Can Parentage Analysis Facilitate Breeding Activities in Root and Tuber Crops?

 ,
,  ,
,
Abstract
:1. Introduction
2. Overview of Parentage Analysis in Breeding Programs
3. The Concept and Application of Parentage Analysis
4. Factors Affecting Parentage Analysis
4.1. Sampling Techniques
4.2. Molecular Marker Systems
4.3. Analysis Method
4.3.1. Exclusion Technique
4.3.2. Categorical Allocation Technique
4.3.3. Fractional Allocation Technique
4.3.4. Full Probability Parentage Analysis Technique
4.3.5. Parental Reconstruction Technique
4.3.6. Sibship Reconstruction Technique
4.4. Genotyping Errors, Mutations and Null Alleles
4.5. Family Structure of Candidate Parents
4.6. Species Effect
4.7. Pollen and Seed Dispersal Mechanism
5. Potential of Parentage Analysis in Root and Tuber Breeding Programs
6. Current Application of Parentage Analysis in Root and Tuber Crops
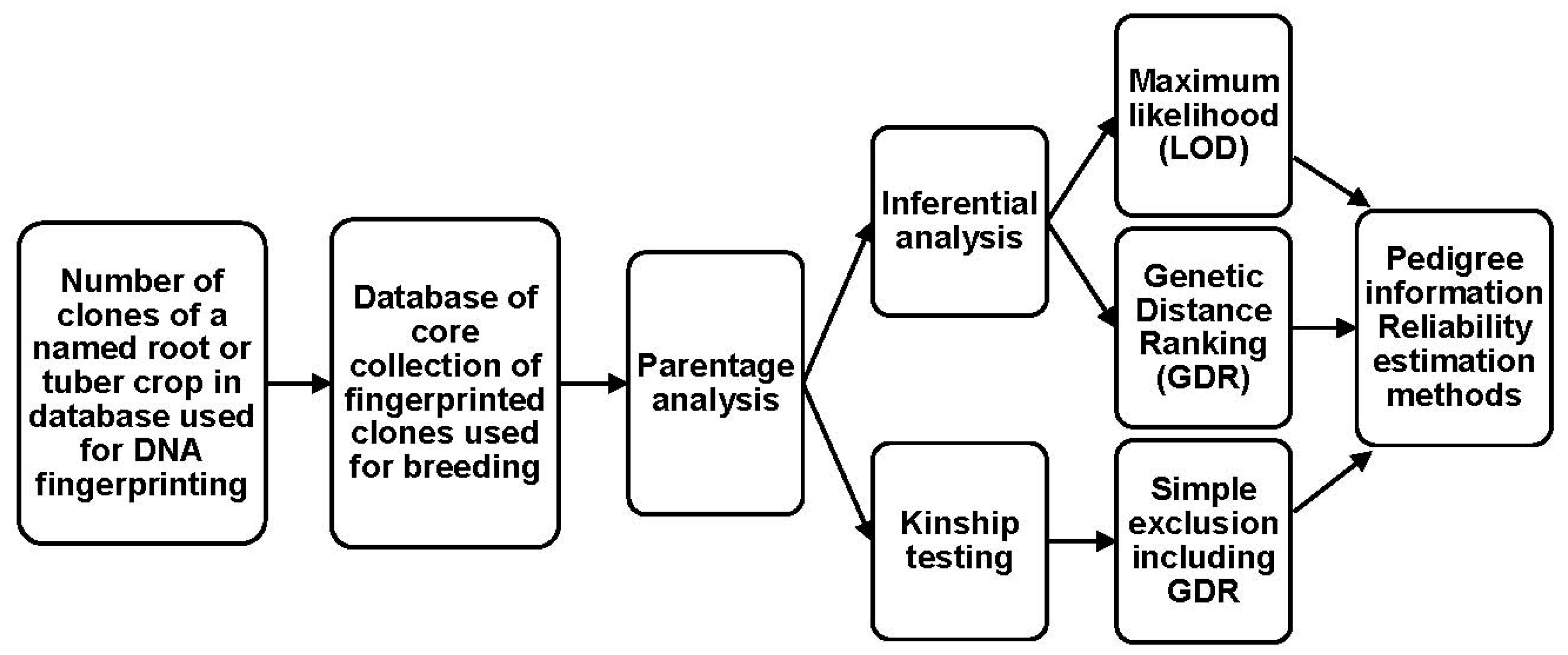
7. Generic Framework of Parentage Analysis in Root and Tuber Crops
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Akanbi, W.B.; Adeboye, C.O.; Togun, A.O.; Ogunride, J.O.; Adeyeye, S.A. Growth, herbage and seed yield and quality of Telfairia occidentalis as influenced by cassava peel compost and mineral fertilizer. World J. Agric. Sci. 2007, 3, 508–516. [Google Scholar]
- Iyagba, A.G. A review on root and tuber crop production and their weed management among small scale farmers in Nigeria. ARPN J. Agric. Biol. Sci. 2010, 5, 52–58. [Google Scholar]
- Okigbo, B.N. New crops for food industry: The roots and tuber in tropical Africa. In New Crops for Food Industry; Weekens, G.E., Haq, N., Day, P., Eds.; Chapman and Wall: London, UK, 1989; pp. 123–134. [Google Scholar]
- Lebot, V. Tropical Root and Tuber Crops Cassava, Sweet Potato, Yams and Aroids; Crop Production Science in Horticulture Series 17; CABI Publishing: Wallingford, UK, 2008. [Google Scholar]
- Berloo, R.; Hutten, R.C.B.; Eck, H.J.; Visser, R.G.F. An online potato pedigree database resource. Potato Res. 2007, 50, 45–57. [Google Scholar] [CrossRef]
- Nybom, H.; Weising, K.; Rotter, B. DNA fingerprinting in botany: Past, present, future. Investig. Genet. 2014, 5, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Tarn, T.R.; Tai, G.C.C.; De Long, H.; Murphy, A.M.; Seabrook, J.E.A. Breeding potatoes for long-day, temperate climates. In Plant Breeding Reviews; Wiley: New York, NY, USA, 1992; pp. 217–332. [Google Scholar]
- Spanoghe, M.; Marique, T.; Rivière, J.; Lanterbecq, D.; Gadenne, M. Investigation and development of potato parentage analysis methods using multiplexed SSR fingerprinting. Potato Res. 2015, 58, 43–65. [Google Scholar] [CrossRef]
- Luo, Z.W.; Hackett, C.A.; Bradshaw, J.E.; McNicol, J.W.; Milbourne, D. Predicting parental genotypes and gene segregation for tetrasomic inheritance. Theor. Appl. Genet. 2000, 100, 1067–1073. [Google Scholar] [CrossRef]
- Bink, M.P.; Uimari, P.; Sillanpaa, J.; Janss, G.; Jansen, C. Multiple QTL mapping in related plant populations via a pedigree-analysis approach. Theor. Appl. Genet. 2002, 104, 751–762. [Google Scholar] [PubMed]
- D’hoop, B.B.; Paulo, M.J.; Kowitwanich, K.; Sengers, M.; Visser, R.G.F.; Van Eck, H.J.; van Eeuwijk, F.A. Population structure and linkage disequilibrium unravelled in tetraploid potato. Theor. Appl. Genet. 2010, 121, 1151–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldwin, S.J.; Dodds, K.G.; Auvray, B.; Genet, R.A.; Macknight, R.C.; Jacobs, J.M.E. Association mapping of cold-induced sweetening in potato using historical phenotypic data. Ann. Appl. Biol. 2011, 158, 248–256. [Google Scholar] [CrossRef]
- Song, Y.S.; Hepting, L.; Schweizer, G.; Hartl, L.; Wenzel, G.; Schwarzfischer, A. Mapping of extreme resistance to PVY (Ry (sto)) on chromosome XII using anther-culture-derived primary dihaploid potato lines. Theor. Appl. Genet. 2005, 111, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Wang-Pruski, G.; Mayich, M.; Jong, H. RAPD and pedigree-based genetic diversity estimates in cultivated diploid potato hybrids. Theor. Appl. Genet. 2003, 107, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Wenzel, G. Molecular analysis of genetic variation in potato (Solanum tuberosum L.). I. German cultivars and advanced clones. Potato Res. 2004, 47, 81–92. [Google Scholar] [CrossRef]
- Gjedrem, T. The first family-based breeding program in aquaculture. Rev. Aquac. 2010, 2, 2–15. [Google Scholar] [CrossRef]
- Demeke, T.; Lynch, D.R.; Kawchuk, L.M.; Kozub, G.C.; Armstrong, J.D. Genetic diversity of potato determined by random amplified polymorphic DNA analysis. Plant Cell Rep. 1996, 15, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Isenegger, D.A.; Taylor, P.W.J.; Ford, R.; Franz, P.; McGregor, G.R.; Hutchinson, J.F. DNA fingerprinting and genetic relationships of potato cultivars (Solanum tuberosum L.) commercially grown in Australia. Aust. J. Agric. Res. 2001, 52, 911–918. [Google Scholar] [CrossRef]
- Vincent, K.; Robert, K.; Morag, F.; Tadeo, K.; Yona, B.; Peter, V. Identification of F1 cassava (Manihot esculenta Crantz) progeny using microsatellite markers and capillary electrophoresis. Am. J. Plant Sci. 2014, 5, 119–125. [Google Scholar] [CrossRef]
- Contreras, R.N.; Ranney, T.G.; Milla-Lewis, S.R.; Yencho, G.C. Investigating parentage and hybridity of three azaleodendrons using amplified fragment length polymorphism analysis. Hortscience 2007, 42, 740–743. [Google Scholar]
- Levine, L.; Asmussen, M.; Olvera, O.; Powell, J.R.; De La Rosa, M.E.; Salceda, V.M.; Gaso, M.I.; Guzman, J.; Anderson, W.W. Population genetics of Mexican Drosophila. V. A high rate of multiple insemination in a natural population of Drosophila pseudoobscura. Am. Nat. 1980, 116, 493–503. [Google Scholar] [CrossRef]
- Ellstrand, N.C. Multiple paternity within the fruits of the wild radish, Raphanus sativus. Am. Nat. 1984, 123, 819–828. [Google Scholar] [CrossRef]
- Gill, P.; Jeffreys, A.J.; Werrett, D.J. Forensic application of DNA ‘fingerprints’. Nature 1985, 318, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, A.J.; Brookfield, J.F.Y.; Semeonoff, R. Positive identification of an immigration test case using DNA fingerprints. Nature 1985, 317, 818–819. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, A.J. Genetic fingerprinting. Nat. Med. 2005, 11, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, H.L.; Patrick, J.; Weatherhead, P.J.; Boag, P.T.; White, B.N.; Tabak, L.M.; Hoysak, D.J. Realized reproductive success of polygynous red-winged blackbirds revealed by DNA markers. Science 1990, 250, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Meagher, T.R.; Thompson, E.A. The relationship between single parent and parent pair genetic likelihoods in genealogy reconstruction. Theor. Popul. Biol. 1986, 29, 87–106. [Google Scholar] [CrossRef]
- Pena, S.D.J.; Chakraborty, R. Paternity testing in the DNA era. Trends Genet. 1994, 10, 204–209. [Google Scholar] [CrossRef]
- Jones, A.G.; Small, C.M.; Paczolt, K.A.; Ratterman, N.L. A practical guide to methods of parentage analysis. Mol. Ecol. Res. 2010, 10, 6–30. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.G.; Ardren, W.R. Methods of parentage analysis in natural populations. Mol. Ecol. 2003, 12, 2511–2523. [Google Scholar] [CrossRef] [PubMed]
- Scarcelli, N.; Daïnou, O.; Agbangla, C.; Tostain, S.; Pham, J.L. Segregation patterns of isozyme loci and microsatellite markers show the diploidy of African yam Dioscorea rotundata (2n = 40). Theor. Appl. Genet. 2005, 111, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Bousalem, M.; Viader, V.; Mariac, C.; Gomez, R.M.; Hochu, I.; Santoni, S.; David, J. Evidence of diploidy in the wild Amerindian yam, a putative progenitor of the endangered species Dioscorea trifida (Dioscoreaceae). Genome 2010, 53, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Gedil, M.; Sartie, A.M. Perspectives on molecular breeding of Africa’s main staple food crops—Cassava and yam. Asp. Appl. Biol. 2010, 96, 123–136. [Google Scholar]
- Sartie, A.; Asiedu, R. Development of mapping populations for genetic analysis in yams (Dioscorea rotundata Poir. and Dioscorea alata L.). Afr. J. Biotechnol. 2011, 10, 3040–3050. [Google Scholar]
- Nemorin, A.; David, J.; Maledon, E.; Nudol, E.; Dalon, J.; Arnau, G. Microsatellite and flow cytometry analysis to help understand the origin of Dioscorea alata polyploids. Ann. Bot. 2013, 112, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Arnau, G.; Maledon, E.; Bachand, I.; Abraham, K. Production of interploid hybrids and molecular markers heterozygosity determination using microsatellite markers in the greater yam, D. alata: Importance for the genetic improvement of the greater yam. In Proceedings of the 14th Triennial Symposium of the International Society for Tropical Roots Crops, Thiruvananthapuram, India, 20–26 November 2006. [Google Scholar]
- Girma, G.; Hyma, K.E.; Asiedu, R.; Mitchell, S.E.; Gedil, M.; Spillane, C. Next-generation se-quencing based genotyping, cytometry and phenotyping for understanding diversity and evolution of guinea yams. Theor. Appl. Genet. 2014, 127, 1783–1794. [Google Scholar] [CrossRef] [PubMed]
- Arnau, G.; Bhattacharjee, R.; Shela, M.N.; Chair, H.; Malapa, R.; Lebot, V.; Abraham, K.; Perrier, X.; Petro, D.; Penet, L.; et al. Understanding the genetic diversity and population structure of yam (Dioscorea alata L.) using microsatellite markers. PLoS ONE 2017, 12, e0174150. [Google Scholar] [CrossRef] [PubMed]
- Tamiru, M.; Natsume, S.; Takagi, H.; White, B.; Yaegashi, H.; Shimizu, M.; Yoshida, K.; Uemura, A.; Oikawa, K.; Abe, A.; et al. Genome sequencing of the staple food crop white Guinea yam enables the development of a molecular marker for sex determination. BMC Biol. 2017, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Mignouna, H.D.; Mank, R.A.; Ellis, T.H.N.; Van den Bosch, N.; Asiedu, R.; Ng, S.Y.C.; Peleman, J. A genetic linkage map of Guinea yam (Dioscorea rotundata L.) based on AFLP markers. Theor. Appl. Genet. 2002, 105, 716–725. [Google Scholar] [PubMed]
- Mignouna, H.D.; Mank, R.A.; Ellis, T.H.N.; Van den Bosch, N.; Asiedu, R.; Abang, M.M.; Peleman, J. A genetic linkage map of water yam (Dioscorea alata L.) based on AFLP markers and QTL analysis for anthracnose resistance. Theor. Appl. Genet. 2002, 105, 726–735. [Google Scholar] [PubMed]
- Cao, J.; Jiang, D.; Zhao, Z.; Yuan, S.; Zhang, Y.; Zhang, T.; Zhong, W.; Yuan, Q.; Huang, L. Development of chloroplast genomic resources in Chinese Yam (Dioscorea polystachya). BioMed Res. Int. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Smyda-Dajmund, P.; Śliwka, J.; Wasilewicz-Flis, I.; Jakuczun, H.; Zimnoch-Guzowska, E. Genetic composition of interspecific potato somatic hybrids and autofused 4x plants evaluated by DArT and cytoplasmic DNA markers. Plant Cell Rep. 2016, 35, 1345–1358. [Google Scholar] [CrossRef] [PubMed]
- Thieme, R.; Darsow, U.; Gavrilenko, T.; Dorokhov, D.; Tiemann, H. Production of somatic hybrids between S. tuberosum L. and late blight resistant Mexican wild potato species. Euphytica 1997, 97, 189–200. [Google Scholar] [CrossRef]
- Bradshaw, J.E.; Hackett, C.A.; Meyer, R.C.; Milbourne, D.; McNicol, J.W.; Phillips, M.S.; Waugh, R. Identification of AFLP and SSR markers associated with quantitative resistance to Globodera pallida (Stone) in tetraploid potato (Solanum tuberosum subsp. tuberosum) with a view to marker-assisted selection. Theor. Appl. Genet. 1998, 97, 202–210. [Google Scholar] [CrossRef]
- Ashkenazi, V.; Chani, E.; Lavi, U.; Levy, D.; Hillel, J.; Veilleux, R.E. Development of microsatellite markers in potato and their use in phylogenetic and fingerprinting analyses. Genome 2001, 44, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Simko, I.; Haynes, K.G.; Jones, R.W. Mining data from potato pedigrees: Tracking the origin of susceptibility and resistance to Verticillium dahliae in North American cultivars through molecular analysis. Theor. Appl. Genet. 2004, 108, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.; Kerr, E.M. A rapid simple sequence repeat (SSR)–based identification method for potato cultivars. Plant Genet. Res. 2007, 5, 7–13. [Google Scholar] [CrossRef]
- Bryan, G.; Lloyd, D.; Bradshaw, J. Understanding and Improving Potato Flavour Characteristics; Project Report; Agriculture and Horticulture Development Board (AHDB): Warwickshire, UK, 2008.
- Côté, M.J.; Leduc, L.; Reid, A. Evaluation of simple sequence repeat (SSR) markers established in Europe as a method for the identification of potato varieties grown in Canada. Am. J. Potato Res. 2013, 90, 340–350. [Google Scholar] [CrossRef]
- Hackett, C.A.; McLean, K.; Bryan, G.J. Linkage analysis and QTL mapping using SNP dosage data in a tetraploid potato mapping population. PLoS ONE 2013, 8, e63939. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.N.; Hirsch, C.D.; Felcher, K.; Coombs, J.; Zarka, D.; Van Deynze, A.; De Jong, W.; Veilleux, R.E.; Jansky, S.; Bethke, P. Retrospective view of North American potato (Solanum tuberosum L.) breeding in the 20th and 21st centuries. Genes Genomes Genet. 2013, 3, 1003–1013. [Google Scholar]
- Sharma, S.K.; Bolser, D.; de Boer, F.; Sønderkær, M.; Amoros, W.; Carboni, M.F.; Amoros, J.M.; de la Cruz, G.; Di Genova, A.; Douches, D.S.; et al. Construction of reference chromosome-scale pseudomolecules for potato: Integrating the potato genome with genetic and physical maps. Genes Genomes Genet. 2013, 3, 2031–2047. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Bhardwaj, V.; Dalamu, D.; Kaushik, S.K.; Singh, B.P.; Sharma, S.; Umamaheshwari, R.; Baswaraj, R.; Kumar, V.; Gebhardt, C. Identification of elite potato genotypes possessing multiple disease resistance genes through molecular approaches. Sci. Hortic. 2014, 179, 204–211. [Google Scholar] [CrossRef]
- Yada, B. Genetic Analysis of Agronomic Traits and Resistance to Sweetpotato Weevil and Sweet Potato Virus Disease in a Bi-Parental Sweetpotato Population. Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2014. [Google Scholar]
- Goyer, A.; Hamlin, L.; Crosslin, J.M.; Buchanan, A.; Chang, J.H. RNA-seq analysis of resistant and susceptible potato varieties during the early stages of potato virus Y infection. BMC Genom. 2015, 16, 472. [Google Scholar] [CrossRef] [PubMed]
- Vos, P.G.; Uitdewilligen, J.G.A.M.L.; Voorrips, R.E.; Visser, R.G.F.; van Eck, H.J. Development and analysis of a 20 K SNP array for potato (Solanum tuberosum): An insight into the breeding history. Theor. Appl. Genet. 2015, 128, 2387–2401. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Potato Variety Identification with a Panel of SNP Markers. Master’s Thesis, Wageningen University, Wageningen, The Netherlands, 1 March 2016. [Google Scholar]
- Endelman, J.B.; Carley, C.A.S.; Douches, D.S.; Coombs, J.J.; Bizimungu, B.; De Jong, W.S.; Holm, K.G.; Holm, D.G.; Creighton Miller, J., Jr.; Novy, R.G.; et al. Pedigree reconstruction with genome-wide markers in potato. Am. J. Potato Res. 2017, 94, 184–190. [Google Scholar] [CrossRef]
- Maras, M.; Sedlar, A.; Reid, A.; Božović, V.; Jovović, Z.; Meglič, V.; Peter Dolničar, P. Genetic diversity and redundancy among potato accessions in the Montenegrin collection as revealed by microsatellite markers. Am. J. Potato Res. 2017, 94, 306–313. [Google Scholar] [CrossRef]
- Roger, A.C.; Jones, R.A.C.; Vincent, S.J. Strain-specific hypersensitive and extreme resistance phenotypes elicited by Potato virus Y among 39 potato cultivars released in three world regions over a 117-year period. Plant Dis. 2018, 102, 185–196. [Google Scholar]
- Stich, B.; Inghelandt, D.V. Prospects and potential uses of genomic prediction of key performance traits in tetraploid potato. Front. Plant Sci. 2018, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Deperi, S.I.; Tagliotti, M.E.; Bedogni, M.C.; Manrique-Carpintero, N.C.; Coombs, J.; Zhang, R.; Douches, M.; Huarte, M.A. Discriminant analysis of principal components and pedigree assessment of genetic diversity and population structure in a tetraploid potato panel using SNPs. PLoS ONE 2018, 13, e0194398. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, D.C.S.; Martins, M.L.L.; Santos, A.S.; da Silva Santos, V.; Alves, A.A.C.; da Silva Ledo, C.A. Obtaining hybrids of cultivars and wild subspecies of cassava. Pesqui. Agropecu. Bras. 2018, 53, 182–188. [Google Scholar] [CrossRef]
- Chavarriaga-Aguirre, P.; Maya, M.M.; Bonierbale, M.W.; Kresovich, S.; Fregene, M.A.; Tohme, J.; Kochert, G. Microsatellites in cassava (Manihot esculenta Crantz): Discovery, inheritance and variability. Theor. Appl. Genet. 1998, 97, 493–501. [Google Scholar] [CrossRef]
- Ceballos, H.; Kawuki, R.S.; Gracen, V.E.; Yencho, G.C.; Hershey, C.H. Conventional breeding, marker-assisted selection, genomic selection and inbreeding in clonally propagated crops: A case study for cassava. Theor. Appl. Genet. 2015, 128, 1647–1667. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, T.R.; Filho, P.S.V.; Gonçalves-Vidigal, M.C.; Galván, M.Z.; Lacanallo, G.F.; da Silva, L.I.; Kvitschal, M.V. Genetic diversity and population structure of sweet cassava using simple sequence repeat (SSR) molecular markers. Afr. J. Biotechnol. 2013, 12, 1040–1048. [Google Scholar]
- De Oliveira, E.J.; de Resende, M.D.V.; da Silva Santos, V.; Ferreira, C.F.; Oliveira, G.A.F.; da Silva, M.S.; Oliveira, L.A.; Aguilar-Vildoso, C.I. Genome-wide selection in cassava. Euphytica 2012, 187, 263–276. [Google Scholar] [CrossRef]
- Vieira, L.D.J.; Tavares Filho, L.F.D.Q.; Souza, F.V.D.; Alves, A.A.C.; De Oliveira, E.J. Development of interspecific hybrids of cassava and paternity analysis with molecular markers. J. Agric. Sci. 2013, 151, 849–861. [Google Scholar] [CrossRef]
- Sakurai, T.; Plata, G.; Rodríguez-Zapata, F.; Seki, M.; Salcedo, A.; Toyoda, A.; Ishiwata, A.; Tohme, J.; Sakaki, Y.; Shinozaki, K.; et al. Sequencing analysis of 20,000 full-length cDNA clones from cassava reveals lineage specific expansions in gene families related to stress response. BMC Plant Biol. 2007, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Mba, R.E.C.; Stephenson, P.; Edwards, K.; Melzer, S.; Mkumbira, J.; Gullberg, U.; Apel, K.; Gale, M.; Tohme, J.; Fregene, M. Simple Sequence Repeat (SSR) Markers Survey of the Cassava (Manihot esculenta Crantz) Genome: Towards an SSR-Based Molecular Genetic Map of Cassava. Theor. Appl. Genet. 2001, 102, 21–31. [Google Scholar] [CrossRef]
- Mohan, C.; Shanmugasundaram, P.; Senthil, N. Identification of true hybrid progenies in cassava using simple sequence repeat (SSR) markers. Bangladesh J. Bot. 2013, 42, 155–159. [Google Scholar] [CrossRef]
- Otti, G.; Ayodele, F.; Ikpan, A.; Melaku, G. Development of genomic tools for verification of hybrids and selfed progenies in cassava (Manihot esculenta). Afr. J. Biotechnol. 2011, 10, 17400–17408. [Google Scholar]
- Olsen, K.M.; Schael, B.A. Evidence on the origin of cassava: Phygeography of Manihot esculenta. Proc. Natl. Acad. Sci. USA 1999, 96, 5586–5591. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.B.; Busack, C.; Britt, J.; Bentzen, P. The aunt and uncle effect: An empirical evaluation of the confounding influence of full sibs of parents on pedigree reconstruction. J. Hered. 2001, 92, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Olsen, K.M. SNPs, SSRs, and inferences on cassava’s origin. Plant Mol. Biol. 2004, 56, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Ouborg, N.J.; Piquot, Y.; Van Groenendael, J.M. Estimating pollen flow using SSR markers and paternity exclusion: Accounting for mistyping. Mol. Ecol. 2005, 14, 3109–3121. [Google Scholar]
- Roa, A.C.; Chavarriaga-Aguirre, P.; Duque, M.C.; Maya, M.M.; Bonierbale, M.W.; Iglesias, C.; Tohme, J. Cross-species amplification of cassava (Manihot esculenta) (Euphorbiaceae) microsatellites: Allelic polymorphism and degree of relationship. Am. J. Bot. 2000, 87, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Rabbi, I.; Hamblin, M.; Gedil, M.; Kulakow, P.; Ferguson, M.; Ikpan, A.S.; Ly, D.; Jannink, J.-L. Genetic mapping using genotyping-by-sequencing in the clonally propagated cassava. Crop Sci. 2014, 54, 1384–1396. [Google Scholar] [CrossRef]
- Rabbi, I.Y.; Kulakow, P.A.; Manu-Aduening, J.A.; Dankyi, A.A.; Asibuo, J.Y.; Parkes, E.Y.; Abdoulaye, T.; Girma, G.; Gedil, M.A.; Ramu, P.; et al. Tracking crop varieties using genotyping by-sequencing markers: A case study using cassava (Manihot esculenta Crantz). BMC Genet. 2015, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Jones, A. Cytological observations and fertility measurements of sweetpotato (Ipomoea batatas (L.) Lam.). Proc. Am. Soc. Hortic. Sci. 1965, 86, 527–537. [Google Scholar]
- Jarret, R.L.; Gawel, N.; Whittensmore, A. Phylogenetic relationships of the sweet potato (Ipomoea batatas (L.) Lam.). J. Am. Soc. Hortic. Sci. 1992, 117, 633–637. [Google Scholar]
- Buteler, M.I. Microsatellite-Based Paternity Analysis in Hexaploid Sweetpotato (Ipomoea batatas (L.) Lam.). Ph.D. Thesis, Louisiana State University, Baton Rouge, LA, USA, 1996. [Google Scholar]
- Buteler, M.I.; Labonte, D.R.; Macchiavelli, R.E. Determining paternity in polyploids: Hexaploid simulation studies. Euphytica 1997, 96, 353–361. [Google Scholar] [CrossRef]
- Buteler, M.I.; Jarret, R.L.; La Bonte, D.R. Sequence characterisation of microsatellites in diploid and polyploid Ipomoea. Theor. Appl. Genet. 1999, 99, 123–132. [Google Scholar] [CrossRef]
- Buteler, M.I.; LaBonte, R.L.; Jarret, D.R.; Macchiavelli, R.E. Microsatellite-based paternity analysis in polyploid sweetpotato. J. Am. Soc. Hortic. Sci. 2002, 127, 392–396. [Google Scholar]
- Lopez-Lavalle, L.A.B.; Orjeda, G. Occurrence and cytological mechanism of 2n pollen formation in a tetraploid accession of Ipomoea batatas (sweet potato). J. Hered. 2002, 93, 185–192. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Tseng, Y.T.; Lo, H.F. Application of simple sequence repeats in determining the genetic relationships of cultivars used in sweet potato polycross breeding in Taiwan. Sci. Hortic. 2002, 93, 215–224. [Google Scholar] [CrossRef]
- Hu, J.; Nakatani, M.; Mizuno, K.; Fujimura, T. Development and characterization of microsatellite markers in sweetpotato. Breed. Sci. 2004, 54, 177–188. [Google Scholar] [CrossRef]
- Mwanga, R.O.M.; Andrade, M.I.; Carey, E.E.; Low, J.W.; Yencho, G.C.; Grüneberg, W.J. Sweetpotato (Ipomoea batatas L.). In Genetic Improvement of Tropical Crops; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Rosmayati, M.S.; Bakti, D. Identification and phylogenetic analysis of local yellow and orange sweet potatoes genotypes in Sumatera Utara. IOP Conf. Ser. Earth Environ. Sci. 2018, 122, 012048. [Google Scholar] [CrossRef]
- Ashley, M.V. Plant parentage, pollination, and dispersal: How DNA microsatellites have altered the landscape. Crit. Rev. Plant Sci. 2010, 29, 148–161. [Google Scholar] [CrossRef]
- Caron, G.E.; Leblanc, R. Pollen contamination in a small black spruce seedling seed orchard for three consecutive years. For. Ecol. Manag. 1992, 53, 245–261. [Google Scholar] [CrossRef]
- Greenwood, M.S. Gene exchange in loblolly pine: The relation between pollination mechanism, female receptivity and pollen availability. Am. J. Bot. 1986, 73, 1443–1451. [Google Scholar] [CrossRef]
- Linhart, Y.B.; Busby, W.H.; Beach, J.H.; Feinsinger, P. Forager behavior, pollen dispersal, and inbreeding in two species of humming bird pollinated plants. Evolution 1987, 41, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Parra, V.; Vargas, C.F.; Eguiarte, L.E. Reproductive biology, pollen and seed dispersal, and neighborhood size in the hummingbird-pollinated Echeveria gibbiflora (Crassulaceae). Am. J. Bot. 1993, 80, 153–159. [Google Scholar] [CrossRef]
- Walther-Hellwig, K.; Frankl, R. Foraging distances of Bombus muscorum, Bombus lapidarius, and Bombus terrestris (Hymenoptera, Apidae). J. Insect Behav. 2000, 13, 239–246. [Google Scholar] [CrossRef]
- Sunnucks, P. Efficient genetic markers for population biology. Trends Ecol. Evol. 2000, 15, 199–203. [Google Scholar] [CrossRef]
- Cercueil, A.; Bellemain, E.; Manel, S. PARENTE: Computer program for parentage analysis. J. Hered. 2002, 93, 458–459. [Google Scholar] [CrossRef] [PubMed]
- Coltman, D.W. Male reproductive success in a promiscuous mammal: Behavioural estimates compared with genetic paternity. Mol. Ecol. 1999, 8, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, P.N.M.; Pemberton, J.; Komers, P.; Malarky, G. Genetic and behavioural evidence of monogamy in a mammal, Kirk’s dik–dik (Madoqua kirkii). Proc. R. Soc. Lond. B Biol. Sci. 1997, 264, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Goossens, B.; Graziani, L.; Waits, L. Extra-pair paternity in the monogamous Alpine marmot revealed by nuclear DNA microsatellite analysis. Behav. Ecol. Sociobiol. 1998, 43, 281–288. [Google Scholar] [CrossRef]
- Clapham, P.J.; Palsboll, P.J. Molecular analysis of paternity shows promiscuous mating in female humpback whales (Megaptera novaeangliae Borowski). Proc. R. Soc. Lond. B Biol. Sci. 1997, 264, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Howitt, C.A.; Miskelly, A. Identification of grain variety and quality type. In Cereal Grains: Assessing and Managing Quality, 2nd ed.; Wrigley, C., Batey, I., Miskelly, D., Eds.; Woodhead Publishing: Cambridge, UK, 2017; pp. 311–341. [Google Scholar]
- Pemberton, J.M. Wild pedigrees: The way forward. Proc. R. Soc. Lond. B Biol. Sci. 2009, 275, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, S.; Bernasconi, G. High prevalence of multiple paternity within fruits in natural populations of Silene latifolia, as revealed by microsatellite DNA analysis. Mol. Ecol. 2007, 16, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.; Fowler, S.; Burgoyne, L.A.; Scott, A.C.; Harding, H.W.J. Repetitive deoxyribonucleic acid (DNA) and human genome variation: A concise review relevant to forensic biology. J. Forensic Sci. 1988, 33, 1111–1126. [Google Scholar]
- Jacob, H.J.; Lindpaintner, K.; Lincoln, S.E.; Kusumi, K.; Bunker, R.K.; Mao, Y.P.; Ganten, D.; Dzau, V.J.; Lander, E.S. Genetic mapping of a gene causing hypertension in the stroke-prone spontaneously hypertensive rat. Cell 1991, 67, 213–224. [Google Scholar] [CrossRef]
- Walsh, P.S.; Fildes, N.J.; Reynolds, R. Sequence analysis and characterization of stutter products at the tetranucleotide repeat locus vWA. Nucleic Acids Res. 1996, 24, 2807–2812. [Google Scholar] [CrossRef] [PubMed]
- Smulders, M.J.M.; Bredemeijer, G.; Rus-Kortekaas, W.; Arens, P.; Vosman, B. Use of short microsatellites from database sequences to generate polymorphisms among Lycopersicon esculentum cultivars and accessions of other Lycopersicon species. Theor. Appl. Genet. 1997, 94, 264–272. [Google Scholar] [CrossRef]
- Park, Y.-J.; Lee, J.K.; Kim, N.-S. Simple sequence repeat polymorphisms (SSRPs) for evaluation of molecular diversity and germplasm classification of minor crops. Molecules 2009, 14, 4546–4569. [Google Scholar] [CrossRef] [PubMed]
- Grattapaglia, D.; Kirst, M. Eucalyptus applied genomics: From gene sequences to breeding tools. New Phytol. 2008, 179, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Morin, P.A.; Luikart, G.; Wayne, R.K. SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 2004, 19, 208–216. [Google Scholar] [CrossRef]
- Meudt, H.M.; Clarke, A.C. Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci. 2007, 12, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.C.; Garza, J.C. The power of single-nucleotide polymorphisms for large-scale parentage inference. Genetics 2006, 172, 2567–2582. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.I.; Bi, I.V.; Schroeder, S.C.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S. Evolution: The effects of artificial selection on the maize genome. Science 2005, 308, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Batley, J.; Barker, G.; O’Sullivan, H.; Edwards, K.J.; Edwards, D. Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data. Plant Physiol. 2003, 132, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Tingey, S.V.; Morgante, M. Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res. 2001, 11, 1660–1676. [Google Scholar] [CrossRef] [PubMed]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jaccoud, D.; Peng, K.; Feinstein, D.; Kilian, A. Diversity arrays: A solid state technology for sequence information independent genotyping. Nucleic Acids Res. 2001, 29, e25. [Google Scholar] [CrossRef] [PubMed]
- Lezar, S.; Myburg, A.A.; Berger, D.K.; Wingfield, M.J.; Wingfield, B.D. Development and assessment of microarray-based DNA fingerprinting in Eucalyptus grandis. Theor. Appl. Genet. 2004, 109, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Kilian, A.; Huttner, E.; Wenzl, P.; Jaccoud, D.; Carling, J.; Caig, V.; Evers, M.; Heller-Uszynska, K.; Cayla, C.; Patarapuwadol, S.; et al. The fast and the cheap: SNP and DarT-based whole genome profiling for crop improvement. In Proceedings of the International Congress in the Wake of the Double Helix: From the Green Revolution to the Gene Revolution, Bologna, Italy, 27–31 May 2003; Tuberosa, R., Phillips, R.L., Gale, M., Eds.; Oxford University Press: Oxiford, UK; pp. 443–461. [Google Scholar]
- Akbari, M.; Wenzl, P.; Caig, V.; Carling, J.; Xia, L.; Yang, S.; Uszynski, G.; Mohler, V.; Lehmensiek, A.; Kuchel, H.; et al. Diversity arrays technology (DArT) for high-throughput profiling of the hexaploid wheat genome. Theor. Appl. Genet. 2006, 113, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- James, K.E.; Schneider, H.; Ansell, S.W.; Evers, M.; Robba, L.; Uszynski, G.; Pedersen, N.; Newton, A.E.; Russell, S.J.; Vogel, J.C.; et al. Diversity arrays technology (DArT) for pan-genomic evolutionary studies of non-model organisms. PLoS ONE 2008, 3, e1682. [Google Scholar] [CrossRef] [PubMed]
- Semagn, K.; Bjørnstad, Å.; Ndjiondjop, M.N. An overview of molecular marker methods for plants. Afr. J. Biotechnol. 2006, 5, 2540–2568. [Google Scholar]
- Poland, J.A.; Rife, T.W. Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, J.C.; Rhodes, O.E.; DeWoody, J.A. Prospects for inferring pairwise relationships with single nucleotide polymorphisms. Mol. Ecol. 2003, 12, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.; Walsh, D.; Werner, L.; Fiumera, A. Using blocks of linked single nucleotide polymorphisms as highly polymorphic genetic markers for parentage analysis. Mol. Ecol. Res. 2009, 9, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.G.; Arguello, J.R.; Arnold, S.J. Validation of Bateman’s principles: A genetic study of mating patterns and sexual selection in newts. Proc. R. Soc. Lond. B Biol. Sci. 2002, 269, 2533–2539. [Google Scholar] [CrossRef] [PubMed]
- DeWoody, J.A.; Walker, D.; Avise, J.C. Genetic parentage in large half-sib clutches: Theoretical estimates and empirical appraisals. Genetics 2000, 154, 1907–1912. [Google Scholar] [PubMed]
- DeWoody, J.A.; DeWoody, Y.D.; Fiumera, A.C.; Avise, J.C. On the number of reproductives contributing to a half-sib progeny array. Genet. Res. 2000, 75, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Neff, B.D.; Pitcher, T.E.; Repka, J. A Bayesian model for assessing the frequency of multiple mating in nature. J. Hered. 2002, 93, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sefc, K.M.; Koblmuller, S. Assessing parent numbers from offspring genotypes: The importance of marker polymorphism. J. Hered. 2009, 100, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Marshall, T.C.; Slate, J.; Kruuk, L.E.B.; Pemberton, J.M. Statistical confidence for likelihood-based paternity inference in natural populations. Mol. Ecol. 1998, 7, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.I.; Amos, W. Microsatellite genotyping errors: Detection approaches, common sources and consequences for paternal exclusion. Mol. Ecol. 2005, 14, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L. Sibship reconstruction from genetic data with typing errors. Genetics 2004, 166, 1963–1979. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Hadfield, J.D.; Sefc, K.M.; Sturmbauer, C. Pedigree reconstruction in wild fish populations. Mol. Ecol. 2008, 17, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Dakin, E.E.; Avise, J.C. Microsatellite null alleles in parentage analysis. Heredity 2004, 93, 504–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Creel, S. Using DNA from noninvasive samples to identify individuals and census populations: An evidential approach tolerant of genotyping errors. Conserv. Genet. 2006, 7, 319–329. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Wagner, A.P.; Taper, M.L. ML-RELATE: A computer program for maximum likelihood estimation of relatedness and relationship. Mol. Ecol. Notes 2006, 6, 576–579. [Google Scholar] [CrossRef]
- Grant, V. Plant Speciation, 1st ed.; Columbia University Press: New York, NY, USA, 1971. [Google Scholar]
- Double, M.C.; Cockburn, A.; Barry, S.C.; Smouse, P.E. Exclusion probabilities for single-locus paternity analysis when related males compete for matings. Mol. Ecol. 1997, 6, 1155–1166. [Google Scholar] [CrossRef]
- Olsen, K.M.; Schael, B.A. Microsatellite variation in cassava (Manihot esculenta, Euphorbiaceae) and its wild relatives: Further evidence for a southern Amazonian origin of domestication. Am. J. Bot. 2001, 88, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Mattila, D.K.; Clapham, P.J.; Palsboll, P.J. Statistical approaches to paternity analysis in natural populations and applications to the North Atlantic humpback whale. Genetics 2001, 157, 1673–1682. [Google Scholar] [PubMed]
- Duchesne, P.; Meldgaard, T.; Berrebi, P. Parentage analysis with few contributing breeders: Validation and improvement. J. Hered. 2008, 99, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Thompson, E.A.; Meagher, T.R. Parental and sib likelihoods in genealogy reconstruction. Biometrics 1987, 43, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, G.L. Chromsomal Evolution in Higher Plants; Addison-Wesley: Reading, MA, USA, 1971. [Google Scholar]
- Ashley, M.V.; Wilk, J.A.; Styan, S.M.N.; Craft, K.J.; Jones, K.L.; Feldheim, K.A.; Lewers, K.S.; Ashman, T.L. High variability and disomic segregation of microsatellites in the octoploid Fragaria virginiana Mill. (Rosaceae). Theor. Appl. Genet. 2003, 107, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Hanson, T.; Brunsfeld, S.; Finegan, B.; Waits, L. Conventional and genetic measures of seed dispersal for Dipteryx panamensis (Fabaceae) in continuous and fragmented Costa Rican rain forest. J. Trop. Ecol. 2007, 23, 635–642. [Google Scholar] [CrossRef]
- Hanson, T.R.; Brunsfeld, S.J.; Finegan, B.; Waits, L.P. Pollen dispersal and genetic structure of the tropical tree Dipteryx panamensis in a fragmented Costa Rican landscape. Mol. Ecol. 2008, 17, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Scribner, K.T. Parentage and sibship inference from markers in polyploids. Mol. Ecol. Resour. 2014, 14, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Dick, C.W.; Hardy, O.J.; Jones, F.A.; Petit, R.J. Spatial scales of pollen and seed-mediated gene flow in tropical rain forest trees. Trop. Plant Biol. 2008, 1, 20–33. [Google Scholar] [CrossRef]
- Aguilar, R.; Quesada, M.; Ashworth, L.; Herrerias-Diego, Y.; Loco, J. Genetic consequences of habitat fragmentation in plant populations: Susceptible signals in plant traits and methodological approaches. Mol. Ecol. 2008, 17, 5177–5188. [Google Scholar] [CrossRef] [PubMed]
- Bacles, C.F.E.; Ennos, R.A. Paternity analysis of pollen mediated gene flow for Fraxinus excelsior L. in a chronically fragmented landscape. Heredity 2008, 101, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Burczyk, J.; DiFazio, S.P.; Adams, W.T. Gene flow in forest trees: How far do genes really travel? For. Genet. 2004, 11, 1–14. [Google Scholar]
- Bittencourt, J.M.; Sebbenn, A.M. Patterns of pollen and seed dispersal in a small fragmented population of a wind pollinated Araucaria angustifolia in southern Brazil. Heredity 2007, 99, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Hardesty, B.D.; Hubbell, S.P.; Bermingham, E. Genetic evidence of frequent long-distance recruitment in a vertebrate dispersed tree. Ecol. Lett. 2006, 9, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Slavov, G.T.; Leonardi, S.; Burczyk, J.; Adams, W.T.; Strauss, S.H.; Difazio, S.P. Extensive pollen flow in two ecologically contrasting populations of Populus trichocarpa. Mol. Ecol. 2009, 18, 357–373. [Google Scholar] [CrossRef] [PubMed]
- Hamadina, E.I.; Craufurd, P.Q.; Asiedu, R. Flowering intensity in White yam (Dioscorea rotundata). J. Agric. Sci. 2009, 147, 469–477. [Google Scholar] [CrossRef]
- Sadik, S.; Okereke, O.U. Flowering, pollen grain germination, fruiting, seed germination and seedling development of white yam, Dioscorea rotundata Poir. Ann. Bot. 1975, 39, 597–604. [Google Scholar] [CrossRef]
- Orkwor, G.C.; Asiedu, R.; Ekanayake, I.J. Food Yams: Advances in Research; IITA: Ibadan, Nigeria, 2000. [Google Scholar]
- Govaerts, R.; Wilkin, P.; Saunders, R.M.K. World Checklist of Dioscoreales: Yams and Their Allies; The Board of Trustees of the Royal Botanic Gardens: London, UK, 2007; pp. 1–65.
- Halsey, M.K.; Olsen, K.M.; Taylor, H.J.; Chavarriaga-Aguirre, P. Reproductive biology of cassava (Manihot esculenta Crantz) and isolation of experimental field trials. Crop Sci. 2008, 48, 49–58. [Google Scholar] [CrossRef]
- Taxonomy of Sweet Potato Batata (Ipomoea batatas L.) Sec. il Cronquist System. Available online: http://sperimentazione.altervista.org/Sweetpotato.html (accessed on 6 July 2015).
- Scurrah, M.; Celis-Gamboa, C.; Chumbiauca, S.; Salas, A.; Visser, R.G.F. Hybridization between wild and cultivated potato species in the Peruvian Andes and biosafety implications for deployment of GM potatoes. Euphytica 2008, 164, 881–892. [Google Scholar] [CrossRef] [Green Version]
- Petti, C.; Meade, C.; Downes, M.; Mullins, E. Facilitating co-existence by tracking gene dispersal in conventional potato systems with microsatellite markers. Environ. Biosaf. Res. 2007, 6, 223–236. [Google Scholar] [CrossRef] [PubMed]
- White, J.W. Pollination of potatoes under natural conditions. CIP Circ. 1983, 11, 1–2. [Google Scholar]
- Canadian Food Inspection Agency (CFIA). The Biology of Solanum tuberosum (L.) (Potatoes). Available online: http://www.inspection.gc.ca/plants/plants-with-novel-traits/applicants/directive-94-08/biology-documents/solanum-tuberosum-l-/eng/1330982063974/1330982145930 (accessed on 21 April 2016).
- Hawkes, J.G. The evolution of cultivated potatoes and their tuber-bearing wild relatives. Die Kulturpflanze 1988, 36, 189–208. [Google Scholar] [CrossRef]
- Lian, C.; Goto, S.; Kubo, T.; Takahashi, Y.; Nakagawa, M.; Hogetsu, T. Nuclear and chloroplast microsatellite analysis of Abies sachalinensis regeneration on fallen logs in a subboreal forest in Hokkaido, Japan. Mol. Ecol. 2008, 17, 2948–2962. [Google Scholar] [CrossRef] [PubMed]
- Ouborg, N.J.; Piquot, Y.; Van Groenendael, J.M. Population genetics, molecular markers and the study of dispersal in plants. J. Ecol. 1999, 87, 551–568. [Google Scholar] [CrossRef] [Green Version]
- Garcia, C.; Jordano, P.; Godoy, J.A. Contemporary pollen and seed dispersal in a Prunus mahaleb population: Patterns in distance and direction. Mol. Ecol. 2007, 16, 1947–1955. [Google Scholar] [CrossRef] [PubMed]
- Terakawa, M.; Isagi, Y.; Matsui, K.; Yumoto, T. Microsatellite analysis of the maternal origin of Myrica rubra seeds in the feces of Japanese macaques. Ecol. Res. 2009, 24, 663–670. [Google Scholar] [CrossRef]
- Grivet, D.; Smouse, P.E.; Sork, V.L. A novel approach to an old problem: Tracking dispersed seeds. Mol. Ecol. 2005, 14, 3585–3595. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.A.; Chen, J.; Weng, G.J.; Hubbell, S.P. A genetic evaluation of seed dispersal in the neotropical tree Jacaranda copaia (Bignoniaceae). Am. Nat. 2005, 166, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Iwaizumi, M.G.; Takahashi, M.; Watanabe, A.; Ubukata, M. Simultaneous evaluation of paternal and maternal immigrant gene flow and the implications for the overall genetic composition of Pinus densiflora dispersed seeds. J. Hered. 2009, 101, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Bacles, C.F.E.; Lowe, A.; Ennos, R. Effective seed dispersal across a fragmented landscape. Science 2006, 311, 628. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, M.; Haffray, P. Parentage assignment with genomic markers: A major advance for understanding and exploiting genetic variation of quantitative traits in farmed aquatic animals. Front. Genet. 2014, 5, 432. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Woolliams, J.A.; Smith, D.; Williams, J.L. Estimation of pedigree errors in the UK dairy population using microsatellite markers and the impact on selection. J. Dairy Sci. 2002, 85, 2368–2375. [Google Scholar] [CrossRef]
- Muñoz, P.R.; Resende, M.F.R., Jr.; Huber, D.A.; Quesada, T.; Resende, M.D.V.; Neale, D.B.; Wegrzyn, J.L.; Kirst, M.; Peter, G.F. Genomic relationship matrix for correcting pedigree errors in breeding populations: Impact on genetic parameters and genomic selection accuracy. Crop Sci. 2014, 54, 1115–1123. [Google Scholar] [CrossRef]
- Douches, D.S.; Ludlam, K.; Freyre, R. Isozyme and plastid DNA assessment of pedigrees of nineteenth century potato cultivars. Theor. Appl. Genet. 1991, 82, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P.; Hansey, C.N.; Whitty, B.R.; Stoffel, K.; Massa, A.N.; Van Deynze, A.; De Jong, W.S.; Douches, D.S.; Buell, C.R. Single nucleotide polymorphism discovery in elite north American potato germplasm. BMC Genom. 2011, 12, 302. [Google Scholar] [CrossRef] [PubMed]
- Felcher, K.J.; Coombs, J.J.; Massa, A.N.; Hansey, C.N.; Hamilton, J.P.; Veilleux, R.E.; Buell, C.R.; Douches, D.S. Integration of two diploid potato linkage maps with the potato genome sequence. PLoS ONE 2012, 7, e36347. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Crop | Type | Database | Number of Studies | Reference |
|---|---|---|---|---|
| Yam (Dioscorea spp.) | Tuber | Google Scholar, Research Gate, BioMed Central | 12 | [31,32,33,34,35,36,37,38,39,40,41,42] |
| Potato (Solanum tuberosum L.) | Tuber | PubMed, Google Scholar Springer Link, Springer, | 24 | [5,15,19,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63] |
| Cassava (Manihot esculentus Crantz) | Root | Google Scholar, BioMed Central, Research Gate | 18 | [20,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80] |
| Sweet potato (Ipomoea batatas L.) | Root | Google Scholar, Springer Link | 11 | [81,82,83,84,85,86,87,88,89,90,91] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norman, P.E.; Asfaw, A.; Tongoona, P.B.; Danquah, A.; Danquah, E.Y.; Koeyer, D.D.; Asiedu, R. Can Parentage Analysis Facilitate Breeding Activities in Root and Tuber Crops? Agriculture 2018, 8, 95. https://doi.org/10.3390/agriculture8070095
Norman PE, Asfaw A, Tongoona PB, Danquah A, Danquah EY, Koeyer DD, Asiedu R. Can Parentage Analysis Facilitate Breeding Activities in Root and Tuber Crops? Agriculture. 2018; 8(7):95. https://doi.org/10.3390/agriculture8070095
Chicago/Turabian StyleNorman, Prince Emmanuel, Asrat Asfaw, Pangirayi Bernard Tongoona, Agyemang Danquah, Eric Yirenkyi Danquah, David De Koeyer, and Robert Asiedu. 2018. "Can Parentage Analysis Facilitate Breeding Activities in Root and Tuber Crops?" Agriculture 8, no. 7: 95. https://doi.org/10.3390/agriculture8070095