Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications


 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
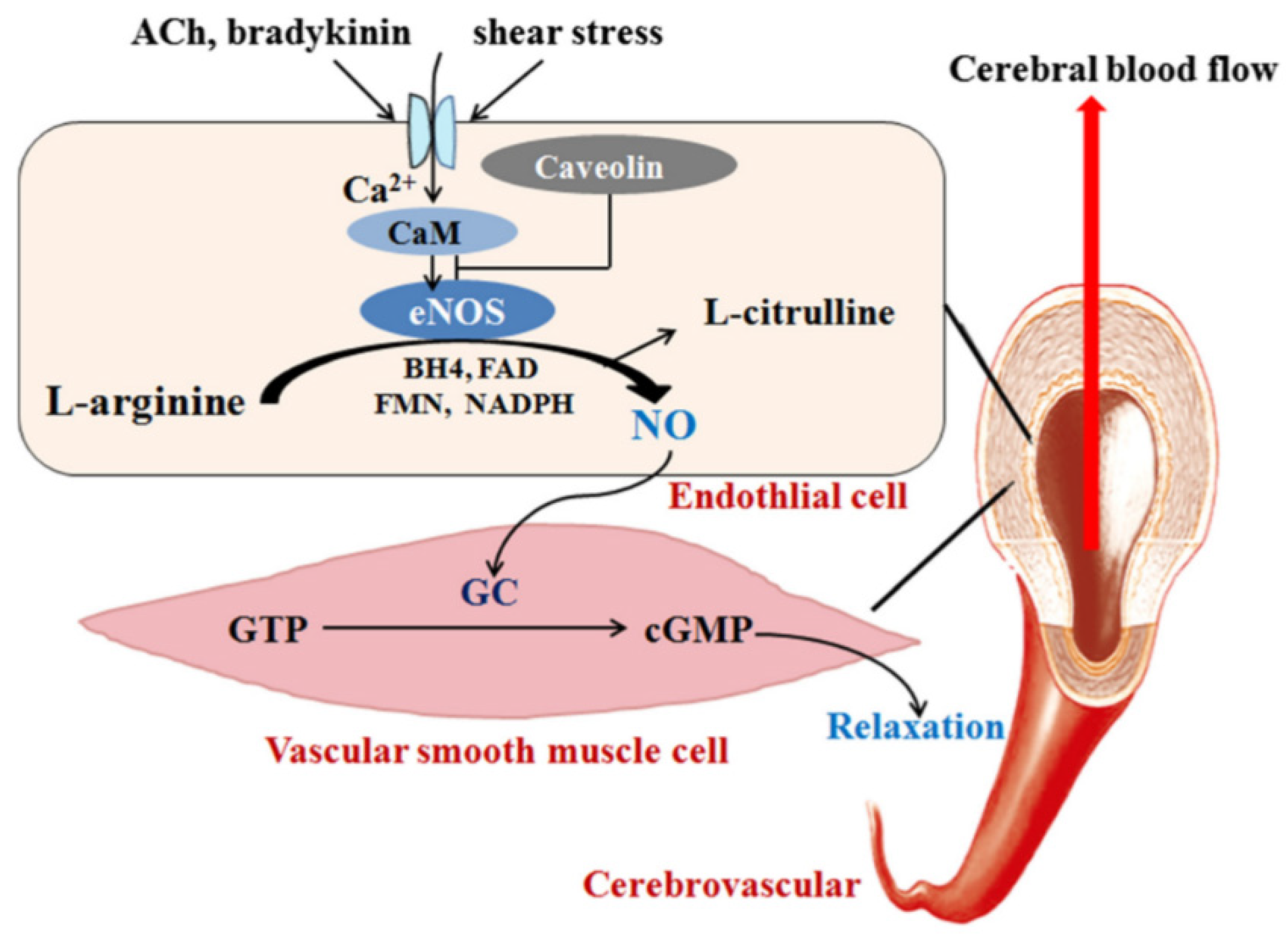
2. Signaling Pathways and Molecules Mediating the Effect of Statins on NO Production
3. Therapeutic Implications of Statin-Induced Elevation of NO
3.1. Heart Failure
3.2. Hypertension
3.3. Atherosclerosis
3.4. Sepsis
3.5. Cirrhosis
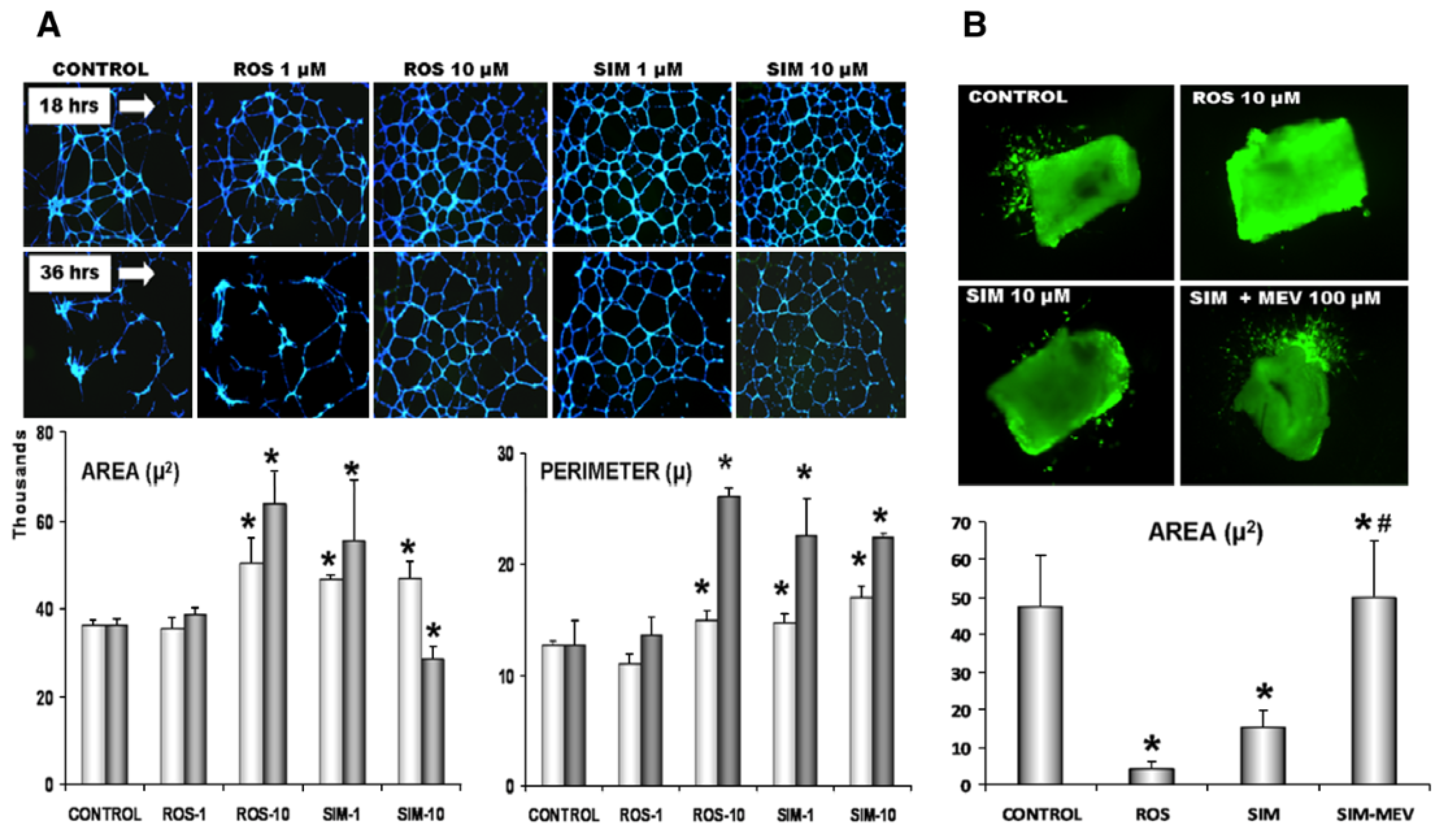
3.6. Therapeutic Induction of Angiogenesis and Vasculogenesis
3.7. Neuroprotection
3.8. Cancer Treatment
4. NO-Releasing Statins to Augment the NO-Mediated Therapeutic Effects of Statin
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HMG | Hydroxymethylglutaryl |
| NO | Nitric Oxide |
| eNOS | Endothelial NO Synthase |
| ROS | Reactive Oxygen Species |
| ROCK | Rho-Associated Protein Kinase |
| PI3k | Phosphoinositide 3-kinase |
| Akt | Protein kinase B |
| AMPK | AMP-Activated Protein Kinase |
| GGPP | Geranylgeranyl Pyrophosphate |
| EPC | Endothelial Progenitor Cells |
| HUVECs | Human Umbilical Vein Endothelial Cells |
| Nox | NADPH Oxidase |
| HO-1 | Heme Oxygenase 1 |
| BH4 | Tetrahydrobiopterin |
| GTP | Guanosine-5-triphosphate |
| GTPCH | GTP Cyclohydrolase |
| CRP | C Reactive Protein |
| Cav-1 | Caveolin-1 |
| SDF-1α | Stromal Cell-Derived Factor 1 α |
| CXCR-4 | CXC Chemokine Receptor-4 |
| LV | Left Ventricle |
| CHF | Chronic Heart Failure |
| MI | Myocardial Infarction |
| cGMP | Cyclic 3,5-guanosine Monophosphate |
| ApoE | Apolipoprotein E |
| MSCs | Mesenchymal Stem Cells |
| mTOR | Mammalian Target of Rapamycin |
| BDNF | Brain-Derived Neurotrophic Factor |
| GDNF | Glial Cell Line-Derived Neurotrophic Factor |
| iNOS | Inducible NOS |
| VEGF | Vascular Endothelial Growth Factor |
References
- Banach, M.; Aronow, W.S.; Serban, C.; Sahabkar, A.; Rysz, J.; Voroneanu, L.; Covic, A. Lipids, blood pressure and kidney update 2014. Pharmacol. Res. 2015, 95–96, 111–125. [Google Scholar] [CrossRef]
- Sahebkar, A.; Watts, G.F. New therapies targeting apoB metabolism for high-risk patients with inherited dyslipidaemias: what can the clinician expect? Cardiovasc Drugs Ther. 2013, 27, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Yu, X.; Zhang, B.; Zhang, H.; Fang, Y.; Liu, S.; Liu, T.; Ding, X. Atorvastatin improves survival of implanted stem cells in a rat model of renal ischemia-reperfusion injury. Am. J. Nephrol. 2014, 39, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Aktas, O.; Albrecht, P.; Hartung, H.P. Optic neuritis as a phase 2 paradigm for neuroprotection therapies of multiple sclerosis: Update on current trials and perspectives. Curr. Opin. Neurol. 2016, 29, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Chruściel, P.; Sahebkar, A.; Rembek-Wieliczko, M.; Serban, M.C.; Ursoniu, S.; Mikhailidis, D.P.; Jones, S.R.; Mosteoru, S.; Blaha, M.J.; Martin, S.S.; et al. Impact of statin therapy on plasma adiponectin concentrations: A systematic review and meta-analysis of 43 randomized controlled trial arms. Atherosclerosis 2016, 253, 194–208. [Google Scholar] [CrossRef]
- Parizadeh, S.M.R.; Azarpazhooh, M.R.; Moohebati, M.; Nematy, M.; Ghayour-Mobarhan, M.; Tavallaie, S.; Rahsepar, A.A.; Amini, M.; Sahebkar, A.; Mohammadi, M.; et al. Simvastatin therapy reduces prooxidant-antioxidant balance: Results of a placebo-controlled cross-over trial. Lipids 2011, 46, 333–340. [Google Scholar] [CrossRef]
- Sahebkar, A.; Kotani, K.; Serban, C.; Ursoniu, S.; Mikhailidis, D.P.; Jones, S.R.; Ray, K.K.; Blaha, M.J.; Rysz, J.; Toth, P.P.; et al. Statin therapy reduces plasma endothelin-1 concentrations: A meta-analysis of 15 randomized controlled trials. Atherosclerosis 2015, 241, 433–442. [Google Scholar] [CrossRef]
- Sahebkar, A.; Serban, C.; Mikhailidis, D.P.; Undas, A.; Lip, G.Y.H.; Muntner, P.; Bittner, V.; Ray, K.K.; Watts, G.F.; Hovingh, G.K.; et al. Association between statin use and plasma d-dimer levels: A systematic review and meta-analysis of randomised controlled trials. Thromb. Haemost. 2015, 114, 546–557. [Google Scholar] [CrossRef]
- Kabaklic, A.; Fras, Z. Moderate-dose atorvastatin improves arterial endothelial function in patients with angina pectoris and normal coronary angiogram: A pilot study. Arch. Med. Sci. 2017, 13, 827–836. [Google Scholar] [CrossRef]
- Mehta, J.L.; Li, D.Y.; Chen, H.J.; Joseph, J.; Romeo, F. Inhibition of LOX-1 by statins may relate to upregulation of eNOS. Biochem. Biophys. Res. Commun. 2001, 289, 857–861. [Google Scholar] [CrossRef]
- Kosmidou, I.; Moore, J.P.; Weber, M.; Searles, C.D. Statin treatment and 3’ polyadenylation of eNOS mRNA. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2642–2649. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Gertz, K.; Huang, P.; Nickenig, G.; Bohm, M.; Dirnagl, U.; Endres, M. Atorvastatin upregulates type III nitric oxide synthase in thrombocytes, decreases platelet activation, and protects from cerebral ischemia in normocholesterolemic mice. Stroke 2000, 31, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Liao, J.K. Rho GTPases, statins, and nitric oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Endres, M.; Custodis, F.; Gertz, K.; Nickenig, G.; Liao, J.K.; Böhm, M. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of Rho GTPase gene transcription. Circulation 2000, 102, 3104–3110. [Google Scholar] [CrossRef]
- Geng, J.; Xu, H.; Yu, X.; Xu, G.; Cao, H.; Lin, G.; Sui, D. Rosuvastatin protects against oxidized lowdensity lipoproteininduced endothelial cell injury of atherosclerosis in vitro. Mol. Med. Rep. 2019, 19, 432–440. [Google Scholar] [CrossRef]
- Meda, C.; Plank, C.; Mykhaylyk, O.; Schmidt, K.; Mayer, B. Effects of statins on nitric oxide/cGMP signaling in human umbilical vein endothelial cells. Pharmacol. Rep. 2010, 62, 100–112. [Google Scholar] [CrossRef]
- Rossoni, L.V.; Wareing, M.; Wenceslau, C.F.; Al-Abri, M.; Cobb, C.; Austin, C. Acute simvastatin increases endothelial nitric oxide synthase phosphorylation via AMP-activated protein kinase and reduces contractility of isolated rat mesenteric resistance arteries. Clin. Sci. 2011, 121, 449–458. [Google Scholar] [CrossRef]
- Lee, T.-S.; Chang, C.-C.; Zhu, Y.; Shyy, J. Simvastatin induces heme oxygenase-1 a novel mechanism of vessel protection. Circulation 2004, 110, 1296–1302. [Google Scholar] [CrossRef]
- Piechota-Polanczyk, A.; Kopacz, A.; Kloska, D.; Zagrapan, B.; Neumayer, C.; Grochot-Przeczek, A.; Huk, I.; Brostjan, C.; Dulak, J.; Jozkowicz, A. Simvastatin treatment upregulates HO-1 in patients with abdominal aortic aneurysm but independently of Nrf2. Oxidative Med. Cell. Longev. 2018, 2018, 2028936. [Google Scholar] [CrossRef]
- Laufs, U.; Liao, J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J. Biol. Chem. 1998, 273, 24266–24271. [Google Scholar] [CrossRef]
- Downward, J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr. Opin. Cell Biol. 1998, 10, 262–267. [Google Scholar] [CrossRef]
- Llevadot, J.; Murasawa, S.; Kureishi, Y.; Uchida, S.; Masuda, H.; Kawamoto, A.; Walsh, K.; Isner, J.M.; Asahara, T. HMG-CoA reductase inhibitor mobilizes bone marrow–derived endothelial progenitor cells. J. Clin. Investig. 2001, 108, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.; Kitajima, I.; Wang, Z. Effects of different statins on endothelial nitric oxide synthase and AKT phosphorylation in endothelial cells. Int. J. Cardiol. 2008, 127, 33–39. [Google Scholar] [CrossRef]
- Harris, M.B.; Blackstone, M.A.; Sood, S.G.; Li, C.; Goolsby, J.M.; Venema, V.J.; Kemp, B.E.; Venema, R.C. Acute activation and phosphorylation of endothelial nitric oxide synthase by HMG-CoA reductase inhibitors. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H560–H566. [Google Scholar] [CrossRef]
- Wolfrum, S.; Dendorfer, A.; Rikitake, Y.; Stalker, T.J.; Gong, Y.; Scalia, R.; Dominiak, P.; Liao, J.K. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1842–1847. [Google Scholar] [CrossRef]
- Brouet, A.; Sonveaux, P.; Dessy, C.; Moniotte, S.; Balligand, J.-L.; Feron, O. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide; mediated effects of statins. Circ. Res. 2001, 89, 866–873. [Google Scholar] [CrossRef]
- Sun, W.; Lee, T.S.; Zhu, M.; Gu, C.; Wang, Y.; Zhu, Y.; Shyy, J.Y. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation 2006, 114, 2655–2662. [Google Scholar] [CrossRef]
- Kamel, W.A.-E.; Sugihara, E.; Yamaguchi, S.I.; Nobusue, H.; Maki, K.; Muto, A.; Saya, H.; Shimizu, T. Statins induce apoptosis in osteosarcoma cells by activation of Ampk and p38-MAPK via suppression of mevalonate pathway. Cancer Res. 2016, 76, 4182. [Google Scholar] [CrossRef]
- Wang, J.-C.; Li, X.-X.; Sun, X.; Li, G.-Y.; Sun, J.-L.; Ye, Y.-P.; Cong, L.-L.; Li, W.-M.; Lu, S.-Y.; Feng, J.; et al. Activation of AMPK by simvastatin inhibited breast tumor angiogenesis via impeding HIF-1α-induced pro-angiogenic factor. Cancer Sci. 2018, 109, 1627–1637. [Google Scholar] [CrossRef]
- Dong, Q.; Yang, Y.; Song, L.; Qian, H.; Xu, Z. Atorvastatin prevents mesenchymal stem cells from hypoxia and serum-free injury through activating amp-activated protein kinase. Int. J. Cardiol. 2011, 153, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Yang, Y.J.; Dong, Q.T.; Qian, H.Y.; Xu, H.; Meng, X.M.; Tang, Y. Atorvastatin protects swine bone marrow mesenchymal stem cells from apoptosis through AMPK but not PI3K/Akt pathway. Zhonghua Xin Xue Guan Bing Za Zhi 2011, 39, 1033–1038. [Google Scholar] [PubMed]
- Yu, B.; Liu, D.; Zhang, H.; Xie, D.; Nie, W.; Shi, K.; Yang, P. Anti-hypertrophy effect of atorvastatin on myocardium depends on AMPK activation-induced miR-143-3p suppression via Foxo1. Biomed Pharmacother. 2018, 106, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Cerda, A.; Fajardo, C.M.; Basso, R.G.; Hirata, M.H.; Hirata, R.D.C. Role of microRNAs 221/222 on statin induced nitric oxide release in human endothelial cells. Arq. Bras. Cardiol. 2015, 104, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, H.; Ren, J.; Song, J.; Zhang, F.; Zhang, J.; Lee, C.; Li, S.; Geng, Q.; Cao, C.; et al. Effects of statin on circulating microRNAome and predicted function regulatory network in patients with unstable angina. BMC Med. Genom. 2015, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davignon, J.; Jacob, R.F.; Mason, R.P. The antioxidant effects of statins. Coron. Artery Dis. 2004, 15, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Moon, G.J.; Kim, S.J.; Cho, Y.H.; Ryoo, S.; Bang, O.Y. Antioxidant effects of statins in patients with atherosclerotic cerebrovascular disease. J. Clin. Neurol. 2014, 10, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Zhang, X.; Meng, X.; Lu, L.; Mai, D.; Qu, S. Simvastatin inhibits activation of NADPH oxidase/p38 MAPK pathway and enhances expression of antioxidant protein in parkinson disease models. Front. Mol. Neurosci. 2018, 11, 165. [Google Scholar] [CrossRef] [Green Version]
- Wassmann, S.; Laufs, U.; Bäumer, A.T.; Müller, K.; Konkol, C.; Sauer, H.; Böhm, M.; Nickenig, G. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: Involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol. Pharmacol. 2001, 59, 646–654. [Google Scholar] [CrossRef]
- Takeno, A.; Kanazawa, I.; Tanaka, K.; Notsu, M.; Yokomoto-Umakoshi, M.; Sugimoto, T. Simvastatin rescues homocysteine-induced apoptosis of osteocytic MLO-Y4 cells by decreasing the expressions of NADPH oxidase 1 and 2. Endocr. J. 2016, 63, 389–395. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleda, S.; De Haro, J.; Florez, A.; Varela, C.; Esparza, L.; Acin, F. Long-term pleiotropic effect of statins upon nitric oxide and C-reactive protein levels in patients with peripheral arterial disease. Heart Asia 2011, 3, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Martínez Aguilar, E.; De Haro Miralles, J.; Flórez González, A.; Varela Casariego, C.; Bleda Moreno, S.; Acín García, F. In vivo confirmation of the Role of statins in reducing nitric oxide and C-reactive protein levels in peripheral arterial disease. Eur. J. Vasc. Endovasc. Surg. 2009, 37, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plenz, G.A.; Hofnagel, O.; Robenek, H. Differential modulation of caveolin-1 expression in cells of the vasculature by statins. Circulation 2004, 109, e7–e8. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.H.; Murray, F.; Insel, P.A. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 359–391. [Google Scholar] [CrossRef] [Green Version]
- Stillwell, W. Long-range membrane properties. In An Introduction to Biological Membranes; Stillwell, W., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 221–245. [Google Scholar]
- Gratton, J.-P.; Bernatchez, P.; Sessa, W.C. Caveolae and caveolins in the cardiovascular system. Circ. Res. 2004, 94, 1408–1417. [Google Scholar] [CrossRef]
- Feron, O.; Dessy, C.; Moniotte, S.; Desager, J.P.; Balligand, J.L. Hypercholesterolemia decreases nitric oxide production by promoting the interaction of caveolin and endothelial nitric oxide synthase. J. Clin. Investig. 1999, 103, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Pelat, M.; Dessy, C.; Massion, P.; Desager, J.P.; Feron, O.; Balligand, J.L. Rosuvastatin decreases caveolin-1 and improves nitric oxide-dependent heart rate and blood pressure variability in apolipoprotein E-/- mice in vivo. Circulation 2003, 107, 2480–2486. [Google Scholar] [CrossRef] [Green Version]
- Mottaghi, S.; Larijani, B.; Sharifi, A.M. Atorvastatin: An efficient step forward in mesenchymal stem cell therapy of diabetic retinopathy. Cytotherapy 2013, 15, 263–266. [Google Scholar] [CrossRef]
- Cai, A.; Qiu, R.; Li, W.; Zheng, D.; Dong, Y.; Yu, D.; Huang, Y.; Rao, S.; Zhou, Y.; Mai, W. Atorvastatin treatment of rats with ischemia-reperfusion injury improves adipose-derived mesenchymal stem cell migration and survival via the SDF-1α/CXCR-4 axis. PLoS ONE 2013, 8, e79100. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Gyurko, R.; Han, F.; Scherrer-Crosbie, M.; Aretz, T.H.; Hajjar, R.; Picard, M.H.; Huang, P.L. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 2001, 104, 448–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strazzullo, P.; Kerry, S.M.; Barbato, A.; Versiero, M.; D’Elia, L.; Cappuccio, F.P. Do statins reduce blood pressure: A meta-analysis of randomized, controlled trials. Hypertension 2007, 49, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Golomb, B.A.; Dimsdale, J.E.; White, H.L.; Ritchie, J.B.; Criqui, M.H. Reduction in blood pressure with statins: Results from the UCSD Statin Study, a randomized trial. Arch. Intern. Med. 2008, 168, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Cebova, M.; Rehakova, R.; Kosutova, M.; Pechanova, O. Simvastatin does not affect nitric oxide generation increased by sesame oil in obese Zucker rats. Oxid. Med. Cell. Longev. 2018, 2018, 5413423. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T. The role of the nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Weis, M.; Heeschen, C.; Glassford, A.J.; Cooke, J.P. Statins have biphasic effects on angiogenesis. Circulation 2002, 105, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Bauersachs, J.; Galuppo, P.; Fraccarollo, D.; Christ, M.; Ertl, G. Improvement of left ventricular remodeling and function by hydroxymethylglutaryl coenzyme a reductase inhibition with cerivastatin in rats with heart failure after myocardial infarction. Circulation 2001, 104, 982–985. [Google Scholar] [CrossRef] [Green Version]
- Hayashidani, S.; Tsutsui, H.; Shiomi, T.; Suematsu, N.; Kinugawa, S.; Ide, T.; Wen, J.; Takeshita, A. Fluvastatin, a 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitor, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2002, 105, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Hua, P.; Liu, J.; Tao, J.; Zou, R.; Lin, X.; Zhang, D.; Yang, S. Efficacy and mechanism of preoperative simvastatin therapy on myocardial protection after extracorporeal circulation. BioMed Res. Int. 2017, 2017, 6082430. [Google Scholar] [CrossRef] [Green Version]
- Winzer, E.B.; Gaida, P.; Höllriegel, R.; Fischer, T.; Linke, A.; Schuler, G.; Adams, V.; Erbs, S. Impact of Rosuvastatin Treatment on HDL-Induced PKC-βII and eNOS Phosphorylation in Endothelial Cells and Its Relation to Flow-Mediated Dilatation in Patients with Chronic Heart Failure. Cardiol. Res. Pract. 2016, 2016, 48261021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmadbouh, I.; Mansour, M.; Nabeh, M.; Faried, W.; Abdelsabour, A.; Omar, A. Atorvastatin improves cardiac function and remodeling in chronic non-ischemic heart failure: A clinical and pre-clinical study. Egypt. Heart J. 2015, 67, 289–298. [Google Scholar] [CrossRef] [Green Version]
- An, L.; An, S.; Jia, Z.; Wang, H.; Yang, Z.; Xu, C.; Teng, X.; Wang, J.; Liu, X.; Cao, Q.; et al. Atorvastatin improves left ventricular remodeling and cardiac function in rats with congestive heart failure by inhibiting RhoA/Rho kinase-mediated endothelial nitric oxide synthase. Exp. Ther. Med. 2019, 17, 960–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Haehling, S.; Anker, S.D.; Bassenge, E. Statins and the role of nitric oxide in chronic heart failure. Heart Fail. Rev. 2003, 8, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Laufs, U.; Huang, Z.; Nakamura, T.; Huang, P.; Moskowitz, M.A.; Liao, J.K. Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1998, 95, 8880–8885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szyguła-Jurkiewicz, B.; Szczurek, W.; Król, B.; Zembala, M. The role of statins in chronic heart failure. Kardiochirurgia Torakochirurgia Polska 2014, 11, 301–305. [Google Scholar] [CrossRef]
- Westman, P.C.; Lipinski, M.J. The use of statins in patients with heart failure: More questions than answers. J. Thorac. Dis. 2015, 7, 1687–1690. [Google Scholar] [CrossRef]
- Bielecka-Dabrowa, A.; Fabis, J.; Mikhailidis, D.P.; von Haehling, S.; Sahebkar, A.; Rysz, J.; Banach, M. Prosarcopenic Effects of Statins May Limit Their Effectiveness in Patients with Heart Failure. Trends Pharmacol. Sci. 2018, 39, 331–353. [Google Scholar] [CrossRef]
- Lorkowska, B.; Chlopicki, S. Statins as coronary vasodilators in isolated bovine coronary arteries--involvement of PGI2 and NO. ProstaglandinsLeukot. Essent. Fat. Acids 2005, 72, 133–138. [Google Scholar] [CrossRef]
- Koh, K.K. Effects of statins on vascular wall: Vasomotor function, inflammation, and plaque stability. Cardiovasc. Res. 2000, 47, 648–657. [Google Scholar] [CrossRef]
- Vladimirova-Kitova, L.G.; Kitov, S.I. Resistance of statin therapy, and methods for its influence. In Hypercholesterolemia; Kumar, S.A., Ed.; IntechOpen: London, UK, 2015. [Google Scholar]
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting early atherosclerosis: A focus on oxidative stress and inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef] [PubMed]
- Zaric, B.; Obradovic, M.; Trpkovic, A.; Banach, M.; Mikhailidis, D.P.; Isenovic, E. Endothelial dysfunction in dyslipidaemia: Molecular mechanisms and clinical implications. Curr. Med. Chem. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Csiszar, A.; Huang, A.; Kaminski, P.M.; Wolin, M.S.; Koller, A. High pressure induces superoxide production in isolated arteries via protein kinase c–dependent activation of NAD (P) H oxidase. Circulation 2003, 108, 1253–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD (P) H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, E.C.; Bertolami, M.C.; Faludi, A.A.; Salem, M.; Bersch, D.; Abdalla, D.S.P. Effects of simvastatin and l -arginine on vasodilation, nitric oxide metabolites and endogenous NOS inhibitors in hypercholesterolemic subjects. Free Radic. Res. 2003, 37, 529–536. [Google Scholar] [CrossRef]
- Lee, S.-E.; Chang, H.-J.; Sung, J.M.; Park, H.-B.; Heo, R.; Rizvi, A.; Lin, F.Y.; Kumar, A.; Hadamitzky, M.; Kim, Y.J.; et al. Effects of statins on coronary atherosclerotic plaques. JACC Cardiovasc. Imaging 2018, 11, 1475–1484. [Google Scholar] [CrossRef]
- Bittencourt, M.S.; Cerci, R.J. Statin effects on atherosclerotic plaques: Regression or healing? BMC Med. 2015, 13, 260. [Google Scholar] [CrossRef] [Green Version]
- Heeba, G.; Moselhy, M.E.; Hassan, M.; Khalifa, M.; Gryglewski, R.; Malinski, T. Anti-atherogenic effect of statins: Role of nitric oxide, peroxynitrite and haem oxygenase-1. Br. J. Pharm. 2009, 156, 1256–1266. [Google Scholar] [CrossRef] [Green Version]
- Rasmusen, C.; Cynober, L.; Couderc, R. Arginine and statins: Relationship between the nitric oxide pathway and the atherosclerosis development. Ann. Biol. Clin. 2005, 63, 443–455. [Google Scholar]
- McGown, C.C.; Brookes, Z.L.S. Beneficial effects of statins on the microcirculation during sepsis: The role of nitric oxide. Br. J. Anaesth. 2007, 98, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafra, C.; Abraldes, J.G.; Turnes, J.; Berzigotti, A.; Fernández, M.; García-Pagán, J.C.; Rodés, J.; Bosch, J. Simvastatin enhances hepatic nitric oxide production and decreases the hepatic vascular tone in patients with cirrhosis. Gastroenterology 2004, 126, 749–755. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Rodríguez-Vilarrupla, A.; Graupera, M.; Zafra, C.; García-Calderó, H.; García-Pagán, J.C.; Bosch, J. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J. Hepatol. 2007, 46, 1040–1046. [Google Scholar] [CrossRef]
- Trebicka, J.; Hennenberg, M.; Laleman, W.; Shelest, N.; Biecker, E.; Schepke, M.; Nevens, F.; Sauerbruch, T.; Heller, J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007, 46, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.E. The Use of Statins in Patients with Cirrhosis. Gastroenterol. Hepatol. 2018, 14, 485–487. [Google Scholar]
- Chu, H.; Wang, Y. Therapeutic angiogenesis: Controlled delivery of angiogenic factors. Ther. Deliv. 2012, 3, 693–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Cheng, M.; Liao, Y.-H.; Hu, Y.; Wu, M.; Wang, Q.; Qin, B.; Wang, H.; Zhu, Y.; Gao, X.-M.; et al. Rosuvastatin Enhances Angiogenesis via eNOS-Dependent Mobilization of Endothelial Progenitor Cells. PLoS ONE 2013, 8, e63126. [Google Scholar] [CrossRef] [Green Version]
- Urbich, C.; Dernbach, E.; Zeiher, A.M.; Dimmeler, S. Double-edged role of statins in angiogenesis signaling. Circ. Res. 2002, 90, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Llevadot, J.; Asahara, T. Effects of statins on angiogenesis and vasculogenesis. Rev. Esp. Cardiol. 2002, 55, 838–844. [Google Scholar] [CrossRef]
- Yu, W.-L.; Sun, T.-W.; Qi, C.; Zhao, H.-K.; Ding, Z.-Y.; Zhang, Z.-W.; Sun, B.-B.; Shen, J.; Chen, F.; Zhu, Y.-J.; et al. Enhanced osteogenesis and angiogenesis by mesoporous hydroxyapatite microspheres-derived simvastatin sustained release system for superior bone regeneration. Sci. Rep. 2017, 7, 44129. [Google Scholar] [CrossRef]
- Liu, H.; Li, W.; Liu, C.; Tan, J.; Wang, H.; Hai, B.; Cai, H.; Leng, H.-J.; Liu, Z.-J.; Song, C.-L. Incorporating simvastatin/poloxamer 407 hydrogel into 3D-printed porous Ti6Al4V scaffolds for the promotion of angiogenesis, osseointegration and bone ingrowth. Biofabrication 2016, 8, 045012. [Google Scholar] [CrossRef] [PubMed]
- Khaidakov, M.; Wang, W.; Khan, J.A.; Kang, B.Y.; Hermonat, P.L.; Mehta, J.L. Statins and angiogenesis: Is it about connections? Biochem. Biophys. Res. Commun. 2009, 387, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Zemankova, L.; Varejckova, M.; Dolezalova, E.; Fikrova, P.; Jezkova, K.; Rathouska, J.; Cerveny, L.; Botella, L.M.; Bernabeu, C.; Nemeckova, I.; et al. Atorvastatin-induced endothelial nitric oxide synthase expression in endothelial cells is mediated by endoglin. J. Physiol. Pharm. 2015, 66, 403–413. [Google Scholar]
- Zhang, J.; Wang, H.; Ye, P. Effect of atorvastatin on eNOS synthesis in organs of aging rats with myocardial ischemia-reperfusion. Nan Fang Yi Ke Da Xue Xue Bao 2012, 32, 1708–1712. [Google Scholar]
- Yang, Y.; Yin, D.; Wang, F.; Hou, Z.; Fang, Z. In situ eNOS/NO up-regulation—A simple and effective therapeutic strategy for diabetic skin ulcer. Sci. Rep. 2016, 6, 30326. [Google Scholar] [CrossRef] [Green Version]
- Gao, K.; Wang, G.; Wang, Y.; Han, D.; Bi, J.; Yuan, Y.; Yao, T.; Wan, Z.; Li, H.; Mei, X. Neuroprotective effect of simvastatin via inducing the autophagy on spinal cord injury in the rat model. BioMed Res. Int. 2015, 2015, 260161. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.; Prabhakaran, S. Statins for neuroprotection after acute ischemic stroke. Stroke 2017, 48, 2922–2923. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-Q.; Mou, R.-T.; Feng, D.-X.; Wang, Z.; Chen, G. The role of nitric oxide in stroke. Med. Gas Res. 2017, 7, 194–203. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Song, W.; Li, L.; Fan, X. Endothelial nitric oxide synthase: A potential therapeutic target for cerebrovascular diseases. Mol. Brain 2016, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Zheng, H.; Nie, B.; Gong, W.; Cui, X. Statin use and risk of liver cancer: An update meta-analysis. BMJ Open 2014, 4, e005399. [Google Scholar] [CrossRef]
- Fukumura, D.; Kashiwagi, S.; Jain, R.K. The role of nitric oxide in tumour progression. Nat. Rev. Cancer 2006, 6, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Wei, D.; Huang, S.; Lancaster, J.R.; Xie, K. Nitric oxide synthase II suppresses the growth and metastasis of human cancer regardless of its up-regulation of protumor factors. Proc. Natl. Acad. Sci. USA 2005, 102, 8758–8763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, J.-I.; Kuo, P.-C.; Hsu, T.-S. Down-regulation of survivin in nitric oxide-induced cell growth inhibition and apoptosis of the human lung carcinoma cells. J. Biol. Chem. 2004, 279, 20267–20276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotamraju, S.; Williams, C.L.; Kalyanaraman, B. Statin-induced breast cancer cell death: Role of inducible nitric oxide and arginase-dependent pathways. Cancer Res. 2007, 67, 7386–7394. [Google Scholar] [CrossRef] [Green Version]
- Momi, S.; Monopoli, A.; Alberti, P.F.; Falcinelli, E.; Corazzi, T.; Conti, V.; Miglietta, D.; Ongini, E.; Minuz, P.; Gresele, P. Nitric oxide enhances the anti-inflammatory and anti-atherogenic activity of atorvastatin in a mouse model of accelerated atherosclerosis. Cardiovasc. Res. 2012, 94, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongini, E.; Impagnatiello, F.; Bonazzi, A.; Guzzetta, M.; Govoni, M.; Monopoli, A.; Del Soldato, P.; Ignarro, L.J. Nitric oxide (NO)-releasing statin derivatives, a class of drugs showing enhanced antiproliferative and antiinflammatory properties. Proc. Natl. Acad. Sci. USA 2004, 101, 8497–8502. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, S.; Raurell, I.; Torres-Arauz, M.; García-Lezana, T.; Genescà, J.; Martell, M. A Nitric Oxide-Donating Statin Decreases Portal Pressure with a Better Toxicity Profile than Conventional Statins in Cirrhotic Rats. Sci. Rep. 2017, 7, 40461. [Google Scholar] [CrossRef] [Green Version]
 : Molecule has become unavailable or inactivate or the signaling pathway is suppressed,
: Molecule has become unavailable or inactivate or the signaling pathway is suppressed,  : Increase or decrease in the amount,
: Increase or decrease in the amount,  : Molecule is activated. GGPP: geranylgeranyl pyrophosphate, ROCK: Rho-associated protein kinase, PI3k: Phosphoinositide 3-kinase, Akt: Protein kinase B, AMPK: AMP-activated protein kinase, miR: micro RNA, HO-1: Heme oxygenase 1, BH4: tetrahydrobiopterin, Nox: NADPH oxidase, Cav-1: caveolin-1, GTPCH: guanosine-5-triphosphate cyclohydrolase, CRP: C reactive protein, NO: nitric oxide, eNOS: endothelial NO synthase, ROS: reactive oxygen species. It is noteworthy that Rac1 is actually a downstream signal for GGPP and the statin-mediated inactivation of GGPP reduces Rac1 level but as statins exert a direct inductive effect on Rac1, the up-regulation of Rac1 and its downstream signaling are shown separately here.
: Molecule has become unavailable or inactivate or the signaling pathway is suppressed, : Increase or decrease in the amount, : Molecule is activated. GGPP: geranylgeranyl pyrophosphate, ROCK: Rho-associated protein kinase, PI3k: Phosphoinositide 3-kinase, Akt: Protein kinase B, AMPK: AMP-activated protein kinase, miR: micro RNA, HO-1: Heme oxygenase 1, BH4: tetrahydrobiopterin, Nox: NADPH oxidase, Cav-1: caveolin-1, GTPCH: guanosine-5-triphosphate cyclohydrolase, CRP: C reactive protein, NO: nitric oxide, eNOS: endothelial NO synthase, ROS: reactive oxygen species. It is noteworthy that Rac1 is actually a downstream signal for GGPP and the statin-mediated inactivation of GGPP reduces Rac1 level but as statins exert a direct inductive effect on Rac1, the up-regulation of Rac1 and its downstream signaling are shown separately here.
: Molecule is activated. GGPP: geranylgeranyl pyrophosphate, ROCK: Rho-associated protein kinase, PI3k: Phosphoinositide 3-kinase, Akt: Protein kinase B, AMPK: AMP-activated protein kinase, miR: micro RNA, HO-1: Heme oxygenase 1, BH4: tetrahydrobiopterin, Nox: NADPH oxidase, Cav-1: caveolin-1, GTPCH: guanosine-5-triphosphate cyclohydrolase, CRP: C reactive protein, NO: nitric oxide, eNOS: endothelial NO synthase, ROS: reactive oxygen species. It is noteworthy that Rac1 is actually a downstream signal for GGPP and the statin-mediated inactivation of GGPP reduces Rac1 level but as statins exert a direct inductive effect on Rac1, the up-regulation of Rac1 and its downstream signaling are shown separately here.
: Molecule has become unavailable or inactivate or the signaling pathway is suppressed, : Increase or decrease in the amount, : Molecule is activated. GGPP: geranylgeranyl pyrophosphate, ROCK: Rho-associated protein kinase, PI3k: Phosphoinositide 3-kinase, Akt: Protein kinase B, AMPK: AMP-activated protein kinase, miR: micro RNA, HO-1: Heme oxygenase 1, BH4: tetrahydrobiopterin, Nox: NADPH oxidase, Cav-1: caveolin-1, GTPCH: guanosine-5-triphosphate cyclohydrolase, CRP: C reactive protein, NO: nitric oxide, eNOS: endothelial NO synthase, ROS: reactive oxygen species. It is noteworthy that Rac1 is actually a downstream signal for GGPP and the statin-mediated inactivation of GGPP reduces Rac1 level but as statins exert a direct inductive effect on Rac1, the up-regulation of Rac1 and its downstream signaling are shown separately here.


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorabi, A.M.; Kiaie, N.; Hajighasemi, S.; Banach, M.; Penson, P.E.; Jamialahmadi, T.; Sahebkar, A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. J. Clin. Med. 2019, 8, 2051. https://doi.org/10.3390/jcm8122051
Gorabi AM, Kiaie N, Hajighasemi S, Banach M, Penson PE, Jamialahmadi T, Sahebkar A. Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications. Journal of Clinical Medicine. 2019; 8(12):2051. https://doi.org/10.3390/jcm8122051
Chicago/Turabian StyleGorabi, Armita Mahdavi, Nasim Kiaie, Saeideh Hajighasemi, Maciej Banach, Peter E. Penson, Tannaz Jamialahmadi, and Amirhossein Sahebkar. 2019. "Statin-Induced Nitric Oxide Signaling: Mechanisms and Therapeutic Implications" Journal of Clinical Medicine 8, no. 12: 2051. https://doi.org/10.3390/jcm8122051