Individualised Multimodal Treatment Strategies for Anaplastic and Poorly Differentiated Thyroid Cancer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Patient-Derived Human Tumor Tissue (PDTT)
2.2. PDTT Cells and HF Culture Condition
2.3. Compounds Tested
2.4. Real-Time Cell Viability Analysis
2.5. RNA Isolation and Quantitative Real-Time RT-PCR
2.6. Protein Isolation and Western Blotting
2.7. In Vitro Radioiodine Up-Take
2.8. Statistical Analysis
2.9. Ethical Approval
3. Results
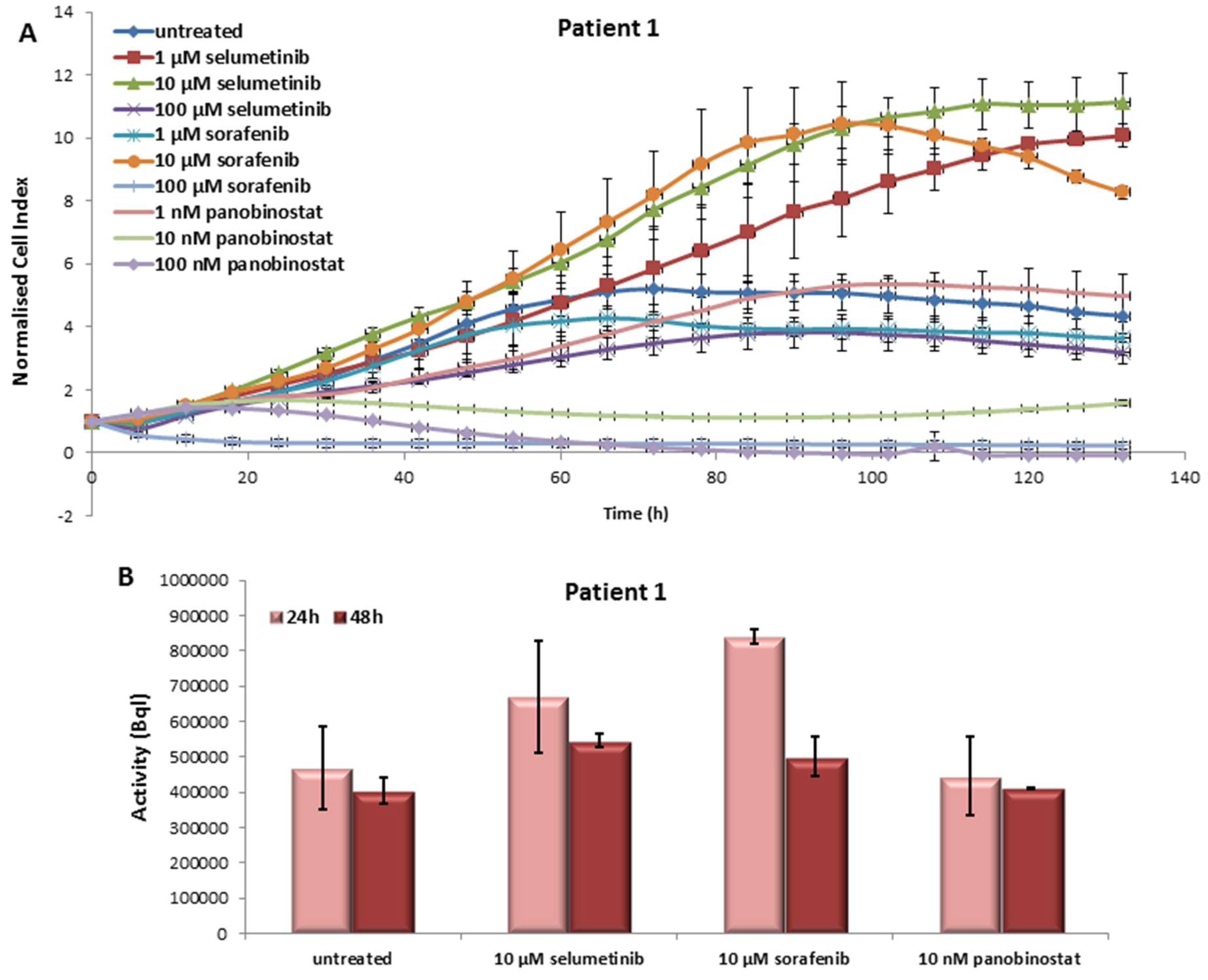
3.1. In Vitro Results of the First Patient
3.1.1. Effects on Cell Viability of the Individual Tumor Cells Caused by Treatment with Sorafenib, Panobinostat and Selumetinib
3.1.2. Radioiodine Up-Take after Treatment with Sorafenib, Panobinostat and Selumetinib
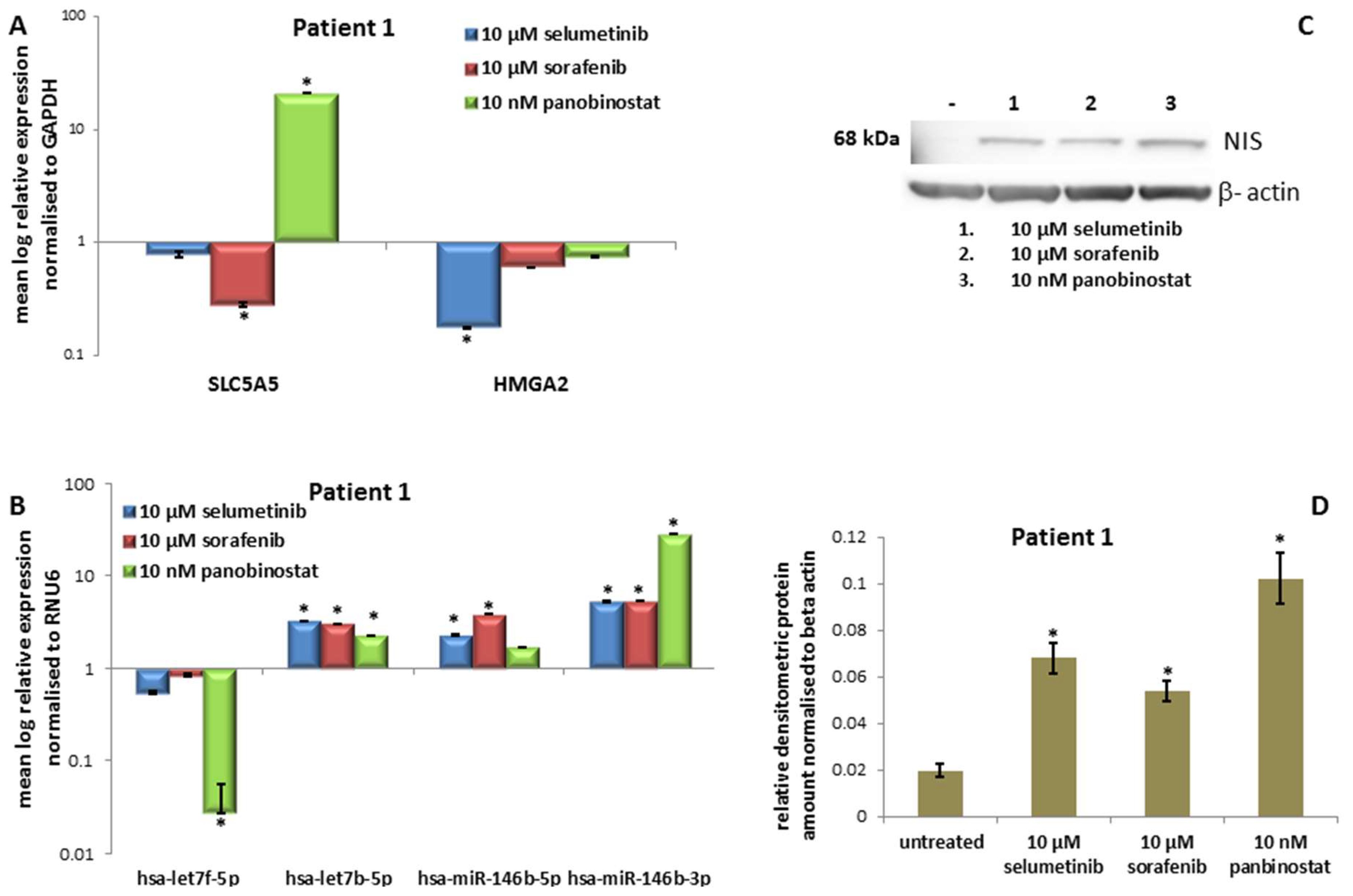
3.1.3. Expression of SLC5A5 (NIS), HMGA2 and miRNAs
3.1.4. Expression of NIS Protein Level
3.2. In Vitro Results of the Second Patient
3.2.1. Effects on Cell Viability of the Individual Tumor Cells Caused by Treatment with Sorafenib, Panobinostat and Selumetinib
3.2.2. Radioiodine Up-Take after Treatment with Sorafenib, Panobinostat and Selumetinib
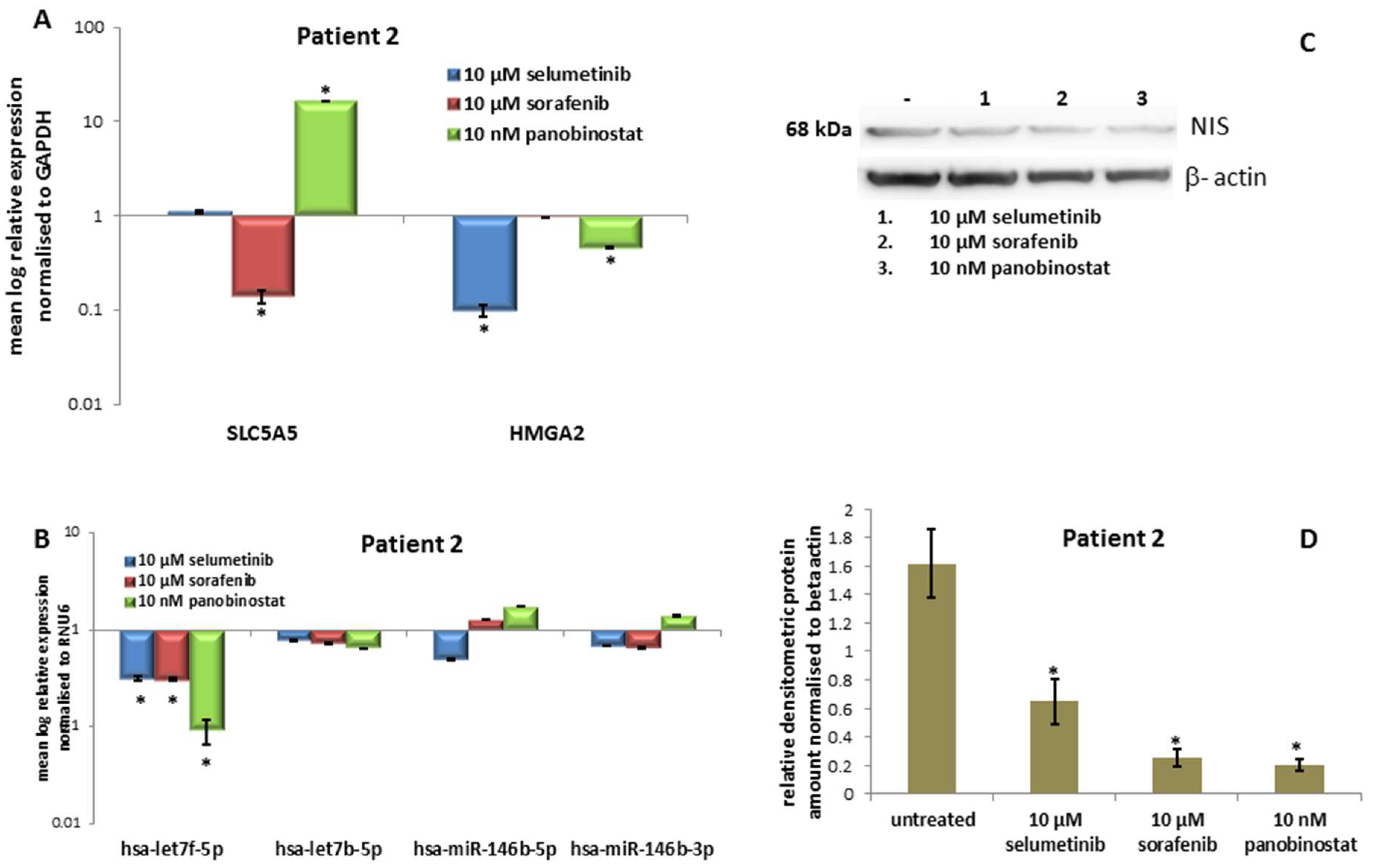
3.2.3. Expression of SLC5A5 (NIS), HMGA2 and Their Regulatory miRNAs
3.2.4. Expression of NIS Protein Level
3.3. In Vitro Results of the Third Patient
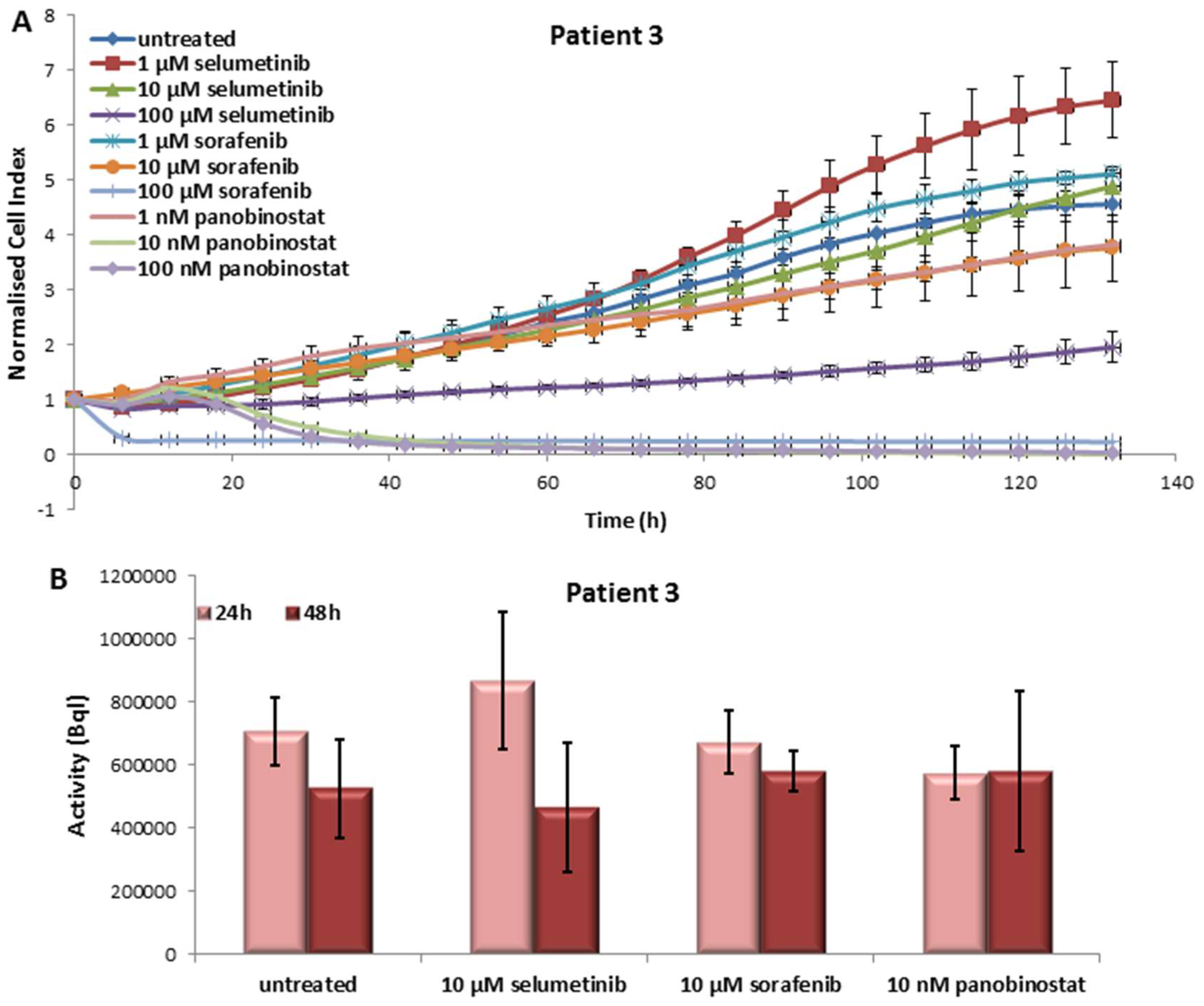
3.3.1. Effects on Cell Viability of the Individual Tumor Cells Caused by Treatment with Sorafenib, Panobinostat and Selumetinib
3.3.2. Radioiodine Up-Take after Treatment with Sorafenib, Panobinostat and Selumetinib
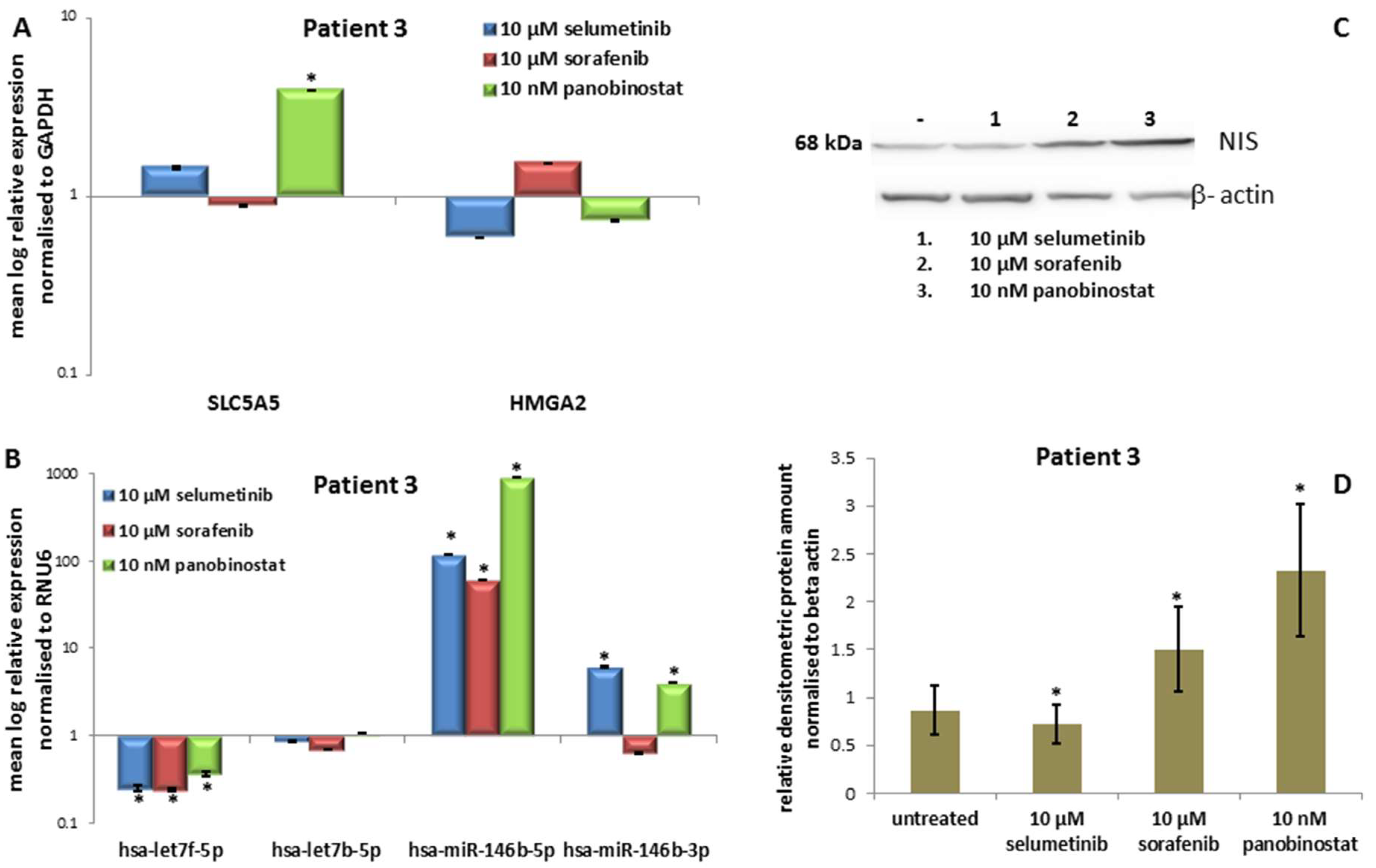
3.3.3. Expression of SLC5A5 (NIS), HMGA2 and miRNAs
3.3.4. Expression of NIS Protein Level
3.4. In Vitro Results of the Fourth Patient
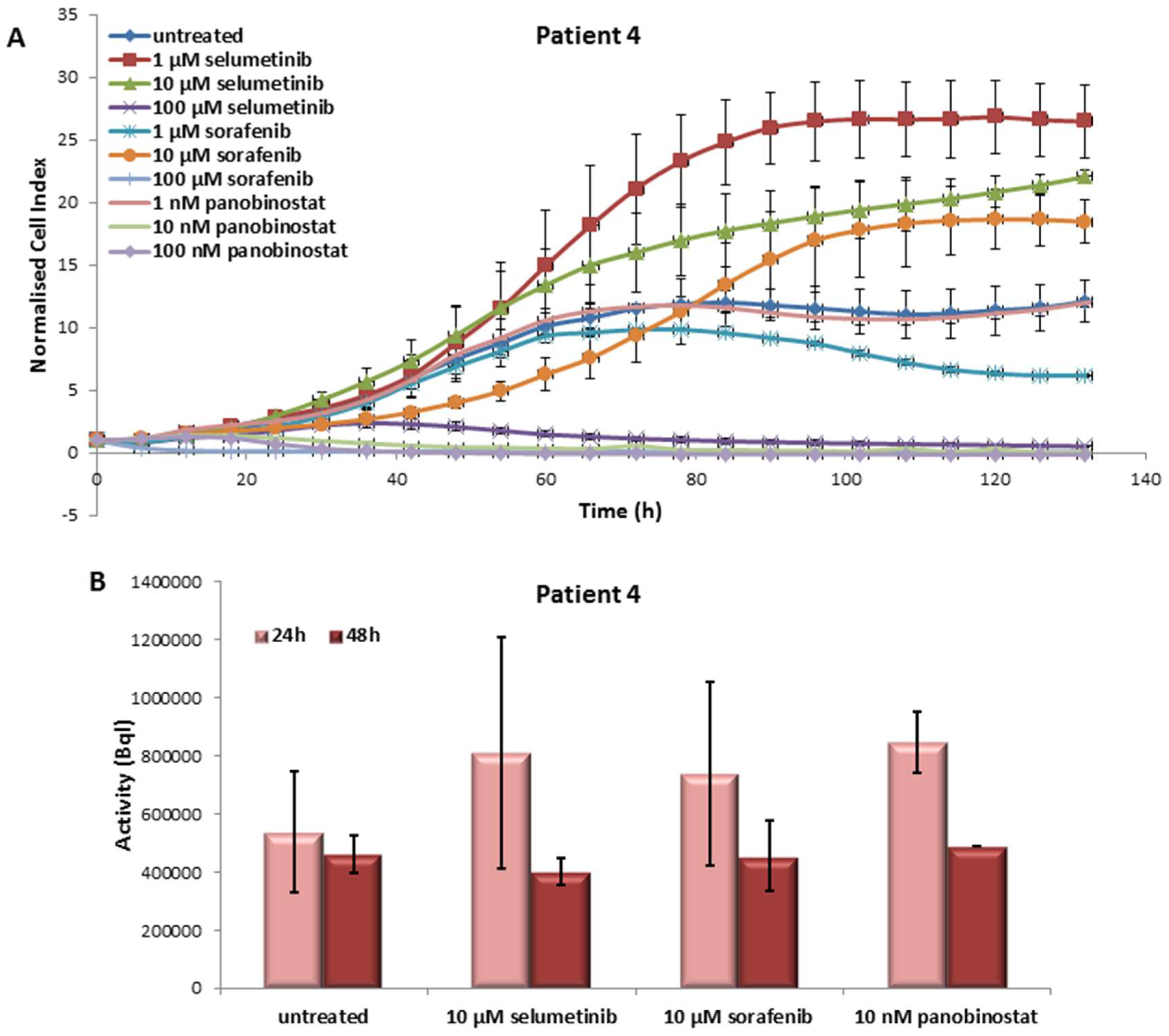
3.4.1. Effects on Cell Viability of the Individual Tumor Cells Caused by Treatment with Sorafenib, Panobinostat and Selumetinib
3.4.2. Radioiodine Up-Take after Treatment with Sorafenib, Panobinostat and Selumetinib
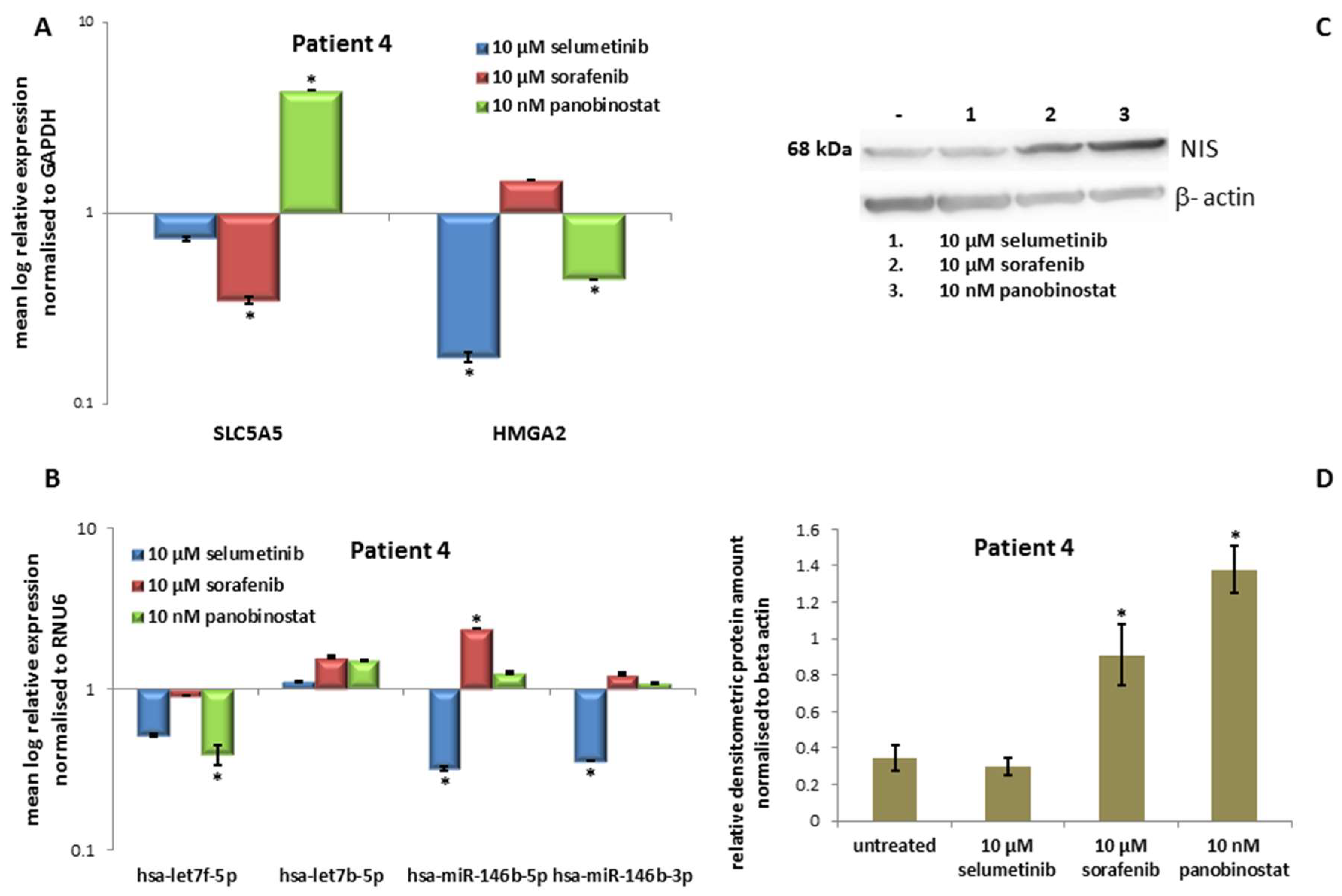
3.4.3. Expression of SLC5A5 (NIS), HMGA2 and miRNAs
3.4.4. Expression of NIS Protein Level
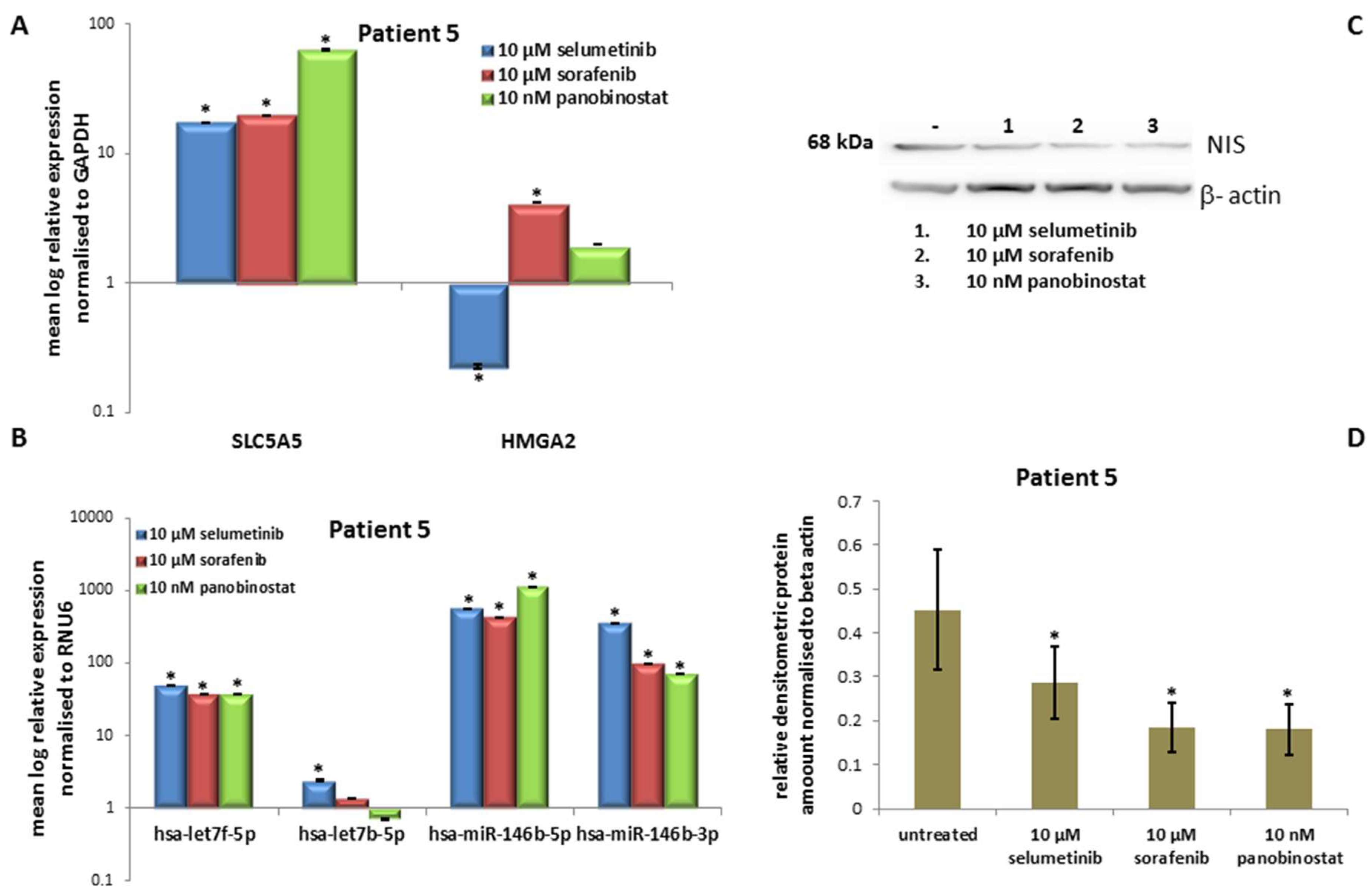
3.5. In Vitro Results of the Fifth Patient
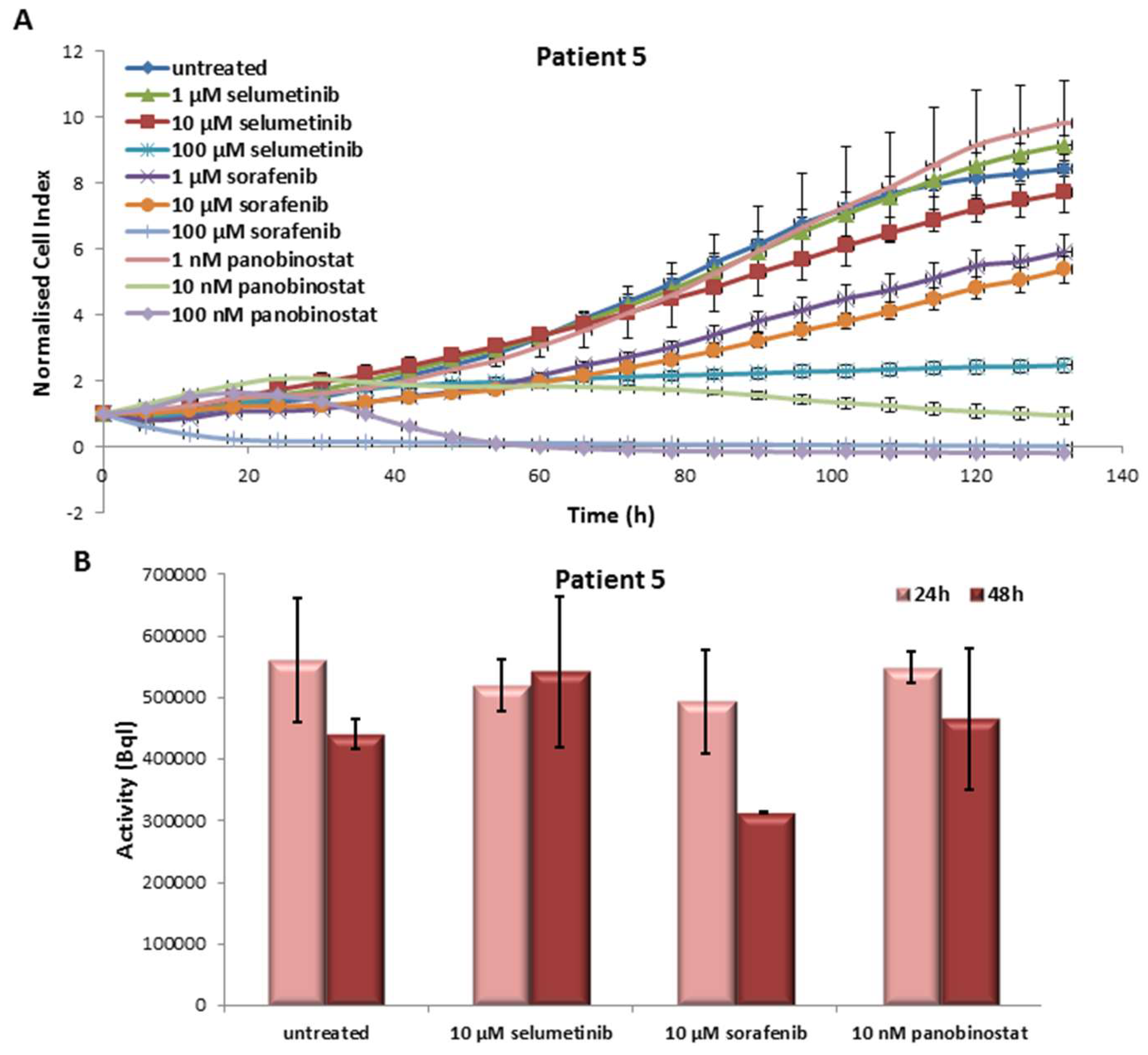
3.5.1. Effects on Cell Viability of the Individual Tumor Cells Caused by Treatment with Sorafenib, Panobinostat and Selumetinib
3.5.2. Radioiodine Up-Take after Treatment with Sorafenib, Panobinostat and Selumetinib
3.5.3. Expression of SLC5A5 (NIS), HMGA2 and miRNAs
3.5.4. Expression of NIS Protein Level
3.6. Human Foreskin Fibroblast
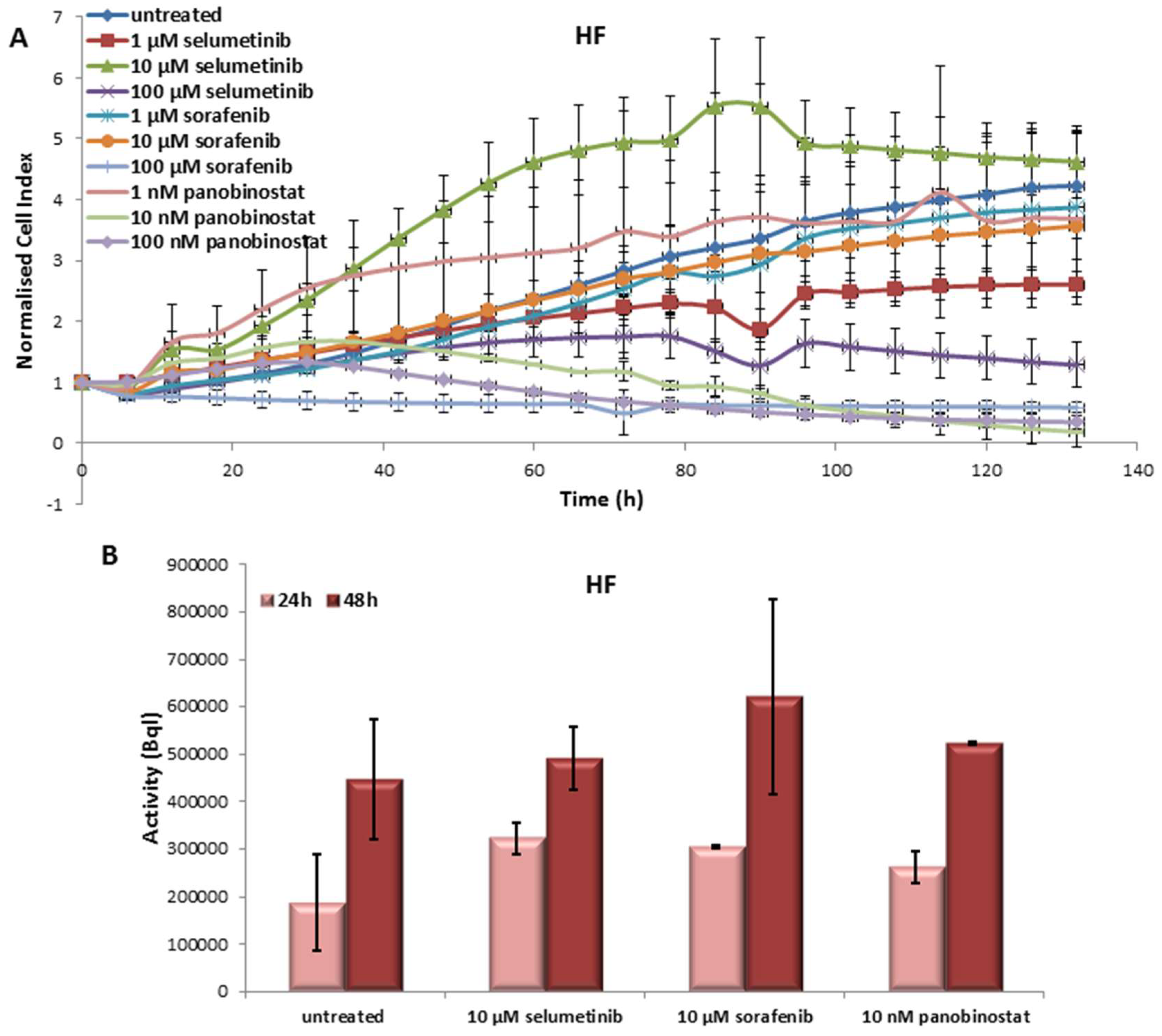
3.6.1. Effects on Cell Viability of Human Fibroblast Caused by Treatment with Sorafenib, Panobinostat and Selumetinib
3.6.2. Radioiodine Up-Take after Treatment with Sorafenib, Panobinostat and Selumetinib
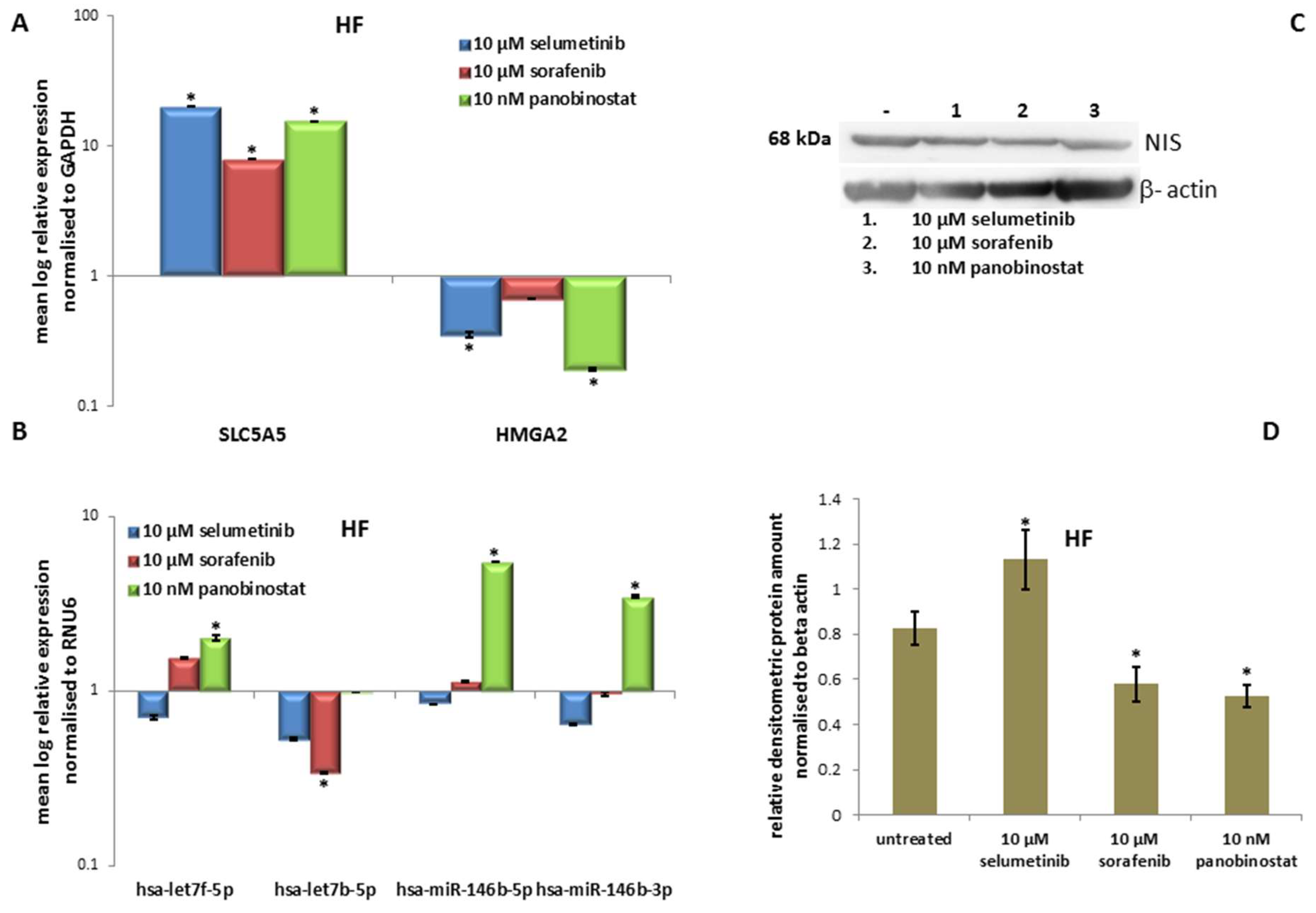
3.6.3. Expression of SLC5A5 (NIS), HMGA2 and miRNAs
3.6.4. Expression of NIS Protein Level
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Nagaiah, G.; Hossain, A.; Mooney, C.J.; Parmentier, J.; Remick, S.C. Anaplastic thyroid cancer: A review of epidemiology, pathogenesis, and treatment. J. Oncol. 2011, 2011, 542358. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.H.-H.; Lo, C.-Y. Surgical options in undifferentiated thyroid carcinoma. World J. Surg. 2007, 31, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Viola, D.; Valerio, L.; Molinaro, E.; Agate, L.; Bottici, V.; Biagini, A.; Lorusso, L.; Cappagli, V.; Pieruzzi, L.; Giani, C.; et al. Treatment of advanced thyroid cancer with targeted therapies: Ten years of experience. Endocr. Relat. Cancer 2016, 23, R185–R205. [Google Scholar] [CrossRef] [PubMed]
- Yaish, P.; Gazit, A.; Gilon, C.; Levitzki, A. Blocking of EGF-dependent cell proliferation by EGF receptor kinase inhibitors. Science 1988, 242, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, S.; Hoffmann, S.; Damanakis, A.I.; Di Fazio, P.; Pfestroff, A.; Luster, M.; Wunderlich, A.; Bartsch, D.K. Individualized multimodal treatment strategy for anaplastic thyroid carcinoma—Case report of long-term remission and review of literature. Int. J. Surg. Case Rep. 2016, 25, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Lirov, R.; Worden, F.P.; Cohen, M.S. The treatment of advanced thyroid cancer in the age of novel targeted therapies. Drugs 2017, 77, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Sasanakietkul, T.; Murtha, T.D.; Javid, M.; Korah, R.; Carling, T. Epigenetic modifications in poorly differentiated and anaplastic thyroid cancer. Mol. Cell Endocrinol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [PubMed]
- Dohán, O.; De la Vieja, A.; Paroder, V.; Riedel, C.; Artani, M.; Reed, M.; Ginter, C.S.; Carrasco, N. The sodium/iodide symporter (NIS): Characterization, regulation, and medical significance. Endocr. Rev. 2003, 24, 48–77. [Google Scholar] [CrossRef] [PubMed]
- Wapnir, I.L.; van de Rijn, M.; Nowels, K.; Amenta, P.S.; Walton, K.; Montgomery, K.; Greco, R.S.; Dohán, O.; Carrasco, N. Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J. Clin. Endocrinol. Metab. 2003, 88, 1880–1888. [Google Scholar] [CrossRef] [PubMed]
- Damanakis, A.I.; Eckhardt, S.; Wunderlich, A.; Roth, S.; Wissniowski, T.T.; Bartsch, D.K.; Di Fazio, P. MicroRNAs let7 expression in thyroid cancer: Correlation with their deputed targets HMGA2 and SLC5A5. J. Cancer Res. Clin. Oncol. 2016, 142, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lv, B.; Chen, B.; Guan, M.; Sun, Y.; Li, H.; Zhang, B.; Ding, C.; He, S.; Zeng, Q. Inhibition of miR-146b expression increases radioiodine-sensitivity in poorly differential thyroid carcinoma via positively regulating NIS expression. Biochem. Biophys. Res. Commun. 2015, 462, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.; Wojcicka, A.; Kotlarek, M.; Zhang, X.; Jazdzewski, K.; Jhiang, S.M. microRNA-339-5p modulates Na+/I- symporter-mediated radioiodide uptake. Endocr. Relat. Cancer 2015, 22, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Fuziwara, C.S.; Kimura, E.T. MicroRNA deregulation in anaplastic thyroid cancer biology. Int. J. Endocrinol. 2014, 2014, 743450. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Visone, R.; Ferracin, M.; Ferraro, A.; Berlingieri, M.T.; Troncone, G.; Chiappetta, G.; Liu, C.G.; Santoro, M.; Negrini, M. MicroRNA deregulation in human thyroid papillary carcinomas. Endocr. Relat. Cancer 2006, 13, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Swierniak, M.; Wojcicka, A.; Czetwertynska, M.; Stachlewska, E.; Maciag, M.; Wiechno, W.; Gornicka, B.; Bogdanska, M.; Koperski, L.; de la Chapelle, A.; et al. In-depth characterization of the microRNA transcriptome in normal thyroid and papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E1401–E1409. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.M.; Fuziwara, C.S.; Yamashita, A.S.; Rezende, E.; da-Silva, M.J.; Kimura, E.T. Effects of let-7 microRNA on cell growth and differentiation of papillary thyroid cancer. Transl. Oncol. 2009, 2, 236–241. [Google Scholar] [PubMed]
- Geraldo, M.V.; Fuziwara, C.S.; Friguglieti, C.U.M.; Costa, R.B.; Kulcsar, M.A.l.V.; Yamashita, A.S.; Kimura, E.T. MicroRNAs miR-146-5p and let-7f as prognostic tools for aggressive papillary thyroid carcinoma: A case report. Arq Bras Endocrinol. Metab. 2012, 56, 552–557. [Google Scholar] [CrossRef]
- Chou, C.-K.; Yang, K.D.; Chou, F.-F.; Huang, C.-C.; Lan, Y.-W.; Lee, Y.-F.; Kang, H.Y.; Liu, R.T. Prognostic implications of miR-146b expression and its functional role in papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2013, 98, E196–E205. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wu, B.; Xiao, K.; Kang, J.; Xie, J.; Zhang, X.; Fan, Y. MiR-146b-5p promotes metastasis and induces epithelial-mesenchymal transition in thyroid cancer by targeting ZNRF3. Cell. Physiol. Biochem. 2015, 35, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Pallante, P.; Sepe, R.; Puca, F.; Fusco, A. High mobility group a proteins as tumor markers. Front. Med. 2015, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Palmieri, D.; Fusco, A. HMGA2: A pituitary tumour subtype-specific oncogene? Mol. Cell. Endocrinol. 2010, 326, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Fedele, M.; Visone, R.; De Martino, I.; Troncone, G.; Palmieri, D.; Battista, S.; Ciarmiello, A.; Pallante, P.; Arra, C.; Melillo, R.M.; et al. HMGA2 induces pituitary tumorigenesis by enhancing E2F1 activity. Cancer Cell 2006, 9, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Di Fazio, P.; Montalbano, R.; Neureiter, D.; Alinger, B.; Schmidt, A.; Merkel, A.L.; Quint, K.; Ocker, M. Downregulation of HMGA2 by the pan-deacetylase inhibitor panobinostat is dependent on hsa-let-7b expression in liver cancer cell lines. Exp. Cell Res. 2012, 318, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.G.; Pugliese, M.; Gargantini, E.; Grange, C.; Bussolati, B.; Asioli, S.; Bosco, O.; Poli, R.; Compagnone, A.; Bandino, A.; et al. Cytotoxic activity of the histone deacetylase inhibitor panobinostat (LBH589) in anaplastic thyroid cancer in vitro and in vivo. Int. J. Cancer 2012, 130, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.; Zheng, Y.; Tyner, J.W.; Chng, W.J.; Chien, W.W.; Gery, S.; Leong, G.; Braunstein, G.D.; Koeffler, H.P. Belinostat and panobinostat (HDACI): In vitro and in vivo studies in thyroid cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Wächter, S.; Damanakis, A.; Elxnat, M.; Roth, S.; Wunderlich, A.; Verburg, F.; Fellinger, S.; Bartsch, D.; Di Fazio, P. Epigenetic modifications in thyroid cancer cells restore NIS and radio-iodine uptake and promote cell death. J. Clin. Med. 2018, 7, 61. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, M.; Fortunati, N.; Germano, A.; Asioli, S.; Marano, F.; Palestini, N.; Frairia, R.; Boccuzzi, G.; Catalano, M.G. Histone deacetylase inhibition affects sodium iodide symporter expression and induces 131I cytotoxicity in anaplastic thyroid cancer cells. Thyroid 2013, 23, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.M.; Ahn, B.-C. Redifferentiation of radioiodine refractory differentiated thyroid cancer for reapplication of I-131 Therapy. Front. Endocrinol. 2017, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Furuya, F.; Shimura, H.; Suzuki, H.; Taki, K.; Ohta, K.; Haraguchi, K.; Onaya, T.; Endo, T.; Kobayashi, T. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology 2004, 145, 2865–2875. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Hardin, H.; Montemayor-Garcia, C.; Asioli, S.; Righi, A.; Maletta, F.; Lloyd, R.V. In situ hybridization analysis of miR-146b-5p and miR-21 in thyroid nodules: Diagnostic implications. Endocr. Pathol. 2015, 26, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Fortunati, N.; Catalano, M.G.; Marano, F.; Mugoni, V.; Pugliese, M.; Bosco, O.; Mainini, F.; Boccuzzi, G. The pan-DAC inhibitor LBH589 is a multi-functional agent in breast cancer cells: Cytotoxic drug and inducer of sodium-iodide symporter (NIS). Breast Cancer Res. Treat. 2010, 124, 667–675. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | 1 | 2 | 3 |
|---|---|---|---|
| Date of diagnosis (month/year) | April/2015 | December/2016 | September/2012 |
| Gender | Male | Male | Male |
| Age at diagnosis (years) | 83 | 81 | 42 |
| Histopathology 1 | ATC | ATC | ATC |
| Tumor origin | Thyroid tissue | Thyroid tissue | Thyroid tissue |
| Tumor stage at diagnosis 2 | UICC Stage: IVC on the right side | UICC Stage: IVB | UICC Stage: IVB |
| TNM-stage | pT4a pN1a (21/32) pM1 | pT4b pN1b (2/5) pM0 | pT4b pN0 (0/36) pM0 |
| Mutation-status | BRAFV600E+ | BRAFV600E− | BRAFV600E− |
| Aerodigestive infiltration | Trachea | - | - |
| Primary surgery | Thyroidectomy + bilateral cervico-central + unilateral cervico-lateral lymphadenectomy | Hemi-thyroidectomy + cervicocentral lymphadenectomy+ resection of the recurrent laryngeal nerve on one side | Thyroidectomy + bilateral cervicocentral + cervicolateral lymphadenectomy |
| Resection status 3 | R2 | R2 | R0 |
| External beam radiation | - | - | Yes |
| Chemotherapy | - | - | Docetaxel + Cisplatin |
| Radioiodine therapy | - | - | - |
| Follow–up | - | - | June/2013: pulmonary metastasis -> video-assisted thoracoscopic wedge resection |
| Individual treatment design | No | No | Sorafenib -> adverse events: polyneuropathy, pain in muscles and bones |
| Palliative treatment | Yes | Yes | - |
| Overall survival after diagnosis (weeks) | 2 | 8 | 282 |
| Patient | 4 | 5 |
|---|---|---|
| Date of diagnosis (month/year) | May/2010 | November/2012 |
| Gender | Male | Female |
| Age at diagnosis (years) | 73 | 44 |
| Histopathology 1 | PDTC | PDTC |
| Tumor origin | Lymph node metastasis | Lymph node metastasis |
| Tumor stage at diagnosis 2 | UICC Stage: I | UICC Stage: I |
| TNM-stage | pT2 pN0 pM0 | pT3 pN0 (0/19) pM0 |
| Mutation-status | BRAFV600E− | BRAFV600E− |
| Aerodigestive infiltration | - | - |
| Primary surgery | Thyroidectomy + bilateral cervico-central lymphadenectomy | Thyroidectomy + bilateral cervicocentral + cervicolateral lymphadenectomy |
| Resection status 3 | R0 | R1 |
| External beam radiation | - | Yes |
| Chemotherapy | - | Docetaxel + Cisplatin |
| Radioiodine therapy | Yes (three times between 2010 and 2011) | Yes (one time in 2012) |
| Follow–up | September/2011: cervical, osseus and pulmonary metastases -> treatment with sorafenib -> due to adverse events chemotherapy with carboplatin + etoposid September/2014: reexploration of the neck with tumordebulking to establish primary cell culture -> again therapy with sorafenib | November/2013: bone and pulmonary metastases -> surgical resection July/2014: osseus, hepatic and pulmonary progress -> re-exploration of the neck with resection of cervical lymphnode metastases to establish primary cell cultures October/2014: progress of metastases -> treatment with sorafenib April/2015: progress of metastases -> treatment with Lenvatinib |
| Individual treatment design | Sorafenib -> adverse events: polyneuropathy, dry skin, sore throat | Sorafenib -> adverse events: polyneuropathy, diarrhea, sore throat Lenvatinib -> adverse events |
| Palliative treatment | - | Yes (since September/2015) |
| Overall survival after diagnosis (weeks) | 396 | 174 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wächter, S.; Wunderlich, A.; Roth, S.; Mintziras, I.; Maurer, E.; Hoffmann, S.; Verburg, F.A.; Fellinger, S.A.; Holzer, K.; Bartsch, D.K.; et al. Individualised Multimodal Treatment Strategies for Anaplastic and Poorly Differentiated Thyroid Cancer. J. Clin. Med. 2018, 7, 115. https://doi.org/10.3390/jcm7050115
Wächter S, Wunderlich A, Roth S, Mintziras I, Maurer E, Hoffmann S, Verburg FA, Fellinger SA, Holzer K, Bartsch DK, et al. Individualised Multimodal Treatment Strategies for Anaplastic and Poorly Differentiated Thyroid Cancer. Journal of Clinical Medicine. 2018; 7(5):115. https://doi.org/10.3390/jcm7050115
Chicago/Turabian StyleWächter, Sabine, Annette Wunderlich, Silvia Roth, Ioannis Mintziras, Elisabeth Maurer, Sebastian Hoffmann, Frederik A. Verburg, Sebastian A. Fellinger, Katharina Holzer, Detlef K. Bartsch, and et al. 2018. "Individualised Multimodal Treatment Strategies for Anaplastic and Poorly Differentiated Thyroid Cancer" Journal of Clinical Medicine 7, no. 5: 115. https://doi.org/10.3390/jcm7050115