
Acute Kidney Injury in Heart Failure Revisited—The Ameliorating Impact of “Decongestive Diuresis” on Renal Dysfunction in Type 1 Acute Cardiorenal Syndrome: Accelerated Rising Pro B Naturetic Peptide Is a Predictor of Good Renal Prognosis

 ,
,
Abstract
:1. Introduction
Heart Failure, Renal Failure and the Role of Renal Venous Hypertension in Cardiorenal Syndrome: A Review of the Current Literature
2. Case Reports
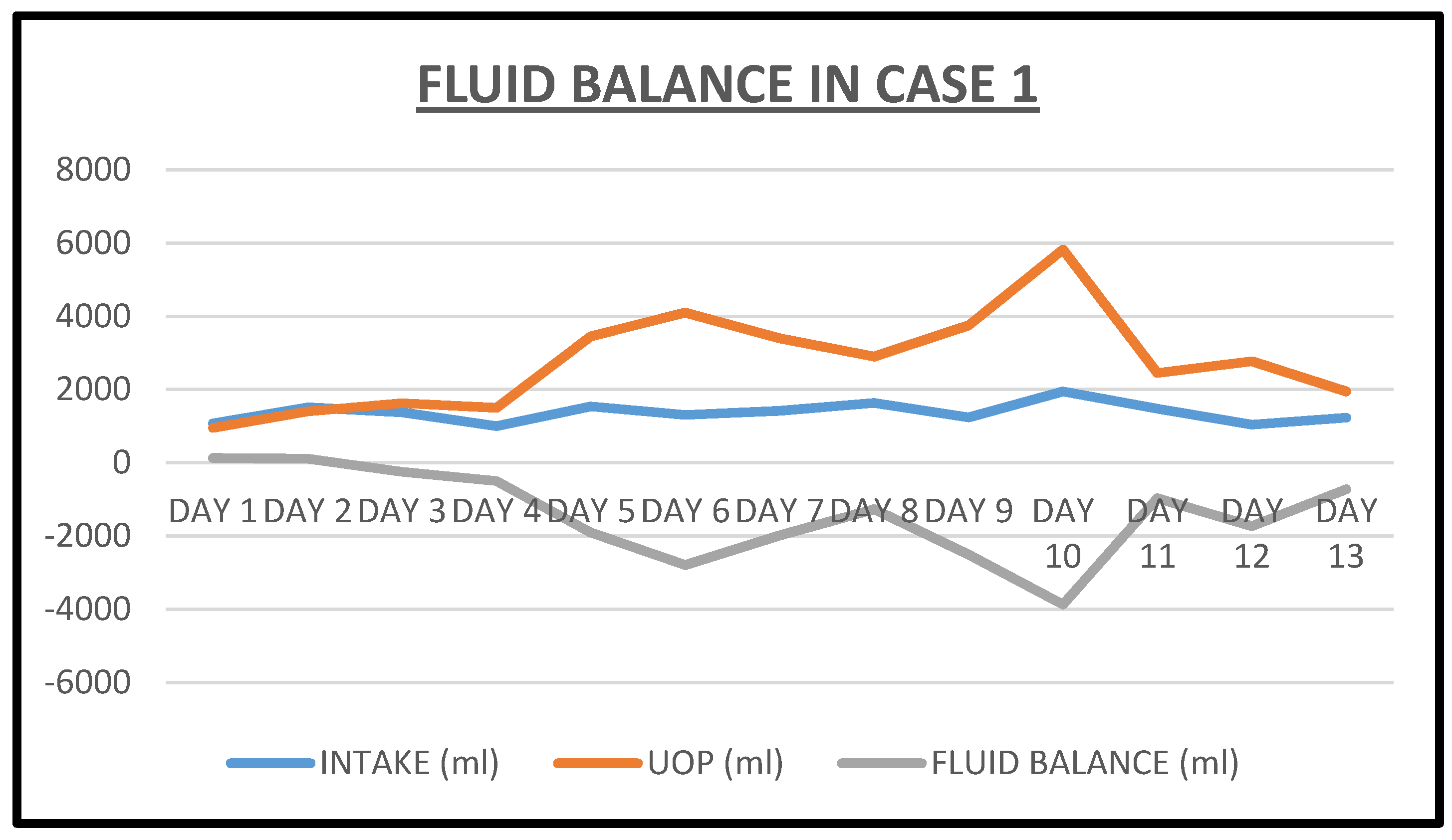
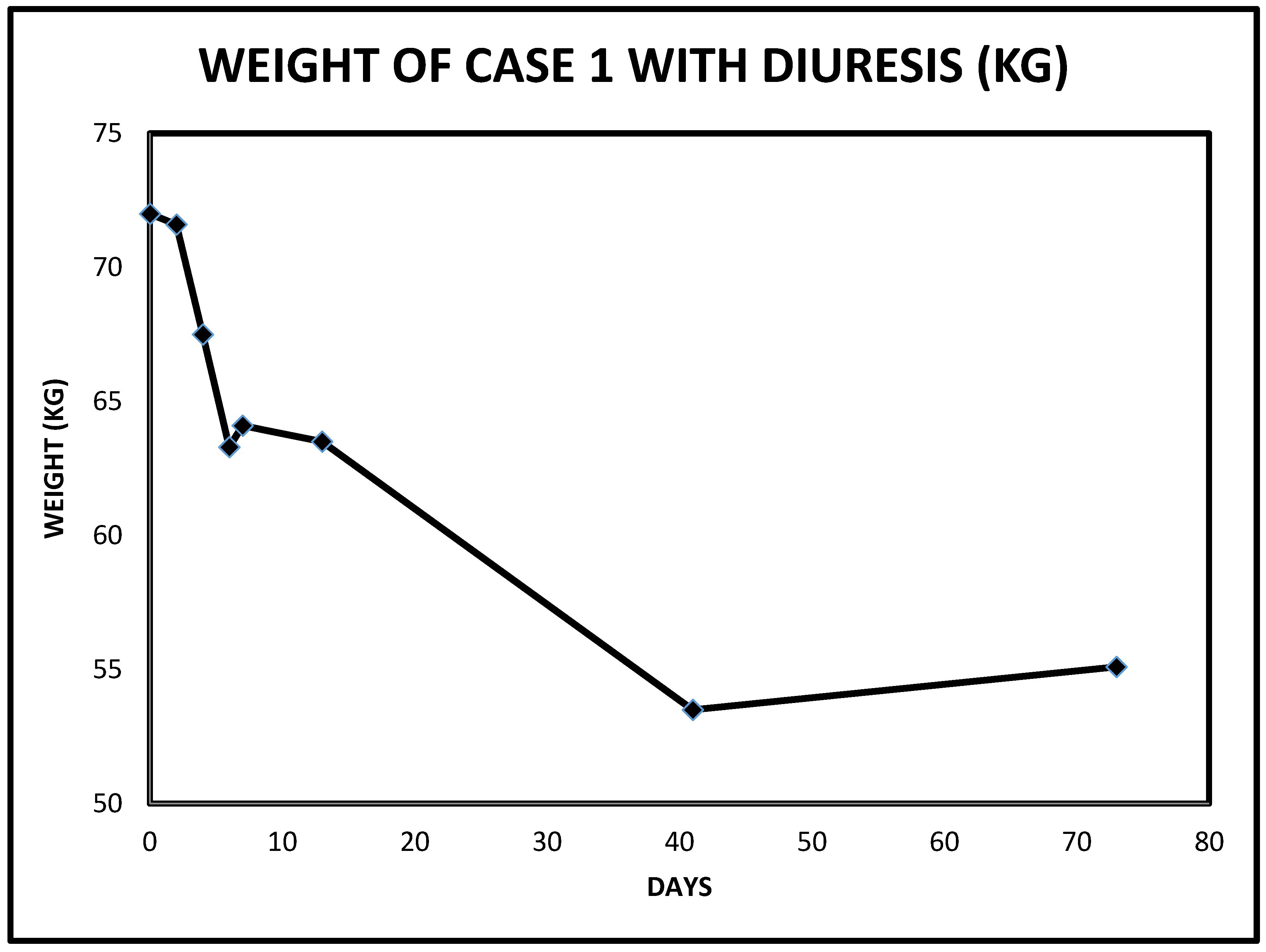
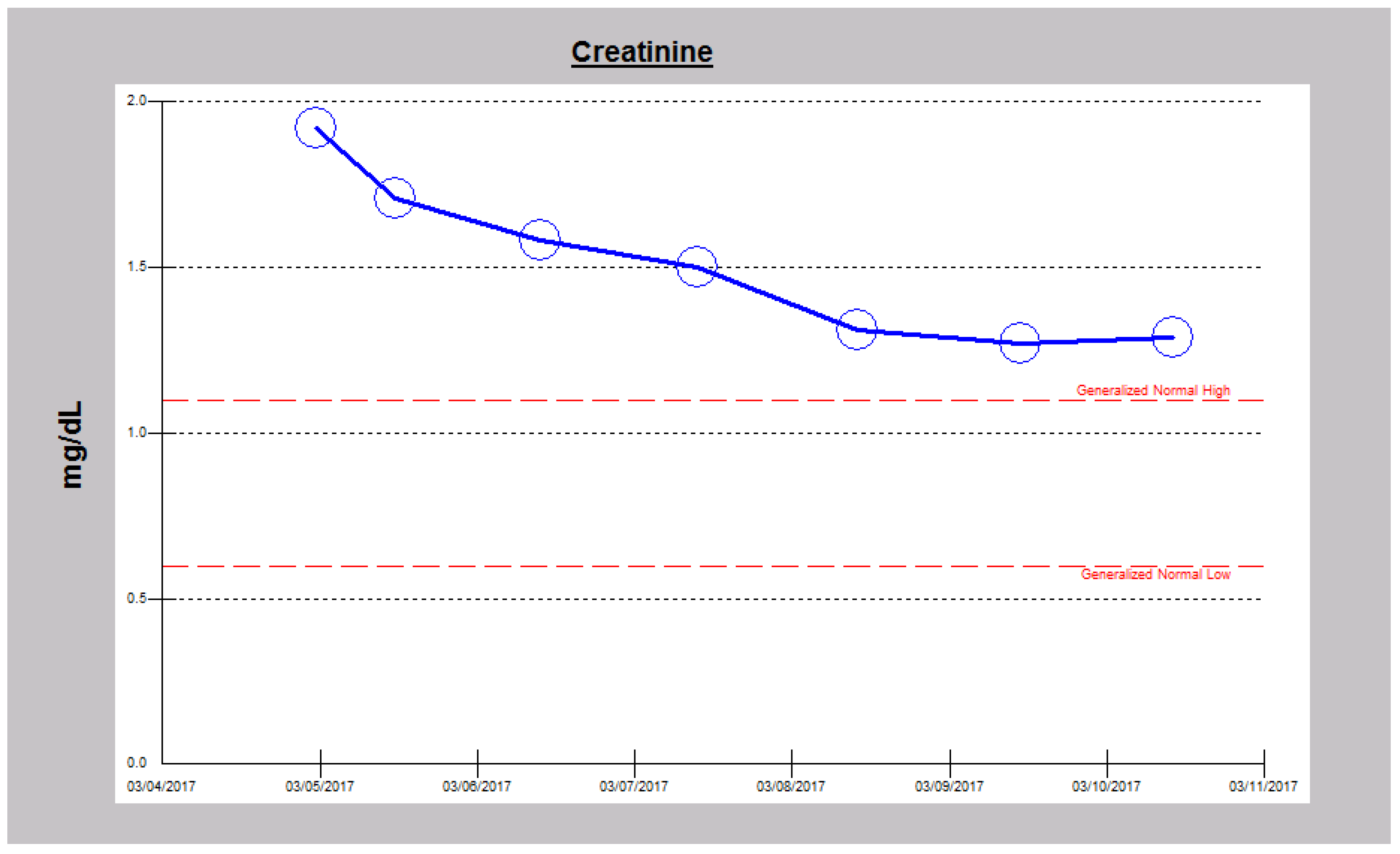
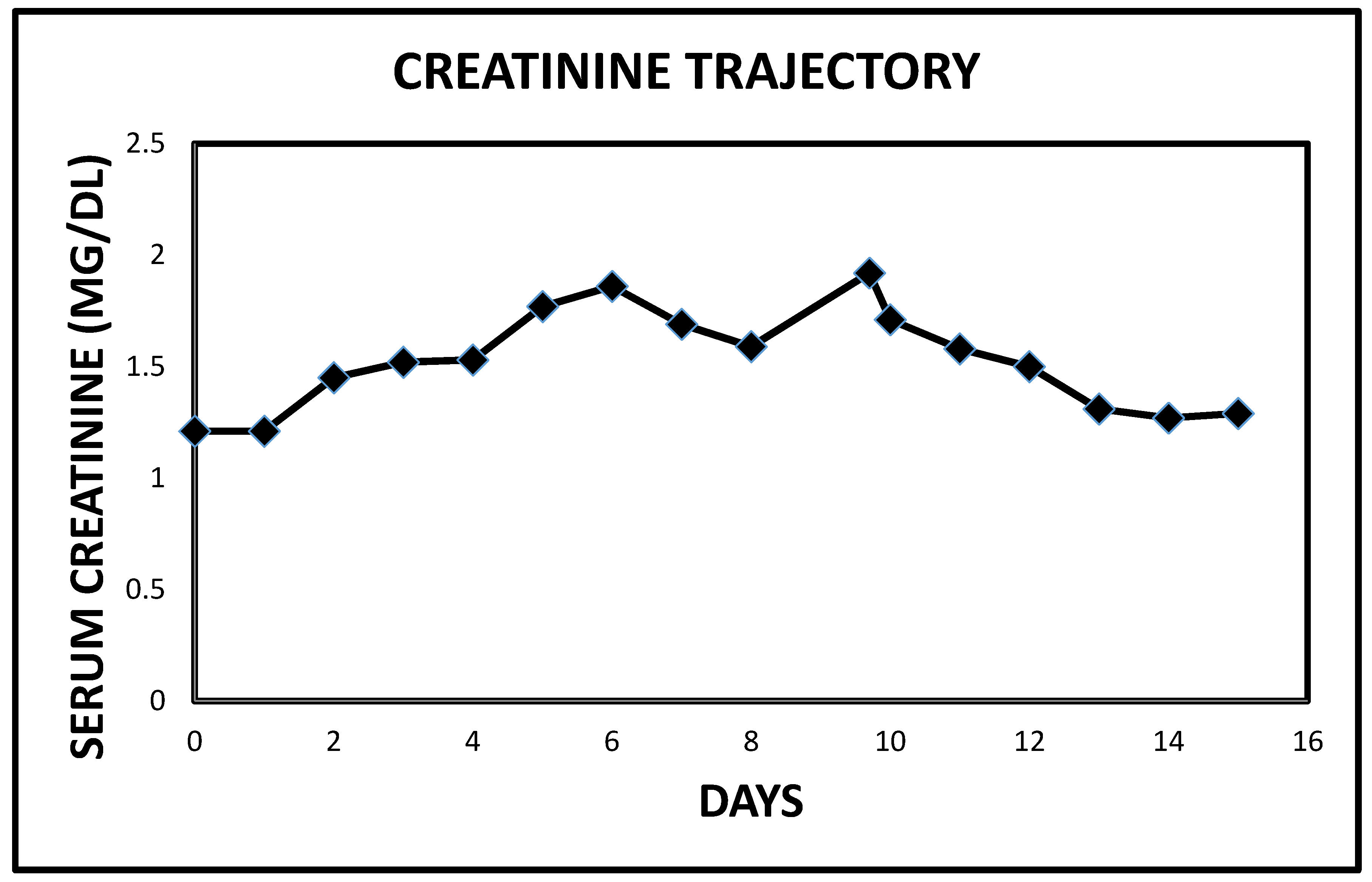
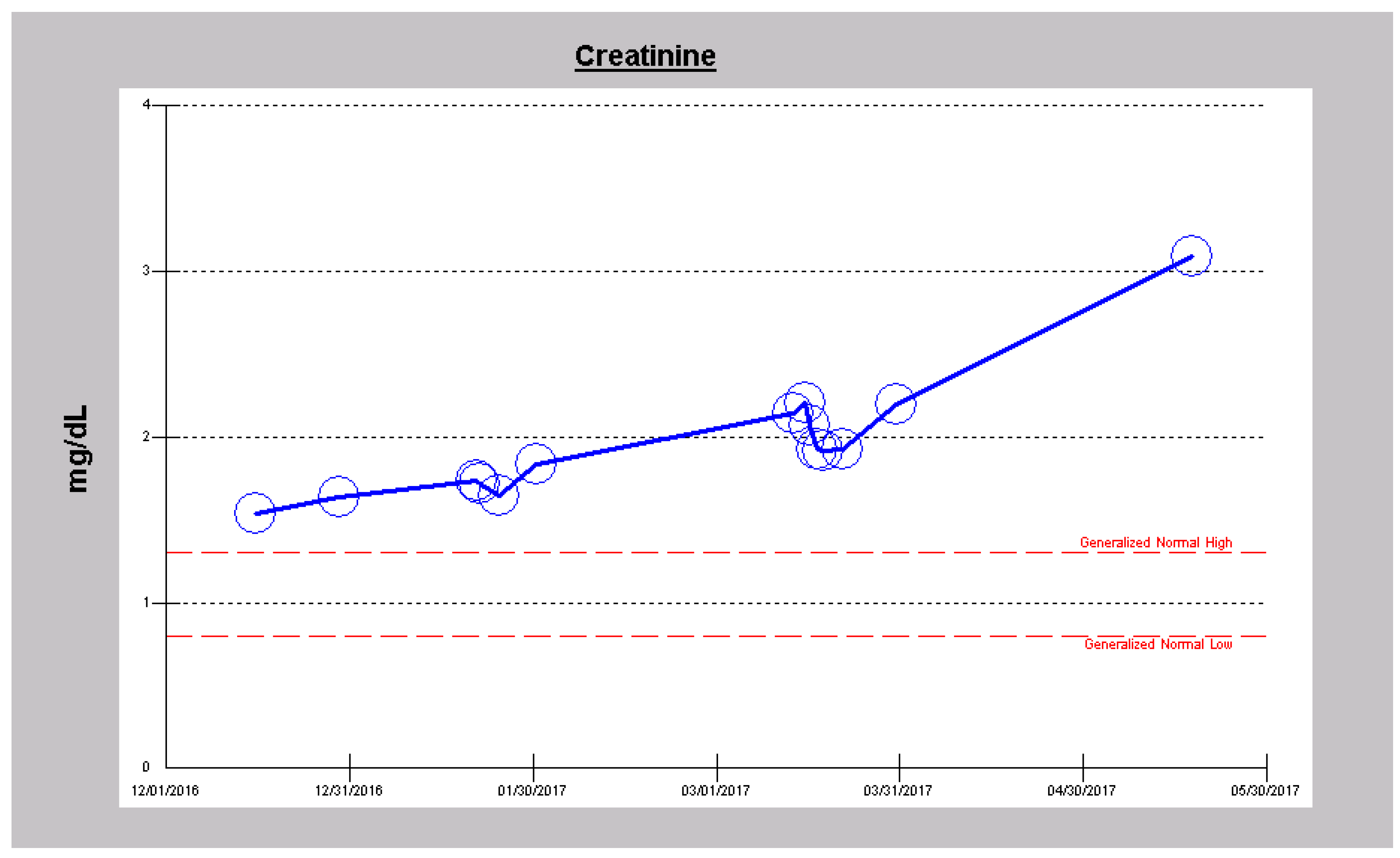
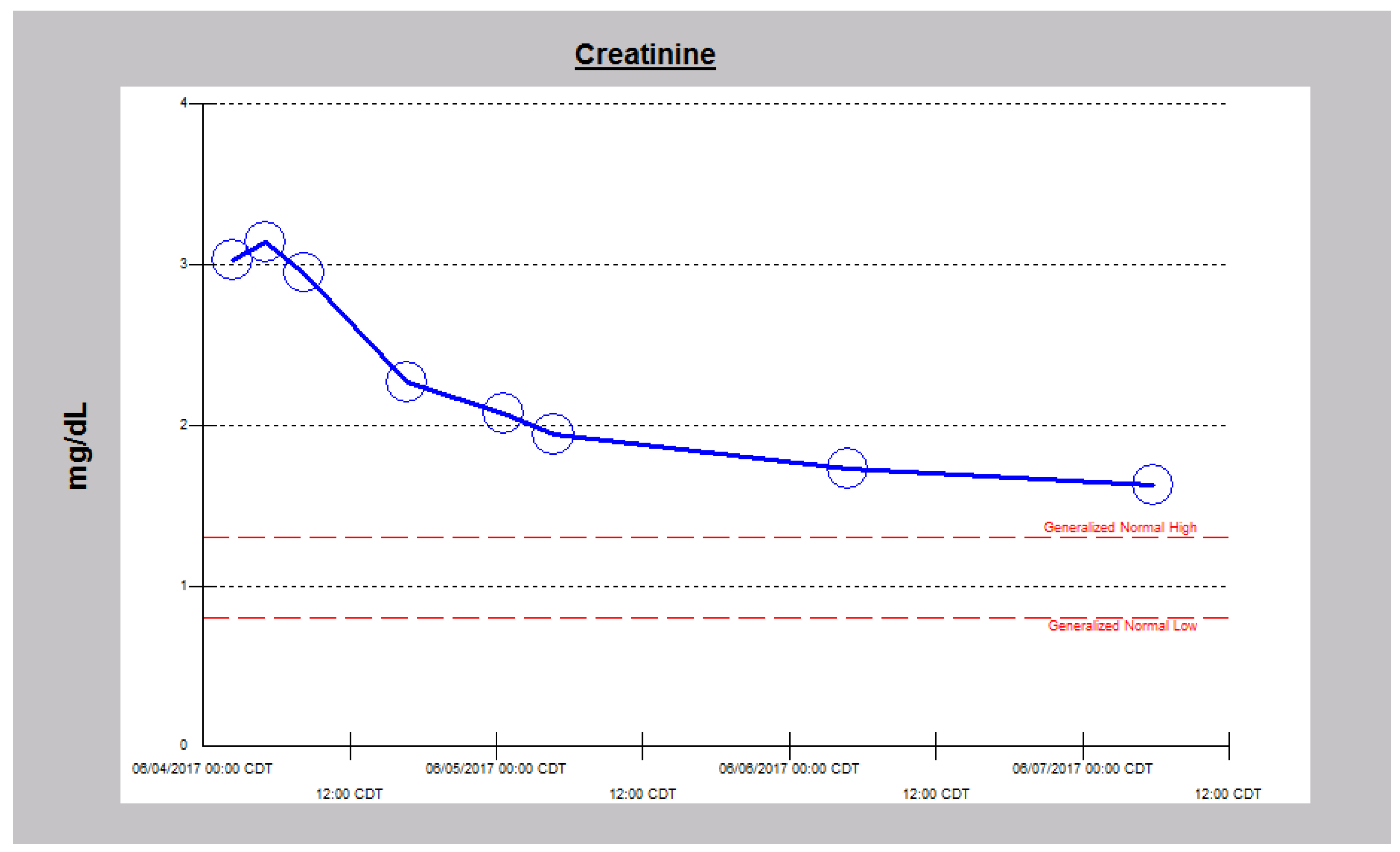
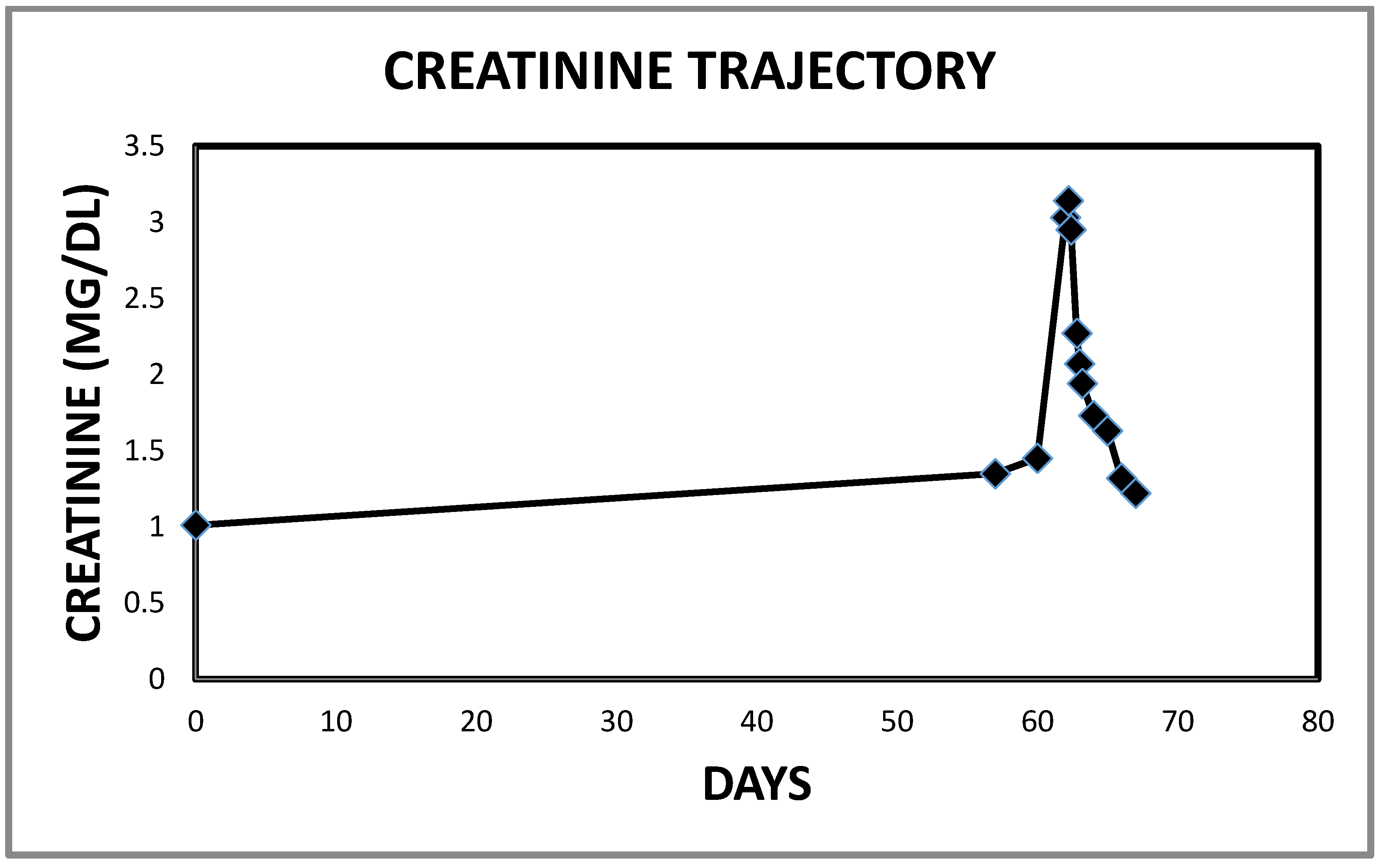
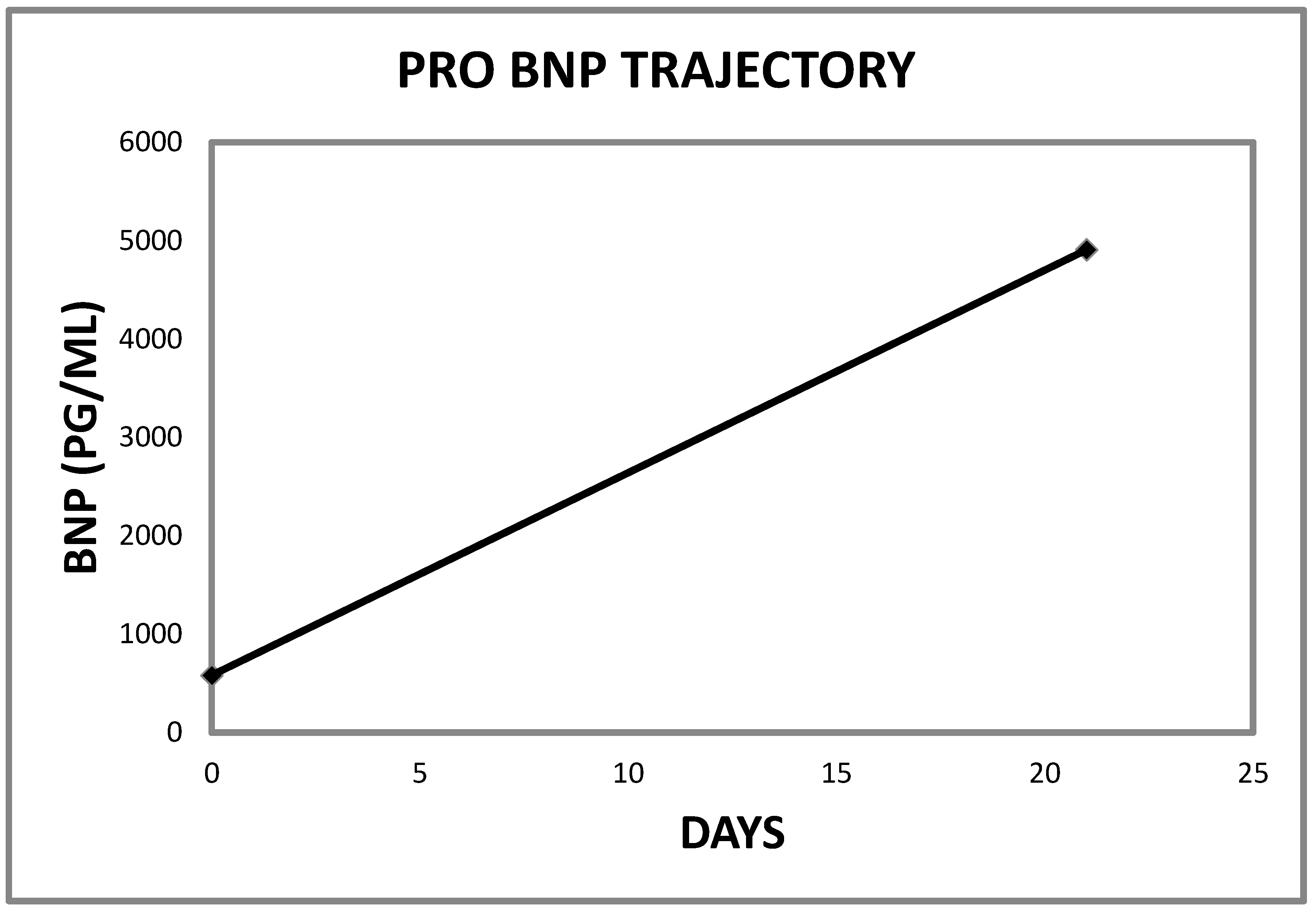
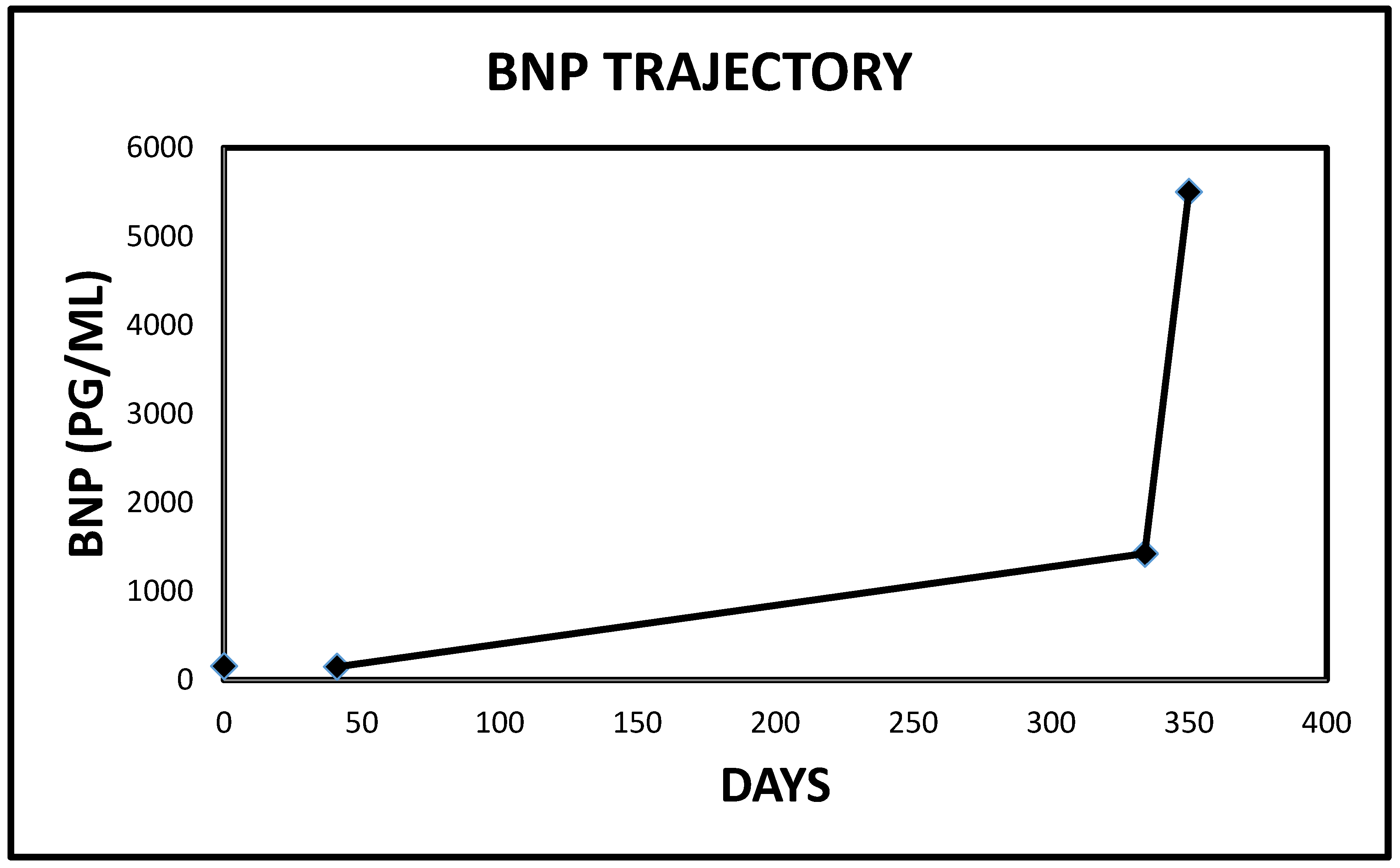
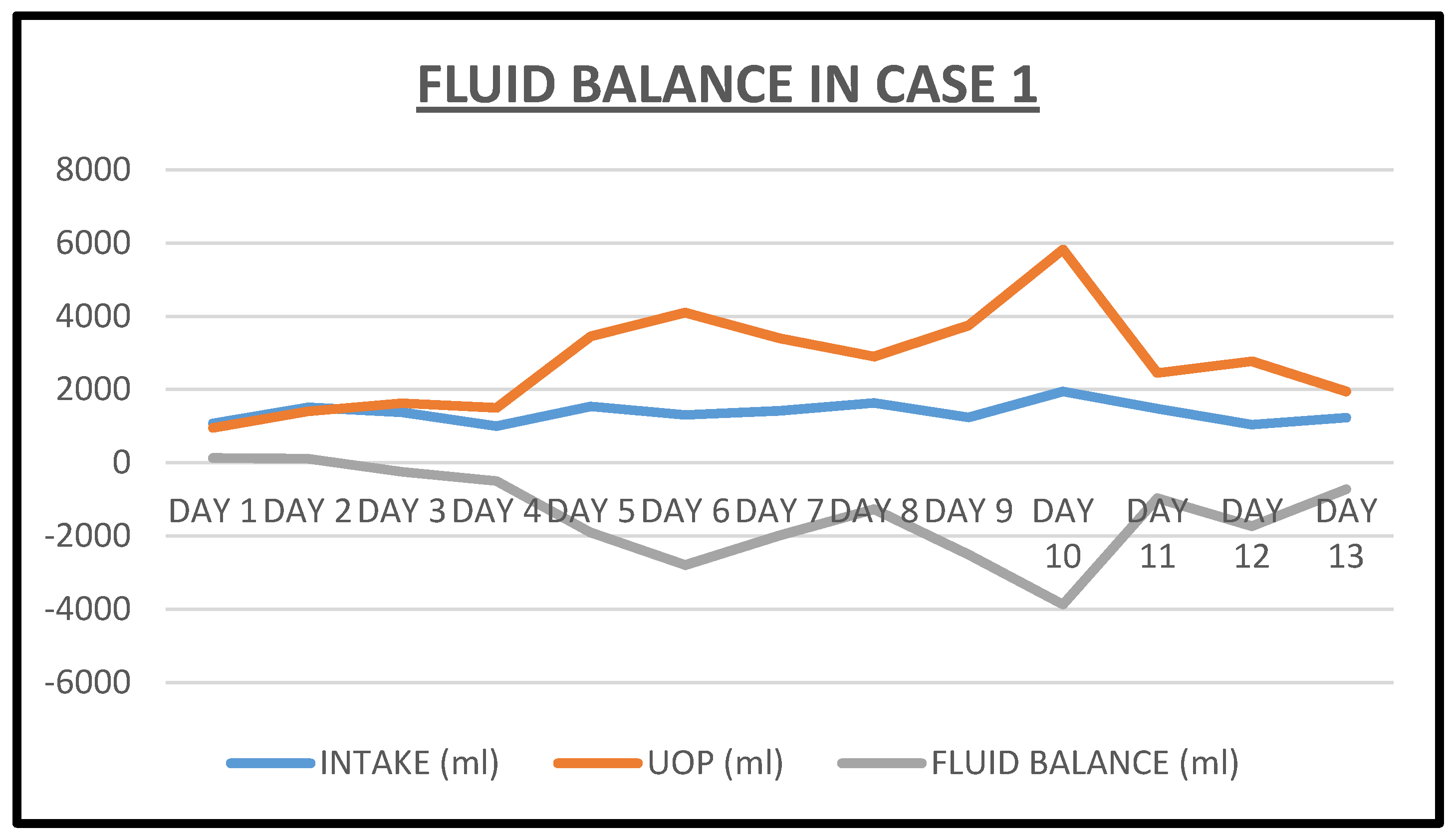
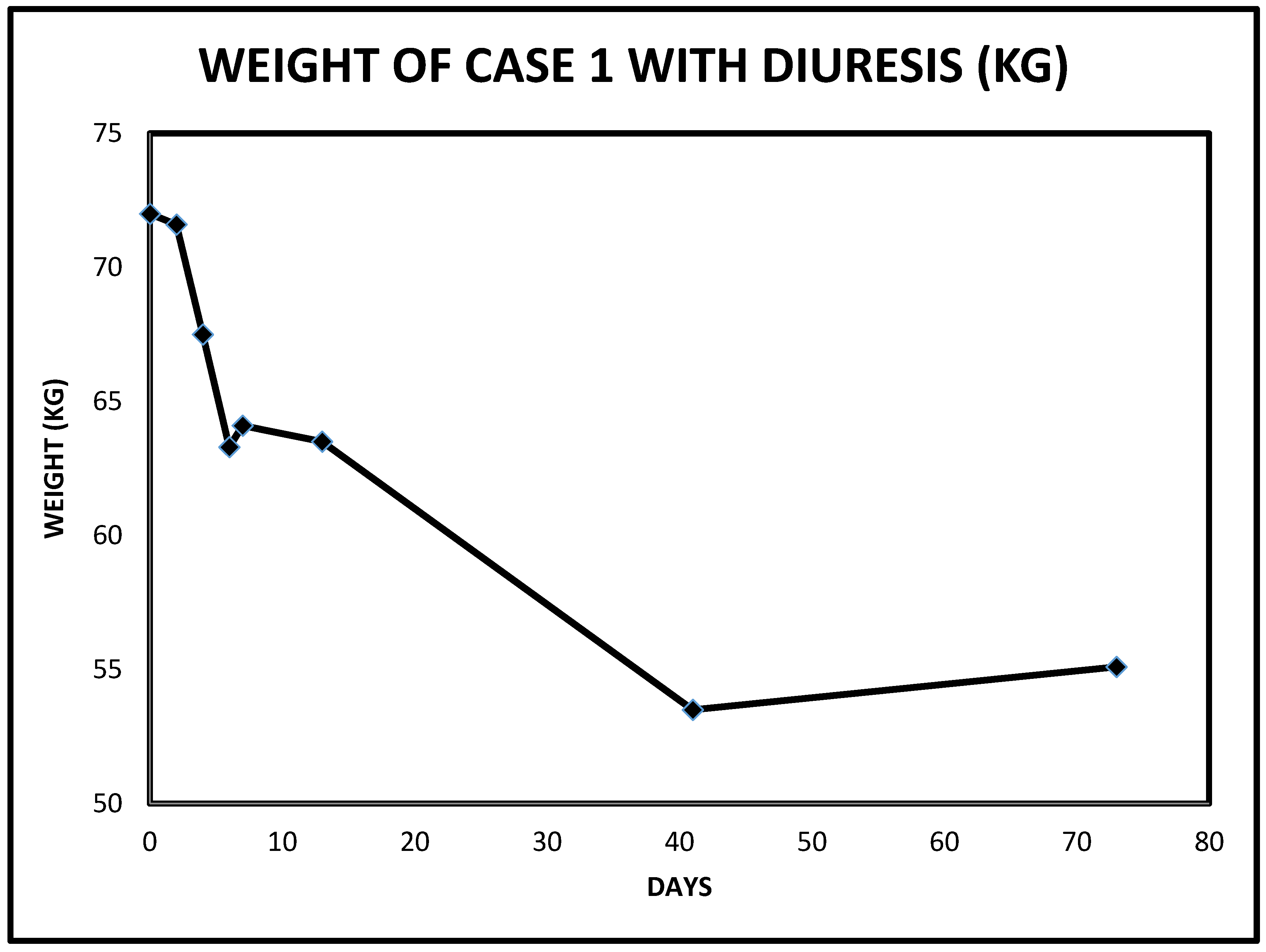
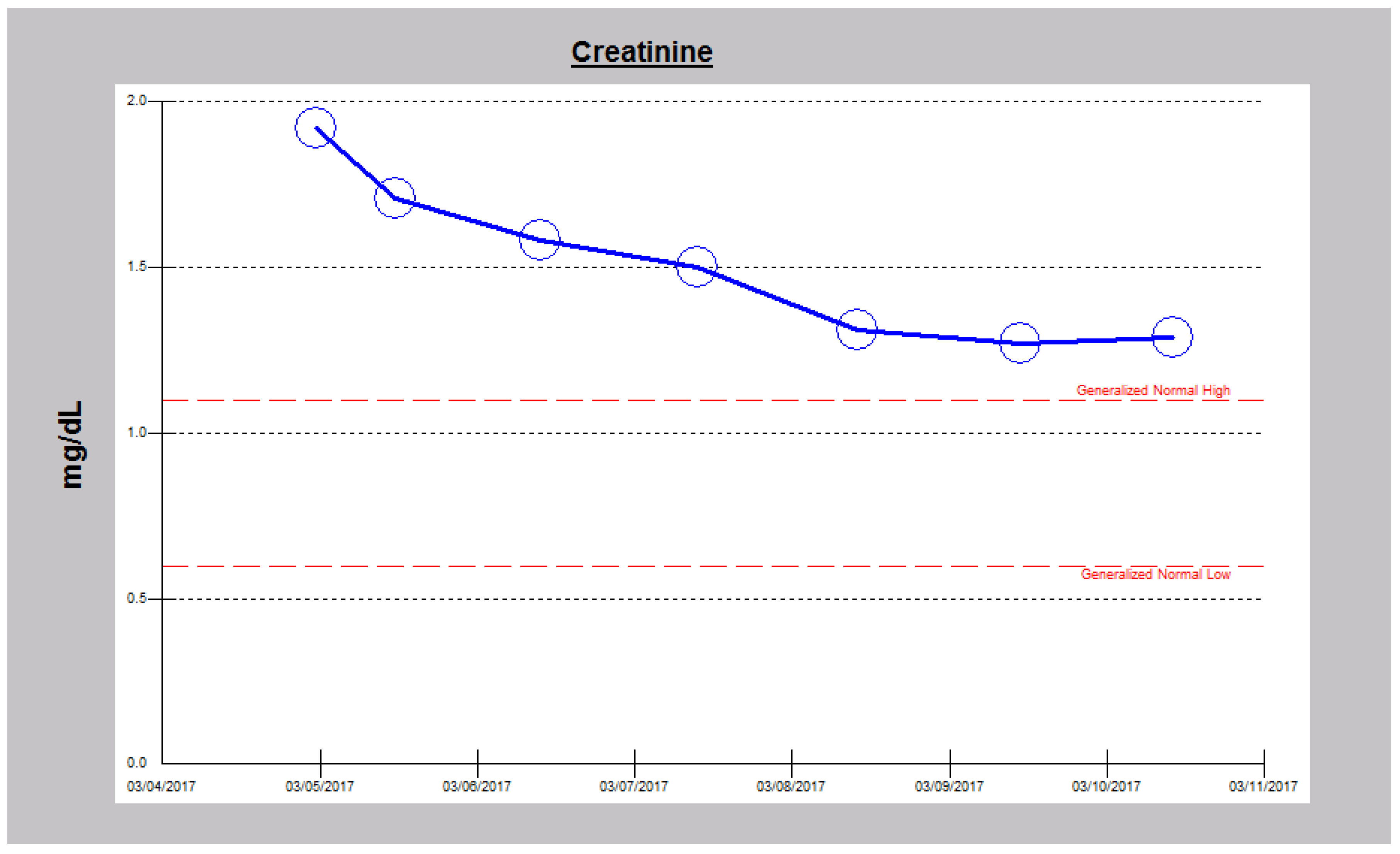
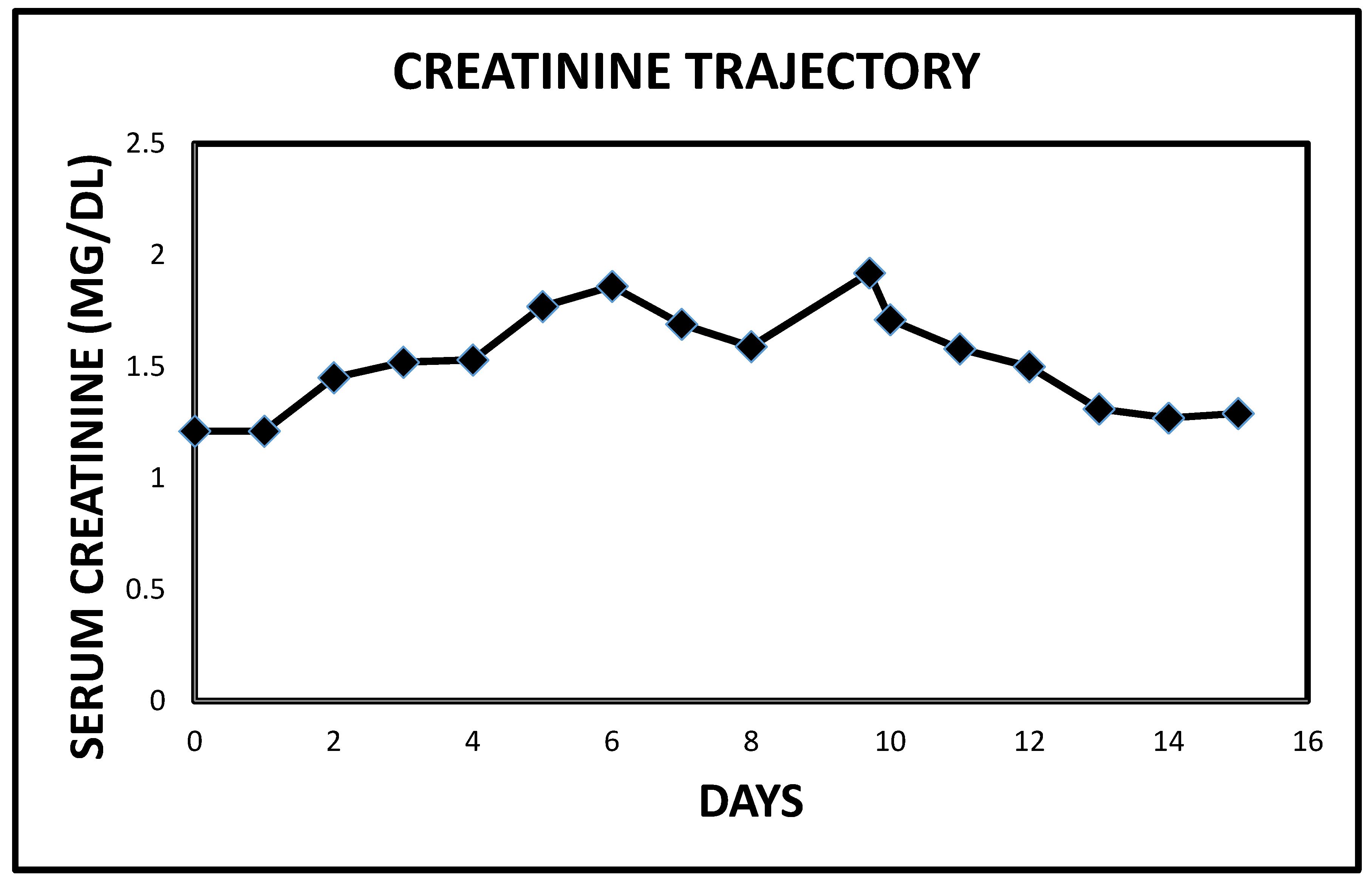
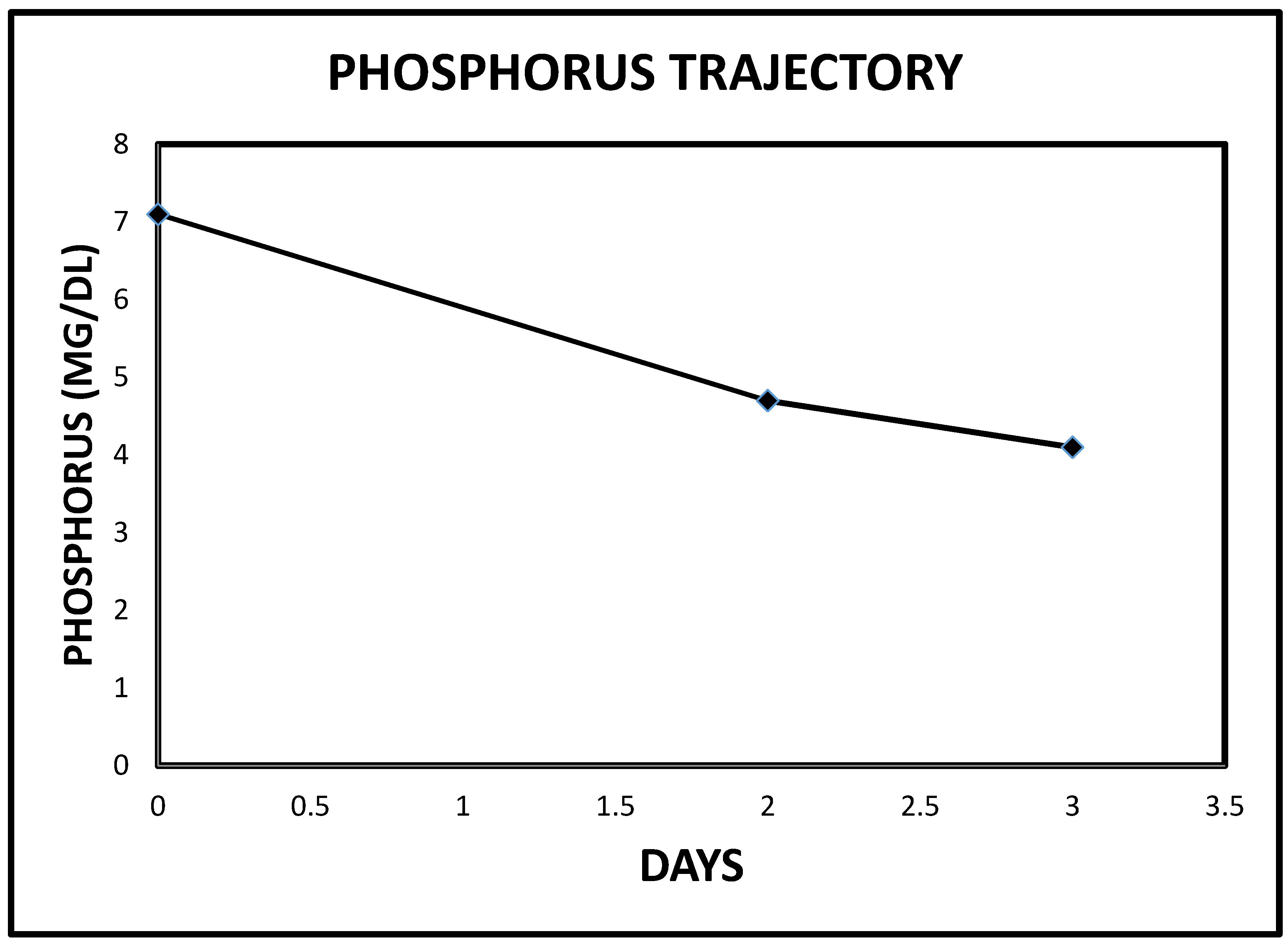
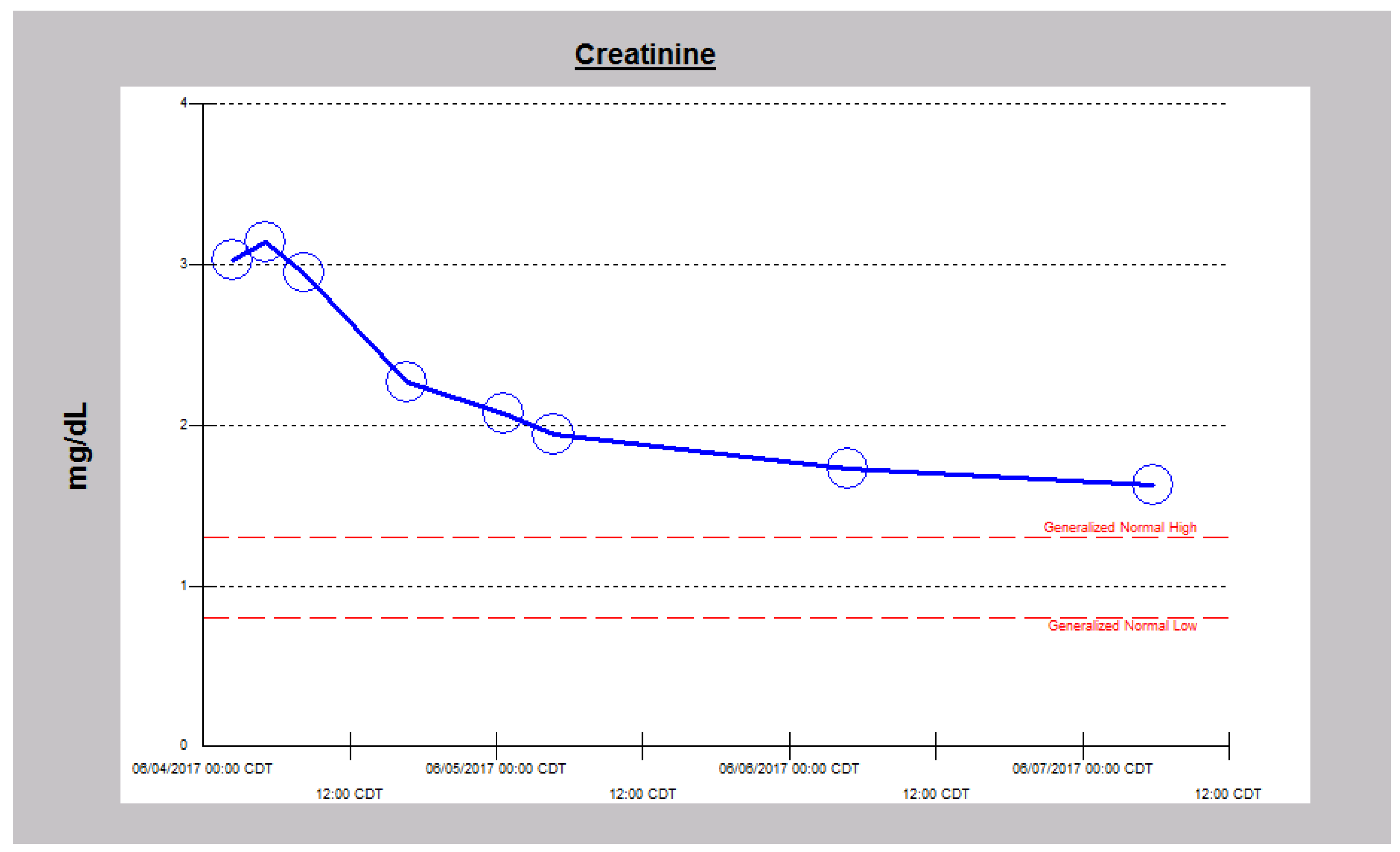
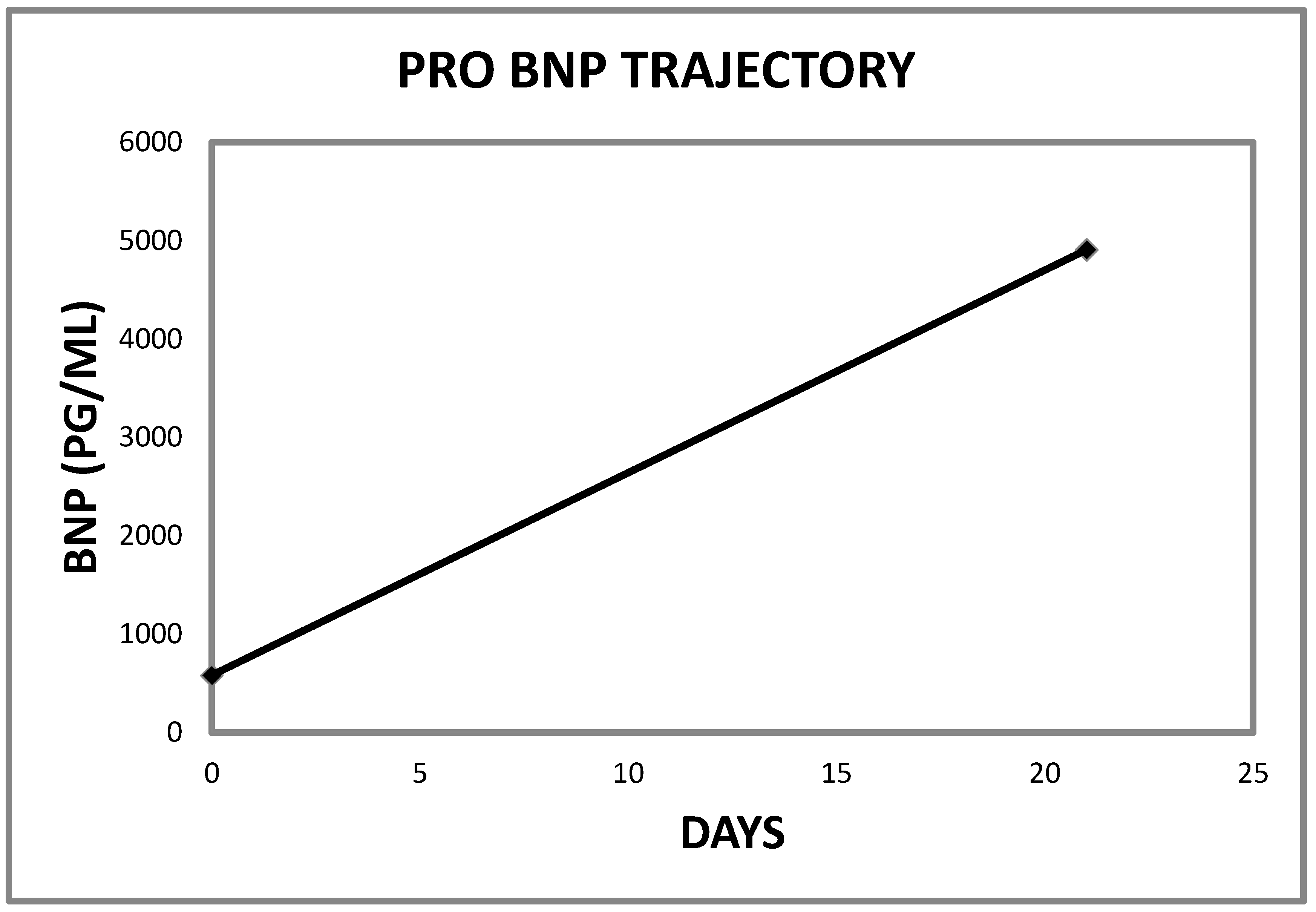
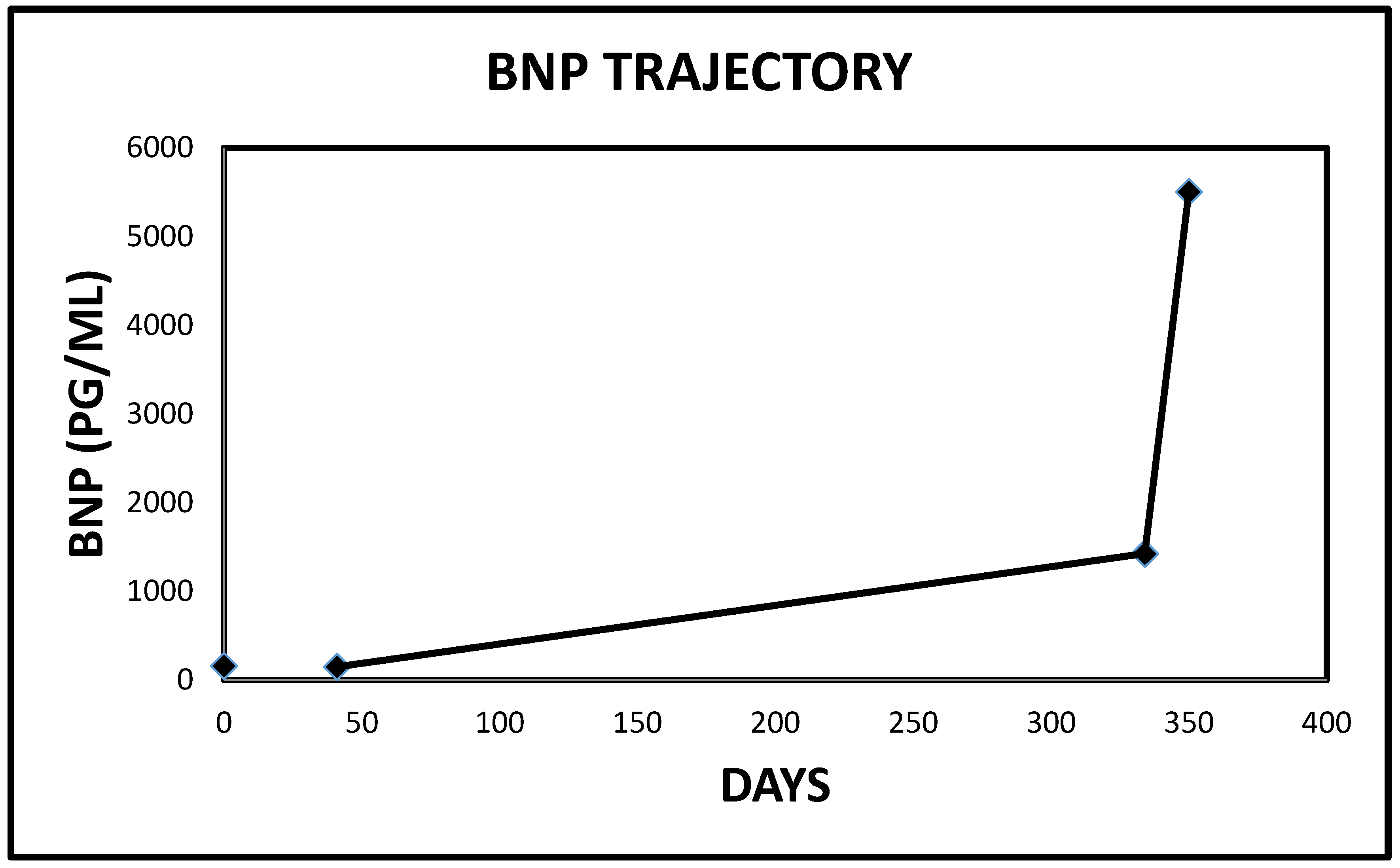
2.1. Case 1
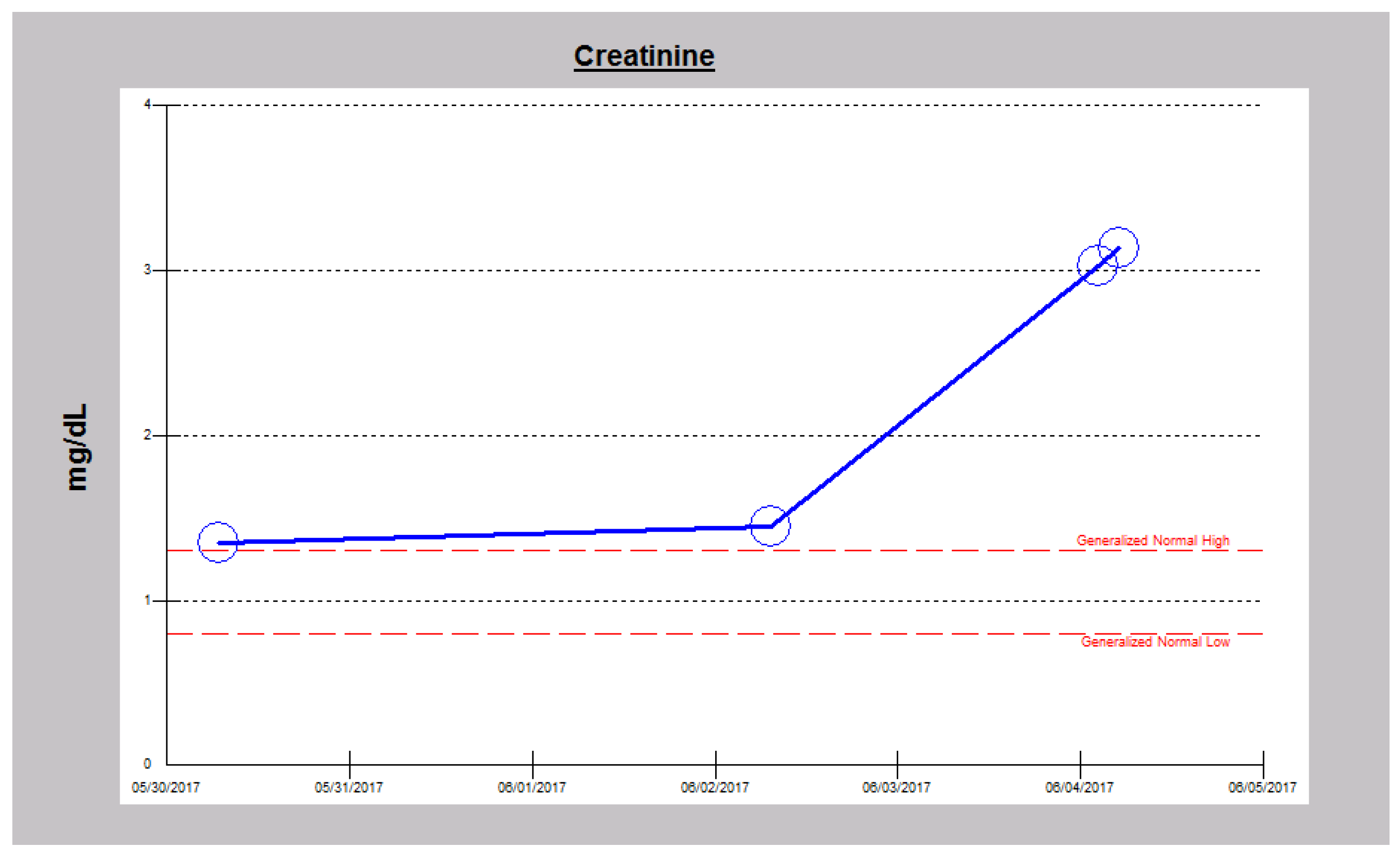
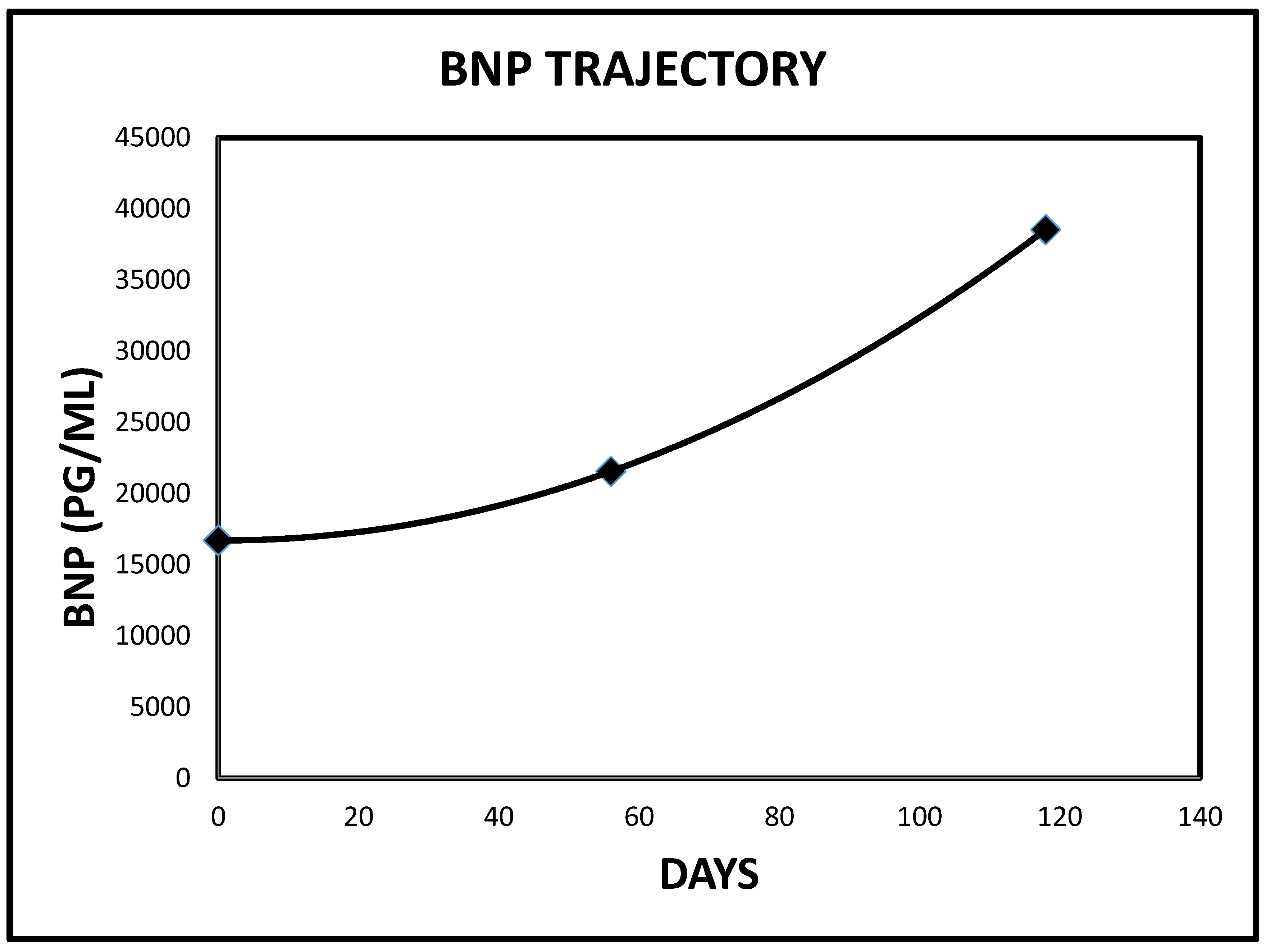
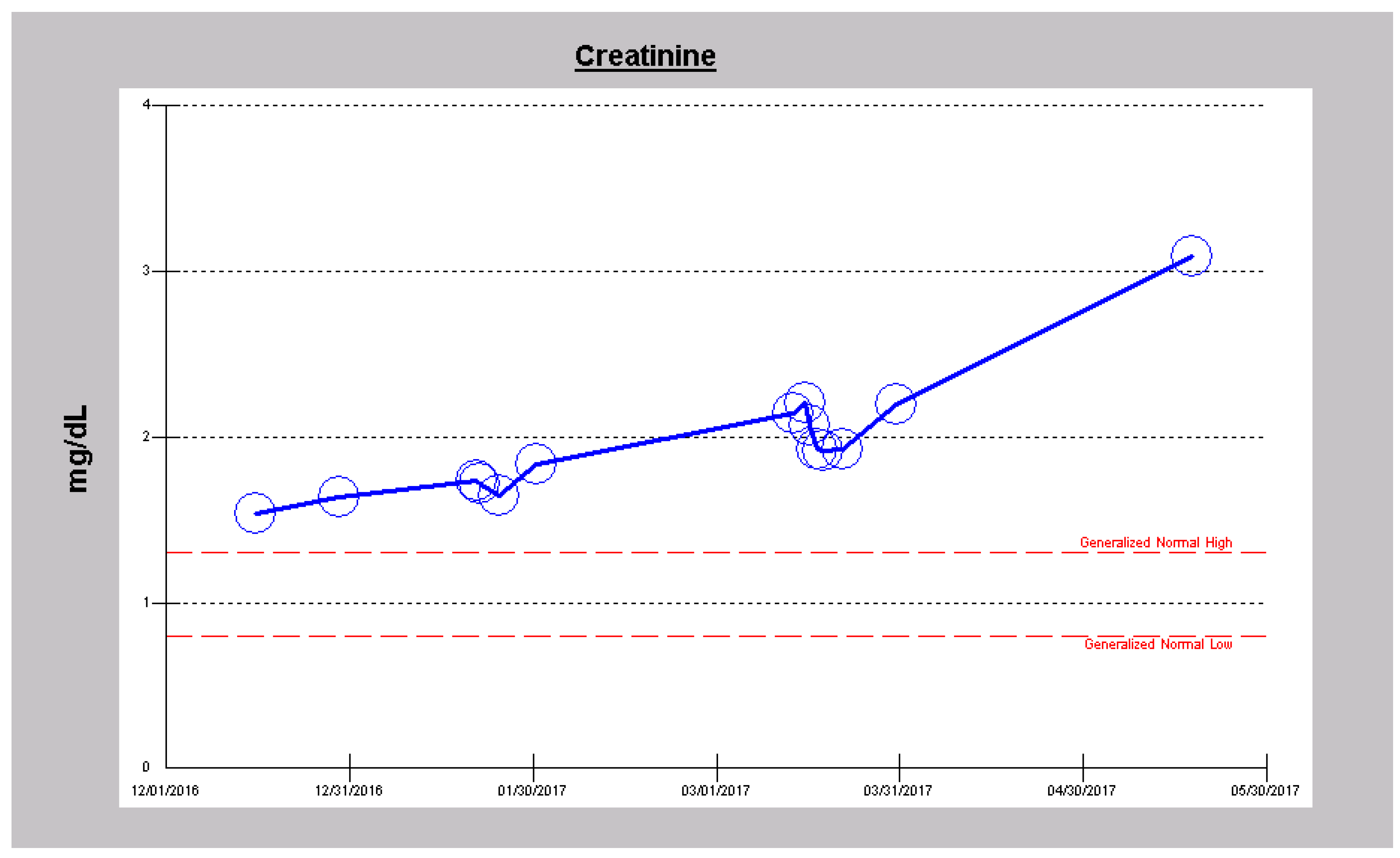
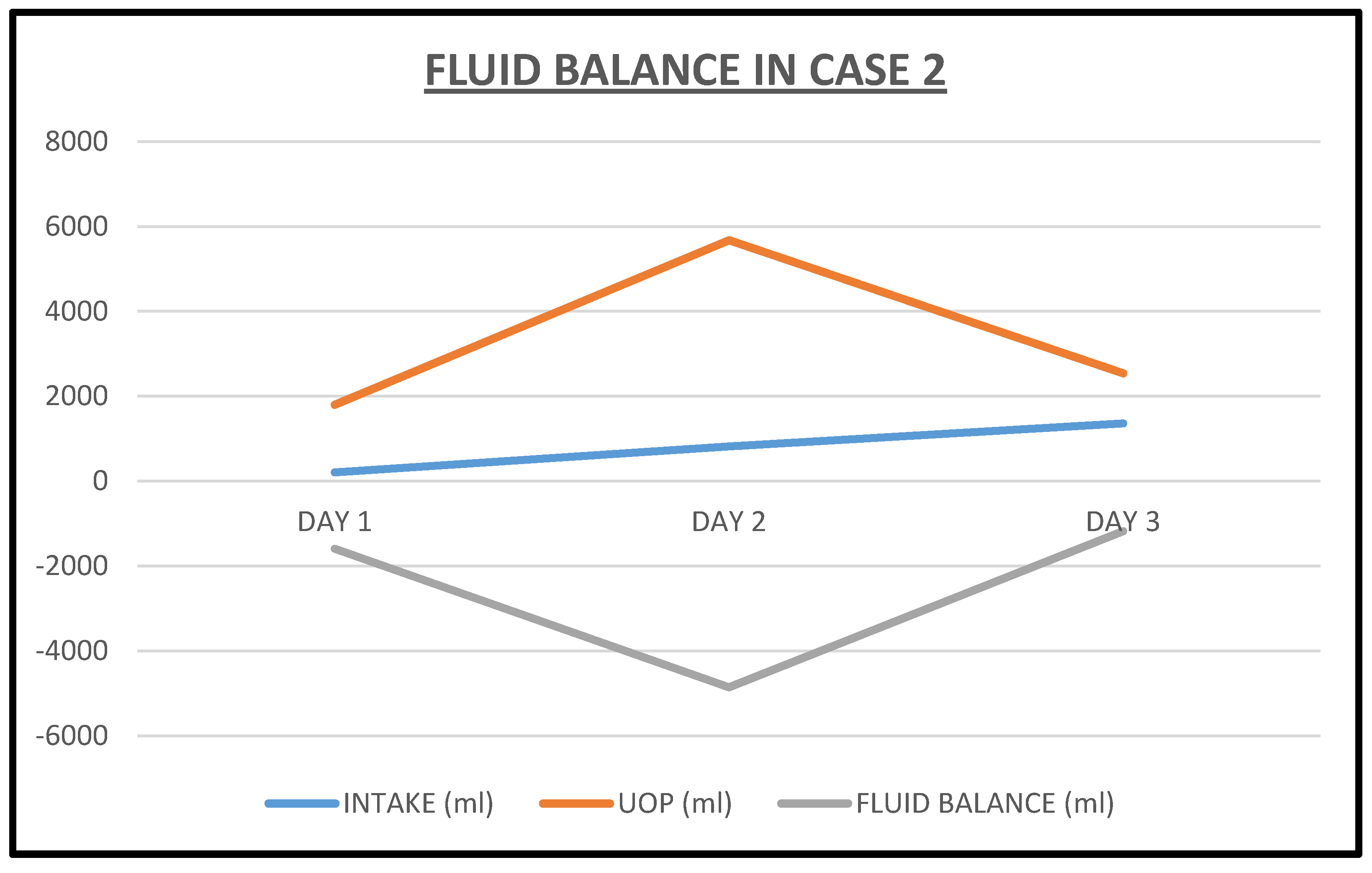
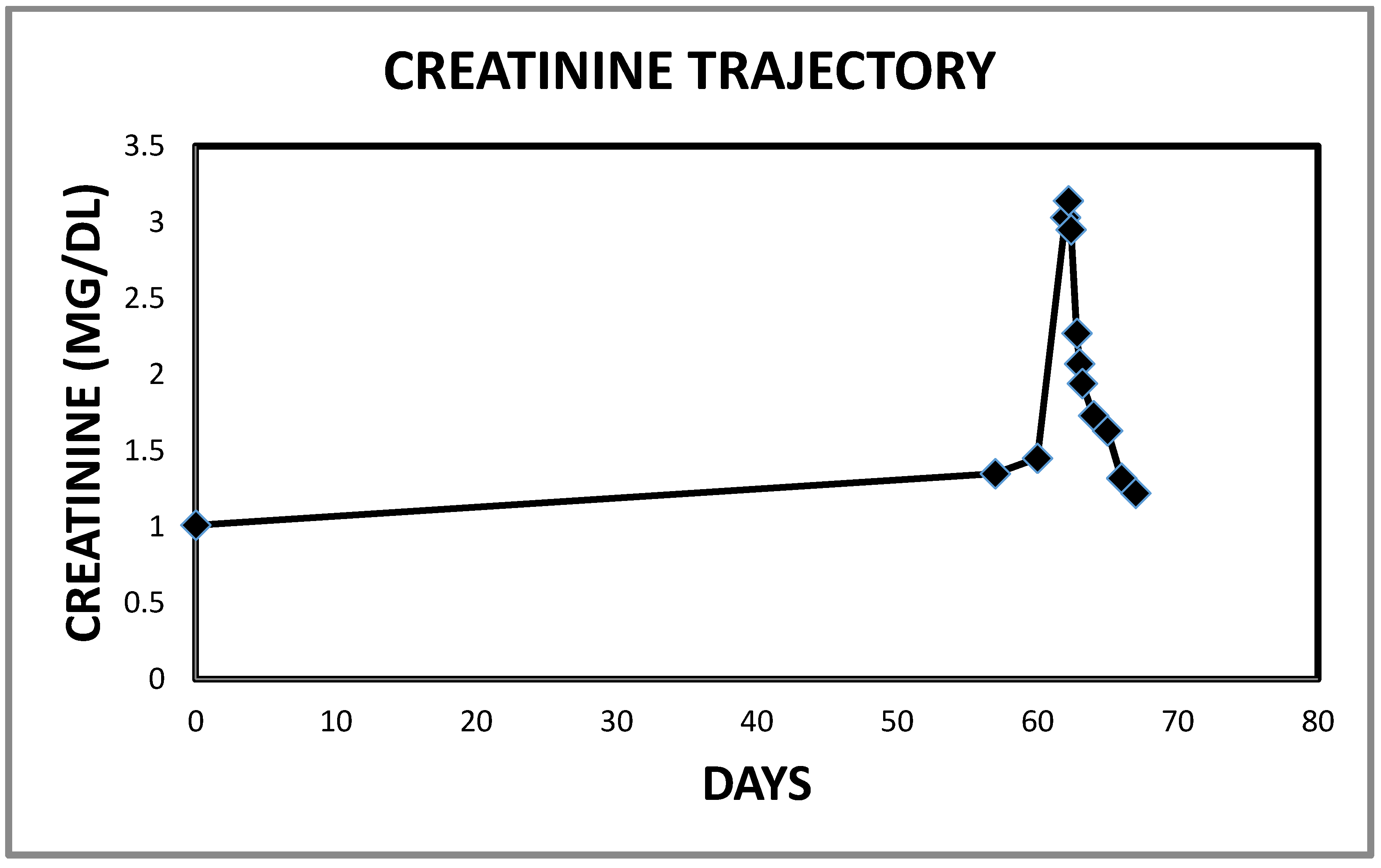
2.2. Case 2
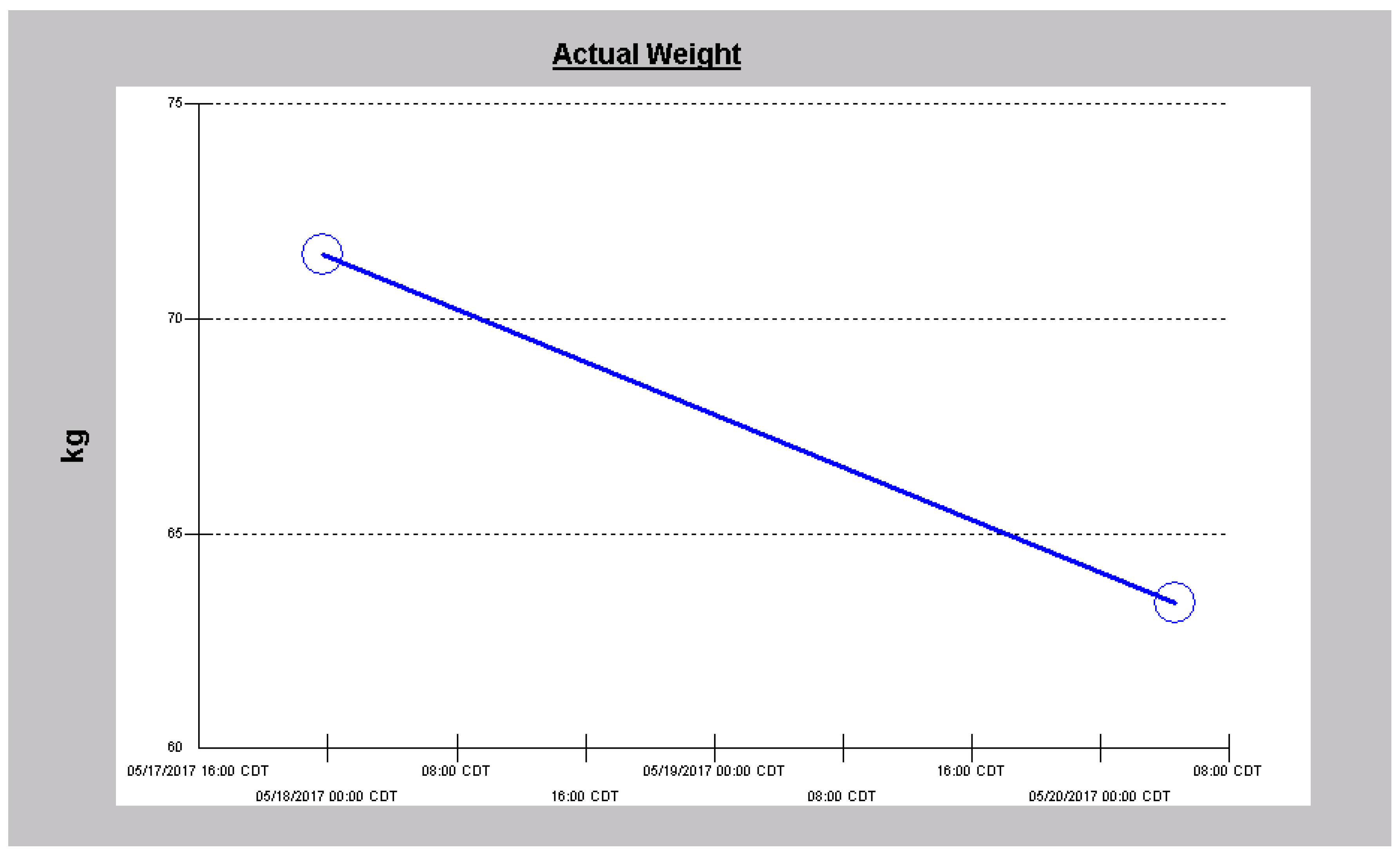
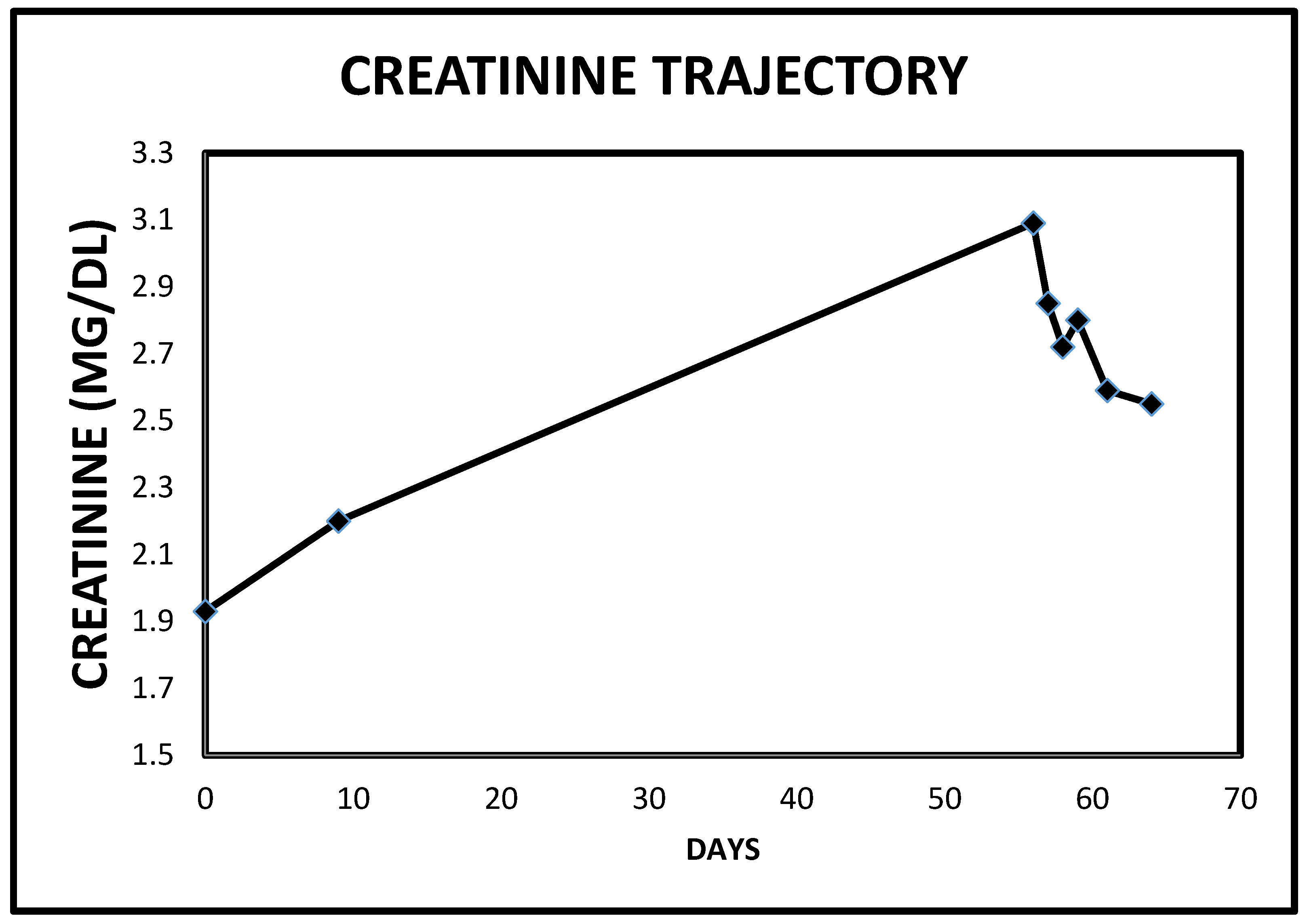
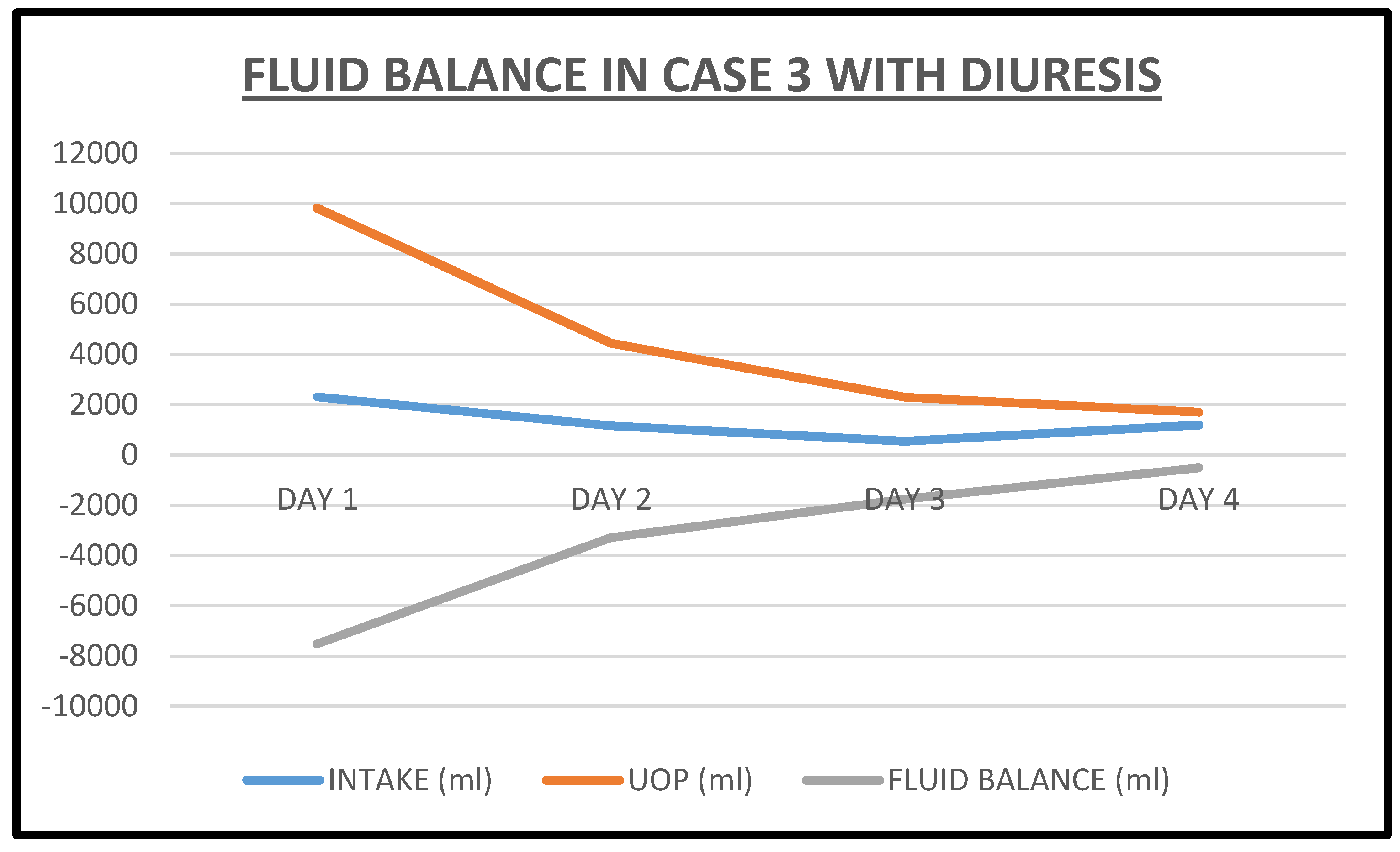
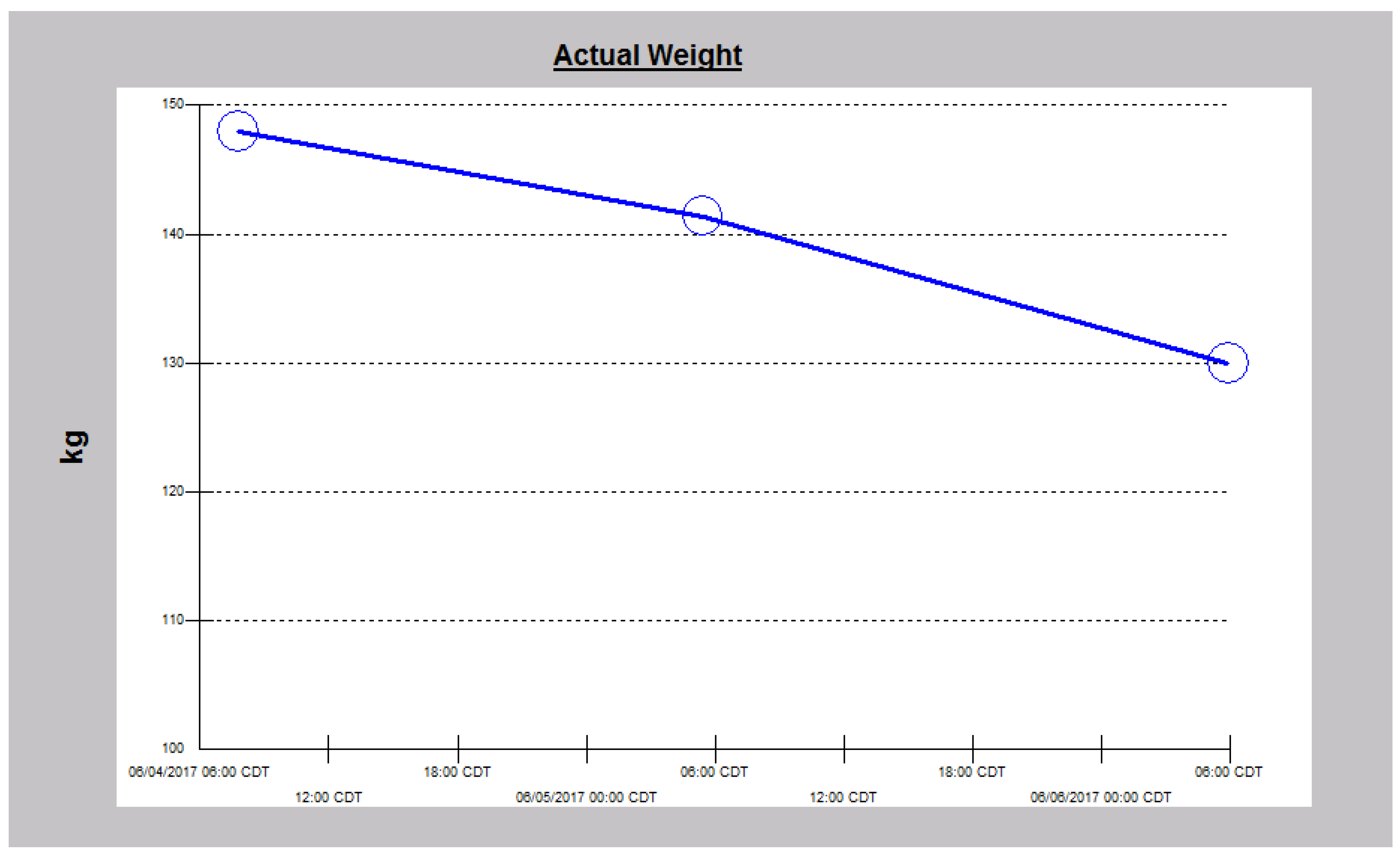
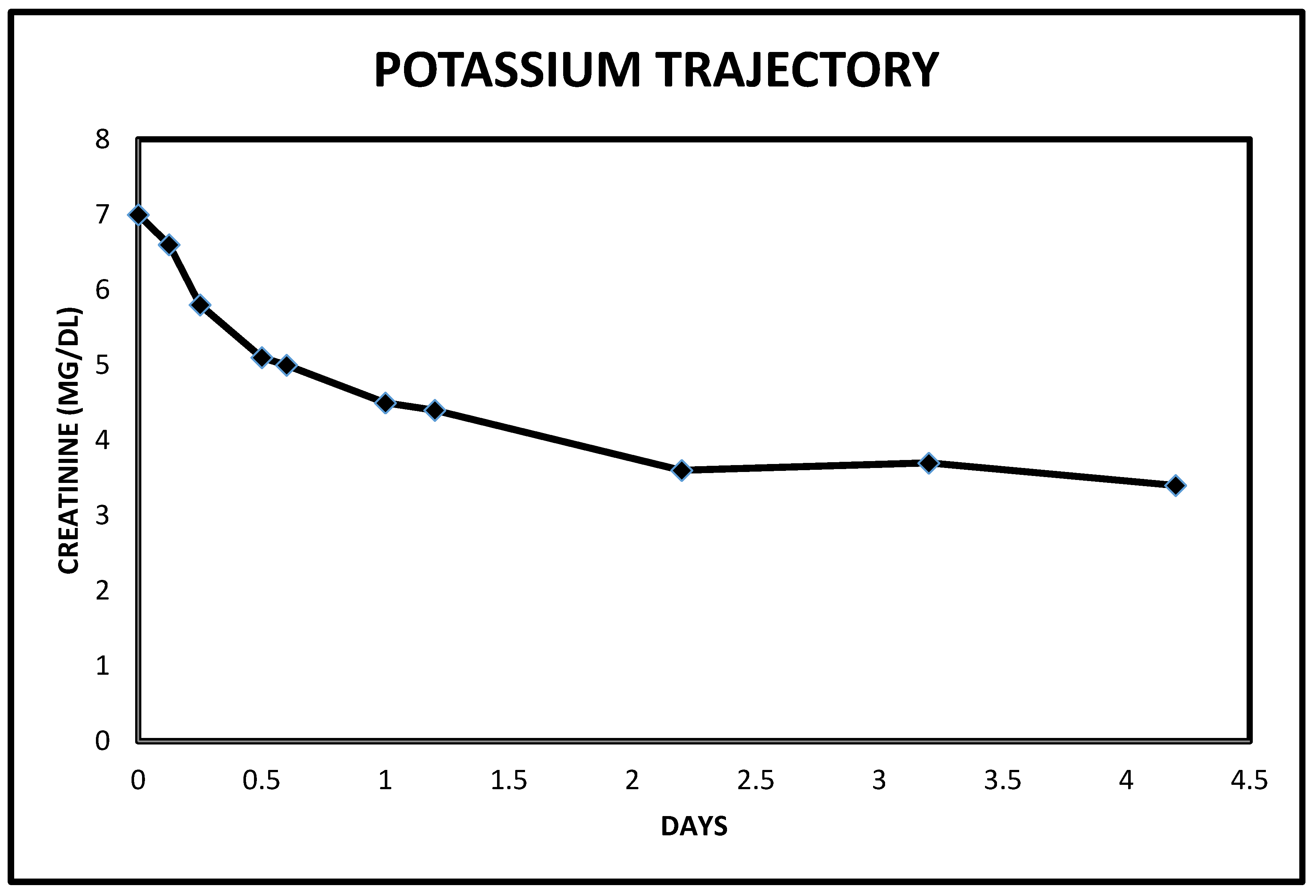
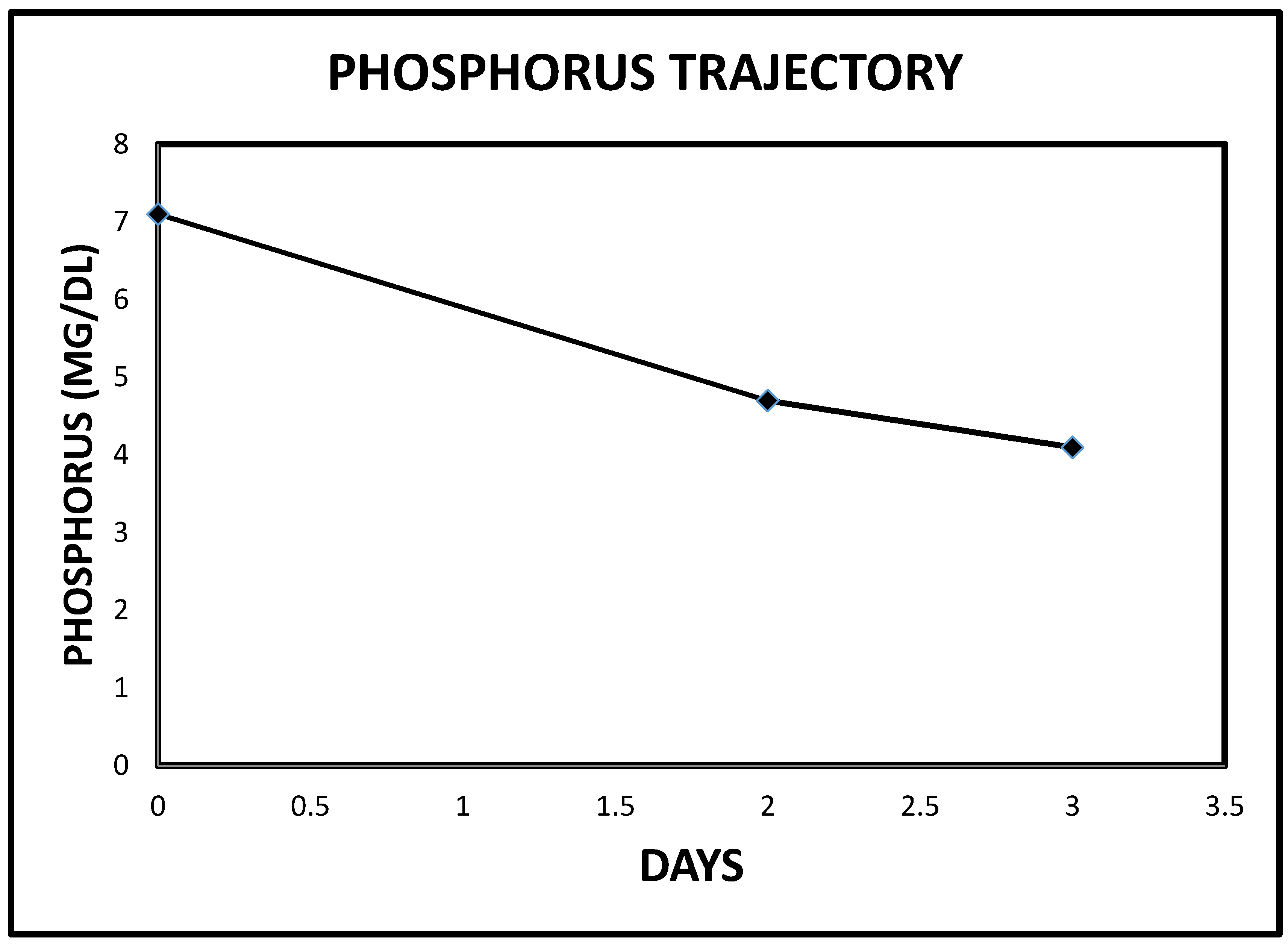
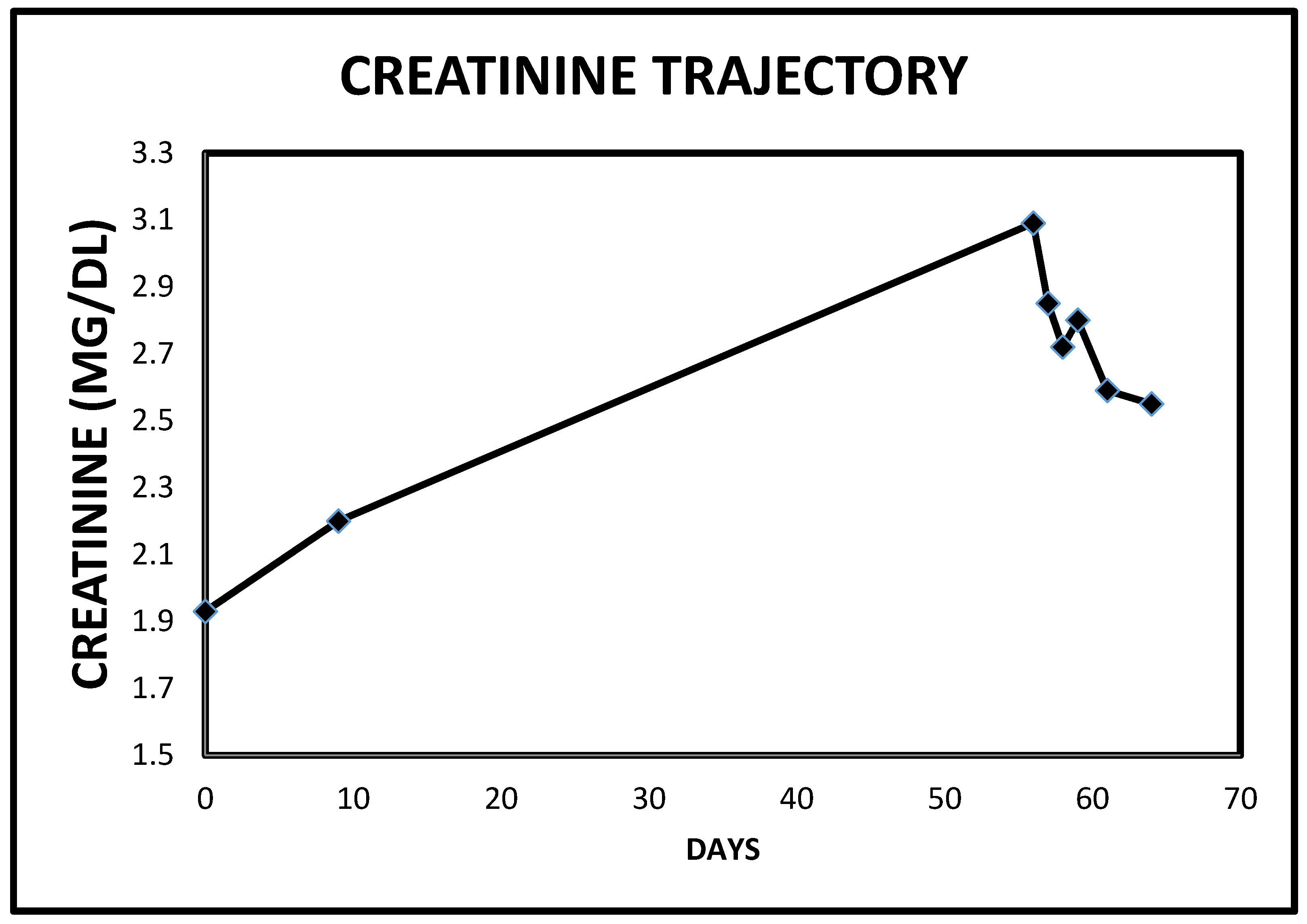
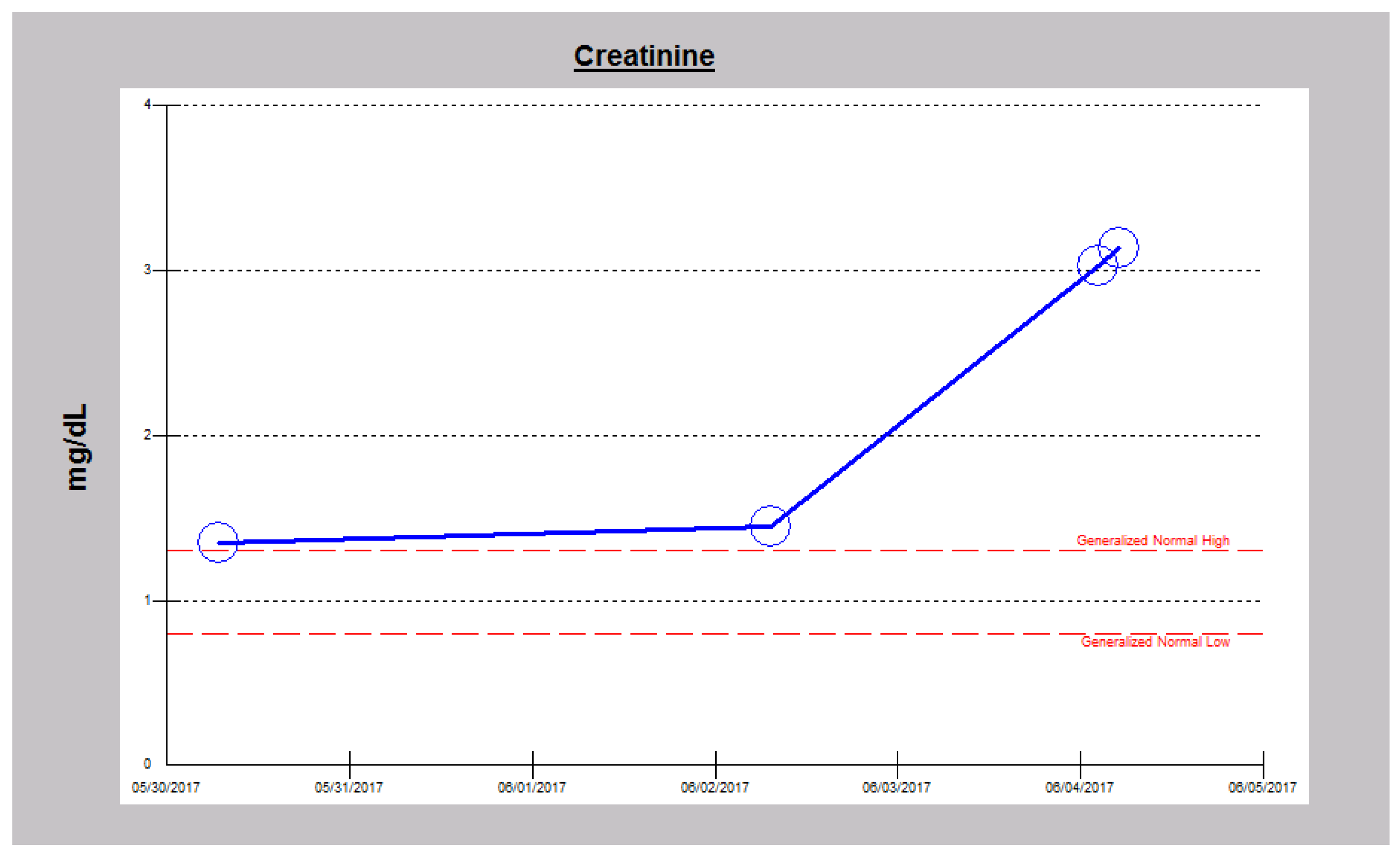
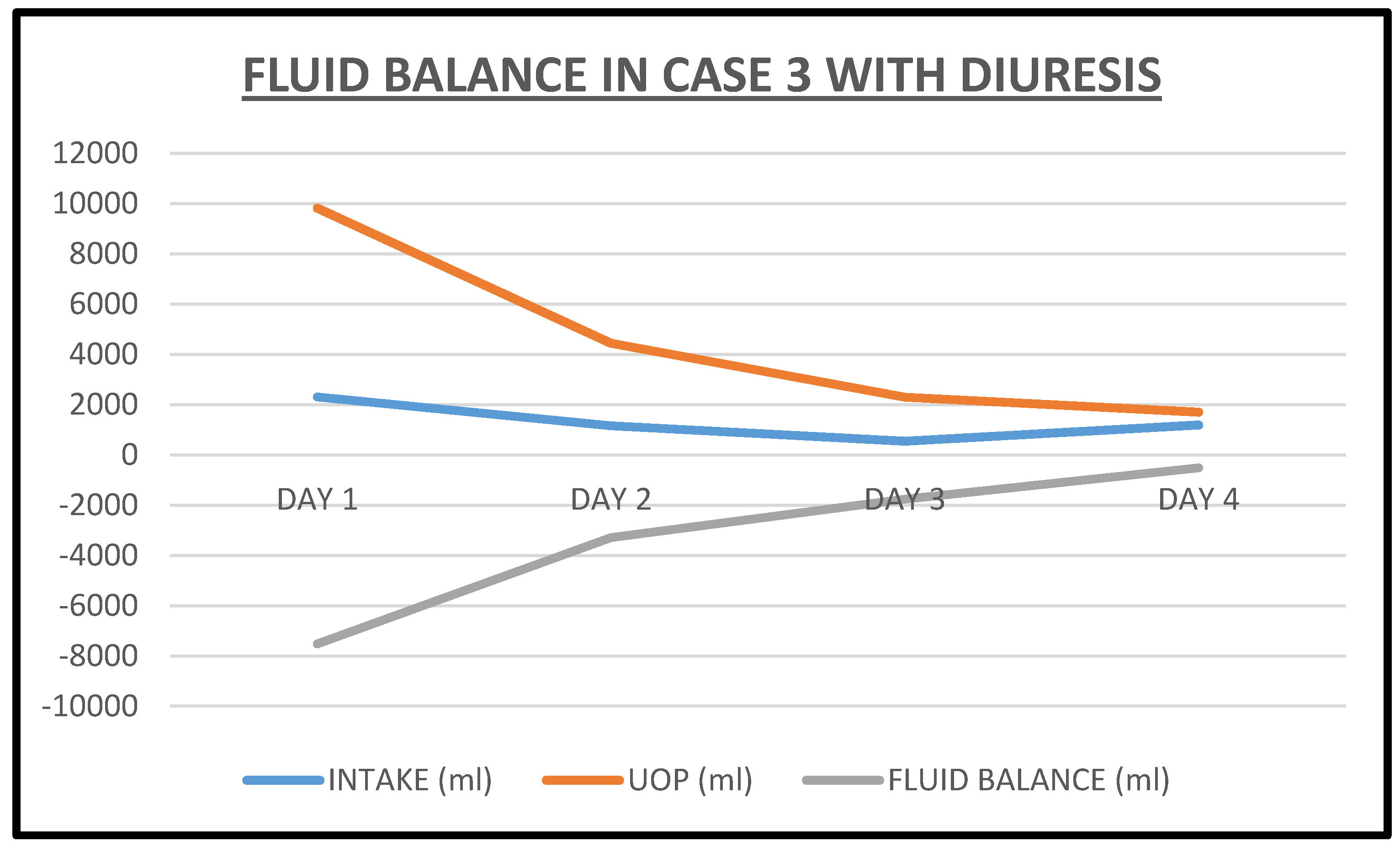
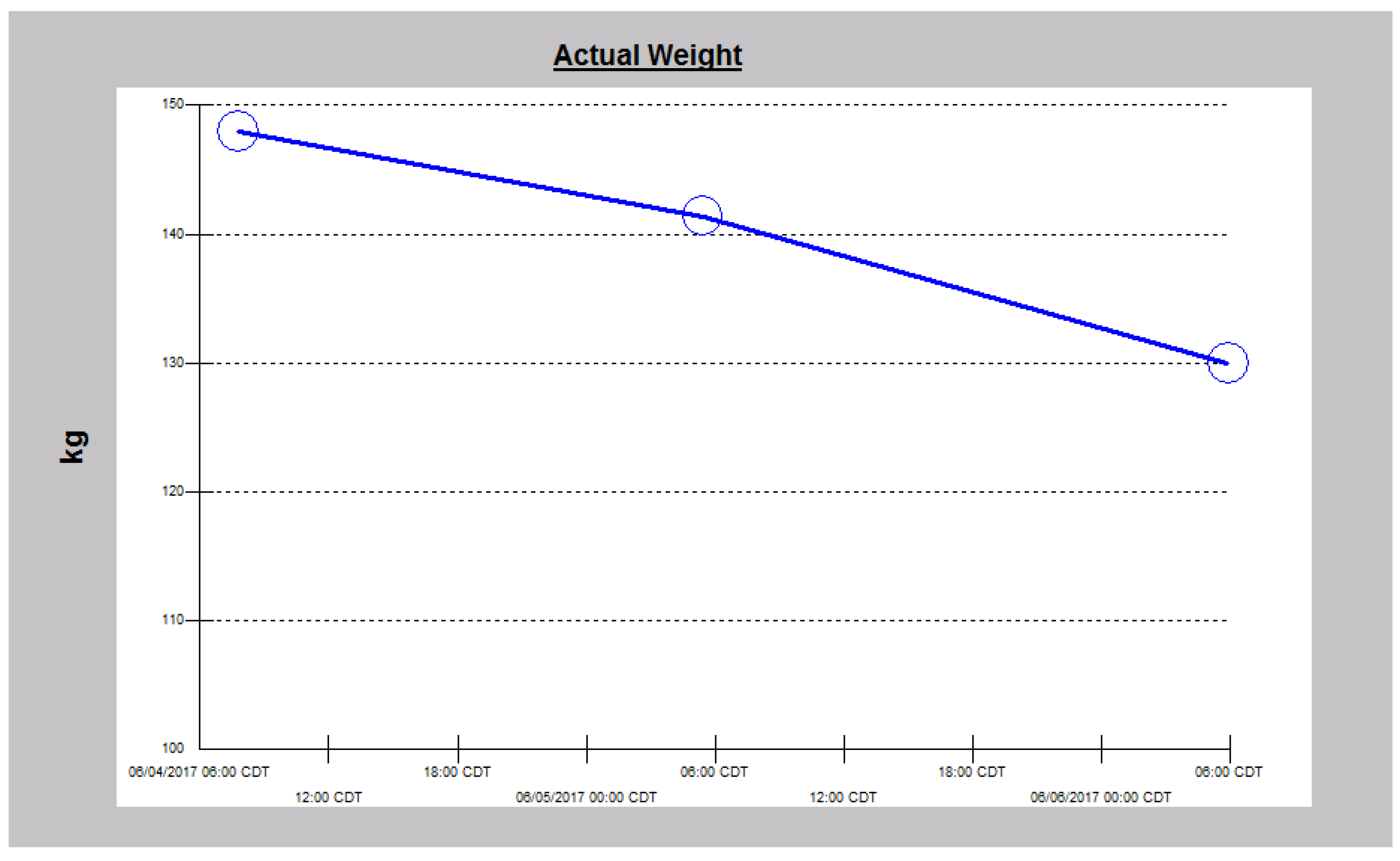
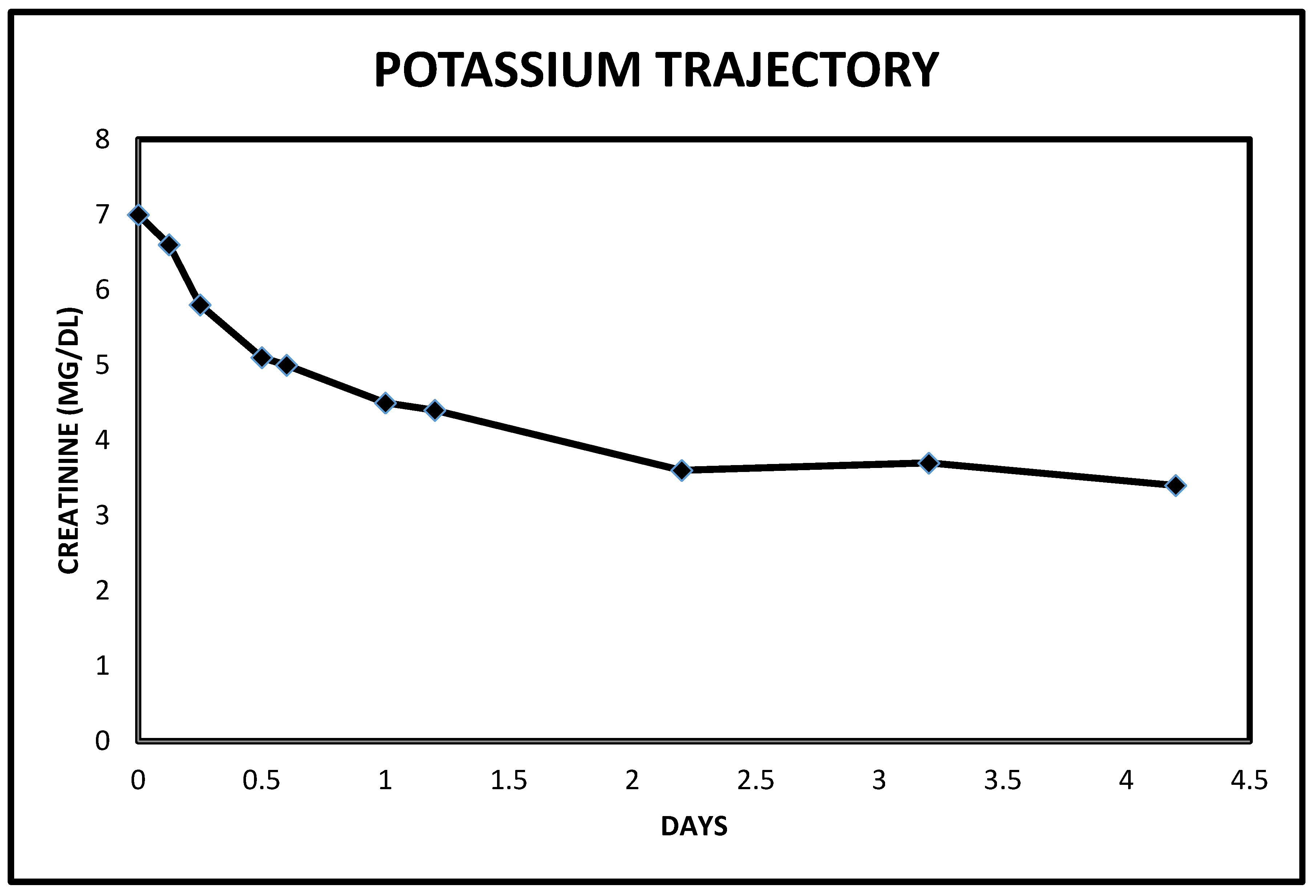
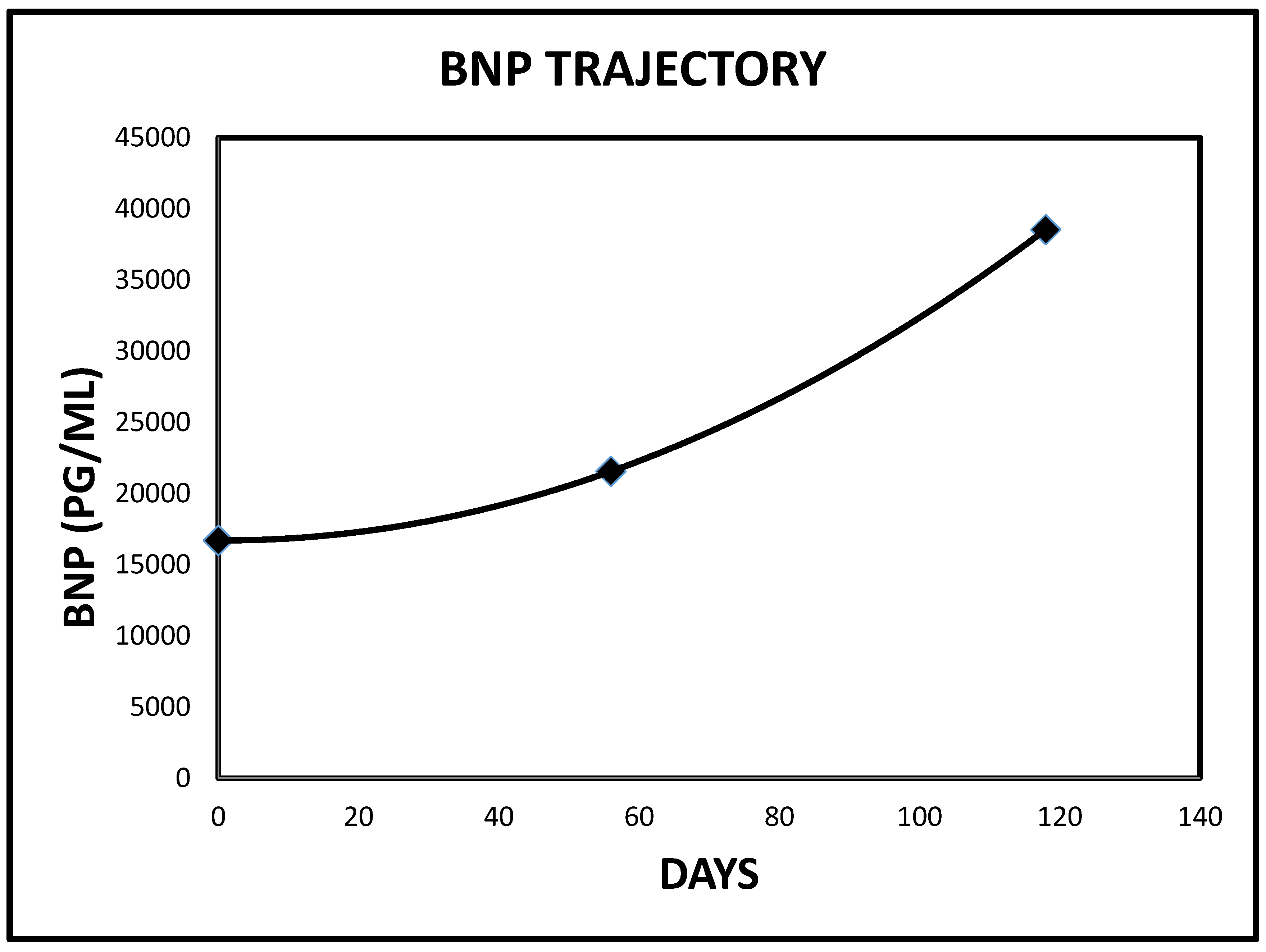
2.3. Case 3
3. Conclusions
3.1. The Natriuretric Peptides, Heart Failure, Renal Dysfunction and Cardiovascular Disease
3.2. Diuretic Resistance in Cardiorenal Syndrome
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ross, E.A. Congestive renal failure: The pathophysiology and treatment of renal venous hypertension. J. Card. Fail. 2012, 18, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Heywood, J.T.; Fonarow, G.C.; Costanzo, M.R.; Mathur, V.S.; Wigneswaran, J.R.; Wynne, J. ADHERE Scientific Advisory Committee and Investigators. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: A report from the ADHERE database. J. Card. Fail. 2007, 13, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Nohria, A.; Hasselblad, V.; Stebbins, A.; Pauly, D.F.; Fonarow, G.C.; Shah, M.; Yancy, C.W.; Califf, R.M.; Stevenson, L.W.; Hill, J.A. Cardiorenal interactions: Insights from the ESCAPE trial. J. Am. Coll. Cardiol. 2008, 51, 1268–1274. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; van Deursen, V.M.; Navis, G.; Voors, A.A.; van Veldhuisen, D.J.; Hillege, H.L. Increased central venous pressure is associated with impaired renal function and mortality in a broad spectrum of patients with cardiovascular disease. J. Am. Coll. Cardiol. 2009, 53, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Damman, K.; Voors, A.A.; Hillege, H.L.; Navis, G.; Lechat, P.; van Veldhuisen, D.J.; Dargie, H.J. CIBIS-2 Investigators and Committees. Congestion in chronic systolic heart failure is related to renal dysfunction and increased mortality. Eur. J. Heart Fail. 2010, 12, 974–982. [Google Scholar] [CrossRef] [PubMed]
- Guglin, M.; Rivero, A.; Matar, F.; Garcia, M. Renal dysfunction in heart failure is due to congestion but not low output. Clin. Cardiol. 2011, 34, 113–136. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.T.; Holst, D.P.; Kaye, D.M. Tricuspid regurgitation contributes to renal dysfunction in patients with heart failure. J. Card. Fail. 2008, 14, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Rafique, Z.; Weir, M.R.; Onuigbo, M.; Pitt, B.; Lafayette, R.; Butler, J.; Lopes, M.; Farnum, C.; Peacock, W.F. Expert Panel Recommendations for the Identification and Management of Hyperkalemia and Role of Patiromer in Patients with Chronic Kidney Disease and Heart Failure. J. Manag. Care Spec Pharm. 2017, 23, S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Calò, L.A.; Maiolino, G.; Zoccali, C. Ultrafiltration for the treatment of congestion: A window into the lung for a better caress to the heart. Nephrol. Dial. Transplant. 2014, 29, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Mullens, W.; Tang, W.H. Management of Cardio-Renal Syndrome and Diuretic Resistance. Curr. Treat. Opt. Cardiovasc. Med. 2016, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Grodin, J.L.; Stevens, S.R.; de Las Fuentes, L.; Kiernan, M.; Birati, E.Y.; Gupta, D.; Bart, B.A.; Felker, G.M.; Chen, H.H.; Butler, J.; et al. Intensification of Medication Therapy for Cardiorenal Syndrome in Acute Decompensated Heart Failure. J. Card. Fail. 2016, 22, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Dupont, M.; Bertrand, P.B.; Nijst, P.; Penders, J.; Dens, J.; Werhaert, D.; Vandervoort, P.; Wilson Tang, W.H.; Mullens, W. Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol. 2015, 70, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Corboy, J.C.; Walker, R.J.; Simmonds, M.B.; Wilkins, G.T.; Richards, A.M.; Espiner, E.A. Plasma natriuretic peptides and cardiac volume during acute changes in intravascular volume in haemodialysis patients. Clin. Sci. 1994, 87, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Van de Pol, A.C.; Frenken, L.A.; Moret, K.; Baumgarten, R.; van der Sande, F.M.; Beerenhout, C.M.; Kooman, J.P.; Leunissen, K.M. An evaluation of blood volume changes during ultrafiltration pulses and natriuretic peptides in the assessment of dry weight in hemodialysis patients. Hemodial. Int. 2007, 11, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Tapolyai, M.; Uysal, A.; Maeweathers, G.; Bahta, E.; Dossabhoy, N.R. B-type natriuretic peptide-directed ultrafiltration improves care in acutely hospitalized dialysis patients. Congest. Heart Fail. 2009, 15, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, J.; Iimori, S.; Kuwahara, M.; Sasaki, S.; Tsukamoto, Y. Diagnostic value of B-type natriuretic peptide for estimating left atrial size and its usefulness for predicting all-cause mortality and cardiovascular events among chronic haemodialysis patients. Nephrology 2014, 19, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Rubattu, S.; Sciarretta, S.; Valenti, V.; Stanzione, R.; Volpe, M. Natriuretic peptides: An update on bioactivity, potential therapeutic use, and implication in cardiovascular diseases. Am. J. Hypertens. 2008, 21, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Testani, J.M.; Damman, K.; Brisco, M.A.; Chen, S.; Laur, O. A combined-biomarker approach to clinical phenotyping renal dysfunction in heart failure. J. Card. Fail. 2014, 20, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Holditch, S.J.; Schreiber, C.A.; Nini, R.; Tonne, J.M.; Peng, K.W.; Geurts, A.; Jacob, H.J.; Burnett, J.C.; Cataliotti, A.; Ikeda, Y. B-Type Natriuretic Peptide Deletion Leads to Progressive Hypertension, Associated Organ Damage, and Reduced Survival: Novel Model for Human Hypertension. Hypertension 2015, 66, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.E.; Feridooni, T.; Hotchkiss, A.; Kruzliak, P.; Pasumarthi, K.B. Mechanisms of renal hyporesponsiveness to BNP in heart failure. Can. J. Physiol. Pharmacol. 2015, 93, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Onuigbo, M.A. RAAS inhibition and cardiorenal syndrome. Curr. Hypertens. Rev. 2014, 10, 107–111. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
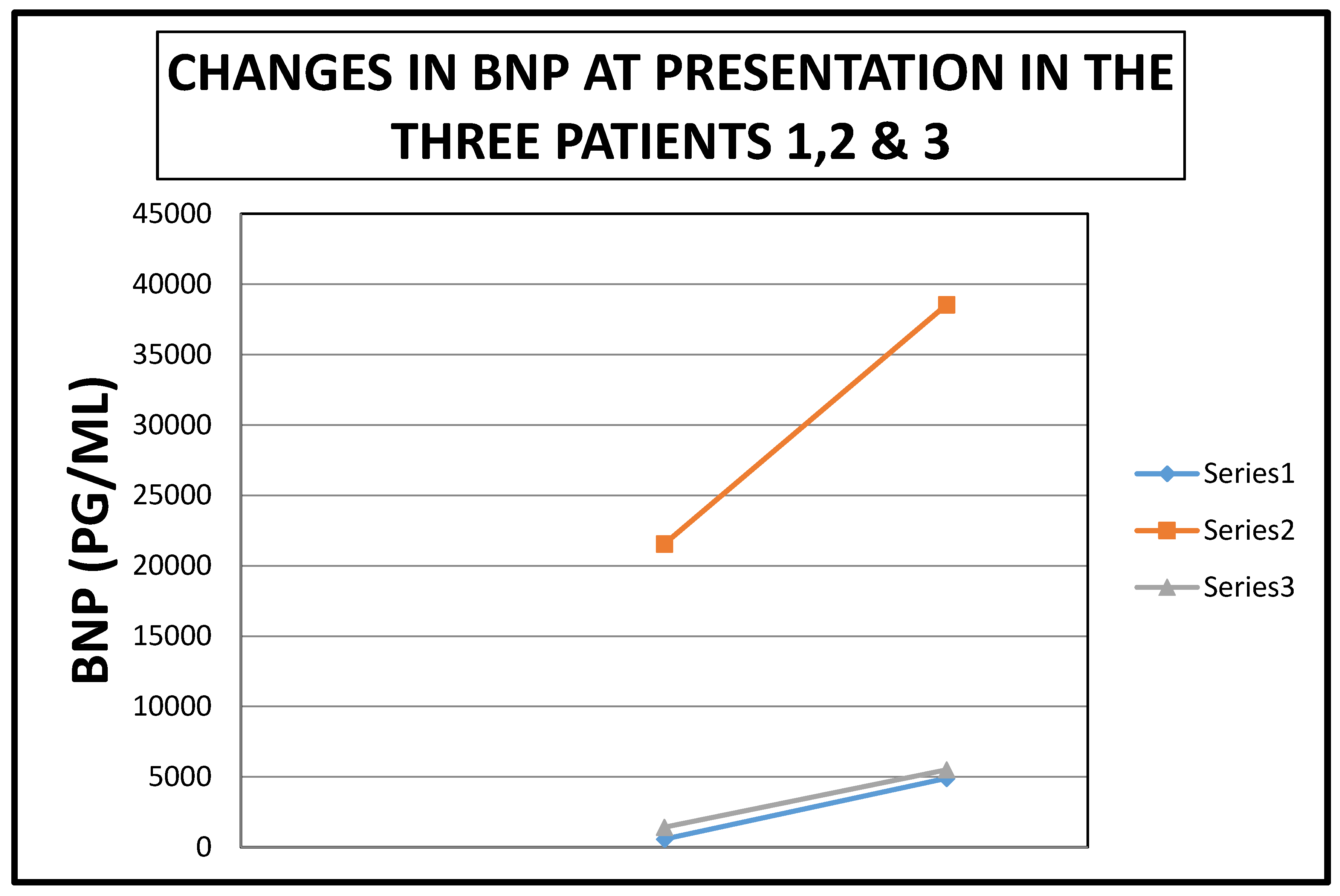
| Patient | BNP Level at Presentation | Penultimate BNP Level | Reference Range | Interval between BNP Tests (Days) |
|---|---|---|---|---|
| 1 | 4907 | 581 | <450 PG/ML | 21 |
| 2 | 38,547 | 21,557 | <450 PG/ML | 63 |
| 3 | 5503 | 1426 | <125 PG/ML | 16 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onuigbo, M.A.C.; Agbasi, N.; Sengodan, M.; Rosario, K.F. Acute Kidney Injury in Heart Failure Revisited—The Ameliorating Impact of “Decongestive Diuresis” on Renal Dysfunction in Type 1 Acute Cardiorenal Syndrome: Accelerated Rising Pro B Naturetic Peptide Is a Predictor of Good Renal Prognosis. J. Clin. Med. 2017, 6, 82. https://doi.org/10.3390/jcm6090082
Onuigbo MAC, Agbasi N, Sengodan M, Rosario KF. Acute Kidney Injury in Heart Failure Revisited—The Ameliorating Impact of “Decongestive Diuresis” on Renal Dysfunction in Type 1 Acute Cardiorenal Syndrome: Accelerated Rising Pro B Naturetic Peptide Is a Predictor of Good Renal Prognosis. Journal of Clinical Medicine. 2017; 6(9):82. https://doi.org/10.3390/jcm6090082
Chicago/Turabian StyleOnuigbo, Macaulay Amechi Chukwukadibia, Nneoma Agbasi, Mohan Sengodan, and Karen Flores Rosario. 2017. "Acute Kidney Injury in Heart Failure Revisited—The Ameliorating Impact of “Decongestive Diuresis” on Renal Dysfunction in Type 1 Acute Cardiorenal Syndrome: Accelerated Rising Pro B Naturetic Peptide Is a Predictor of Good Renal Prognosis" Journal of Clinical Medicine 6, no. 9: 82. https://doi.org/10.3390/jcm6090082