Updates on the Role of Molecular Alterations and NOTCH Signalling in the Development of Neuroendocrine Neoplasms

 ,
,  ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuroendocrine Neoplasms and Molecular Heterogeneity
3. Common Genetic Alterations and Molecular Pathways in the Development of Neuroendocrine Neoplasms
3.1. Heritable Genetic Traits in Neuroendocrine Neoplasms
3.2. Genetic Alterations and Tumour Mutation Burden in NENs
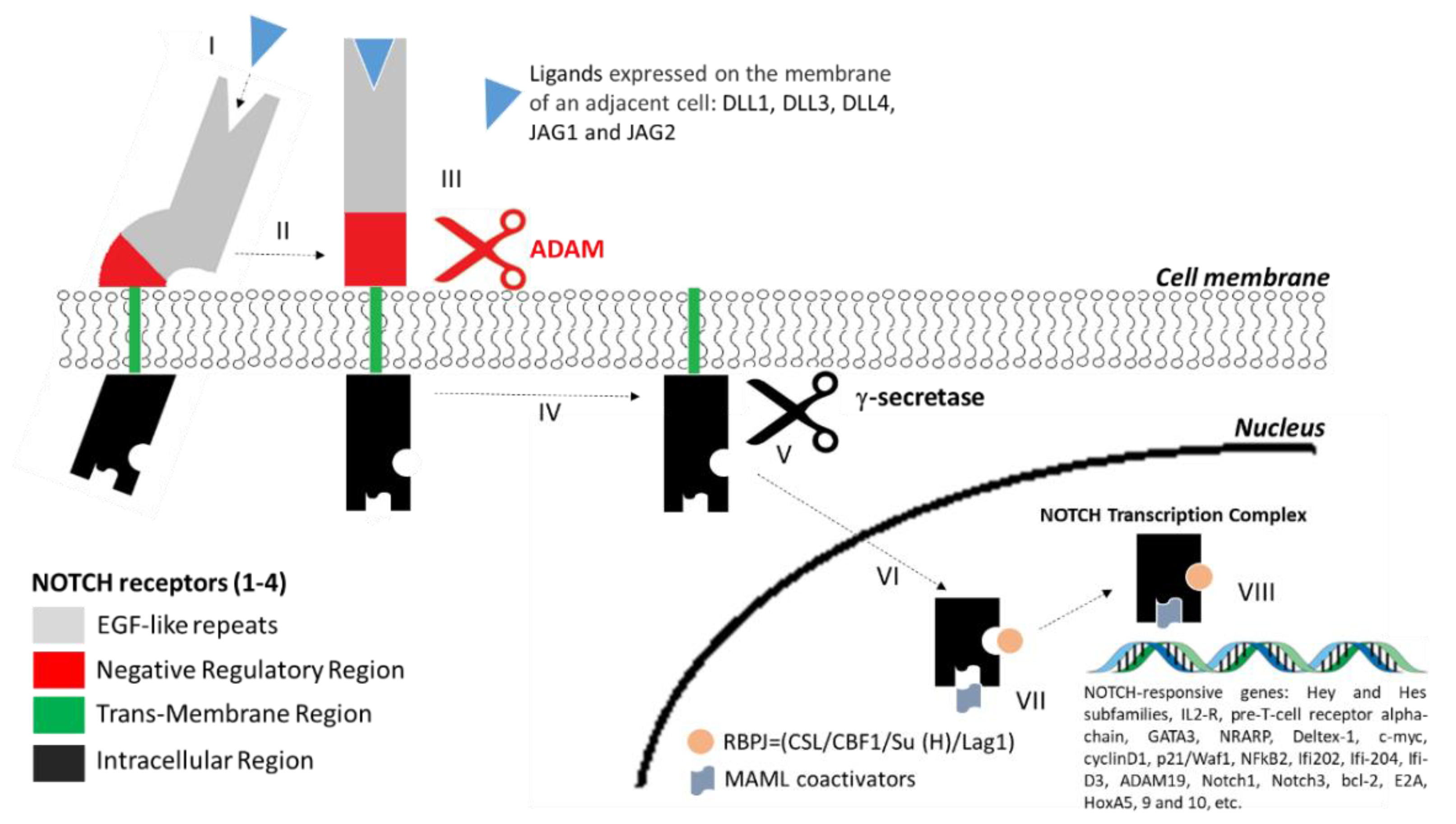
4. Structure of NOTCH Receptors and the NOTCH Signalling Pathway
5. The Role of NOTCH Signalling in NENs
5.1. NOTCH in NENs: The Epigenetic Implications
5.2. Role of NOTCH in Neuroendocrine Tumour of the Lung
5.3. Role of NOTCH in Neuroendocrine Gastro-Entero-Pancreatic Neuroendocrine Neoplasms (GEP-NENs)
5.4. Role of NOTCH in Medullary Thyroid Cancer
5.5. Role of NOTCH in Malignant Castration-Resistant Prostatic Cells
6. Therapeutic Approach Targeting NOTCH in NENs
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.P.; Kopp, J.L.; Sandhu, M.; Dubois, C.L.; Seymour, P.A.; Grapin-Botton, A.; Sander, M. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development 2012, 139, 2488–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.; Galoian, K. Molecular challenges of neuroendocrine tumors. Oncol. Lett. 2018, 15, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- Chikara, S.; Reindl, K.M. NOTCH signaling: A hero or villain in the war against cancer? Transl. Lung Cancer Res. 2013, 2, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Carter, Y.; Jaskula-Sztul, R.; Chen, H.; Mazeh, H. Signaling pathways as specific pharmacologic targets for neuroendocrine tumor therapy: RET, PI3K, MEK, growth factors, and NOTCH. Neuroendocrinology 2013, 97, 57–66. [Google Scholar] [CrossRef]
- Hassan, W.A.; Yoshida, R.; Kudoh, S.; Hasegawa, K.; Niimori-Kita, K.; Ito, T. Notch1 controls cell invasion and metastasis in small cell lung carcinoma cell lines. Lung Cancer 2014, 86, 304–310. [Google Scholar] [CrossRef]
- Krausch, M.; Kroepil, F.; Lehwald, N.; Lachenmayer, A.; Schott, M.; Anlauf, M.; Cupisti, K.; Knoefel, W.T.; Raffel, A. NOTCH 1 tumor expression is lacking in highly proliferative pancreatic neuroendocrine tumors. Endocrine 2013, 44, 182–186. [Google Scholar] [CrossRef]
- Kunnimalaiyaan, M.; Chen, H. Tumor suppressor role of notch-1 signaling in neuroendocrine tumors. Oncologist 2007, 12, 535–542. [Google Scholar] [CrossRef]
- Kunnimalaiyaan, M.; Yan, S.; Wong, F.; Zhang, Y.W.; Chen, H. Hairy enhancer of split-1 (HES-1), a Notch1 effector, inhibits the growth of carcinoid tumor cells. Surgery 2005, 138, 1137–1142. [Google Scholar] [CrossRef]
- Banck, M.S.; Kanwar, R.; Kulkarni, A.A.; Boora, G.K.; Metge, F.; Kipp, B.R.; Zhang, L.; Thorland, E.C.; Minn, K.T.; Tentu, R.; et al. The genomic landscape of small intestine neuroendocrine tumors. J. Clin. Investig. 2013, 123, 2502–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, G.; Bianchi, F.; Hofman, P.; Pattini, L.; Ströbel, P.; Calabrese, F.; Naheed, S.; Holden, C.; Cave, J.; Bohnenberger, H.; et al. Recent advances in the molecular landscape of lung neuroendocrine tumors. Expert Rev. Mol. Diagn. 2019, 19, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.L.; Yang, K.C.; Shen, Y.; Zhao, E.Y.; Loree, J.M.; Kennecke, H.F.; Kalloger, S.E.; Karasinska, J.M.; Lim, H.J.; Mungall, A.J.; et al. Molecular characterization of metastatic pancreatic neuroendocrine tumors (PNETs) using whole-genome and transcriptome sequencing. Cold Spring Harb. Mol. Case Stud. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Beltran, H.; Demichelis, F.; Rubin, M.A. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med. 2017, 23, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1 and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management and controversies. Cancer 2008, 113, 1807–1843. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.M.; Kiezun, A.; Ramos, A.H.; Serra, S.; Pedamallu, C.S.; Qian, Z.R.; Banck, M.S.; Kanwar, R.; Kulkarni, A.A.; Karpathakis, A.; et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat. Genet. 2013, 45, 1483–1486. [Google Scholar] [CrossRef]
- Singhi, A.D.; Liu, T.C.; Roncaioli, J.L.; Cao, D.; Zeh, H.J.; Zureikat, A.H.; Tsung, A.; Marsh, J.W.; Lee, K.K.; Hogg, M.E.; et al. Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin. Cancer Res. 2017, 23, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergia, N.; Boland, P.M.; Handorf, E.; Gustafson, K.S.; Gong, Y.; Cooper, H.S.; Sheriff, F.; Astsaturov, I.; Cohen, S.J.; Engstrom, P.F. Molecular profiling of neuroendocrine malignancies to identify prognostic and therapeutic markers: A Fox Chase Cancer Center Pilot Study. Br. J. Cancer 2016, 115, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Öberg, K. The Genetics of neuroendocrine tumors. Semin. Oncol. 2013, 40, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.J. Recent topics around multiple endocrine neoplasia type. J. Clin. Endocrinol. Metab. 2018, 103, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Frost, M.; Lines, K.E.; Thakker, R.V. Current and emerging therapies for PNETs in patients with or without MEN1. Nat. Rev. Endocrinol. 2018, 14, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepe, S.; Korbonits, M.; Iacovazzo, D. Germline and mosaic mutations causing pituitary tumours: Genetic and molecular aspects. J. Endocrinol. 2019, 240, R21–R45. [Google Scholar] [CrossRef] [PubMed]
- Khatami, F.; Tavangar, S.M. Multiple endocrine neoplasia syndromes from genetic and epigenetic Perspectives. Biomark. Insights 2018, 13. [Google Scholar] [CrossRef]
- Ren, F.; Xu, H.W.; Hu, Y.; Yan, S.H.; Wang, F.; Su, B.W.; Zhao, Q. Expression and subcellular localization of menin in human cancer cells. Exp. Ther. Med. 2012, 3, 1087–1091. [Google Scholar] [CrossRef]
- Petr, E.J.; Else, T. Genetic predisposition to endocrine tumors: Diagnosis, surveillance and challenges in care. Semin. Oncol. 2016, 43, 582–590. [Google Scholar] [CrossRef]
- Plaza-Menacho, I. Structure and function of RET in multiple endocrine neoplasia type 2. Endocr. Relat. Cancer 2018, 25, T79–T90. [Google Scholar] [CrossRef] [Green Version]
- Wells, S.A.; Pacini, F.; Robinson, B.G.; Santoro, M. Multiple Endocrine Neoplasia Type 2 and Familial Medullary Thyroid Carcinoma: An Update. J. Clin. Endocrinol. Metab. 2013, 98, 3149–3164. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Höfler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef]
- Varshney, N.; Kebede, A.A.; Owusu-Dapaah, H.; Lather, J.; Kaushik, M.; Bhullar, J.S. A review of von hippel-lindau syndrome. J. Kidney Cancer VHL 2017, 4, 20–29. [Google Scholar] [CrossRef] [PubMed]
- López-Jiménez, E.; Gómez-López, G.; Leandro-García, L.J.; Muñoz, I.; Schiavi, F.; Montero-Conde, C.; De Cubas, A.A.; Ramires, R.; Landa, I.; Leskela, S.; et al. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-Related pheochromocytomas. Mol. Endocrinol. 2010, 24, 2382–2391. [Google Scholar] [CrossRef] [PubMed]
- Dahia, P.L.; Ross, K.N.; Wright, M.E.; Hayashida, C.Y.; Santagata, S.; Barontini, M.; Kung, A.L.; Sanso, G.; Powers, J.F.; Tischler, A.S.; et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005, 1, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Suzuki, T.; Pan, X.; Chen, G.; Pan, S.; Bartman, T.; Whitsett, J.A. CUL2 Is Required for the activity of hypoxia-inducible factor and vasculogenesis. J. Boil. Chem. 2008, 283, 16084–16092. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, Y.; Kato, M.; Tokunaga, F.; Iwai, K. The COP9/signalosome increases the efficiency of von hippel-lindau protein ubiquitin ligase-mediated hypoxia-inducible factor-ubiquitination. J. Boil. Chem. 2008, 283, 16622–16631. [Google Scholar] [CrossRef] [PubMed]
- Ku, Y.H.; Ahn, C.H.; Jung, C.H.; Lee, J.E.; Kim, L.K.; Kwak, S.H.; Jung, H.S.; Park, K.S.; Cho, Y.M. A novel mutation in the von hippel-lindau tumor suppressor gene identified in a patient presenting with gestational diabetes mellitus. Endocrinol. Metab. 2013, 28, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Lodish, M.B.; Stratakis, C.A. Endocrine tumours in neurofibromatosis type 1, tuberous sclerosis and related syndromes. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 439–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Bottillo, I.; Dasdia, M.C.; Morella, A.; Lanari, V.; Bernardini, L.; Divona, L.; Giustini, S.; Sinibaldi, L.; Novelli, A.; et al. Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J. Med. Genet. 2007, 44, 800–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachdi, L.; Balcazar, N.; Osorio-Duque, F.; Elghazi, L.; Weiss, A.; Gould, A.; Chang-Chen, K.J.; Gambello, M.J.; Bernal-Mizrachi, E. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc. Natl. Acad. Sci. USA 2008, 105, 9250–9255. [Google Scholar] [CrossRef] [PubMed]
- Willenberg, H.; Lehwald, N.; Hafner, D.; Knoefel, W.T.; Krausch, M.; Raffel, A.; Anlauf, M.; Schott, M.; Cupisti, K.; Eisenberger, C.F. Loss of PTEN expression in neuroendocrine pancreatic tumors. Horm. Metab. Res. 2011, 43, 865–871. [Google Scholar]
- Lefebvre, M.; Foulkes, W.D. Pheochromocytoma and paraganglioma syndromes: Genetics and management update. Curr. Oncol. 2014, 21, e8–e17. [Google Scholar] [CrossRef] [PubMed]
- Ricketts, C.J.; Forman, J.R.; Rattenberry, E.; Bradshaw, N.; Lalloo, F.; Izatt, L.; Cole, T.R.; Armstrong, R.; Kumar, V.K.; Morrison, P.J.; et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum. Mutat. 2010, 31, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Brière, J.J.; Libé, R.; Vescovo, L.; Rivière, J.; Tissier, F.; Jouanno, E.; Jeunemaitre, X.; Bénit, P.; Tzagoloff, A.; et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X.; et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar]
- Hao, H.X.; Khalimonchuk, O.; Schraders, M.; Dephoure, N.; Bayley, J.P.; Kunst, H.; Devilee, P.; Cremers, C.W.; Schiffman, J.D.; Bentz, B.G.; et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science 2009, 325, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R.; et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef]
- Niemann, S.; Müller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef]
- Niemann, S.; Müller, U.; Engelhardt, D.; Lohse, P. Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Hum. Genet. 2003, 113, 92–94. [Google Scholar]
- Baysal, B.E. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Ugarte-Camara, M.; Fernandez-Prado, R.; Lorda, I.; Rossello, G.; Gonzalez-Enguita, C.; Cannata-Ortiz, P.; Ortiz, A. Positive/retained SDHB immunostaining in renal cell carcinomas associated to germline SDHB-deficiency: Case report. Diagn. Pathol. 2019, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenkamp, K.; Osinga, T.E.; Links, T.P.; Horst-Schrivers, A.N.; Van Der Horst-Schrivers, A.N. Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Crona, J.; Skogseid, B. GEP—NETS UPDATE: Genetics of neuroendocrine tumors. Eur. J. Endocrinol. 2016, 174, R275–R290. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Vakiani, E.; White, C.M.; Zhong, Y.; Saunders, T.; Morgan, R.; De Wilde, R.F.; Maitra, A.; Hicks, J.; DeMarzo, A.M.; et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am. J. Surg. Pathol. 2012, 36, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Basturk, O.; Yang, Z.; Tang, L.H.; Hruban, R.H.; Adsay, N.V.; McCall, C.M.; Krasinskas, A.M.; Jang, K.T.; Frankel, W.L.; Balci, S.; et al. The high grade (Who G3) pancreatic neuroendocrine tumor category is morphologically and biologically heterogeneous and includes both well differentiated and poorly differentiated neoplasms. Am. J. Surg. Pathol. 2015, 39, 683–690. [Google Scholar] [CrossRef]
- Scarpa, A.; Mantovani, W.; Capelli, P.; Beghelli, S.; Boninsegna, L.; Bettini, R.; Panzuto, F.; Pederzoli, P.; Fave, G.D.; Falconi, M. Pancreatic endocrine tumors: Improved TNM staging and histopathological grading permit a clinically efficient prognostic stratification of patients. Mod. Pathol. 2010, 23, 824–833. [Google Scholar] [CrossRef]
- Vélayoudom-Céphise, F.L.; Duvillard, P.; Foucan, L.; Hadoux, J.; Chougnet, C.N.; Leboulleux, S.; Malka, D.; Guigay, J.; Goere, D.; Debaere, T.; et al. Are G3 ENETS neuroendocrine neoplasms heterogeneous? Endocr. Relat. Cancer 2013, 20, 649–657. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lü, X.; Fernandez-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Makuuchi, R.; Terashima, M.; Kusuhara, M.; Nakajima, T.; Serizawa, M.; Hatakeyama, K.; Ohshima, K.; Urakami, K.; Yamaguchi, K. Comprehensive analysis of gene mutation and expression profiles in neuroendocrine carcinomas of the stomach. Biomed. Res. 2017, 38, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, N.; Ohishi, Y.; Hirahashi, M.; Takahashi, S.; Nakamura, K.; Tanaka, M.; Oki, E.; Takayanagi, R.; Oda, Y. Molecular characteristics of colorectal neuroendocrine carcinoma; similarities with adenocarcinoma rather than neuroendocrine tumor. Hum. Pathol. 2015, 46, 1890–1900. [Google Scholar] [CrossRef]
- Ali, A.S.; Grönberg, M.; Federspiel, B.; Scoazec, J.Y.; Hjortland, G.O.; Grønbæk, H.; Ladekarl, M.; Langer, S.W.; Welin, S.; Vestermark, L.W.; et al. Expression of p53 protein in high-grade gastroenteropancreatic neuroendocrine carcinoma. PLoS ONE 2017, 12, e0187667. [Google Scholar] [CrossRef] [PubMed]
- Sahnane, N.; Furlan, D.; Monti, M.; Romualdi, C.; Vanoli, A.; Vicari, E.; Solcia, E.; Capella, C.; Sessa, F.; La Rosa, S. Microsatellite unstable gastrointestinal neuroendocrine carcinomas: A new clinicopathologic entity. Endocr. Relat. Cancer. 2015, 22, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Olevian, D.C.; Nikiforovag, M.N.; Chiosea, S.; Sun, W.; Bahary, N.; Kuan, S.F.; Pai, R.K. Colorectal poorly differentiated neuroendocrine carcinomas frequently exhibit BRAF mutations and are associated with poor overall survival. Hum. Pathol. 2016, 49, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Jesinghaus, M.; Konukiewitz, B.; Keller, G.; Kloor, M.; Steiger, K.; Reiche, M.; Penzel, R.; Endris, V.; Arsenic, R.; Hermann, G.; et al. Colorectal mixed adenoneuroendocrine carcinomas and neuroendocrine carcinomas are genetically closely related to colorectal adenocarcinomas. Mod. Pathol. 2017, 30, 610–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijioka, S.; Hosoda, W.; Mizuno, N.; Hara, K.; Imaoka, H.; Bhatia, V.; Mekky, M.A.; Tajika, M.; Tanaka, T.; Ishihara, M.; et al. Does the WHO 2010 classification of pancreatic neuroendocrine neoplasms accurately characterize pancreatic neuroendocrine carcinomas? J. Gastroenterol. 2015, 50, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Woischke, C.; Schaaf, C.W.; Yang, H.M.; Vieth, M.; Veits, L.; Geddert, H.; Märkl, B.; Stömmer, P.; Schaeffer, D.F.; Frölich, M.; et al. In-depth mutational analyses of colorectal neuroendocrine carcinomas with adenoma or adenocarcinoma components. Mod. Pathol. 2017, 30, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Karkouche, R.; Bachet, J.B.; Sandrini, J.; Mitry, E.; Penna, C.; Côté, J.F.; Blons, H.; Penault-Llorca, F.; Rougier, P.; André, J.P.S.; et al. Colorectal neuroendocrine carcinomas and adenocarcinomas share oncogenic pathways. A clinico-pathologic study of 12 cases. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1430–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vortmeyer, A.O.; Lubensky, I.A.; Merino, M.J.; Wang, C.Y.; Pham, T.; Furth, E.E.; Zhuang, Z. Concordance of genetic alterations in poorly differentiated colorectal neuroendocrine carcinomas and associated adenocarcinomas. J. Natl. Cancer Inst. 1997, 89, 1448–1453. [Google Scholar] [CrossRef]
- Tang, L.H.; Basturk, O.; Sue, J.J.; Klimstra, D.S. A practical approach to the classification of WHO grade 3 (G3) well differentiated neuroendocrine tumor (WD-NET) and poorly differentiated neuroendocrine carcinoma (PD-NEC) of the pancreas. Am. J. Surg. Pathol. 2016, 40, 1192–1202. [Google Scholar] [CrossRef]
- Mafficini, A.; Scarpa, A. genetics and epigenetics of gastroenteropancreatic neuroendocrine neoplasms. Endocr. Rev. 2019, 40, 506–536. [Google Scholar] [CrossRef]
- Ohmoto, A.; Rokutan, H.; Yachida, S. Pancreatic neuroendocrine neoplasms: Basic biology, current treatment strategies and prospects for the future. Int. J. Mol. Sci. 2017, 18, 143. [Google Scholar] [CrossRef] [PubMed]
- Lamberti, G.; Brighi, N.; Maggio, I.; Manuzzi, L.; Peterle, C.; Ambrosini, V.; Ricci, C.; Casadei, R.; Campana, D. The role of mTOR in neuroendocrine tumors: future cornerstone of a winning strategy? Int. J. Mol. Sci. 2018, 19, 747. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Boora, G.K.; Kanwar, R.; Kulkarni, A.A.; Pleticha, J.; Ames, M.; Schroth, G.; Beutler, A.S.; Banck, M.S.; Gilbert, J.A. Exome-level comparison of primary well-differentiated neuroendocrine tumors and their cell lines. Cancer Genet. 2015, 208, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.H.; Jia, D.; Deng, W.M. Cis-interactions between Notch and its ligands block ligand-independent Notch activity. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Wöltje, K.; Jabs, M.; Fischer, A. Serum induces transcription of Hey1 and Hey2 genes by Alk1 but not Notch signaling in endothelial cells. PLoS ONE 2015, 10, e0120547. [Google Scholar] [CrossRef] [PubMed]
- Brzozowa-Zasada, M.; Piecuch, A.; Michalski, M.; Segiet, O.; Kurek, J.; Harabin-Słowińska, M.; Wojnicz, R. Notch and its oncogenic activity in human malignancies. Eur. Surg. 2017, 49, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Zou, B.; Zhou, X.L.; Lai, S.Q.; Liu, J.C. Notch signaling and non-small cell lung cancer. Oncol. Lett. 2018, 15, 3415–3421. [Google Scholar] [CrossRef] [PubMed]
- Fukusumi, T.; Califano, J. The NOTCH pathway in head and neck squamous cell carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kikuchi, H.; Shoji, T.; Takashima, Y.; Kikuchi, E.; Kikuchi, J.; Kinoshita, I.; Dosaka-Akita, H.; Sakakibara-Konishi, J. DLL3 regulates the migration and invasion of small cell lung cancer by modulating Snail. Cancer Sci. 2019, 110, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Jaskula-Sztul, R.; Eide, J.; Tesfazghi, S.; Dammalapati, A.; Harrison, A.D.; Yu, X.M.; Scheinebeck, C.; Winston-McPherson, G.; Kupcho, K.R.; Robers, M.B.; et al. Tumor-suppressor role of Notch3 in medullary thyroid carcinoma revealed by genetic and pharmacological induction. Mol. Cancer Ther. 2015, 14, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Lobry, C.; Oh, P.; Mansour, M.R.; Look, A.T.; Aifantis, I. Notch signaling: Switching an oncogene to a tumor suppressor. Blood 2014, 123, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, J.S.; Singleton, C.S.; Miele, L. Notch Signaling in neuroendocrine tumors. Front. Oncol. 2016, 6, 345. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Sato, Y.; Ikeda, H.; Hsü, M.; Igarashi, S.; Nakanuma, Y. Notch1-Hes1 signalling axis in the tumourigenesis of biliary neuroendocrine tumours. J. Clin. Pathol. 2013, 66, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The varied roles of notch in cancer. Annu. Rev. Pathol. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the mast kinase and notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef]
- Stoeck, A.; Lejnine, S.; Truong, A.; Pan, L.; Wang, H.; Zang, C.; Yuan, J.; Ware, C.; MacLean, J.; Garrett-Engele, P.W.; et al. Discovery of biomarkers predictive of GSI response in triple negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014, 4, 1154–1167. [Google Scholar] [CrossRef]
- Parr, C.; Watkins, G.; Jiang, W.G. The possible correlation of Notch-1 and Notch-2 with clinical outcome and tumour clinicopathological parameters in human breast cancer. Int. J. Mol. Med. 2004, 14, 779–786. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.; Li, X.; Wang, H.; et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a-secretase inhibitor. Clin. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G.; et al. Alterations of the Notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 22293–22298. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wang, J.; Shi, Z.; Franklin, J.L.; Deane, N.G.; Coffey, R.J.; Beauchamp, R.D.; Zhang, B. Deciphering genomic alterations in colorectal cancer through transcriptional subtype-based network analysis. PLoS ONE 2013, 8, e79282. [Google Scholar] [CrossRef] [PubMed]
- Mao, L. NOTCH Mutations: Multiple faces in human malignancies. Cancer Prev. Res. 2015, 8, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.; Zheng, G.; Schoolmeester, J.K.; Li, Z.; Pallavajjala, A.; Haley, L.; Conner, M.G.; Vang, R.; Hung, C.F.; Wu, T.C.; et al. Next-generation sequencing reveals recurrent somatic mutations in small cell neuroendocrine carcinoma of the uterine cervix. Am. J. Surg. Pathol. 2018, 42, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Derks, J.L.; Leblay, N.; Lantuejoul, S.; Dingemans, A.M.C.; Speel, E.J.M.; Fernandez-Cuesta, L.; Speel, E. New insights into the molecular characteristics of pulmonary carcinoids and large cell neuroendocrine carcinomas, and the impact on their clinical management. J. Thorac. Oncol. 2018, 13, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Cuesta, L.; Peifer, M.; Lu, X.; Sun, R.; Ozretić, L.; Seidal, D.; Zander, T.; Leenders, F.; George, J.; Müller, C.; et al. Frequent mutations in chromatin-remodelling genes in pulmonary carcinoids. Nat. Commun. 2014, 5, 3518. [Google Scholar] [CrossRef] [PubMed]
- Cives, M.; Simone, V.; Rizzo, F.M.; Silvestris, F. NETs: Organ-related epigenetic derangements and potential clinical applications. Oncotarget 2016, 7, 57414–57429. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Li, J.; Jang, E.R.; Gulhati, P.; Rychahou, P.G.; Napier, D.L.; Wang, C.; Weiss, H.L.; Lee, E.Y.; Anthony, L.; et al. Deregulation of Wnt/β-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis 2013, 34, 953–961. [Google Scholar] [CrossRef]
- Graves, C.A.; Jones, A.; Reynolds, J.; Stuart, J.; Pirisi, L.; Botrous, P.; Wells, J. Neuroendocrine Merkel cell carcinoma is associated with mutations in key DNA repair, epigenetic and apoptosis pathways: A case-based study using targeted massively parallel sequencing. Neuroendocrinology 2015, 101, 112–119. [Google Scholar] [CrossRef]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci. Signal 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Abraham, K.J.; Zhang, X.; Vidal, R.; Paré, G.C.; Feilotter, H.E.; Tron, V.A. Roles for miR-375 in neuroendocrine differentiation and tumor suppression via Notch pathway suppression in Merkel cell carcinoma. Am. J. Pathol. 2016, 186, 1025–1035. [Google Scholar] [CrossRef]
- Arvidsson, Y.; Rehammar, A.; Bergström, A.; Andersson, E.; Altiparmak, G.; Swärd, C.; Wängberg, B.; Kristiansson, E.; Nilsson, O. miRNA profiling of small intestinal neuroendocrine tumors defines novel molecular subtypes and identifies miR-375 as a biomarker of patient survival. Mod. Pathol. 2018, 31, 1302–1317. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Zahnow, C.A.; Ahuja, N.; Baylin, S.B. Combining epigenetic and immunotherapy to combat cancer. Cancer Res. 2016, 76, 1683–1689. [Google Scholar] [CrossRef] [PubMed]
- Meder, L.; König, K.; Ozretić, L.; Schultheis, A.M.; Ueckeroth, F.; Ade, C.P.; Albus, K.; Boehm, D.; Rommerscheidt-Fuss, U.; Florin, A.; et al. NOTCH, ASCL1, p53 and RB alterations define an alternative pathway driving neuroendocrine and small cell lung carcinomas. Int. J. Cancer 2016, 38, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Saunders, L.R.; Bankovich, A.J.; Anderson, W.C.; Aujay, M.A.; Bheddah, S.; Black, K.; Desai, R.; Escarpe, P.A.; Hampl, J.; Laysang, A.; et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, Y.; Fernandez-Del Castillo, C.; Yilmaz, O.; Deshpande, V. Heterogeneity in signaling pathways of gastroenteropancreatic neuroendocrine tumors: A critical look at notch signaling pathway. Mod. Pathol. 2013, 26, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, L.; Avila, J.L.; Demarest, R.M.; Troutman, S.; Allen, M.; Ratti, F.; Rustgi, A.K.; Stanger, B.Z.; Radtke, F.; Adsay, V.; et al. Notch1 functions as a tumor suppressor in a model of K-ras-induced pancreatic ductal adenocarcinoma. Cancer Res. 2010, 70, 4280–4286. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, Y.; Maitra, A.; Ghosh, B.; Zechner, U.; Argani, P.; Iacobuzio-Donahue, C.A.; Sriuranpong, V.; Iso, T.; Meszoely, I.M.; Wolfe, M.S.; et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003, 3, 565–576. [Google Scholar] [CrossRef]
- Maggi, E.C.; Trillo-Tinoco, J.; Struckhoff, A.P.; Vijayaraghavan, J.; Del Valle, L.; Crabtree, J.S. Retinoblastoma-binding protein 2 (RBP2) is frequently expressed in neuroendocrine tumors and promotes the neoplastic phenotype. Oncogenesis 2016, 5, e257. [Google Scholar] [CrossRef]
- Sippel, R.S.; Carpenter, J.E.; Kunnimalaiyaan, M.; Chen, H. The role of human achaete-scute homolog-1 in medullary thyroid cancer cells. Surgery 2003, 134, 866–871. [Google Scholar] [CrossRef]
- Kunnimalaiyaan, M.; Vaccaro, A.M.; Ndiaye, M.A.; Chen, H. Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. J. Biol. Chem. 2006, 281, 39819–39830. [Google Scholar] [CrossRef]
- Somnay, Y.R.; Yu, X.M.; Lloyd, R.V.; Leverson, G.; Aburjania, Z.; Jang, S.; Jaskula-Sztul, R.; Chen, H. Notch3 expression correlates with thyroid cancer differentiation, induces apoptosis, and predicts disease prognosis. Cancer 2017, 123, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Danza, G.; Di Serio, C.; Rosati, F.; Lonetto, G.; Sturli, N.; Kacer, D.; Pennella, A.; Ventimiglia, G.; Barucci, R.; Piscazzi, A.; et al. Notch signaling modulates hypoxia-induced neuroendocrine differentiation of human prostate cancer cells. Mol. Cancer Res. 2012, 10, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Gavyert, K.; Sailer, V.; Conteduca, V.; Dardenne, E.; Sigouros, M.; Isse, K.; Kearney, M.; Vosoughi, A.; Fernandez, L.; et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.; Jin, H.; Roy, M.; Ma, A.L.; Gong, S.; Jaskula-Sztul, R.; Chen, H. Antineoplastic effects of histone deacetylase inhibitors in neuroendocrine cancer cells are mediated through transcriptional regulation of Notch1 by activator protein. Cancer Med. 2017, 6, 2142–2452. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, T.A.; Holen, K.D.; Jaskula-Sztul, R.; Mulkerin, D.; Lubner, S.J.; Schelman, W.R.; Eickhoff, J.; Chen, H.; LoConte, N.K. A Pilot Phase II Study of valproic acid for treatment of low-grade neuroendocrine carcinoma. Oncologist 2011, 16, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Qian, Q.; Sun, G.; Mackey, L.V.; Fuselier, J.A.; Coy, D.H.; Yu, C.Y. Valproic acid induces NET cell growth arrest and enhances tumor suppression of the receptor-targeted peptide-drug conjugate via activating somatostatin receptor type II. J. Drug Target. 2016, 24, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, Y.; Wu, W.; Gordon, W.R.; Chang, D.W.; Lu, M.; Scoggin, S.; Fu, T.; Vien, L.; Histen, G.; et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J. Biol. Chem. 2008, 283, 8046–8054. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.C.; Fischer, M.M.; Axelrod, F.; Bond, C.; Cain, J.; Cancilla, B.; Henner, W.R.; Meisner, R.; Sato, A.; Shah, J.; et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (Tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin. Cancer Res. 2015, 21, 2084–2095. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Messersmith, W.A.; Mikulski, S.M.; Papadopoulos, K.P.; Kwak, E.L.; Gibbon, D.G.; Patnaik, A.; Falchook, G.S.; Dasari, A.; Shapiro, G.I.; et al. Phase I study of RO4929097, a gamma secretase inhibitor of notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J. Clin. Oncol. 2012, 30, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Morgensztern, D.; Le Moulec, S.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Dowlati, A.; Rudin, C.M.; Lally, S.; et al. Efficacy and safety of rovalpituzumab tesirine (Rova-TTM) in patients with DLL3-expressing, ≥3rd line small cell lung cancer: Results from the phase 2 TRINITY study. J. Clin. Oncol. 2018, 36 (Suppl. S15), 8507. [Google Scholar] [CrossRef]
- Phase 3 Trial of Rova-T as Second-Line Therapy for Advanced Small-Cell Lung Cancer (Tahoe Study) Halted. 2018. Available online: https://bit.ly/2KZcyVl (accessed on 6 December 2018).
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., III; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef]
- Puca, L.; Sailor, V.; Gavyert, K.; Dardenne, E.; Isse, K.; Sigouros, M.; Nanus, D.M.; Tagawa, S.T.; Mosquera, J.M.; Saunders, L.; et al. Abstract 1947: Rovalpituzumab tesirine as a therapeutic agent for neuroendocrine prostate cancer. Exp. Mol. Ther. 2018, 78, 1947. [Google Scholar]
- Chiu, M.L.; Gilliland, G.L. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016, 38, 163–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramonov, V.M.; Desai, D.; Mamaeva, V.; Rosenholm, J.; Rivero-Müller, A.; Sahlgren, C. Mesoporous silica nanoparticles for somatostatin targeted Notch activation in animal model of pancreatic neuroendocrine cancer. Endocr. Abstr. 2016, 40. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Yu, Y.; Liu, S.; Li, T.; Lin, F.; Fan, X.; Shen, Y.; Ding, M.; Tang, Y.; et al. EGFR/Notch antagonists enhance the response to inhibitors of the PI3K-Akt pathway by decreasing tumor-initiating cell frequency. Clin. Cancer Res. 2019, 25, 2835–2847. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Arx, C.; Capozzi, M.; López-Jiménez, E.; Ottaiano, A.; Tatangelo, F.; Di Mauro, A.; Nasti, G.; Tornesello, M.L.; Tafuto, S. Updates on the Role of Molecular Alterations and NOTCH Signalling in the Development of Neuroendocrine Neoplasms. J. Clin. Med. 2019, 8, 1277. https://doi.org/10.3390/jcm8091277
von Arx C, Capozzi M, López-Jiménez E, Ottaiano A, Tatangelo F, Di Mauro A, Nasti G, Tornesello ML, Tafuto S. Updates on the Role of Molecular Alterations and NOTCH Signalling in the Development of Neuroendocrine Neoplasms. Journal of Clinical Medicine. 2019; 8(9):1277. https://doi.org/10.3390/jcm8091277
Chicago/Turabian Stylevon Arx, Claudia, Monica Capozzi, Elena López-Jiménez, Alessandro Ottaiano, Fabiana Tatangelo, Annabella Di Mauro, Guglielmo Nasti, Maria Lina Tornesello, and Salvatore Tafuto. 2019. "Updates on the Role of Molecular Alterations and NOTCH Signalling in the Development of Neuroendocrine Neoplasms" Journal of Clinical Medicine 8, no. 9: 1277. https://doi.org/10.3390/jcm8091277