Molecular Dynamics Simulation of High Density DNA Arrays


1
Department of Physics, Faculty of Mathematics and Physics, University of Ljubljana, Jadranska 19, SI-1000 Ljubljana, Slovenia
2
Department of Theoretical Physics, J. Stefan Institute, Jamova c. 39, SI-1000 Ljubljana, Slovenia
3
Computational Science & Engineering Laboratory, ETH Zurich, Clausiusstrasse 33, CH-8092 Zurich, Switzerland
4
Laboratory for Molecular Modeling, National Institute of Chemistry, Hajdrihova 19, SI-1001 Ljubljana, Slovenia
*
Author to whom correspondence should be addressed.
Computation 2018, 6(1), 3; https://doi.org/10.3390/computation6010003
Submission received: 15 December 2017
/
Revised: 4 January 2018
/
Accepted: 5 January 2018
/
Published: 8 January 2018
(This article belongs to the Special Issue Computation in Molecular Modeling)
Abstract
:Densely packed DNA arrays exhibit hexagonal and orthorhombic local packings, as well as a weakly first order transition between them. While we have some understanding of the interactions between DNA molecules in aqueous ionic solutions, the structural details of its ordered phases and the mechanism governing the respective phase transitions between them remains less well understood. Since at high DNA densities, i.e., small interaxial spacings, one can neither neglect the atomic details of the interacting macromolecular surfaces nor the atomic details of the intervening ionic solution, the atomistic resolution is a sine qua non to properly describe and analyze the interactions between DNA molecules. In fact, in order to properly understand the details of the observed osmotic equation of state, one needs to implement multiple levels of organization, spanning the range from the molecular order of DNA itself, the possible ordering of counterions, and then all the way to the induced molecular ordering of the aqueous solvent, all coupled together by electrostatic, steric, thermal and direct hydrogen-bonding interactions. Multiscale simulations therefore appear as singularly suited to connect the microscopic details of this system with its macroscopic thermodynamic behavior. We review the details of the simulation of dense atomistically resolved DNA arrays with different packing symmetries and the ensuing osmotic equation of state obtained by enclosing a DNA array in a monovalent salt and multivalent (spermidine) counterions within a solvent permeable membrane, mimicking the behavior of DNA arrays subjected to external osmotic stress. By varying the DNA density, the local packing symmetry, and the counterion type, we are able to analyze the osmotic equation of state together with the full structural characterization of the DNA subphase, the counterion distribution and the solvent structural order in terms of its different order parameters and consequently identify the most important contribution to the DNA-DNA interactions at high DNA densities.
Keywords:
multiscale modeling and simulation; molecular dynamics; adaptive resolution; osmotic isobaric ensemble; dielectric properties; orientational order parameters; densely packed DNA arraysPACS:
02.70.Ns; 05.70.Fh; 87.14.gk; 87.10.Tf; 87.15.A-; 87.15.ap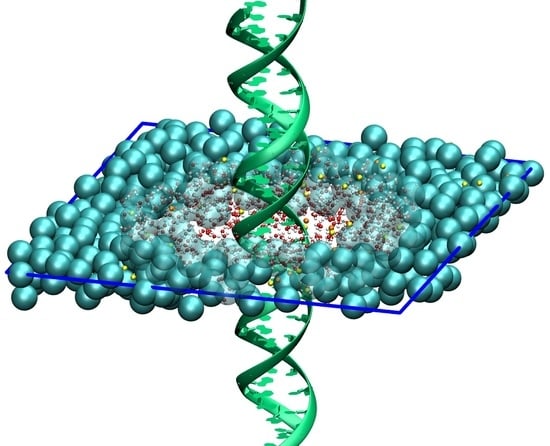
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The properties of dilute (double-stranded) dsDNA solutions do not really suffice in order to understand their behavior in vivo [1,2]. The internal environment of eukaryotic nuclei, bacterial nucleoids and/or viral capsids, is anything but dilute and the properties of DNA solutions in these environments can only be characterized by specifically invoking the fine details of molecular interactions between them at small intersurface spacings [3]. The most direct method to probe the molecular interactions between DNA molecules at high densities is the osmotic stress method as introduced by Parsegian and coworkers [4,5], that yields the thermodynamic osmotic equation of state (EoS), connecting the DNA concentration, as measured e.g., by SAXS, and the DNA osmotic pressure. The detailed dependence of the EoS on the concentration of DNA then allows one to deconvolute the fundamental interactions between DNA molecular surfaces as a function of their separation and different bathing solution conditions, such as the ionic strength of the bathing salt solution, and the identity of the counterions [6]. For monovalent salt solutions, the EoS exhibits a monotonic behavior as a function of the concentration of DNA, characterized by the hydration and fluctuation enhanced electrostatic regimes [3,7,8]. It was only recently, through high resolution control of the osmotic pressure based on its known temperature variation [9,10], that a small discontinuous DNA density change became discernible, between the cholesteric and hexatic phases and between the hexatic and orthorhombic phases for long fragment DNA [10]. The exact sequence and identity of the phases actually depends on the DNA length [11,12] and is apparent also from a sudden discontinuous change in the corresponding order parameters [13,14]. The detailed sequence of mesophase ordering on increase of the DNA density has been identified as consisting of isotropic (I) → cholesteric (Ch) → line hexatic (LH) (or hexagonal columnar H) → orthorhombic (OR) phase [3,11,15,16,17,18], with many details, including the complete DNA length dependence and the demarcation between the line hexatic and hexagonal order, still remaining to be systematically investigated [19]. For multivalent counterions at concentrations above a critical value depending on their identity [20], the EoS exhibits pronounced van der Waals-like density discontinuities that are in general larger then in the case of monovalent salt solutions, signalling a buildup of attractive interactions [21,22], whose details can be inferred from complementary experiments [23], that eventually lead to DNA condensation [24].
The effect of monovalent and most of divalent salts on the EoS can be usually rationalized in terms of a screening electrolyte medium for electrostatic repulsion between highly charged dsDNAs, modified by an additional specific interaction with the DNA surface [25,26]. The only exception to the screening effect are ions at an elevated temperature that actually behave much more like multivalent ions and their effect on the interaction cannot be rationalized within the screening paradigm [21,22]. Counter to the screening effects, the trivalent cations such as and/or at sufficiently large concentrations can lead to dsDNA condensation by turning electrostatic repulsion into either a counterion mediated or solvent mediated attraction, despite DNA’s high negative charge [21,22,24,27,28,29,30], thus enforcing a phase separation from the bathing electrolyte. While multivalent counterions are a necessary condition for attractive interaction, it is not exhaustive as, e.g., CoHex is more efficient as a condensing agent then spermidine, despite both of them being equally charged [25]. On the other hand, monovalent cations such as can act as condensing agents for quadruple stranded DNA [31], while some divalent ions such as and condense the dsDNA, other divalent salts, e.g., cation, act plainly just as a screening electrolyte medium for dsDNA, but as a condensing agent for the triple stranded DNA [32]. While it is generally agreed that purely mean-field electrostatic theories based on the Poisson-Boltzmann phenomenology [33] cannot predict anything but repulsion [28], venturing beyond this framework can be pursued on different levels. By concentrating on the non-mean-field correlation effects [34], the Wigner crystal approach [35] and the strong coupling approach [36] as well as their varieties, such as the charge density wave [37] and ion bridging model [38,39], all single out the universal features of higher valency counterions as responsible for the attractions that they mediate. On the other hand, the tight counterion binding approach [25] was the first to introduce non-electrostatic interactions as governing the distribution of the counterions on the surface of DNA, while preserving the electrostatics as the mechanism mediating the interactions between thus decorated DNAs. The furthest deviation from the predominant role of electrostatics was the proposition by Rau and Parsegian [21,22] that water-mediated interactions present the most important source of DNA attractions and that the way to understand them is to abolish the implicit dielectric continuum model for the solvent.
2. Simulating DNA Arrays
Since direct experiments probing the interactions and/or the associated molecular order can provide only a limited insight into the structural details that would allow an unequivocal deconvolution of the main physical mechanisms underlying these interactions, theoretical interpolations are therefore necessary [40]. While the continuum approaches which mostly underpin the theoretical endeavors described in the paragraph above led to important insights, e.g., the significance of counterion correlations in reversing the sign of the electrostatic interactions between charged helices, backed up by detailed coarse grained simulations [41,42,43,44,45] the molecular details of the DNA solution exposed surface, the granularity of the molecular solvent, and the complicated interactions between both and the mobile charges in solution, preclude the understanding of all the relevant details. As a consequence, detailed all-atom molecular dynamics (MD) simulations appear to be the only vehicle that can bring forth a deeper understanding of the mechanisms and the relevant couplings between them [46], leading to the experimentally observed interaction and ordering phenomenology of DNA in high density mesophases [47,48,49,50,51]. On the downside, all-atom MD simulations of such complicated systems require huge computational resources and the majority of simulations aiming to describe details of the DNA solution phenomenology consist of 1-3 DNA molecules, where the focus is the counterion binding [48,52] and azimuthal dependence of DNA-DNA interaction [49]. Only recently it became feasible to set up an all-atom MD simulation for a larger set of DNA molecules [51,53] characterized by a single packing geometry with only a partial characterization of the DNA countercharge and solvent ordering. This approach has been later extended by the applications of the multiscale MD technique AdResS (Adaptive Resolution Scheme) [54,55,56,57,58,59,60,61,62,63,64,65,66,67], which has been already successfully applied to various biological systems [68,69,70,71,72,73,74,75,76,77], enabling a concurrent and consistent coupling between the atomistic (AT) and the coarse-grained (CG) representations with a key feature of allowing molecules to freely move not only in real space but also in the resolution space across different regions and change their resolution on the fly according to their position in the computational domain.
2.1. Modeling the Solvent Grand Canonical (Osmotic Isobaric) Ensemble
In order to capture the salient properties of high density DNA arrays and their corresponding EoS, any simulation should address the problem of defining and maintaining the osmotic isobaric ensemble, i.e., to maintain the simulated system at a set osmotic pressure, or equivalently at a fixed chemical potential of water, equivalent to the solvent grand canonical ensemble with exchangeable water and solution ions with bulk reservoirs. Fixing the chemical potential of water can be accomplished generally in different ways. In the context of hydrated multilamellar lipid membrane assemblies, the constant water chemical potential is maintained either by including a large water reservoir [78], by direct grand-canonical simulation [79,80], or by the recently developed thermodynamic extrapolation method [81]. The simulation setup corresponds either to two parallel planar lipid monolayers facing the water phase in the middle [82], or a single periodically replicated planar lipid bilayer in the multilamellar liquid-crystalline phase [83]. The extrapolation method then allows for efficient simulations at prescribed chemical potential and yields the interaction forces between membranes with high precision. This enables also a quantitative comparison with measured hydration interactions between two overall charge-neutral lipid membranes in water at small separations and mimics the interactions in a densely packed phospholipid bilayer stack where the undulation forces are suppressed [84,85]. While this type of modeling can be pursued for a one dimensional array of phospholipid membranes, it is nevertheless inapplicable for a two dimensional array of dsDNA molecules. In fact, contrary to the planar membrane case, the interactions between dsDNAs have a strong orientational component [16,25] that fundamentally affects the collective packing symmetry of the ordered array [13,17]. Simulating just two dsDNA molecules in a bathing solution would entirely miss the ensuing orientational frustrations which lead to collective, non-pairwise additive effects. In addition to an implementation of the osmotic isobaric ensemble, simulation of a full array composed of as large a number of DNAs as possible is, therefore, a requirement. Applying the large water reservoir simulation methodology in order to implement the osmotic isobaric ensemble, the DNA subphase can then be segregated from the large water reservoir by way of either a cylindrical [51] or a combined orthorhombic-hexagonal semipermeable boundary [73]. Changing the symmetry of the boundary confining the DNA subphase is a convinient approach to capture the phase transitions with a change of packing symmetry, such as the cholesteric (Ch) → line hexatic (LH) (or hexagonal columnar H) and/or the line hexatic → orthorhombic (OR) phase transitions [3,11,15,18].
2.2. Bathing Solution
Since the effects of monovalent and most of the divalent salts on the EoS, rationalized in terms of a generic screening electrolyte medium, is very different from the effects of multivalent cations, one needs to simulate at least two cases of the bathing solution composition: pure monovalent or divalent salt such as and at a fixed concentration, and a mixed solution composed of a monvalent salt background in addition to , or counterions that are known to exhibit strong electrostatic correlation attractions between equally charged apposed DNA surfaces. In the two most recent simulations, the parametrizations were chosen such as to be close to experimental conditions in the osmotic stress experiments [4,23], with either 250 mM or 1 M pure [73], or with 200/50 mM - ionic mixture [51], leading in both cases to the pronounced screening of the electrostatic interactions. On the other hand, the strongly coupled ions were simulated either as a combination of 1 M and spermidine , where 6 molecules are used per each DNA molecule [73] or sub-mM concentration of and 200 mM [51]. In both cases, the system was neutralized by pure Na counterions or a combination of pure Na counterions and multivalent cations. The monovalent salt concentrations correspond to Debye screening length of for 1 M and to for 200 mM .
The pertaining EoS phenomenology was investigated for a finite orientationally ordered array of 64 vs. 16 explicit atom-resolved DNAs within a solution-permeable membrane, modeled as a confining cylinder [51] or as a confining polygon allowing for hexagonal and/or orthorhombic packing symmetries [73]. In the latter case, the modeling of the semipermeable membrane allows for comparison of the osmotic pressure for two high density phases with different packing symmetries. Characterization of the positional and angular orientational order of the DNA sub-phase together with the Lindemann criterion allows one to extract the osmotic pressure of the phase transitions between the phases of different order and symmetry, all concurrently with the distribution of counterions and positional, orientational and local tetrahedral coordination order of the water molecules.
2.3. Model Interaction Potential Parametrization in DNA Arrays
One of the challenges regarding the molecular simulations of DNA molecules is the reliability of the force fields. On the atomistic scale, simulations are most commonly performed employing the GROMOS [86], AMBER [87], CHARMM [88] or OPLS [89] force fields. The comparison of the two for the Dickerson-Drew dodecamer showed that the AMBER force field was more stable [90]. On the CG level, the DNA molecule is typically modeled with three sites per nucleotide [50,91,92,93,94]. The interactions between CG beads can be obtained either with a top-down approach, i.e., fitted to reproduce key experimental data, especially thermodynamic properties, or with the bottom-up approach, employing methods such as inverse Monte Carlo [95], iterative Boltzmann inversion (IBI) [96], force matching [97,98,99], and relative entropy [100,101,102]. Recently, also the popular MARTINI force field [103] was extended to DNA [104]. On an even coarser level, the DNA can be modeled as a cylinder with charge located on the sites corresponding to the phosphate group in the B-form of DNA [41,42,105]. These models were primarily used early on in Monte Carlo simulations of DNA arrays and were able to reproduce some essential features of the counterion-mediated DNA attraction.
As already stated above, the DNA systems are highly sensitive to the bathing solution. Therefore, the calibration of the force field is not only relevant for the DNA molecule but also for the water and salt of the bathing solution. In a recent paper by Yoo et al. [53], the standard AMBER 03 parameters for the Na and Cl ions were actually found ill parametrized for simulations of DNA arrays. In particular, the DNA molecules deviated from their initial lattice positions and formed clusters, which is in disagreement with the experimental results. Better agreement with the experiments was found for the Joung and Cheatham salt parameters [106], chosen such as to reproduce the relevant experimental osmotic pressure of DNA arrays [53]. In the context of AdResS, where any two AT and CG salt solution models can be coupled, the structure-based coarse-graining approaches, i.e., the Iterative Boltzmann Inversion method [96] that is incorporated into the STOCK web toolkit [68,107], are more convenient, as they allow a direct structural comparison with the underlying AT model.
2.4. Structural Characterization of the High Density DNA Subphase
Extensive MD simulations of DNA arrays with different local packing symmetries allow for a detailed structural characterization of the molecular order in, e.g., the H and OR phases and to identify the driving mechanism(s) of the phase transition between them. The EoS is obtained from multiple simulations with varied DNA-DNA densities corresponding to different average interaxial spacings for each bathing solution composition, either at lower densities [51] or high DNA densities [73] that are relevant for the H-OR phase transition. To conduct the simulations efficiently, the multiscale AdResS was adapted to the simulated system [73]; see the next sections.
The phase transition between the H and OR phases was identified and characterized by invoking the general Lindemann criterion as a proxy for the transition point [73]. Resorting to the Lindemann criterion is an additional approximation and could be avoided if the phase transition were determined more precisely and reliably from the computed free-energy landscape bypassed by enhanced sampling techniques [108,109,110,111]. However, this approach would require much more computational time and computer resources. The computed EoS in the high density DNA density regime in the presence of mono (Na) and multivalent (spermidine) counterions differ typically in the sense that the later exhibits a smaller osmotic pressure at the same background salt and DNA conditions, indicating the existence of an effective attractive interaction, possibly related to the bridging configurations of the multivalent ion between the vicinal DNA surfaces. In addition to the positional and orientational order of DNA molecules, the related local order parameters of water, its local dielectric response profile and the occupancy and the residence times of the ions and water can be obtained from the same simulations. The behavior of the local order parameters of water are then seen to be consistent with a predominant contribution of hydration forces to the DNA EoS at high densities as has been argued all along on purely experimental grounds by Parsegian, Rau and coworkers [26].
3. Adaptive Resolution Simulations of a DNA Molecule Solvated in Salt Solution
In previous sections, we have underlined the importance of performing DNA simulations in the solvent grand canonical ensemble that allows for the exchange of ions and water with the environment. Here, we start to describe our efforts to achieve this goal by employing AdResS, most straightforwardly implementable for a single DNA molecule embedded in a multiscale salt solution [70,71]. We focus on (spatially varying) dielectric properties of solvent and the DNA molecule itself. In simulations, implementing the salt solution environment of DNA demands vast numbers of ions and associated large simulation box sizes. Hence, full-blown AT simulations of such systems are computationally demanding. A possible way to deal with this issue is to use CG models, while at the same time one has to keep in mind the fact, that the dielectric and solvent density profiles display bulk behavior only beyond the second solvent coordination shell of the DNA molecule [70]. Therefore, it would be computationally advantageous to use a simplified CG representation for the salt solution in the “bulk” domain, whereas resorting to the AT level of detail for the proximal solvent. Multiscale simulations that concurrently couple the AT and CG resolutions provide the most efficient way to deal with the implementation of this varying simulation resolution, as they circumvent the limitations of both the AT and CG simulations by providing not only computational benefits but also enhanced physical insight [55].
3.1. AdResS—Single DNA
AdResS [54,55] seamlessly couples two or more levels of detail in the system. In AdResS, recapitulated below, the different resolutions are interlinked via a force interpolation with the total force on a given molecule (or a bundle in the case where a supramolecular mapping is used) defined as
The continuous weighting function w, which governs the transition between different resolution domains, depends on the position of a given molecule (bundle) in the simulation box and is defined such that corresponds to the AT, to the CG, and to the hybrid (HY) domains, respectively. The HY region is sandwiched in between the AT and CG regions and enables an AT molecule to gradually find its energetically favorable orientation with respect to its neighboring molecules. A local thermostat, e.g., the Langevin or Dissipative Particle Dynamics (DPD) thermostat [112], is employed to supply or remove the free-energy associated with the switched on/off molecular degrees of freedom in the HY domain. AdResS allows for various geometric boundaries between the resolution regions. These boundaries can be, for example, one directional [113], cylindrical [70], spherical [69], or even self-adjusting [75]. The relative vector is then a one, two or three dimensional vector, respectively, from the molecular position to the center of the AT domain . The AT region can be centered at either a fixed or a mobile point, as for instance, in a simulation of a protein where it coincides with the protein’s center of mass (CoM). Such setup ensures that the macromolecule and the proximal solvent are always simulated at the AT resolution.
AT and CG molecular models can differ substantially in their thermodynamic properties such as pressure and chemical potential. Pressure (chemical potential) differences at the resolution boundaries lead to density undulations across the HY domain. To mitigate this artefact external thermodynamic (TD) forces opposing the chemical potential gradient are applied to water molecules (bundles) and ions in the HY region [56,114,115]. Employing the TD force, the AdResS methodology can be applied to couple, in principle, any AT and CG models. The structure-based CG approaches (see Section 2.3) are routinely used as they allow a direct structural comparison with the underlying AT model. On the other hand, top-down approaches such as MARTINI [103] focusing on thermodynamic properties can also be applied. However, due to the reduction of degrees of freedom associated with coarse graining, the CG interactions are softer leading to reduced viscosity [55,57,115]. As a result, the CG models frequently display accelerated dynamics. In some cases, the speedup in dynamics is advantageous and can facilitate a more efficient equilibration of complex systems [116]. If required, the transport properties in the CG and AT parts of the system can be matched. This can be accomplished using local thermostats [117,118] or via the Mori-Zwanzig projection formalism [119,120,121,122,123,124] incorporating memory effects in the generalized Langevin equation. CG potentials are also density and temperature dependent [55,125,126,127]. In the context of biomolecular simulations employing AdResS, this does not represent a major drawback as the simulations are manly performed at a fixed temperature of 300 K. Furthermore, the CG region is considered as an external buffer to the region of interest, i.e., the AT region.
3.2. Dielectric Properties
It is known and also confirmed by our simulations that a DNA molecule substantionally influences the surrounding aqueous solvent, i.e., water is considerably ordered and has a slower dynamics owing to the motional restrictions caused by the DNA surface molecular groups and the solution ions. The dielectric constant is hence expected to be much lower than in the bulk [70]. To validate this, we computed the spatially varying dielectric constant of water around a single DNA molecule, employing the Kirkwood’s theory, in which the dielectric constant is related to the average vector sum of the dipole moments of a water molecule centered in a spherical free volume S inside the solvent continuum, with , i.e., [70,128],
is the external dipole moment of the water molecule, is the Boltzmann constant, T the temperature, and the excluded volume due to DNA atoms and ions, respectively. The dipole density is calculated as . Here, V is the volume of a Kirkwood sphere S, which is centered on a given water molecule i, with the number of water molecules with indices in the Kirkwood sphere S around the reference water molecule i. denotes an average over all water molecules in the set, and the angle between dipoles of water molecules i and j.
The spatially varying profile of is computed by discretizing distances from the DNA molecule into bins and calculating the average , in Equation (2), over water molecules that belong to a particular bin. We found that the dielectric constant is increasing with increased distance from the DNA’s CoM and reaches the bulk value at ∼1.7 nm. We also studied the dielectric behavior of water in specific domains around the DNA, namely, the backbone and the minor and major grooves. In agreement with experimental [129,130] and simulation data [128] we observe that (phosphate backbone) > (major groove) > (minor groove) .
4. Simulating Osmotic Isobaric Ensemble: Densely Packed DNA Arrays
To study more complex systems, composed of several DNA molecules one could in principle replicate the one-DNA molecular systems, presented in the previous section. However, in the densely packed DNA arrays, the DNA molecules are positioned so close to each other that there is not enough space to place the different resolution domains in between them. Therefore, to mimic the solvent grand canonical simulations at dense DNA packing, we have embedded the whole DNA array within a reservoir of CG water molecules and ions, see Figure 3. The DNA array and reservoir are separated by a semi-permeable membrane that allows the water molecules and ions (but not the DNA molecules) to freely pass between the two domains [73].
4.1. Computation of Osmotic Pressure
The osmotic stress methodology to determine the osmotic pressure in the DNA subphase relies on the equivalence between the mechanical and the osmotic forces acting on a semipermeable membrane, the latter controlled by a water soluble polymer (usually polyethyleneglycol—PEG) [131], that exerts a uniform compression on the DNA subphase [22]. In MD simulations, the fixed osmotic stress of the pure water and salt solution on the DNA subphase can be implemented via a semipermeable membrane [132,133] surrounding a set of DNA molecules. The osmotic pressure of the DNA array is then determined by measuring the force exerted by the DNA subphase on the confining membrane. The force is computed via a repulsive part of a 10-4 Lennard-Jones interaction between the semipermeable membrane and DNA backbone of the following form [73]
where x is a perpendicular distance to the wall, and are the 12-6 Lennard-Jones parameters and is the wall density. The interaction acts only between the DNA backbone atoms and the membrane to prevent a given DNA molecule from crossing it. The aqueous solution molecules (water, Na, and Cl ions) can nevertheless freely pass through. Thus, the system inside the semipermeable membrane is open, i.e., semi grand canonical, even though the total system is clearly closed. The osmotic pressure of the DNA array can then be computed as , where is the mean force on the semipermeable membrane and A is its area.
For low DNA concentrations, we found that the osmotic pressure is low and does not depend on a specific type or valency of the counterions present. The observed independence of the osmotic pressure on the ion type at low DNA densities is connected with the pronounced ionic screening of electrostatic interactions at 1 M salt conditions beyond the DNA intersurface separation of ∼0.3 nm and is consistent with experiments [134]. In the opposite regime of high DNA densities, the osmotic pressure difference between two different ion types is substantial for both lattices, while the presence of multivalent Spd results in pronounced lowering of the osmotic pressure. At small DNA separations, our quantitative results compare quite well with the experimentally observed values at 2 mM Spd [23] for the hex lattice. At larger separations, however, the pressure obtained with simulations while small, is usually overestimated and attributable to the small size of the simulated array as well as to the possible inaccuracies in the force field. We also observed that the initial lattice in the simulated array is not preserved at the lowest examined DNA concentrations. Hence, it is questionable whether they can be meaningfully compared to the experimental data in this density regime.
4.2. Hexagonal Columnar (H) → Orthorhombic (OR) Phase Transition
Interactions between dsDNAs have a strong orientational component [16,25] that can affect the collective packing symmetry of the ordered array [13,17]. The ensuing azymuthal rotational correlations between DNA molecules appear to be of crucial importance in OR lattices and the loss of azimuthal ordering is found at approximately the same DNA concentrations as the H → OR phase transition. Therefore, the rotational analog of the Lindemann empirical criterion [73] is a good proxy to establish the point of H → OR phase transition. We estimated [73] this transition to occur roughly at DNA concentrations between 700–800 and 500–700 mg/mL for the Na and Na-Spd counterions, respectively, which also compares favorably with the experimental estimations, i.e., ≈670 mg/mL [10,11]. A more consistent determination of the phase transition based on no additional assumptions or approximations could proceed from the free-energy landscape computed by enhanced sampling techniques. While this is an enticing direction it is hampered by much heavier demands on the computational time and resources.
4.3. Orientational Order Parameters
The pronounced orientational dependence of the hydrogen bonding interactions between water molecules and the consequent ordering of the molecular solvent orientations can be quantified by introducing various orientational order parameters. Three different types of these order parametrers, , have been investigated in related contexts. They are defined as
where is the relative angle between a chosen direction and the local orientation of the water molecule. The first two order parameters measure the average orientation of the dipole and the uniaxial quadrupole moments, being equal to for the random orientation in the bulk. The third order parameter quantifies the total quadrupole moment and has a nonzero value in the bulk.
These order parameters can be used to examine the local ordering of interstitial water molecules residing between the DNA molecular surfaces [73]. We have computed all three order parameters along the axis of a given DNA pair for water molecules located in a cuboid (defined by DNA-DNA interaxial spacing, DNA diameter, and its long axis) between a given DNA pair. We found that upon the increase of the DNA concentration the local ordering of water molecules, as quantified by the order parameters , shows distinct features as well as a noticeable symmetry. In particular, the are symmetric w.r.t. the midpoint and indicate progressive orientational layering, while is antisymmetric and quantifies the ordering strength of the opposed DNA surfaces. for vicinal water indicates that it is oriented preferentially perpendicular to the DNA surface, while for the interstitial water indicates it exhibits an almost uniform orientation. These conclusions tally favorably also with water ordering and resulting hydration forces between lipid membranes and polar surfaces in general [81,135]. We consequently conclude that the hydration forces contribute essentially also to the osmotic pressure at large DNA concentrations. This conclusion is further supported by comparing the total osmotic pressure in the system consisting of DNA and the aqueous solution, with the one obtained for DNA only, without any contribution of the solvent. Not only is the latter smaller, but can even show an opposite (negative) sign, which indicates that the aqueous solvent adds an essential contribution to the balance of forces in DNA arrays based on the water-mediated hydration interaction, as has been demonstrated in previous experiments on DNA arrays [4,10].
5. Conclusions and Perspectives
In this review, we presented an overview of computational approaches to simulate densely packed DNA arrays. We highlighted the structural characterization of DNA phases using orientational order parameters, the accurate determination of dielectric properties of solvent and DNA molecules, and modeling of the solvent in the grand-canonical ensemble fashion, as key ingredients to adequately study phenomena such a phase transitions between different high-density DNA mesophases, e.g., with hexagonal and orthorhombic local packings. We presented our attempts to tackle these issues using the multiscale MD method AdResS.
To perform a truly open grand-canonical molecular simulations one should take recourse to the open boundary molecular dynamics (OBMD) [57,116,136,137,138] that permits simulations directly in the grand-canonical ensemble. The OBMD method combines features of AdResS and open MD [139,140], which enable the exchange of energy, momentum, and matter with the external environment through the imposed arbitrary time-dependent external pressure tensor. As discussed in this review, water can be considered as a dielectric continuum from the second DNA coordination shell onward, while the fluctuating cloud of ions surrounding the DNA extends to a wider region. Thus, to properly implement the OBMD simulations that include also the ionic component of the bathing solution, one would need to consider larger all-atom regions that would eventually undermine the original idea of an efficient simulation. To circumvent this limitation one could couple the explicit all-atom salt solution, used in the cylindrical layer surrounding a DNA molecule, and the implicit water with explicit ions, used in the outer part of the simulation box. This would allow for efficient molecular simulations of biomolecules solvated in bathing salt solutions at any ionic strength conditions. The work along these lines is currently underway.
Acknowledgments
R.P. and M.P. acknowledge financial support from the Slovenian Research Agency (research core fundings Nos. P1-0002, P1-0055 and the project J1-7435). J.Z. acknowledges financial support as an ETH Zurich Fellow.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Livolant, F.; Leforestier, A. Condensed Phases of DNA: Structures and Phase Transitions. Prog. Polym. Sci. 1996, 21, 1115–1164. [Google Scholar] [CrossRef]
- Bellini, T.; Cerbino, R.; Zanchetta, G. DNA-Based Soft Phases. Top. Curr. Chem. 2011, 318, 225–279. [Google Scholar]
- Podgornik, R.; Strey, H.H.; Gawrisch, K.; Rau, D.C.; Rupprecht, A.; Parsegian, V.A. Bond orientational order, molecular motion, and free energy of high-density DNA mesophases. Proc. Natl. Acad. Sci. USA 1996, 93, 4261–4266. [Google Scholar] [CrossRef] [PubMed]
- Rau, D.C.; Lee, B.; Parsegian, V.A. Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: Hydration forces between parallel DNA double helices. Proc. Natl. Acad. Sci. USA 1984, 81, 2621–2625. [Google Scholar] [CrossRef] [PubMed]
- Parsegian, V.; Rand, R.; Fuller, N.; Rau, D. Osmotic Stress for the Direct Measurement of Intermolecular Forces. Meth. Enzym. 1986, 127, 400–416. [Google Scholar] [PubMed]
- Strey, H.H.; Podgornik, R.; Rau, D.C.; Parsegian, V.A. DNA-DNA interactions. Curr. Opin. Struct. Biol. 1998, 8, 309–313. [Google Scholar] [CrossRef]
- Podgornik, R.; Rau, D.C.; Parsegian, V.A. The action of interhelical forces on the organization of DNA double helixes: Fluctuation-enhanced decay of electrostatic double-layer and hydration forces. Macromolecules 1989, 22, 1780–1786. [Google Scholar] [CrossRef]
- Strey, H.H.; Parsegian, V.A.; Podgornik, R. Equation of state for polymer liquid crystals: Theory and experiment. Phys. Rev. E 1999, 59, 999–1008. [Google Scholar] [CrossRef]
- Yasar, S.; Podgornik, R.; Parsegian, V. Continuity of states in cholesteric—Line hexatic transition in univalent and polyvalent salt DNA solutions. MRS Proc. 2013, 1619. [Google Scholar] [CrossRef]
- Yasar, S.; Podgornik, R.; Valle-Orero, J.; Johnson, M.; Parsegian, V.A. Continuity of states between the cholesteric → line hexatic transition and the condensation transition in DNA solutions. Sci. Rep. 2014, 4, 6877. [Google Scholar] [CrossRef] [PubMed]
- Durand, D.; Doucet, J.; Livolant, F. A study of the structure of highly concentrated phases of DNA by X-ray diffraction. J. Phys. II Fr. 1992, 2, 1769–1783. [Google Scholar] [CrossRef]
- Nakata, M.; Zanchetta, G.; Chapman, B.D.; Jones, C.D.; Cross, J.O.; Pindak, R.; Bellini, T.; Clark, N.A. End-to-End Stacking and Liquid Crystal Condensation of 6-to-20-Base Pair DNA Duplexes. Science 2007, 318, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Lorman, V.; Podgornik, R.; Žekš, B. Positional, reorientational, and bond orientational order in DNA mesophases. Phys. Rev. Lett. 2001, 87, 218101. [Google Scholar] [CrossRef] [PubMed]
- Grason, G.M. Structural transitions and soft modes in frustrated DNA crystals. EPL Europhys. Lett. 2008, 83, 58003. [Google Scholar] [CrossRef]
- Strzelecka, T.E.; Davidson, M.W.; Rill, R.L. Multiple liquid crystal phases of DNA at high concentrations. Nature 1988, 331, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Kornyshev, A.A.; Leikin, S. Electrostatic interaction between helical macromolecules in dense aggregates: An impetus for DNA poly- and meso-morphism. Proc. Natl. Acad. Sci. USA 1998, 95, 13579–13584. [Google Scholar] [CrossRef] [PubMed]
- Harreis, H.; Kornyshev, A.; Likos, C.; Löwen, H.; Sutmann, G. Phase behavior of columnar DNA assemblies. Phys. Rev. Lett. 2002, 89, 018303. [Google Scholar] [CrossRef] [PubMed]
- Mangenot, S.; Leforestier, A.; Durand, D.; Livolant, F. X-Ray Diffraction Characterization of the Dense Phases Formed by Nucleosome Core Particles. Biophys. J. 2003, 84, 2570–2584. [Google Scholar] [CrossRef]
- Zanchetta, G.; Bellini, T.; Nakata, M.; Clark, N. Physical Polymerization and Liquid Crystallization of RNA Oligomers. J. Am. Chem. Soc. 2008, 130, 12864. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Rau, D. Incomplete Ion Dissociation Underlies the Weakened Attraction between DNA Helices at High Spermidine Concentrations. Biophys. J. 2005, 89, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Rau, D.; Parsegian, V. Direct measurement of the intermolecular forces between counterion-condensed DNA double helices. Evidence for long range attractive hydration forces. Biophys. J. 1992, 61, 246–259. [Google Scholar] [CrossRef]
- Rau, D.C.; Parsegian, V.A. Direct measurment of temperature-dependent solvent forces between DNA double helices. Biophys. J. 1992, 61, 260–271. [Google Scholar] [CrossRef]
- Todd, B.A.; Parsegian, V.A.; Shirahata, A.; Thomas, T.J.; Rau, D.C. Attractive forces between cation condensed DNA double helices. Biophys. J. 2008, 94, 4775–4782. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, V. DNA condensation by multivalent cations. Biopolymers 1997, 44, 269–282. [Google Scholar] [CrossRef]
- Kornyshev, A.A.; Lee, D.J.; Leikin, S.; Wynveen, A. Structure and Interactions of Biological Helices. Rev. Mod. Phys. 2007, 79, 943–996. [Google Scholar] [CrossRef]
- Podgornik, R.; Rau, D.; Parsegian, V. Parametrization of direct and soft-undulatory forces between DNA double helical polyelectrolytes in solutions of several different anions and cations. Biophys. J. 1994, 66, 962–971. [Google Scholar] [CrossRef]
- Bloomfield, V. DNA condensation. Curr. Opin. Struct. Biol. 1996, 6, 334–341. [Google Scholar] [CrossRef]
- Gelbart, W.M.; Bruinsma, R.F.; Pincus, P.A.; Parsegian, V.A. DNA-inspired electrostatics. Phys. Today 2000, 53, 38–44. [Google Scholar] [CrossRef]
- Qiu, X.; Rau, D.C.; Parsegian, V.; Fang, L.T.; Knobler, C.M.; Gelbart, W.M. Salt-dependent DNA-DNA spacings in intact bacteriophage λ eflect relative importance of DNA self-repulsion and bending energies. Phys. Rev. Lett. 2011, 106, 028102. [Google Scholar] [CrossRef] [PubMed]
- Kornyshev, A.A. Physics of DNA: Unravelling hidden abilities encoded in the structure of ’the most important molecule’. Phys. Chem. Chem. Phys. 2010, 12, 12352–12378. [Google Scholar] [CrossRef] [PubMed]
- Yasar, S.; Schimelman, J.; Aksoyoglu, M.A.; Steinmetz, N.; French, R.; Parsegian, V.; Podgornik, R. X-ray characterization of mesophases of human telomeric G-quadruplexes and other DNA analogues. Sci. Rep. 2016, 6, 27079. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Rau, D.C.; Parsegian, V. Divalent counterion-induced condensation of triple-strand DNA. Proc. Natl. Acad. Sci. USA 2010, 107, 21482–21486. [Google Scholar] [CrossRef] [PubMed]
- Markovich, T.; Andelman, D.; Podgornik, R. Handbook of Lipid Membranes; Safynia, C., Raedler, J., Eds.; Taylor and Francis: Sausalito, CA, USA, 2018; Chapter 9. [Google Scholar]
- Rouzina, I.; Bloomfield, V.A. Macroion Attraction Due to Electrostatic Correlation between Screening Counterions. 1. Mobile Surface-Adsorbed Ions and Diffuse Ion Cloud. J. Phys. Chem. 1996, 100, 9977–9989. [Google Scholar] [CrossRef]
- Grosberg, A.; Nguyen, T.; Shklovskii, B. Colloquium: The physics of charge inversion in chemical and biological systems. Rev. Mod. Phys. 2002, 74, 329–345. [Google Scholar] [CrossRef]
- Naji, A.; Kanduč, M.; Forsman, J.; Podgornik, R. Perspective: Coulomb fluids—Weak coupling, strong coupling, in between and beyond. J. Chem. Phys. 2013, 139, 150901. [Google Scholar] [CrossRef] [PubMed]
- Angelini, T.; Golestanian, R.; Coridan, R.; Butler, J.; Beraud, A.; Krisch, M.; Sinn, H.; Schweizer, K.; Wong, G. Counterions between charged polymers exhibit liquid-like organization and dynamics. Proc. Natl. Acad. Sci. USA 2006, 103, 7962–7967. [Google Scholar] [CrossRef] [PubMed]
- Raspaud, A.; Olvera de la Cruz, M.; Sikorav, J.L.; Livolant, F. Precipitation of DNA by Polyamines: A Polyelectrolyte Behavior. Biophys. J. 1998, 74, 381–393. [Google Scholar] [CrossRef]
- Kanduč, M.; Dobnikar, J.; Podgornik, R. Counterion-mediated electrostatic interactions between helical molecules. Soft Matter 2009, 5, 868–877. [Google Scholar] [CrossRef]
- Podgornik, R.; Strey, H.H.; Parsegian, V.A. Equation of State for DNA Liquid Crystals: Fluctuation Enhanced Repulsion. Phys. Rev. E 1997, 78, 895–898. [Google Scholar]
- Lyubartsev, A.P.; Nordenskiöld, L. Monte Carlo simulation study of ion distribution and osmotic pressure in hexagonally oriented DNA. J. Phys. Chem. 1995, 99, 10373–10382. [Google Scholar] [CrossRef]
- Allahyarov, E.; Gompper, G.; Löwen, H. DNA condensation and redissolution: Interaction between overcharged DNA molecules. J. Phys. Condens. Matter 2005, 17, 1827–1840. [Google Scholar] [CrossRef]
- Nguyen, T.; Grosberg, A.; Shklovskii, B. Macroions in salty water with multivalent ions: Giant inversion of charge. Phys. Rev. Lett. 2000, 14, 1568–1571. [Google Scholar] [CrossRef] [PubMed]
- Cherstvy, A. Electrostatic interactions in biological DNA-related systems. Phys. Chem. Chem. Phys. 2011, 13, 9942–9968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T. Grand-canonical simulation of DNA condensation with two salts, effect of divalent counterion size. J. Chem. Phys. 2016, 144, 065102. [Google Scholar] [CrossRef] [PubMed]
- Maffeo, C.; Yoo, J.; Comer, J.; Wells, D.B.; Luan, B.; Aksimentiev, A. Close encounters with DNA. J. Phys. Condens. Matter 2014, 26, 413101. [Google Scholar] [CrossRef] [PubMed]
- Guldbrand, L.; Nilsson, L.; Nordenskiold, L. A Monte Carlo simulation study of electrostatic forces between hexagonally packed DNA double helicess. J. Chem. Phys. 1986, 85, 6686–6698. [Google Scholar] [CrossRef]
- Korolev, N.; Lyubartsev, A.P.; Laaksonen, A.; Nordenskiöld, L. A molecular dynamics simulation study of oriented DNA with polyamine and sodium counterions: Diffusion and averaged binding of water and cations. Nucleic Acids Res. 2006, 31, 5971–5981. [Google Scholar] [CrossRef]
- Dai, L.; Mu, Y.; Nordenskiöld, L.; van der Maarel, J.R.C. Molecular dynamics simulation of multivalent-ion mediated attraction between DNA molecules. Phys. Rev. Lett. 2008, 100, 118301. [Google Scholar] [CrossRef] [PubMed]
- Potoyan, D.A.; Savelyev, A.; Papoian, G.A. Recent successes in coarse-grained modeling of DNA. WIREs Comput. Mol. Sci. 2013, 3, 69–83. [Google Scholar] [CrossRef]
- Yoo, J.; Aksimentiev, A. The structure and intermolecular forces of DNA condensates. Nucleic Acids Res. 2016, 44, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Laughton, C.A.; Harris, S.A. The atomistic simulation of DNA. WIREs Comput. Mol. Sci. 2011, 1, 590–600. [Google Scholar] [CrossRef]
- Yoo, J.; Aksimentiev, A. Improved parametrization of Li+, Na+, K+ and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J. Phys. Chem. Lett. 2011, 3, 45–50. [Google Scholar] [CrossRef]
- Praprotnik, M.; Delle Site, L.; Kremer, K. Adaptive resolution molecular-dynamics simulation: Changing the degrees of freedom on the fly. J. Chem. Phys. 2005, 123, 224106. [Google Scholar] [CrossRef] [PubMed]
- Praprotnik, M.; Delle Site, L.; Kremer, K. Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu. Rev. Phys. Chem. 2008, 59, 545–571. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, S.; Poblete, S.; Junghans, C.; Ciccotti, G.; Delle Site, L.; Kremer, K. Adaptive resolution molecular dynamics simulation through coupling to an internal particle reservoir. Phys. Rev. Lett. 2012, 108, 170602. [Google Scholar] [CrossRef] [PubMed]
- Delle Site, L.; Praprotnik, M. Molecular systems with open boundaries: Theory and Simulation. Phys. Rep. 2017, 693, 1–56. [Google Scholar] [CrossRef]
- Wang, H.; Hartmann, C.; Schütte, C.; Delle Site, L. Grand-canonical-like molecular-dynamics simulations by using an adaptive-resolution technique. Phys. Rev. X 2013, 3, 011018. [Google Scholar] [CrossRef]
- Agarwal, A.; Delle Site, L. Path integral molecular dynamics within the grand canonical-like adaptive resolution technique: Simulation of liquid water. J. Chem. Phys. 2015, 143, 094102. [Google Scholar] [CrossRef] [PubMed]
- Delle Site, L. Formulation of Liouville’s theorem for grand ensemble molecular simulations. Phys. Rev. E 2016, 93, 022130. [Google Scholar] [CrossRef] [PubMed]
- Delle Site, L. Grand Canonical Adaptive Resolution Simulation for Molecules with Electrons: A Theoretical Framework based on Physical Consistency. Comput. Phys. Commun. 2017. [Google Scholar] [CrossRef]
- Potestio, R.; Fritsch, S.; Español, P.; Delgado-Buscalioni, R.; Kremer, K.; Everaers, R.; Donadio, D. Hamiltonian adaptive resolution simulation for molecular liquids. Phys. Rev. Lett. 2013, 110, 108301. [Google Scholar] [CrossRef] [PubMed]
- Potestio, R.; Español, P.; Delgado-Buscalioni, R.; Everaers, R.; Kremer, K.; Donadio, D. Monte Carlo adaptive resolution simulation of multicomponent molecular liquids. Phys. Rev. Lett. 2013, 111, 060601. [Google Scholar] [CrossRef] [PubMed]
- Español, P.; Delgado-Buscalioni, R.; Everaers, R.; Potestio, R.; Donadio, D.; Kremer, K. Statistical mechanics of hamiltonian adaptive resolution simulations. J. Chem. Phys. 2015, 142, 064115. [Google Scholar] [CrossRef] [PubMed]
- Tarenzi, T.; Calandrini, V.; Potestio, R.; Giorgetti, A.; Carloni, P. Open Boundary Simulations of Proteins and Their Hydration Shells by Hamiltonian Adaptive Resolution Scheme. J. Chem. Theory Comput. 2017. [Google Scholar] [CrossRef] [PubMed]
- Everaers, R. Thermodynamic translational invariance in concurrent multiscale simulations of liquids. Eur. Phys. J. Spec. Top. 2016, 225, 1483–1503. [Google Scholar] [CrossRef]
- Delgado-Buscalioni, R. Thermodynamics of adaptive molecular resolution. Philos. Trans. R. Soc. A 2016, 374, 20160152. [Google Scholar] [CrossRef] [PubMed]
- Bevc, S.; Junghans, C.; Kremer, K.; Praprotnik, M. Adaptive resolution simulation of salt solutions. New J. Phys. 2013, 15, 105007. [Google Scholar] [CrossRef]
- Zavadlav, J.; Melo, M.N.; Marrink, S.J.; Praprotnik, M. Adaptive resolution simulation of an atomistic protein in MARTINI water. J. Chem. Phys. 2014, 140, 054114. [Google Scholar] [CrossRef] [PubMed]
- Zavadlav, J.; Podgornik, R.; Praprotnik, M. Adaptive resolution simulation of a DNA molecule in salt solution. J. Chem. Theory Comput. 2015, 11, 5035–5044. [Google Scholar] [CrossRef] [PubMed]
- Zavadlav, J.; Podgornik, R.; Melo, M.N.; Marrink, S.J.; Praprotnik, M. Adaptive resolution simulation of an atomistic DNA molecule in MARTINI salt solution. Eur. Phys. J. Spec. Top. 2016, 225, 1595–1607. [Google Scholar] [CrossRef]
- Zavadlav, J.; Marrink, S.J.; Praprotnik, M. Adaptive resolution simulation of supramolecular water: The concurrent making, breaking, and remaking of water bundles. J. Chem. Theory Comput. 2016, 12, 4138–4145. [Google Scholar] [CrossRef] [PubMed]
- Zavadlav, J.; Podgornik, R.; Praprotnik, M. Order and interactions in DNA arrays: Multiscale molecular dynamics simulation. Sci. Rep. 2017, 7, 4775–4786. [Google Scholar] [CrossRef] [PubMed]
- Zavadlav, J.; Bevc, S.; Praprotnik, M. Adaptive resolution simulations of biomolecular systems. Eur. Biophys. J. 2017, 46, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Kreis, K.; Potestio, R.; Kremer, K.; Fogarty, A.C. Adaptive resolution simulations with self-adjusting high-resolution regions. J. Chem. Theory Comput. 2016, 12, 4067–4081. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, A.C.; Potestio, R.; Kremer, K. A multi-resolution model to capture both global fluctuations of an enzyme and molecular recognition in the ligand-binding site. Proteins 2016, 84, 1902–1913. [Google Scholar] [CrossRef] [PubMed]
- Guzman, H.V.; Junghans, C.; Kremer, K.; Stuehn, T. Scalable and fast heterogeneous molecular simulation with predictive parallelization schemes. Phys. Rev. E 2017, 96, 053311. [Google Scholar] [CrossRef]
- Hua, L.; Zangi, R.; Berne, B.J. Hydrophobic Interactions and Dewetting between Plates with Hydrophobic and Hydrophilic Domains. J. Phys. Chem. C 2009, 113, 5244–5253. [Google Scholar] [CrossRef]
- Hayashi, T.; Pertsin, A.J.; Grunze, M. Grand canonical Monte Carlo simulation of hydration forces between nonorienting and orienting structureless walls. J. Chem. Phys. 2002, 117, 6271–6280. [Google Scholar] [CrossRef]
- Pertsin, A.; Fedyanin, I.; Grunze, M. Computer simulation of water-mediated forces between gel-phase phospholipid bilayers. J. Chem. Phys. 2009, 113, 215102. [Google Scholar] [CrossRef] [PubMed]
- Schneck, E.; Sedlmeier, F.; Netz, R.R. Hydration repulsion between biomembranes results from an interplay of dehydration and depolarization. Proc. Natl. Acad. Sci. USA 2012, 109, 14405–14409. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, M.; Schlaich, A.; Schneck, E.; Netz, R. Water-Mediated Interactions between Hydrophilic and Hydrophobic Surfaces. Langmuir 2016, 32, 8767–8782. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, M.; Schneck, E.; Netz, R.R. Hydration Interaction between Phospholipid Membranes: Insight into Different Measure- ment Ensembles from Atomistic Molecular Dynamics Simulations. Langmuir 2013, 29, 9126–9137. [Google Scholar] [CrossRef] [PubMed]
- Pabst, G.; Katsaras, J.; Raghunathan, V.A.; Rappolt, M. Structure and interactions in the anomalous swelling regime of phospholipid bilayers. Langmuir 2003, 19, 1716–1722. [Google Scholar] [CrossRef]
- Lu, B.S.; Gupta, S.; Belicka, M.; Podgornik, R.; Pabst, G. Modulation of elasticity and interactions in charged lipid multibilayers: Monovalent salt solutions. Langmuir 2016, 32, 13546–13555. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.E.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS Force Field for Proteins. Energy Minimizations for Crystals of Cyclic Peptides and Crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.G.; de Andrade, A.S.C.; Mottin, M.; Netz, P.A. Molecular dynamics of DNA: Comparison of force fields and terminal nucleotide definitions. J. Phys. Chem. B 2010, 114, 9882–9893. [Google Scholar] [CrossRef] [PubMed]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Sulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing improved structural properties and salt dependence into a coarse-grained model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejczyk, M.; Spasic, A.; Liwo, A.; Scheraga, H.A. DNA duplex formation with a coarse-grained model. J. Chem. Theory Comput. 2014, 10, 5020–5035. [Google Scholar] [CrossRef] [PubMed]
- Knotts, T.A.I.; Rathore, N.; Schwartz, D.C.; de Pablo, J.J. A coarse grain model for DNA. J. Chem. Phys. 2007, 126, 084901. [Google Scholar] [CrossRef] [PubMed]
- Voltz, K.; Trylska, J.; Tozzini, V.; Kurkal-Siebert, V.; Langowski, J.; Smith, J. Coarse-grained force field for the nucleosome from self-consistent multiscaling. J. Comput. Chem. 2008, 29, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Lyubartsev, A.P.; Laaksonen, A. Calculation of effective interaction potentials from radial distribution functions: A reverse Monte Carlo approach. Phys. Rev. E 1995, 52, 3730–3737. [Google Scholar] [CrossRef]
- Reith, D.; Pütz, M.; Müller-Plathe, F. Deriving effective mesoscale potentials from atomistic simulations. J. Comput. Chem. 2003, 24, 1624–1636. [Google Scholar] [CrossRef] [PubMed]
- Ayton, G.S.; Noid, W.G.; Voth, G.A. Multiscale modeling of biomolecular systems: In serial and in parallel. Curr. Opin. Struct. Biol. 2007, 17, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Mullinax, J.W.; Noid, W.G. Extended ensemble approach for deriving transferable coarse-grained potentials. J. Chem. Phys. 2009, 131, 104110. [Google Scholar] [CrossRef]
- Rudzinski, J.F.; Noid, W.G. Bottom-up coarse-graining of peptide ensembles and helix–coil transitions. J. Chem. Theory Comput. 2015, 11, 1278–1291. [Google Scholar] [CrossRef] [PubMed]
- Shell, M.S. The relative entropy is fundamental to thermodynamic ensemble optimization. J. Chem. Phys. 2008, 129, 144108. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, S.P.; Shell, M.S. A new multiscale algorithm and its application to coarse-grained peptide models for self-assembly. J. Phys. Chem. B 2012, 116, 8383–8393. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.; Shell, M.S.; Noid, W.G. The impact of resolution upon entropy and information in coarse-grained models. J. Chem. Phys. 2015, 143, 243104. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, J.J.; Ingólfsson, H.I.; Akhshi, P.; Tieleman, D.P.; Marrink, S.J. Martini Coarse-Grained Force Field: Extension to DNA. J. Chem. Theory Comput. 2015, 11, 3932–3945. [Google Scholar] [CrossRef] [PubMed]
- Grönbech-Jensen, N.; Mashl, R.J.; Bruinsma, R.F.; Gelbart, W.M. Counterions-induced attraction between rigid polyelectrolytes. Phys. Rev. Lett. 1997, 78, 2477. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E.r. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Bevc, S.; Junghans, C.; Praprotnik, M. Stock: Structure mapper and online coarse-graining kit for molecular simulations. J. Comput. Chem. 2015, 36, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Bolhuis, P.G.; Chandler, D.; Dellago, C.; Geissler, P.L. Transition path sampling: Throwing Ropes over Rough Mountain Passes, in the Dark. Annu. Rev. Phys. Chem. 2002, 53, 291–318. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, M.; Dellago, C. Transition path sampling studies of solid-solid transformations in nanocrystals under pressure. In Trends in Computational Nanomechanics: Transcending Length and Time Scales; Dumitrica, T., Ed.; Springer: Dordrecht, The Netherlands, 2010; pp. 61–84. [Google Scholar]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.Q.; Lu, J.; Abrams, C.F.; Vanden-Eijnden, E. Multiscale implementation of infinite-swap replica exchange molecular dynamics. Proc. Natl. Acad. Sci. USA 2016, 113, 11744–11749. [Google Scholar] [CrossRef] [PubMed]
- Soddemann, T.; Dunweg, B.; Kremer, K. Dissipative particle dynamics: A useful thermostat for equilibrium and nonequilibrium molecular dynamics simulations. Phys. Rev. E 2003, 68, 046702. [Google Scholar] [CrossRef] [PubMed]
- Zavadlav, J.; Melo, M.N.; Cunha, A.V.; de Vries, A.H.; Marrink, S.J.; Praprotnik, M. Adaptive resolution simulation of MARTINI solvents. J. Chem. Theory Comput. 2014, 10, 2591–2598. [Google Scholar] [CrossRef] [PubMed]
- Poblete, S.; Praprotnik, M.; Kremer, K.; Delle Site, L. Coupling different levels of resolution in molecular simulations. J. Chem. Phys. 2010, 132, 114101. [Google Scholar] [CrossRef] [PubMed]
- Praprotnik, M.; Poblete, S.; Kremer, K. Statistical physics problems in adaptive resolution computer simulations of complex fluids. J. Stat. Phys. 2011, 145, 946–966. [Google Scholar] [CrossRef]
- Delgado-Buscalioni, R.; Sablić, J.; Praprotnik, M. Open boundary molecular dynamics. Eur. Phys. J. Spec. Top. 2015, 224, 2331–2349. [Google Scholar] [CrossRef]
- Junghans, C.; Praprotnik, M.; Kremer, K. Transport properties controlled by a thermostat: An extended dissipative particle dynamics thermostat. Soft Matter 2008, 4, 156–161. [Google Scholar] [CrossRef]
- Matysiak, S.; Clementi, C.; Praprotnik, M.; Kremer, K.; Delle Site, L. Modeling diffusive dynamics in adaptive resolution simulation of liquid water. J. Chem. Phys. 2008, 128, 024503. [Google Scholar] [CrossRef] [PubMed]
- Hijon, C.; Español, P.; Vanden-Eijnden, E.; Delgado-Buscalioni, R. Mori-Zwanzig formalism as a practical computational tool. Faraday Discuss. 2010, 144, 301–322. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bian, X.; Caswell, B.; Karniadakis, G.E. Construction of dissipative particle dynamics models for complex fluids via the Mori-Zwanzig formulation. Soft Matter 2014, 10, 8659. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bian, X.; Li, X.; Karniadakis, G.E. Incorporation of memory effects in coarse-grained modeling via the Mori-Zwanzig formalism. J. Chem. Phys. 2015, 143, 243128. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, Y.; Li, Z.; Kinefuchi, I.; Karniadakis, G.E. Construction of non-Markovian coarse-grained models employing the Mori-Zwanzig formalism and iterative Boltzmann inversion. J. Chem. Phys. 2017, 147, 244110. [Google Scholar] [CrossRef] [PubMed]
- Jung, G.; Hanke, M.; Schmid, F. Iterative Reconstruction of Memory Kernels. J. Chem. Theory Comput. 2017. [Google Scholar] [CrossRef] [PubMed]
- Izvekov, S. Microscopic derivation of particle-based coarse-grained dynamics: Exact expression for memory function. J. Chem. Phys. 2017, 146, 124109. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Song, J.; Jeong, C.; Hsu, D.D.; Phelan, F.R.J.; Douglas, J.F.; Keten, S. Energy-Renormalization for Achieving Temperature Transferable Coarse-Graining of Polymer Dynamics. Macromolecules 2017, 50, 8787–8796. [Google Scholar] [CrossRef]
- Haddadian, E.J.; Zhang, H.; Freed, K.F.; Douglas, J.F. Comparative Study of the Collective Dynamics of Proteins and Inorganic Nanoparticles. Sci. Rep. 2017, 7, 41671. [Google Scholar] [CrossRef] [PubMed]
- Louis, A.A. Beware of density dependent pair potentials. J. Phys. Condens. Matter 2002, 14, 9187. [Google Scholar] [CrossRef]
- Young, M.A.; Jayaram, B.; Beveridge, D.L. Local dielectric environment of B-DNA in solution: Results from a 14 ns molecular dynamics trajectory. J. Phys. Chem. B 1998, 102, 7666–7669. [Google Scholar] [CrossRef]
- Jin, R.; Breslauer, K.J. Characterization of the minor groove environment in a drug-DNA complex: Bisbenzimide bound to the poly[d(AT)]poly[d(AT)]duplex. Proc. Natl. Acad. Sci. USA 1988, 85, 8939–8942. [Google Scholar] [CrossRef] [PubMed]
- Barawkar, A.; Ganesh, K.N. Fluorescent d(CGCGAATTCGCG): Characterization of major groove polarity and study of minor groove interactions through a major groove semantophore conjugate. Nucleic Acids Res. 1995, 23, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Podgornik, R.; Hansen, P.L.; Parsegian, V.A. A Phenomenological One-Parameter Equation of State for Osmotic Pressures of PEG and Other Neutral Flexible Polymers in Good Solvents. J. Phys. Chem. B 2009, 113, 3709–3714. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.W.; Li, C.; van der Vegt, N.F.A.; Peter, C. Transferability of coarse grained potentials: Implicit solvent models for hydrated ions. J. Chem. Theory Comput. 2011, 7, 1916–1927. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Roux, B. Simulation of osmotic pressure in concentrated aqueous salt solutions. J. Phys. Chem. Lett. 2010, 1, 183–189. [Google Scholar] [CrossRef]
- Podgornik, R.; Strey, H.H.; Parsegian, V.A. Colloidal DNA. Curr. Opin. Colloid Interface Sci. 1998, 3, 534–539. [Google Scholar] [CrossRef]
- Kanduč, M.; Schlaich, A.; Schneck, E.; Netz, R.R. Hydration repulsion between membranes and polar surfaces: Simulation approaches versus continuum theories. Adv. Colloid Interface Sci. 2014, 208, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Sablić, J.; Praprotnik, M.; Delgado-Buscalioni, R. Open boundary molecular dynamics of sheared star-polymer melts. Soft Matter 2016, 12, 2416–2439. [Google Scholar] [CrossRef] [PubMed]
- Sablić, J.; Praprotnik, M.; Delgado-Buscalioni, R. Deciphering the dynamics of star molecules in shear flow. Soft Matter 2017, 13, 4971–4987. [Google Scholar] [CrossRef] [PubMed]
- Sablić, J.; Delgado-Buscalioni, R.; Praprotnik, M. Application of the Eckart frame to soft matter: Rotation of star polymers under shear flow. Soft Matter 2017, 13, 6988–7000. [Google Scholar] [CrossRef] [PubMed]
- Flekkoy, E.G.; Delgado-Buscalioni, R.; Coveney, P.V. Flux boundary conditions in particle simulations. Phys. Rev. E 2005, 72, 026703. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Buscalioni, R. Tools for multiscale simulation of liquids using open molecular dynamics. In Numerical Analysis of Multiscale Computations. Lecture Notes in Computational Science and Engineering; Engquist, B., Runborg, O., Tsai, Y.H., Eds.; Springer: Dordrecht, The Netherlands, 2012; Volume 82, pp. 145–166. [Google Scholar]
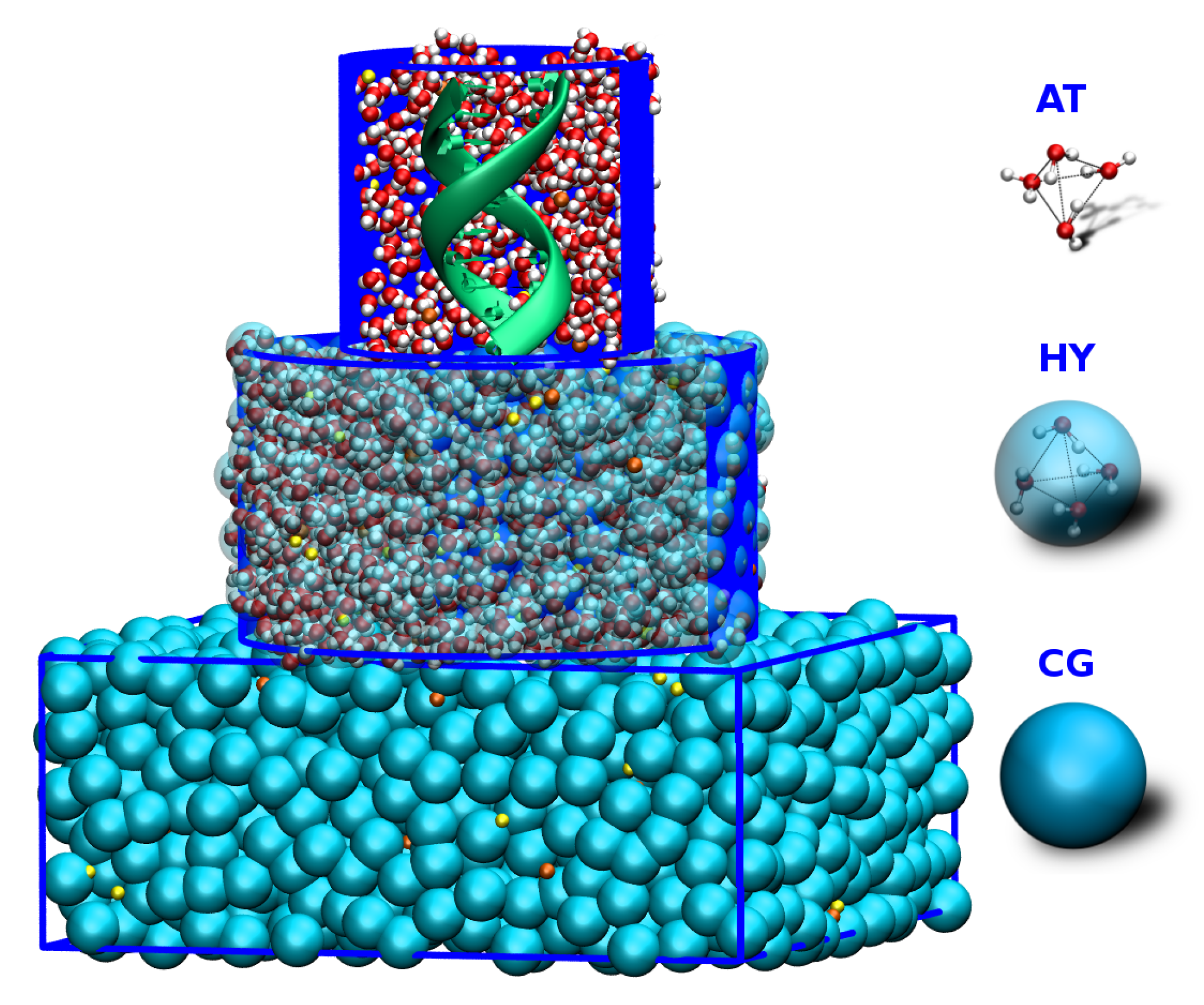
Figure 1.
An atomistic DNA molecule embedded in a dual-resolution salt solution. The proximal solvent around the DNA molecule is modeled using the atomistic level of detail whereas the distal water molecules are represented as grey beads, i.e., one bead corresponds to one water molecule. The salt ions are shown in green (Na) and blue (Cl). Reprinted (adapted) with permission from Ref. [70]. Copyright (2015) American Chemical Society.
Figure 1.
An atomistic DNA molecule embedded in a dual-resolution salt solution. The proximal solvent around the DNA molecule is modeled using the atomistic level of detail whereas the distal water molecules are represented as grey beads, i.e., one bead corresponds to one water molecule. The salt ions are shown in green (Na) and blue (Cl). Reprinted (adapted) with permission from Ref. [70]. Copyright (2015) American Chemical Society.
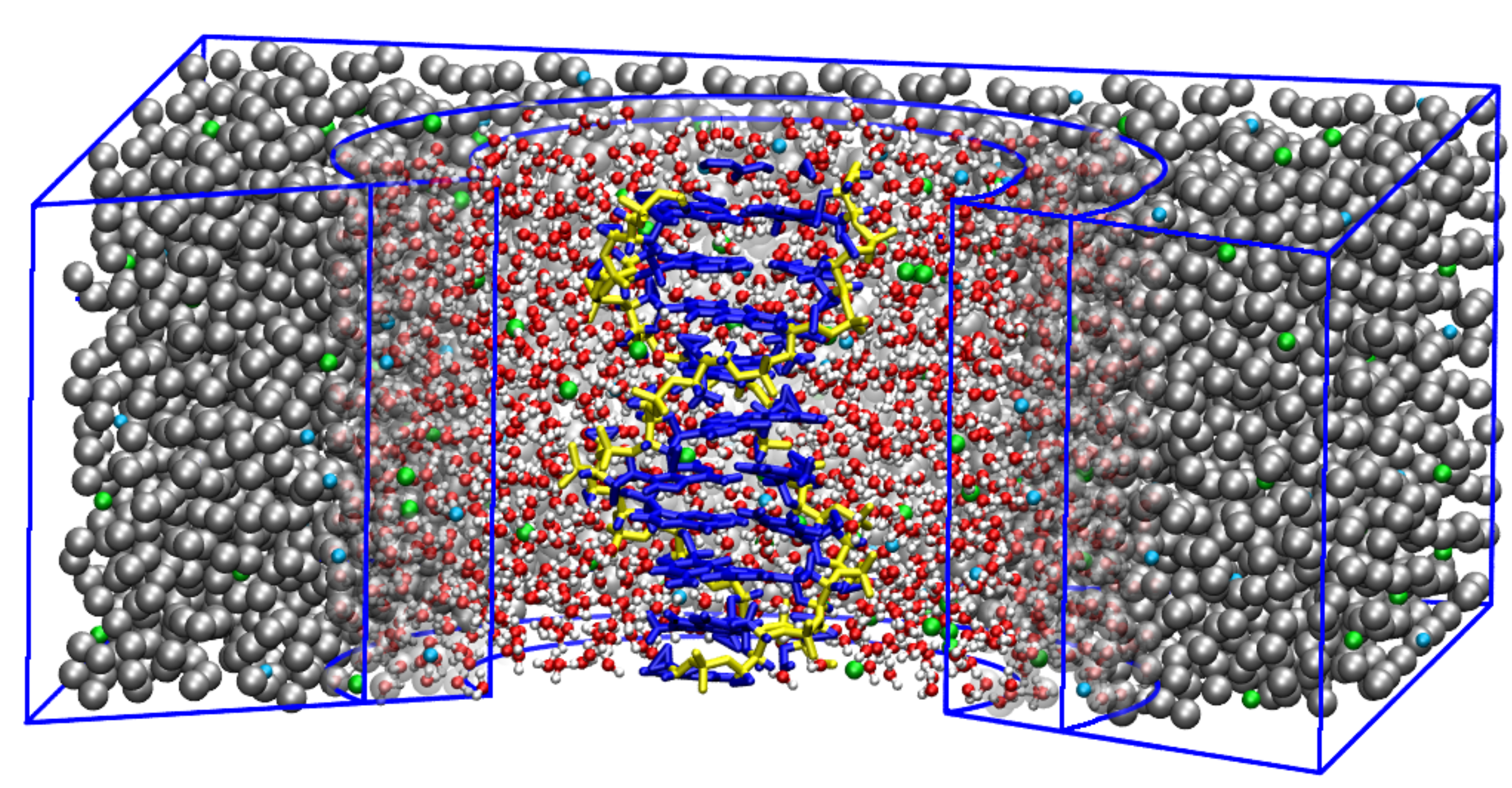
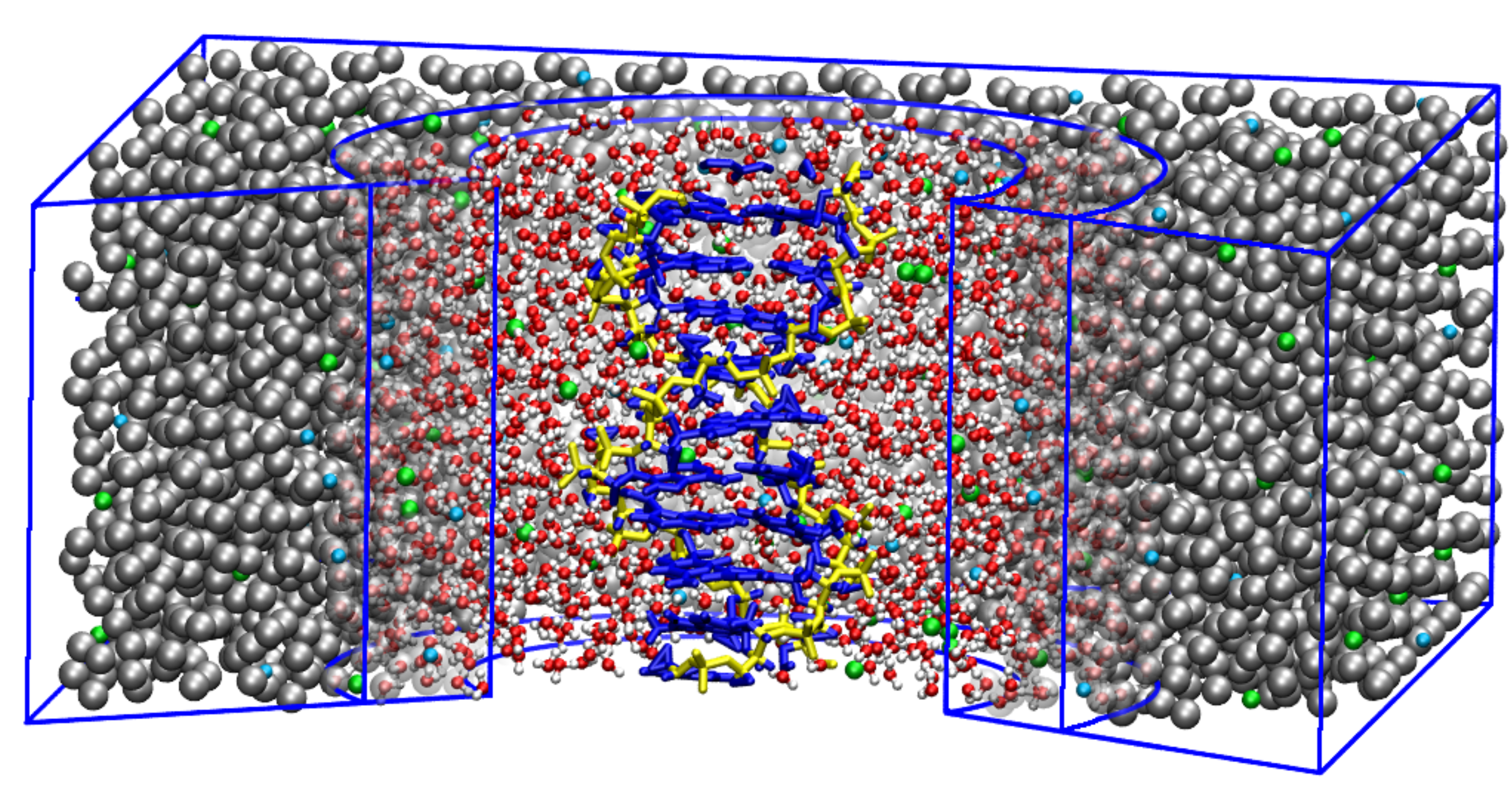
Figure 2.
Schematic depiction of a simulation box with an atomistic DNA in the center surrounded by MARTINI salt solution. Two levels of resolution are used for solvent molecules. AT level of resolution is used for proximal solvent molecules, within a certain radius from the DNA’s center of mass. The distal water molecules are represented as blue MARTINI CG beads, where one CG bead corresponds to four water molecules. Na and Cl ions are shown in orange and yellow, respectively. Adapted from Ref. [71].
Figure 2.
Schematic depiction of a simulation box with an atomistic DNA in the center surrounded by MARTINI salt solution. Two levels of resolution are used for solvent molecules. AT level of resolution is used for proximal solvent molecules, within a certain radius from the DNA’s center of mass. The distal water molecules are represented as blue MARTINI CG beads, where one CG bead corresponds to four water molecules. Na and Cl ions are shown in orange and yellow, respectively. Adapted from Ref. [71].
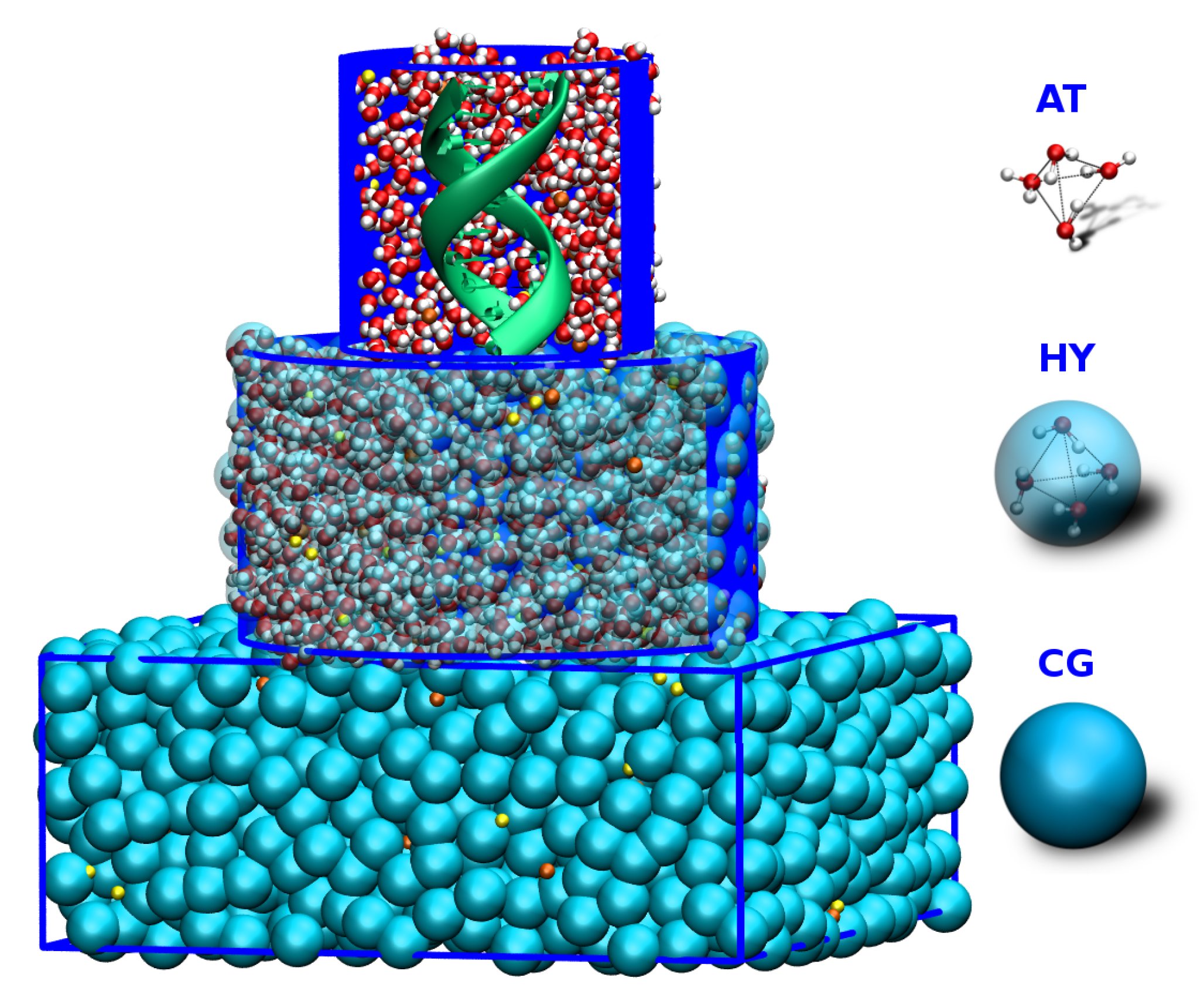
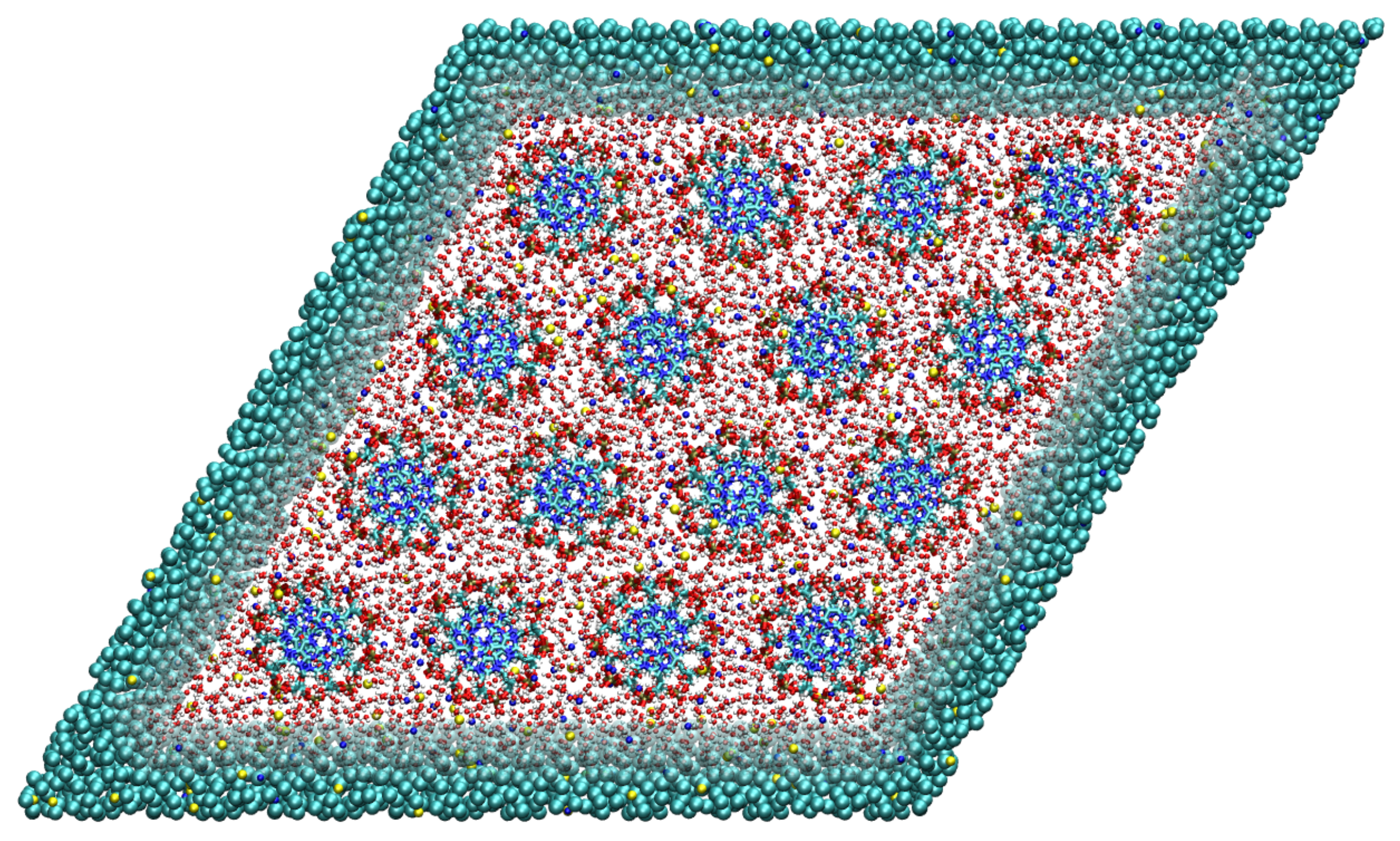
Figure 3.
Top-down view on the simulation box containing 16 DNA molecules arranged on a hexagonal lattice. The DNA molecules are at all times modeled with the high atomistic resolution. For the solvent, we use two levels of resolution, i.e., the atomistic resolution in the rhombic region containing the DNA molecules and the coarse-grained representation outside. Adapted from Ref. [73].
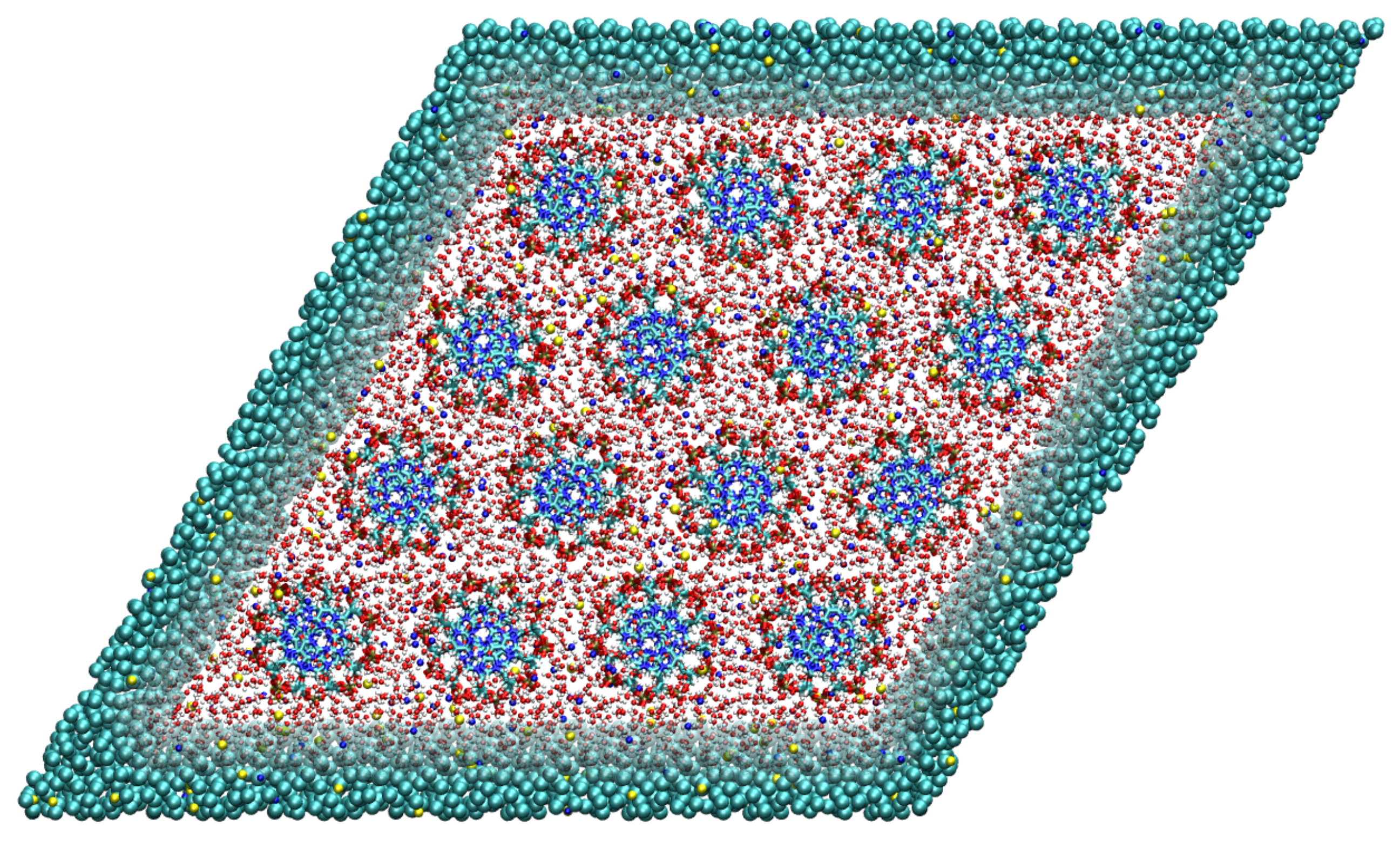
Figure 3.
Top-down view on the simulation box containing 16 DNA molecules arranged on a hexagonal lattice. The DNA molecules are at all times modeled with the high atomistic resolution. For the solvent, we use two levels of resolution, i.e., the atomistic resolution in the rhombic region containing the DNA molecules and the coarse-grained representation outside. Adapted from Ref. [73].
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Podgornik, R.; Zavadlav, J.; Praprotnik, M. Molecular Dynamics Simulation of High Density DNA Arrays. Computation 2018, 6, 3. https://doi.org/10.3390/computation6010003
AMA Style
Podgornik R, Zavadlav J, Praprotnik M. Molecular Dynamics Simulation of High Density DNA Arrays. Computation. 2018; 6(1):3. https://doi.org/10.3390/computation6010003
Chicago/Turabian StylePodgornik, Rudolf, Julija Zavadlav, and Matej Praprotnik. 2018. "Molecular Dynamics Simulation of High Density DNA Arrays" Computation 6, no. 1: 3. https://doi.org/10.3390/computation6010003
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.