaufman, S.; Eisensmith, R.C.; Woo, S.L.C. The phenylalaninemias. In The Metabolic and Molecular Bases of Inherited Disease, 7th ed.; Scriver, C.R., Beaudet, A.L., Valle, D., Sly, W.S., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 1015–1075. [Google Scholar]
- Williams, R.A.; Mamotte, C.D.; Burnett, J.R. Phenylketonuria: An inborn error of phenylalanine metabolism. Clin. Biochem. Rev. 2008, 29, 31–41. [Google Scholar] [PubMed]
- Velema, M.; Boot, E.; Engelen, M.; Hollak, C. Parkinsonism in phenylketonuria: A consequence of dopamine depletion? JIMD Rep. 2015, 20, 35–38. [Google Scholar] [PubMed]
- Krause, W.; Halminski, M.; McDonald, L.; Demure, P.; Salvo, R.; Friedes, S.R.; Elsas, L. Biochemical and neuropsychological effects of elevated plasma phenylalanine in patients with treated phenylketonuria. J. Clin. Investig. 1985, 75, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Ushakova, G.A.; Gubkina, H.A.; Kachur, V.A.; Lepekhin, E.A. Effect of experimental hyperphenylalaninemia on the postnatal rat brain. Int. J. Dev. Neurosci. 1997, 15, 29–36. [Google Scholar] [CrossRef]
- Ercal, N.; Aykin-Burns, N.; Gurer-Orhan, H.; McDonald, J.D. Oxidative stress in a phenylketonuria animal model. Free Radic. Biol. Med. 2002, 32, 906–911. [Google Scholar] [CrossRef]
- Pietz, J. Neurological aspects of adult phenylketonuria. Curr. Opin. Neurol. 1998, 11, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Knowles, J. Behaviour in early treated phenylketonuria: A systematic review. Eur. J. Pediatr. 2000, 159 (Suppl. 2), S89–S93. [Google Scholar] [CrossRef] [PubMed]
- Berry, S.A.; Brown, C.; Grant, M.; Greene, C.L.; Jurecki, E.; Koch, J.; Moseley, K.; Suter, R.; van Calcar, S.C.; Wiles, J.; et al. Newborn screening 50 years later: Access issues faced by adults with PKU. Genet. Med. 2013, 15, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Weglage, J.; Pietsch, M.; Funders, B.; Koch, H.G.; Ullrich, K. Neurological findings in early treated phenylketonuria. Acta Paediatr. 1995, 84, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Dyer, C.A. Comments on the neuropathology of phenylketonuria. Eur. J. Pediatr. 2000, 159 (Suppl. 2), S107–S108. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, P.R. The neuropathology of phenylketonuria: Human and animal studies. Eur. J. Pediatr. 2000, 159 (Suppl. 2), S102–S106. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.C.; Martel, F. Large neutral amino acids supplementation in phenylketonuric patients. J. Inherit. Metab. Dis. 2009, 32, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.M.; Schuck, P.F.; Wenk, G.L.; Ferreira, G.C. Metabolic disturbances in diseases with neurological involvement. Aging Dis. 2013, 5, 238–255. [Google Scholar] [PubMed]
- Sirtori, L.R.; Dutra-Filho, C.S.; Fitarelli, D.; Sitta, A.; Haeser, A.; Barschak, A.G.; Wajner, M.; Coelho, D.M.; Llesuy, S.; Bello-Klein, A.; et al. Oxidative stressvin patients with phenylketonuria. Biochim. Biophys. 2005, 1740, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Sitta, A.; Barschak, A.G.; Deon, M.; Barden, A.T.; Biancini, G.B.; Vargas, P.R.; de Souza, C.F.; Netto, C.; Wajner, M.; Vargas, C.R. Effect of short- and long-term exposition to high phenylalanine blood levels on oxidative damage in phenylketonuric patients. Int. J. Dev. Neurosci. 2009, 27, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Sanayama, Y.; Nagasaka, H.; Takayanagi, M.; Ohura, T.; Sakamato, O.; Ito, T.; Ishige-Wada, M.; Usui, H.; Yoshino, M.; Ohtake, A.; et al. Experimental evidence that phenylalanine is strongly associated to oxidative stress in adolescents and adults with phenylketonuria. Mol. Genet. Metab. 2011, 103, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Schulpis, K.H.; Tsakiris, S.; Traeger-Synodinos, J.; Papassotiriou, I. Low total antioxidant status is implicated with high 8-hydroxy-2-deoxyguanosine serum concentrations in phenylketonuria. Clin. Biochem. 2005, 38, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Sitta, A.; Vanzin, C.S.; Biancini, G.B.; Manfredoni, V.; De Oliviera, A.B.; Wayhs, C.A.Y.; Ribas, G.O.S.; Giuliani, L.; Schwartz, I.V.D.; Bohrer, D.; et al. Evidence that l-carnitine and selenium supplementation reduces oxidative stress in phenylketonuric patients. Cell Mol. Neurobiol. 2011, 31, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, N.; Nakaden, H.; Yamamoto, Y.; Matsuo, S.; Fujikawa, T.; Matsusue, S. Selenium Kinetics and Changes in Glutathione Peroxidase Activities in Patients Receiving Long-Term Parenteral Nutrition and Effects of Supplementation With Selenite. Nutrition 2000, 16, 22–26. [Google Scholar] [CrossRef]
- Kienzle-Hagen, M.E.; Pederzolli, C.D.; Sgaravatti, A.M.; Bridi, R.; Wajner, R.; Wannmacher, C.M.; Wyse, A.T.; Dutra-Filho, C.S. Experimental hyperphenylalaninemia provokes oxidative stress in rat brain. Biochim. Biophys. Acta 2000, 1586, 344–352. [Google Scholar] [CrossRef]
- Sitta, A.; Barschak, A.G.; Deon, M.; de Mari, J.F.; Barden, A.T.; Vanzin, C.S.; Biancini, G.B.; Schwartz, I.V.; Wajner, M.; Vargas, C.R. l-Carnitine blood levels and oxidative stress in treated phenylketonuria patients. Cell Mol. Neurobiol. 2009, 29, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Preissler, T.; Bristot, I.J.; Costa, B.M.L.; Fernandes, E.K.; Rieger, E.; Bortoluzzi, V.T.; de Franceschi, I.D.; Dudra-Filho, C.S.; Moreira, J.C.F.; Wannmacher, C.M.D. Phenylalanine induces oxidative stress and decreases the viability of rat astrocytes: Possible relevance for the pathophysiology of neurodegeneration in phenylketonuria. Metab. Brain Dis. 2016, 31, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P.F.; Malgarin, F.; Cararo, J.H.; Cardoso, F.; Streck, E.L.; Ferreira, G.C. Phenylketonuria pathophysiology: On the role of the metabolic alterations. Aging Dis. 2015, 6, 390–399. [Google Scholar] [CrossRef]
- Castillo, M.; Martinez-Cayuela, M.; Zafra, M.F.; Garcia-Peregrin, E. Effect of phenylalanine derivatives on the main regulatory enzymes of hepatic cholesterogenesis. Mol. Cell Biochem. 1991, 105, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Shefer, S.; Tint, G.S.; Jean-Guillaume, D.; Daikhin, E.; Kendler, A.; Nguyen, L.B.; Yudkoff, M.; Dyer, C.A. Is there a relationship between 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and forebrain pathology in the PKU mouse? J. Neurosci. Res. 2000, 61, 549–563. [Google Scholar] [CrossRef]
- Colome, C.; Artuch, R.; Vilaseca, M.A.; Sierra, C.; Brandi, N.; Cambra, F.J.; Lambruschini, N.; Campistol, J. Ubiquinone-10 content in lymphocytes of phenylketonuric patients. Clin. Biochem. 2002, 35, 81–84. [Google Scholar] [CrossRef]
- Hargreaves, I.P.; Heales, S.J.; Briddon, A.; Land, J.M.; Lee, P.J. Mononuclear cell coenzyme Q (coq) Concentration and mitochondrial respiratory chain succinate cytochrome C reductase (complex li-iii) activity in phenyloketonuric patiens. J. Inher. Metab. Dis. 2002, 25, 18. [Google Scholar] [CrossRef]
- Rech, V.C.; Feksa, L.R.; Dudra-Filho, C.S.; Wyse, A.T.D.S.; Wajner, M.; Wannmacher, C.M.D. Inhibition of the mitochondrial respiratory chain by phenylalanine in rat cerebral; cortex. Neurochem. Res. 2002, 27, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Kyprianou, N.; Murphy, E.; Lee, P.; Hargreaves, I. Assessment of mitochondrial respiratory chain function in hyperphenylalaninemia. J. Inherit. Metab. 2009, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Przyrembel, H.; Bremer, H.J. Nutrition, physical growth, and bone density in treated phenylketonuria. Eur. J. Pediatr. 2000, 159 (Suppl. 2), S129–S135. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, M.; Verduci, E.; Salvatici, E.; Fiori, L.; Riva, E. Phenylketonuria: Dietary and therapeutic challenges. J. Inherit. Metab. Dis. 2007, 30, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Poustie, V.J.; Wildgoose, J. Dietary interventions for phenylketonuria. Cochrane Libr. 2010. [CrossRef]
- McMurry, M.P.; Chan, G.M.; Leonard, C.O.; Ernst, S.L. Bone mineral status in children with phenylketonuria—Relationship to nutritional intake and phenylalanine control. Am. J. Clin. Nutr. 1992, 55, 997–1004. [Google Scholar] [PubMed]
- Wilke, B.C.; Vidailhet, M.; Richard, M.J.; Ducros, V.; Arnaud, J.; Favier, A. Trace elements balance in treated phenylketonuria children. Consequences of selenium deficiency on lipid peroxidation. Arch. Latinoam. Nutr. 1993, 43, 119–122. [Google Scholar] [PubMed]
- Ragsdale, S. Metal-carbon bonds in enzymes and cofactors. Coord. Chem. Rev. 2010, 254, 1948–1949. [Google Scholar] [CrossRef] [PubMed]
- Robert, M.; Rocha, J.C.; van Rijn, M.; Ahring, K.; Bélanger-Quintana, A.; MacDonald, A.; Dokoupil, K.; Ozel, H.G.; Lammardo, A.M.; Goyens, P.; et al. Micronutrient status in phenylketonuria. Mol. Genet. Metab. 2013, 110, S6–S17. [Google Scholar] [CrossRef] [PubMed]
- Bohler, H.; Ulrich, K.; Endres, W.; Behbehani, A.W.; Wendel, U. Inadequate iron availability as a possible cause of low serum carnitine concentrations in patients with phenylketonuria. Eur. J. Pediatr. 1991, 15, 425–428. [Google Scholar] [CrossRef]
- Gullcin, I. Antioxidant and antiradical activities of l-carnitine. Life Sci. 2006, 78, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Ribas, G.S.; Sitta, A.; Wajner, M.; Vargas, C.R. Oxidative stress in phenylketonuria: What is the evidence? Cell Mol. Neurobiol. 2011, 31, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Wai, T.; Leclerc, D.; Wilson, A.; Wu, X.; Dore, C.; Hudson, T.; Rosenblatt, D.S.; Gravel, R.A. Identification of the gene responsible for the cblA complementation group of vitamin B12responsive methylmalonic acidemia based on analysis of prokaryotic gene arrangements. Proc. Natl. Acad. Sci. USA 2002, 99, 15554–15559. [Google Scholar] [CrossRef] [PubMed]
- Fenton, W.A.; Gravel, R.A.A.; Rosenblatt, D.S. Disorders of propionate and methylmalonate metabolism. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sky, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2011; pp. 2165–2193. [Google Scholar]
- Treacy, E.; Arbour, L.; Chessex, P.; Graham, G.; Kasprzak, L.; Casey, K.; Bell, L.; Mamer, O.; Scriver, C.R. Glutathione deficiency as a complication of methylmalonic acidemia: Response to high doses of ascorbate. J. Pediatr. 1996, 129, 445–448. [Google Scholar] [CrossRef]
- Hoffmann, G.F.; Meier-Augenstein, W.; Stocker, S.; Surtees, R.; Rating, D.; Nyhan, W. Physiology and pathophysiology of organic acids in cerebrospinal fluid. J. Inherit. Metab. Dis. 1993, 16, 648–669. [Google Scholar] [CrossRef] [PubMed]
- Manoli, I.; Sloan, J.L.; Venditti, C.P. Isolated methylmalonic acidemia. In Genereviews [Internet]; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 2016; pp. 1993–2017. [Google Scholar]
- Hayasaka, K.; Metoki, K.; Satoh, T.; Narisawa, K.; Tada, K.; Kawakami, T.; Matsuo, N.; Aoki, T. Comparison of cytosolic and mitochondrial enzyme alterations in the livers of propionic or methylmalonic acidemia: A reduction of cytochrome oxidase activity. Tohoku J. Exp. Med. 1982, 137, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.; Alvarez-Barrientos, A.; Perez, B.; Desviat, L.R.; Ugarte, M. Methylmalonic acidaemia leads to increased production reactive oxygen species and induction of apoptosis through the mitocondrial/caspase pathway. J. Pathol. 2007, 213, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, B.; Lindblad, B.S.; Olin, P.; Svanberg, B.; Zetterstrom, R. Methylmalonic academia. A disorder associated with acidosis, hyperlycaemia, and hyperlactatemia. Acta Paediatr. Scand. 1968, 57, 417–424. [Google Scholar] [PubMed]
- Okun, J.C.; Horster, F.; Farkas, L.; Feyh, P.; Hinz, A.; Sauer, S.; Hoffman, G.F.; Unisicker, K.; Mayatepek, E.; Kolker, S. Neurodegeneration in Methylmalonic Aciduria Involves Inhibition of Complex II and the Tricarboxylic Acid Cycle, and Synergistically Acting Excitotoxicity. J. Biol. Chem. 2002, 277, 14674–14680. [Google Scholar] [CrossRef] [PubMed]
- Marisco, P.C.; Ribeiro, M.C.; Bonini, J.S.; Lima, T.T.; Mann, K.C.; Brenner, G.M.; Dutra-Filho, C.S.; Mello, C.F. Ammonia potentiates methylmalonic acid-induced convulsions and TBARS production. Exp. Neurol. 2003, 182, 455–460. [Google Scholar] [CrossRef]
- Krahenbuhl, S.; Chang, M.; Brass, E.P.; Hoppel, C.L. Decreased activities of ubiqunol; ferricytochrome c oxidoreductase (complex III) and ferrocytochrome c: Oxygen oxidoreductase (complex IV) in liver mitochondria from rats with hydroxycobalamin[C-lactam]-induced methylmalonic aciduria. J. Biol. Chem. 1991, 266, 20998–21003. [Google Scholar] [PubMed]
- Pettenuzzo, L.F.; Ferreira, G.D.C.; Schmidt, A.L.; Dutra-Filho, C.S.; Wyse, A.T.S.; Wajner, M. Differential inhibitory effects of methylmalonic acid on respitratory chain complex activities in rat tissues. Int. J. Dev. Neurosci. 2006, 24, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Chandler, R.J.; Zerfas, P.M.; Shanske, S.; Sloan, J.; Hoffman, V.; DiMauro, S.; Venditti, C.P. Mitochondrial dysfunction in mut methylmalonic academia. FASEB J. 2009, 23, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- De Keyzer, Y.; Valayannopoulos, V.; Benoist, J.F.; Batteux, F.; Lacaille, F.; Hubert, L.; Chretien, D.; Chadefeaux-Vekemans, B.; Niaudet, P.; Touati, G.; et al. Multiple OXPHOS deficiency in the liver, kidney, heart, and skeletal muscle of patients with methylmalonic aciduria and propionic aciduria. Pediatr. Res. 2009, 66, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Kashtan, C.E.; Abousedira, M.; Rozen, S.; Manivel, J.C.; McCann, M.; Tuchman, M. Chronic administration of methylmaloic acid (MMA) to rats causes proteinuria and renal tubular injury (abstract). Pediatr. Res. 1998, 43, 309. [Google Scholar] [CrossRef]
- Zsengeller, Z.K.; Alkinovic, N.; Teot, L.A.; Korson, M.; Rodig, N.; Sloan, J.L.; Venditti, C.P.; Berry, G.T.; Rosen, S. Methylmalonic academia: A megamitochondrial disorder affecting the kidney. Pediatr. Nephrol. 2014, 29, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.; Piesowicz, A.T.; Brett, E.M.; Leonard, J.V. Focal changes in the globi pallidi associated with neurological dysfunction in methylmalonic academia. Neuropediatrics 1989, 20, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Brismar, J.; Ozand, P.T. CT and MR of the brain in disorders of propionate and methylmalonate metabolism. Am. J. Neuroradiol. 1994, 15, 1459–1473. [Google Scholar] [PubMed]
- Larnaout, A.; Mongalgi, M.A.; Kaabachi, N.; Khiari, D.; Debbabi, A.; Mebazza, A.; Ben Hamida, M.; Hentati, F. Methylmalonic acidaemia with bilateral globus pallidus involvement: A neuropathological study. J. Inherit. Metab. Dis. 1998, 21, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.J.; van de Vyver, F.L.; Scholte, H.R.; Roodhooft, A.M.; Martin, C.C.; Luyt-Houwen, I.E.J. Defect in succinate oxidation by isolated muscle mitochondria in a patient with symmetrical lesions in the bassel ganglia. J. Neurol. Sci. 1988, 84, 189–200. [Google Scholar]
- Trinh, B.C.; Melhem, E.R.; Barker, P.B. Multi-slice proton MR spectroscopy and diffusion-weighted imaging in methylmalonic acidemia: Report of two cases and review of the literature. Am. J. Neuroradiol. 2001, 22, 831–833. [Google Scholar] [PubMed]
- Heidenreich, R.; Natowicz, M.; Hainline, B.E.; Berman, P.; Kelley, R.I.; Hillman, R.E.; Berry, G.T. Acute extrapyramidal syndrome in methylmalonic acidemia: “Metabolic stroke” involving the globus pallidus. J. Pediatr. 1988, 113, 1022–1027. [Google Scholar] [CrossRef]
- Brusque, A.M.; Borba Rosa, R.; Schuck, P.F.; Dalcin, K.B.; Ribeiro, C.A.J.; Silva, C.G.; Wannmacher, C.M.D.; Dutra-Filho, C.S.; Wyse, A.T.S.; Briones, P.; et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochem. Int. 2002, 40, 593–601. [Google Scholar] [CrossRef]
- Kolker, S.; Okun, J.G. Methylmalonic acid—An endogenous toxin? Cell Mol. Life Sci. 2005, 62, 621–624. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, B.A.; Nelson, D.; Silver, I.A.; Erecinska, M.; Chesselet, M.F. Methylmalonate toxicity in primary neuronal cultures. Neuroscience 1998, 86, 279–290. [Google Scholar] [CrossRef]
- Calabresi, P.; Gubellini, P.; Picconi, B.; Centonze, D.; Pisani, A.; Bonsi, P.; Greengard, P.; Hipskind, R.A.; Borrelli, E.; Bernardi, G. Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J. Neurosci. 2001, 21, 5110–5120. [Google Scholar] [PubMed]
- Royes, L.F.; Fighera, M.R.; Furian, A.F.; Oliveira, M.S.; da Silva, L.G.; Malfatti, C.R.; Schneider, P.H.; Braga, A.L.; Wajner, M.; Mello, C.F. Creatine protects against the convulsive behavior and lactate production elicited by the intrastriatal injection of methylmalonate. Neuroscience 2003, 118, 1079–1090. [Google Scholar]
- Fleck, J.; Ribeiro, M.C.; Schneider, C.M.; Sinhorin, V.D.; Rubin, M.A.; Mello, C.F. Intrastriatal malonate administration induces convulsive behaviour in rats. J. Inherit. Metab. Dis. 2004, 27, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Atkuri, K.R.; Cowan, T.M.; Kwan, T.; Ng, A.; Herzenberg, L.A.; Herzenberg, LA.; Enns, G.M. Inherited disorders affecting mitochondrial function are associated with glutathione deficiency and hypocitrullinemia. Proc. Natl. Acad. Sci. USA 2009, 106, 3941–3944. [Google Scholar] [CrossRef] [PubMed]
- Fontella, F.; Pulronick, V.; Gassen, E.; Wannmacher, C.M.D.; Klein, A.B.; Wajner, M.; Dutra-Filho, C.S. Propionic and l-methylmalonic acids induce oxidative stress in brain of young rats. Neuroreport 2000, 28, 541–544. [Google Scholar] [CrossRef]
- Manoli, I.; Myles, J.G.; Sloan, J.L.; Carrillo-Carrasco, N.; Morava, E.; Strauss, K.A.; Morton, H.; Venditti, C.P. A critical reappraisal of dietary practices in methylmalonic academia raises concerns about the safety of medical foods. Part 2: Cobalamin C deficiency. Genet. Med. 2016, 18, 396–404. [Google Scholar] [PubMed]
- Manoli, I.; Sysol, J.R.; Li, L.; Houillier, P.; Garone, C.; Wang, C.; Zerfas, P.M.; Cusmano-Ozog, K.; Young, S.; Trivedi, N.S.; et al. Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic academia. Proc. Natl. Acad. Sci. USA 2013, 110, 13552–13557. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Sheena, Y.; Land, J.M.; Heales, S.J. Glutathione deficiency in patients with mitochondrial disease: Implications for pathogenesis and treatment. J. Inherit. Metab. Dis. 2005, 28, 1–88. [Google Scholar] [CrossRef] [PubMed]
- Salmi, H.; Leonard, J.; Lapatto, R. Patients with organic acidaemias have an alteredthiol status. Acta Paediatr. 2012, 101, e505–e508. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Niklowitz, P.; Horster, F.; Baumgartner, E.R.; Prasad, C.; Rodenburg, R.J.; Hoffmann, T.; Menke, T.; Okun, J.G. Coenzyme Q10 is decreased in fibroblasts of patients with methylmalonic aciduria but not in mevalonic aciduria. J. Inherit. Metab. Dis. 2009, 4, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. 2003, 40, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Baumgarther, M.R.; Horster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinar-Sueiro, S.; Martinez-Fernondez, R.; Lage-Medines, S.; Aldamiz-Echevarria, L.; Vecino, E. Optic neuropathy in methylmalonic acidemia: The role of neuroprotection. J. Inherit. Metab. Dis. 2010, 3, S199–S203. [Google Scholar] [CrossRef] [PubMed]
- Williams, Z.R.; Hurley, P.E.; Altipamak, V.E.; Feldon, S.E.; Arnold, G.L.; Eggenberger, E.; Mejico, L.J. Late onset optic neuropathy in methylmalonic and propionic academia. Am. J. Ophthalmol. 2009, 147, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Leipnitz, G.; Amaral, A.U.; Fernandes, C.G.; Seminotti, B.; Zanatta, A.; Knebel, L.A.; Vargas, C.R.; Wajner, M. Pristanic acid promotes oxidative stress in brain damage in peroxisomal disorders. Brain Res. 2011, 1382, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Weller, S.; Gould, S.J.; Valle, D. Peroxisomes biogenesis disorders. Ann. Rev. Genom. Hum. Genet. 2003, 4, 165–211. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.M.; Sumpter, R.; Zou, Z.; Sirasanagandia, S.; Wei, Y.; Mishra, P.; Rosewich, H.; Crane, D.I.; Levine, B. Peroxisomal protein PEX13 functions in selective autophagy. EMBO Rep. 2017, 18, 48–60. [Google Scholar] [CrossRef] [PubMed]
- White, A.L.; Modaff, P.; Holland-Morris, P.; Pauli, P.M. Natural history of rhizomelic chondrodysplasia punctate. Am. J. Med. Genet. 2003, 118, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Aubourg, P.; Pujol, A. General aspects and neuropathology of X-linked adrenoleukodystrophy. Brain Pathol. 2010, 20, 817–830. [Google Scholar] [CrossRef] [PubMed]
- Moser, H.; Smith, K.D.; Watkins, P.A.; Powers, J.; Moser, A.B. X-linked adrenoleukodystrophy. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C., Ed.; McGraw-Hill: New-York, NY, USA, 2001; Volume 2, pp. 3257–3301. [Google Scholar]
- Powers, J.M.; DeCiero, D.P.; Ito, M.; Moser, A.B.; Moser, H.W. Adrenomyeloneuropathy: A neuropathologic review featuring its noninflammatory myelopathy. J. Neuropathol. Exp. Neurol. 2000, 59, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; van Roermund, W.T.; Shutgens, R.B.H.; Barth, P.G.; Heymans, H.S.A.; van den Bosch, H.; Tager, J.M. The inborn errors of peroxisomal beta-oxidation. A review. J. Inher. Metab. Dis. 1990, 13, 4–36. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H. Beta oxidation of fatty acids. Biochim. Biophys. Acta 1991, 1081, 109–120. [Google Scholar] [CrossRef]
- Poirier, Y.; Antonenkov, V.D.; Glumoff, T.; Hiltunen, J.K. Peroxisomal beta-oxidation- a metabolic pathway with multiple functions. Biochim. Biophys. Acta 2006, 1763, 1413–1426. [Google Scholar] [CrossRef] [PubMed]
- Angermuller, S.; Bruder, G.; Volkl, A.; Wesch, H.; Fahimi, H.D. Localization of xanthine oxidase in crystalline cores of peroxisomes. A cytochemical and biochemical study. Eur. J. Cell Biol. 1987, 45, 137–144. [Google Scholar] [PubMed]
- Lismont, C.; Nordgen, M.; van Veldhoven, P.P.; Fransen, M. Redox interplay between mitochondria and peroxisomes. Front. Cell Dev. Biol. 2015, 3, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Vargas, C.R.; Wajner, M.; Sirtori, L.R.; Goulart, L.; Chiochetta, M.; Coelho, D.; Latini, A.; Llesuy, S.; Bello-Klein, A.; Giugliani, R.; et al. Evidence that oxidative stress is increased in patients with X-linked adrenoleukodystrophy. Biochim. Biophys. Acta 2004, 1688, 26–32. [Google Scholar] [CrossRef] [PubMed]
- El-Bassyouni, H.T.; Abel Maksoud, S.A.; Salem, F.A.; Badr El-Deen, R.; Abdel Aziz, H.; Thomas, M.M. Evidence of oxidative stress in peroxisomal disorders. Singap. Med. J. 2012, 53, 608. [Google Scholar]
- Schrader, M.; Fahimi, H.D. Mammalian peroxisomes and reactive oxygen species. Histochem. Cell Biol. 2004, 122, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Fransen, M.; Nordgren, M.; Wang, B.; Apanasets, O. Role of peroxisomes in ROS/RNS-metabolism: Implications for human disease. Biochim. Biophys. Acta 2012, 1822, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, C.; Imamure, A.; Hashiguchi, V.; Shimozawa, N.; Suzuki, Y.; Kondo, N.; Imanaka, T.; Tsukamoto, T.; Osumi, T. Catalase-less Peroxisomes: Implication in the milder forms of peroxisome biogenesis disorder. J. Biol. Chem. 2000, 275, 37271–37277. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, E.; Vanhorebeek, I.; Grabenbauer, M.; Borgers, M.; Declercq, P.E.; Fahimi, H.D.; Baes, M. Mitochondrial alterations caused by defective peroxisomal biogenesis in a mouse model of Zellweger syndrome (PEX5 knock out mouse). Am. J. Pathol. 2001, 159, 1477–1494. [Google Scholar] [CrossRef]
- Schrakamp, G.; Schalkwijk, C.G.; Schutgens, R.B.; Wanders, R.J.; Tager, J.M.; van den Bosch, H. Plasmalogen biosynthesis in peroxisomal disorders: Fatty alcohol versus alkylglycerol precursors. J. Lipid Res. 1988, 29, 325–334. [Google Scholar] [PubMed]
- Wood, C.S.; Koepke, J.I.; Teng, H.; Boucher, K.K.; Katz, S.; Chang, P.; Terlecky, L.J.; Papanayotou, I.; Walton, P.A.; Terlecky, S.R. Hypocatalasemic fibroblasts accumulate hydrogen peroxide and display age-associated pathologies. Traffic 2006, 7, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Schutgens, R.B.; Barth, P.G. Peroxisomal disorders: A review. J. Neuropathol. Exp. Neurol. 1995, 54, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Erauskin, J.; Galino, J.; Ruiz, M.; Cuezva, J.M.; Fabregat, I.; Cacabelos, D.; Boada, J.; Martinez, J.; Ferrer, I.; Pamplona, R.; et al. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2013, 22, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.K.; Moser, H.; Kishimoto, Y.; Hamilton, J.A. Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J. Clin. Investig. 1995, 96, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Sarnat, H.B.; Machin, G.; Darwish, H.Z.; Rubin, S.Z. Mitochondrial myopathy of cerebrohepato-renal (Zellweger) syndrome. Can. J. Neurol. Sci. 1983, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Muller-Hocker, J.; Walther, J.R.; Bise, K.; Pongratz, D.; Hubner, G. Mitochondrial myopathy with loosely coupled oxidative phosphorylation in a case of Zellweger syndrome. Virchows Arch. B Cell Pathol. Zell-Pathol. 1984, 45, 125–138. [Google Scholar] [CrossRef]
- Wolff, J.; Nyhanf, W.L.; Powell, H.; Takahashi, D.; Hutzler, J.; Hajra, A.K.; Datta, N.S.; Singh, I.; Moser, H.W. Myopathy in an infant with a fatal peroxisomal disorder. Pediatr. Neurol. 1986, 2, 141. [Google Scholar] [CrossRef]
- Powers, J.M.; Pei, Z.; Heinzer, A.K.; Deering, R.; Moser, A.B.; Moser, H.W.; Watkins, P.A.; Smith, K.D. Adreno-leukodystrophy: Oxidative stress of mice and men. J. Neuropathol. Exp. Neurol. 2005, 64, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, S.; Ruiz, M.; Guilera, C.; Hahnen, E.; Brichta, L.; Naudi, A.; Portero-Otin, M.; Dacremont, G.; Cartier, N.; Wanders, R.; et al. Valproic acid induces antioxidant effects in X-linked adrenoleukodystrophy. Hum. Mol. Genet. 2010, 19, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Phadke, R.; Saggar, A.; Hargreaves, I.P.; Yates, R.; Fokoloros, C.; Mankad, K.; Hertecant, J.; Ruggieri, M.; McCormick, D.; et al. Zellweger syndrome and secondary mitochondrial myopathy. Eur. J. Pediatr. 2015, 174, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Fourcade, S.; Lopez-Erauskin, J.; Galino, J.; Duval, C.; Naudi, A.; Jove, M.; Kemp, S.; Villarroya, F.; Ferrer, I.; Pamplona, R.; et al. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum. Mol. Genet. 2008, 17, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Erauskin, J.; Galino, J.; Bianchi, P.; Fourcade, S.; Andreu, A.L.; Ferrer, I.; Munoz-Pinedo, C.; Pujol, A. Oxidative stress modulates mitochondrial failure and cyclophilin D function in X-linked adrenoleukodystrophy. Brain 2012, 135, 3584–3598. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; Pujol, A. Pathomechanisms underlying X-adrenoleukodystrophy: A three-hit hypothesis. Brain Pathol. 2010, 20, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Galea, E.; Launay, N.; Portero-Otin, M.; Ruiz, M.; Pamplona, R.; Aubourg, P.; Ferrer, I.; Pujol, A. Oxidative stress underlying axonal degeneration in adrenoleukodystrophy: A paradigm for multifactorial neurodegenerative diseases? Biochim. Biophys. Acta 2012, 9, 1475–1488. [Google Scholar] [CrossRef] [PubMed]
- Galino, J.; Ruiz, M.; Fourcade, S.; Schluter, A.; Lopez-Erauskin, J.; Guilera, C.; Jove, M.; Naudi, A.; Garcia-Arumi, E.; Andreu, A.L.; et al. Oxidative damage compromises energy metabolism in the axonal degeneration mouse model of X-adrenoleukodystrophy. Antioxid. Redox Signal. 2011, 15, 2095–2107. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Erauskin, J.; Fourcade, S.; Galino, J.; Ruiz, M.; Schluter, A.; Naudi, A.; Jove, M.; Portero-Otin, M.; Pamplona, R.; Ferrer, I.; et al. Antioxidants halt axonal degeneration in a mouse model of X-adrenoleukodystrophy. Ann. Neurol. 2011, 70, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, D.P.; Donida, B.; Deon, M.; Jacques, C.E.; Jardim, L.B.; Vargas, C.R. In vitro effects of N-acetyl-l-cysteine on glutathione and sulfhryl levels in X-linked adrenoleukodystrophy patients. Clin. Biomed. Res. 2017, 37, 33–37. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Finckh, B.; de Hingh, Y.C.; Stroomer, L.E.; Denis, S.; Kohlschutter, A.; Wanders, R.J. Evidence for increased oxidative stress in peroxisomal d-bifunctional protein deficiency. Mol. Genet. Metab. 2003, 79, 281–287. [Google Scholar] [CrossRef]
- Huang, J.; Liu, X.; Tang, L.L.; Long, J.T.; Zhu, J.; Hua, R.X.; Li, J. XPG gene polymorphisms and cancer susceptibility: Evidence from 47 studies. Oncotarget 2017. [CrossRef] [PubMed]
- Carre, G.; Marelli, C.; Anheim, M.; Geny, C.; Renaud, M.; Rezvani, H.R.; Koenig, M.; Guissart, C.; Tranchant, C. Xeroderma pigmentosum complementation group F: A rare cause of cerebellar ataxia with chorea. J. Neurol. Sci. 2017, 376, 198–201. [Google Scholar]
- Niedernhofer, L.J.; Bohr, V.A.; Sander, M.; Kraemer, K.H. Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: Molecules to patients. Mech. Ageing Dev. 2011, 132, 340–347. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on xeroderma pigmentosum. J. Investig. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.E.; Hanke, C.W.; Michalak, J.A. The molecular basis of xeroderma pigmentosum. Dermatol. Surg. 1997, 23, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Choi, B.; Tang, M.S. Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts. Proc. Natl. Acad. Sci. USA 2010, 107, 12180–12185. [Google Scholar] [CrossRef] [PubMed]
- Pascucci, B.; D’errico, M.; Parlanti, E.; Giovannini, S.; Dogliotti, E. Role of nucleotide excision repair proteins in oxidative DNA damage repair: An updating. Biochemistry 2011, 76, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Itoh, M.; Araki, S.; Kumada, S.; Shioda, K.; Tamagawa, K.; Mizutani, T.; Morimatsu, Y.; Minagawa, M.; Oda, M. Oxidative stress and disturbed glutamate transport in hereditary nucleotide repair disorders. J. Neuropathol. Exp. Neurol. 2001, 60, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Enokido, Y.; Inamura, N.; Yoshino, M.; Nakatsu, Y.; van der Horst, G.T.; Hoeijmakers, J.H.; Tanaka, K.; Hatanaka, H. Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne syndrome Group B DNA repair genes. Proc. Natl. Acad. Sci. USA 2001, 98, 13379–13384. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. The 8,5′-cyclopurine-2′-deoxynucleosides: Candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair 2008, 7, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Ahmad, S.I.; Hanaoka, F. Roles of oxidative stress in xeroderma pigmentosum. Adv. Exp. Med. Biol. 2008, 637, 120–127. [Google Scholar] [PubMed]
- Melis, J.P.; van Steeg, H.; Luijten, M. Oxidative DNA damage and nucleotide excision repair. Antioxid. Redox Signal. 2013, 18, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bales, E.S.; Peterson, C.A.; Legerski, R.J. Characterization of molecular defects in xeroderma pigmentosum group C. Nat. Genet. 1993, 5, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, H.R.; Rossignol, R.; Ali, N.; Bernard, G.; Tang, X.; Yang, H.S.; Jouary, T.; de Verneuil, H.; Taieb, A.; Kim, A.L.; et al. XPC silencing in normal human keratinocytes triggers metabolic alterations through NOX-1 activation-mediated reactive oxygen species. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Mahfouf, W.; Serrano-Sanchez, M.; Raad, H.; Harfouche, G.; Bonneu, M.; Claverol, S.; Mazurier, F.; Rossignol, R.; Taieb, A.; et al. Premature Skin Aging Features Rescued by Inhibition of NADPH Oxidase Activity in XPC-Deficient Mice. J. Investig. Dermatol. 2015, 135, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; Sengupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD(+)/SIRT1 Reduction. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease- links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Reza Rezvani, H.; Taieb, A. Le xeroderma pigmentosum. M/S Med. Sci. 2011, 27, 467. [Google Scholar]
- Liu, J.; Fang, H.; Chi, Z.; Wu, Z.; Wei, D.; Mo, D.; Niu, K.; Balajee, A.S.; Hei, T.K.; Nie, L.; et al. XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucl. Acids Res. 43. [CrossRef] [PubMed]
- Mori, M.P.; Costa, R.P.; Soltys, D.T.; Freire, T.D.S.; Rossato, F.A.; Amigo, I.; Kowaltowski, A.J.; Vercesi, A.E.; de Souza-Pinto, N.C. Lack of XPC leads to a a shift between respiratory complexes I and II but sensitizes cells to mitochondrial stress. Sci. Rep. 2017, 7, 155. [Google Scholar] [CrossRef] [PubMed]
- Rothe, M.; Werner, D.; Thielmann, H.W. Enhanced expression of mitochondrial genes in xeroderma pigmentosum fibroblast strains from various complementation groups. J. Cancer Res. Clin. Oncol. 1993, 119, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Otley, C.C. Skin cancer in organ transplant recipients: Epidemiology, pathogenesis and management. J. Am. Acad. Dermatol. 2002, 47, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organisation, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Quillet, X.; Chevallier-Lagente, O.; Zeng, L.; Calvayrac, R.; Mezzina, M.; Sarasin, A.; Vuillaume, M. Retroviral-mediated correction of DNA repair defect in xeroderma pigmentosa cells is associated with recovery of catalase activity. Mutat. Res. DNA Repair 1997, 385, 235–242. [Google Scholar] [CrossRef]
- Taneka, J.; Nagai, T.; Okada, S. Serum concentration of coenzyme Q in xeroderma pigmentosum. Rinsho Shinkeigaku Clin. Neurol. 1998, 38, 57–59. [Google Scholar]
- Goncalves-Maia, M.; Magnaldo, T. Genetic therapy of xeroderma pigmentosa, analysis of strategies and translation. Exp. Opin. Orphan Drugs 2017, 5, 5–17. [Google Scholar] [CrossRef]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Chertow, G.M.; Burdick, E.; Honour, M.; Bonventre, J.V.; Bates, D.W. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J. Am. Soc. Nephr. 2005, 16, 3365–3370. [Google Scholar] [CrossRef] [PubMed]
- Uchino, S.; Kellum, J.A.; Bellomo, R.; Doig, G.S.; Morimatsu, H.; Morgera, S.; Achetz, M.; Tan, I.; Bouman, C.; Macedo, E.; et al. Acute renal failure in critically ill patients: A multinational, multicenter study. JAMA 2005, 294, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Bagshaw, S.M.; Uchino, S.; Bellomo, R.; Morimatsu, H.; Morgera, S.; Schetz, M.; Tan, I.; Bouman, C.; Macedo, E.; Gibney, N.; et al. Septic acute kidney injury in critically ill patients: Clinical characteristics and outcomes. Clin. J. Am. Soc. Nephrol. 2007, 2, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Keir, I.; Kellum, J.A. Acute kidney injury in severe sepsis: Pathophysiology, diagnosis, and treatment recommendations. J. Veter. Emerg. Crit. Care 2015, 25, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E.; Singer, M. Mechanisms of sepsis-induced organ dysfunction. Crit. Care Med. 2007, 35, 2408–2416. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.S.; Mannino, D.M.; Eaton, S.; Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2000, 348, 1546–1554. [Google Scholar] [CrossRef] [PubMed]
- Weycker, D.; Akhras, K.S.; Edelsberg, J.; Angus, D.C.; Oster, G. Long-term mortality and medical care charges in patients with severe sepsis. Crit. Care Med. 2003, 31, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.C.; Girard, T.D.; Gordon, S.M.; Thompson, J.L.; Shintani, A.K.; Thomason, J.W.W.; Pun, B.T.; Canonico, A.E.; Dunn, J.G.; Bernard, G.R.; et al. Long-term cognitive and psychological outcomes in the awakening and breathing controlled trial. Am. J. Respir. Crit. Care Med. 2010, 182, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA 2010, 304, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Trumbeckaite, S.; Opalka, J.R.; Neuhof, C.; Zierz, S.; Gellerich, F.N. Different sensitivity of a rabbit heart and skeletal muscle to endotoxin-induced impairment of mitochondrial function. Eur. J. Biochem. 2001, 268, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Belcher, E.; Mitchell, J.; Evans, T. Myocardial dysfunction in sepsis: No role for NO. Heart 2000, 87, 507–509. [Google Scholar] [CrossRef]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Landes Biosci. 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Quoilin, C.; Mouithys-Mickalad, A.; Lecart, S.; Fontaine-Aupart, M.P.; Hoebeke, M. Evidence of oxidative stress and mitochondrial respiratory chain dysfunction in an in vitro model of sepsis-induced kidney injury. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Erusalimsky, J.D. Does nitric oxide modulate mitochondrial energy generation and apoptosis? Nat. Rev. Mol. Cell Biol. 2002, 3, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Adrie, C.; Bachelet, M.; Vayssier-Taussat, M.; Russo-Marie, F.; Bouchaert, I.; Adib-Conquy, M.; Cavaillon, J.M.; Pinski, M.R.; Dhainaut, J.F.; Polla, B.S. Mitochondrial membrane potential and apoptosis in peripheral blood monocytes in severe human sepsis. Am. J. Respir. Crit. Care Med. 2001, 164, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Gellerich, F.N.; Trumbeckaite, S.; Opalka, J.R.; Gellerich, J.F.; Chen, Y.; Zierz, S.; Werdan, K.; Neuhof, C.; Redl, H. Mitochondrial dysfunction in sepsis: Evidence from bacteraemic baboons and endotoxaemic rabbits. Biosci. Rep. 2002, 22, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Sener, G.; Toklu, H.; Ercan, F.; Erkanli, G. Protective effect of β-glucan against oxidative organ injury in a rat model of sepsis. Int. Immunopharmacol. 2005, 5, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Lee, S.-M. Vitamins C and E protect hepatic cytochrome P450 dysfunction induced by polymicrobial sepsis. Eur. J. Pharmacol. 2006, 534, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Baines, M.; Perry, S.E.; McLaughlin, P.J.; Carson, J.; Wenstone, R.; Shenkin, A. Effect of selenium supplementation on biochemical markers and outcome in critically ill patients. Clin. Nutr. 2007, 26, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Angstwurm, M.W.; Engelman, L.; Zimmermann, T.; Lehmann, C.; Spes, C.H.; Abel, P.; Strau, R.; Meier-Hellmann, A.; Insel, R.; Radke, J.; et al. Selenium in intensive care (SIC): Results of a prospective randomized, placebo-controlled, multiple-centre study in patients with severe systemic inflammatory response syndrome, sepsis and septic shock. Crit. Care Med. 2007, 35, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F. Oxidative stress and mitochondrial dysfunction in sepsis. Br. J. Anaesth. 2011, 107, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Cassina, A.; Hodara, R.; Quijano, C.; Castro, L. Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med. 2002, 33, 1451–1464. [Google Scholar] [CrossRef]
- Laganiere, S.; Yu, B.P. Modulation of membrane phospholipid fatty acid composition and food restriction. Gerontology 1993, 39, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Hamasaki, N. Mitochondrial oxidative stress and mitochondrial DNA. Clin. Chem. Lab. Med. 2003, 41, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P.; Duncan, A.J.; Wu, L.; Agrawal, A.; Land, J.M.; Heales, S.J. Inhibition of mitochondrial complex IV leads to secondary loss complex II–III activity: Implications for the pathogenesis and treatment of mitochondrial encephalomyopathies. Mitochondrion 2007, 7, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Gasparovic, A.C. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.J.; Heales, S.J.; Mills, K.; Eaton, S.; Land, J.M.; Hargreaves, I.P. Determination of coenzyme Q10 status in blood mononuclear cells, skeletal muscle and plasma by HPLC with a di-propoxy-coenzyme Q10 as an internal standard. Clin. Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Enns, G.M.; Cowan, T.M. Glutathione as a Redox Biomarker in Mitochondrial Disease-Implications for Therapy. J. Clin. Med. 2017, 6, 50. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsma, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Thoolen, M. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Homstrom, K.H.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Abramov, A.Y. NrF2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepien, K.M.; Heaton, R.; Rankin, S.; Murphy, A.; Bentley, J.; Sexton, D.; Hargreaves, I.P. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J. Clin. Med. 2017, 6, 71. https://doi.org/10.3390/jcm6070071
Stepien KM, Heaton R, Rankin S, Murphy A, Bentley J, Sexton D, Hargreaves IP. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. Journal of Clinical Medicine. 2017; 6(7):71. https://doi.org/10.3390/jcm6070071
Chicago/Turabian StyleStepien, Karolina M., Robert Heaton, Scott Rankin, Alex Murphy, James Bentley, Darren Sexton, and Iain P. Hargreaves. 2017. "Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders" Journal of Clinical Medicine 6, no. 7: 71. https://doi.org/10.3390/jcm6070071