Synthesis and Evaluation of a Molecularly Imprinted Polymer for Selective Solid-Phase Extraction of Irinotecan from Human Serum Samples

 ,
,
Abstract
:
1. Introduction


2. Experimental Section
2.1. Chemicals and Standards
2.2. HPLC Analysis
2.3. Synthesis of Polymers
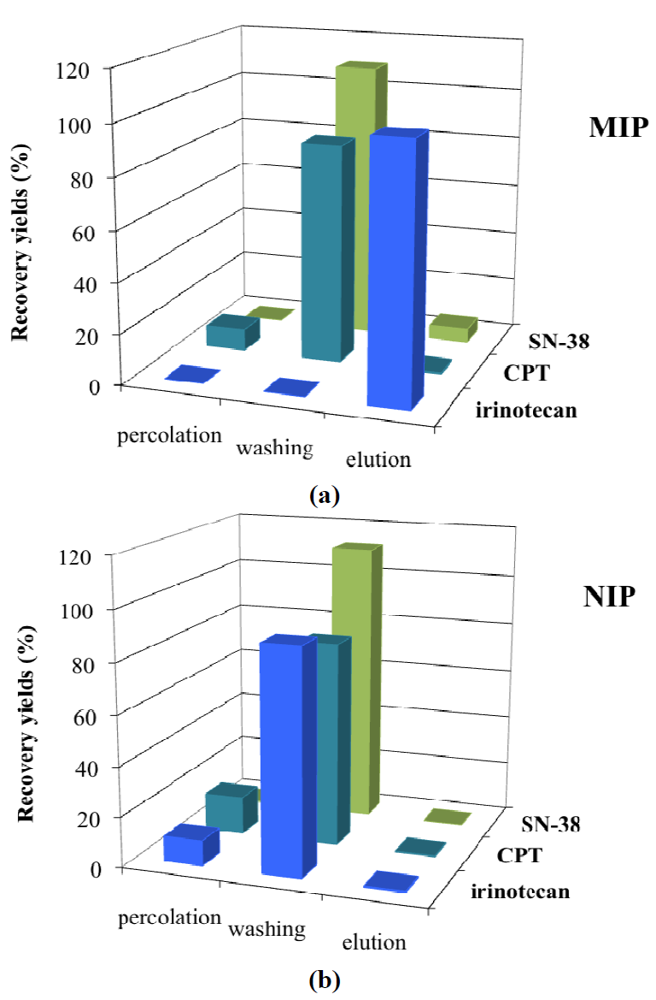
2.4. MISPE Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recovery (%) | ||||||
|---|---|---|---|---|---|---|
| Condition A | Condition B | Condition C | ||||
| MIP | NIP | MIP | NIP | MIP | NIP | |
| Load | 0 | 10.1 | 0 | 0 | 0 | 0 |
| Wash | 0 | 89.2 | 0 | 26.4 | 0 | 94.0 |
| Elute | 99.8 | 0.6 | 107.7 | 83.5 | 100.3 | 5.8 |
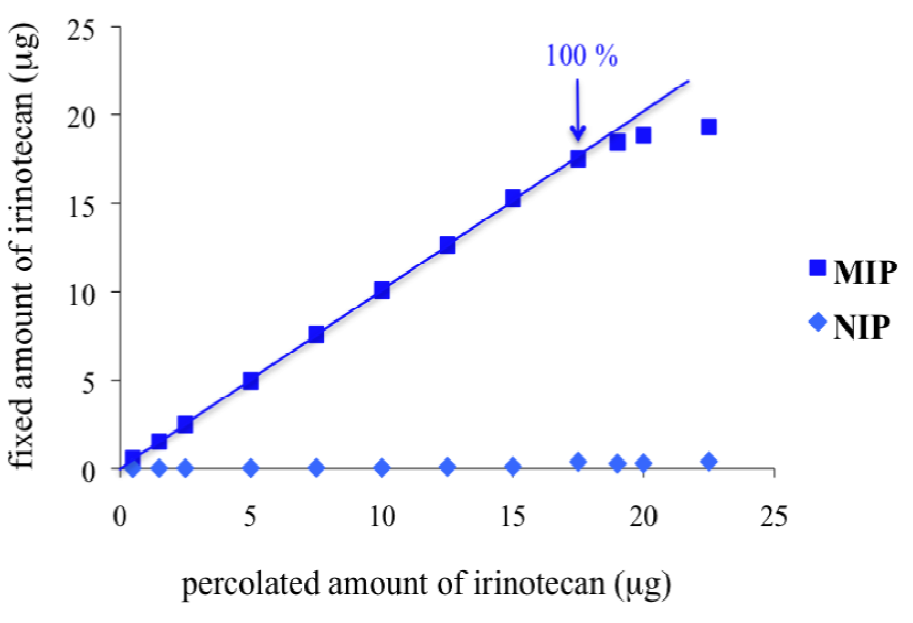
2.5. MIP Loading Capacity
2.6. Study of Cross-Reactivity
2.7. Extraction of Irinotecan from Human Serum Samples
3. Results and Discussion
3.1. Preparation of Molecularly Imprinted Polymer
3.2. Characterization of the Molecularly Imprinted Polymer
3.3. MIP Binding Capacity

3.4. Retention-Behaviour of Structural Analogs on the MIP

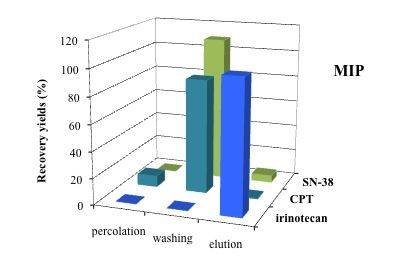
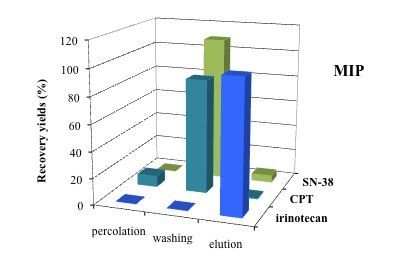
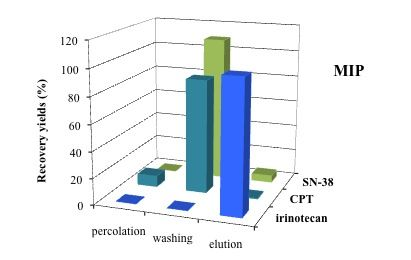
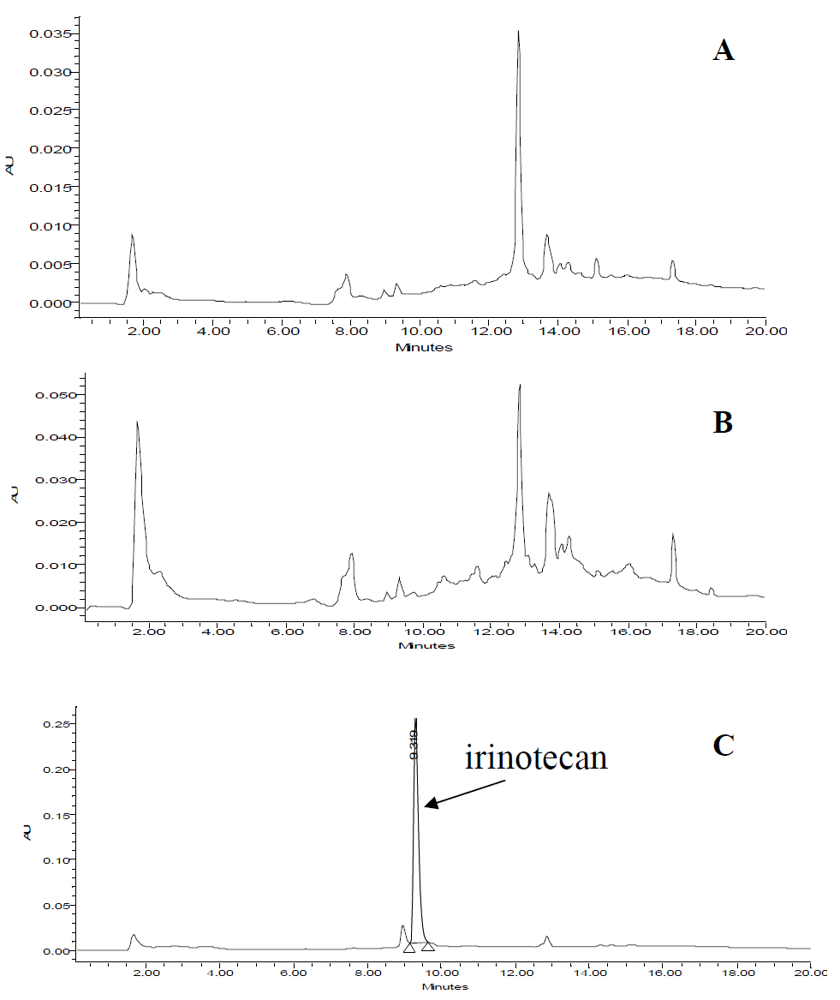
3.5. Application of MISPE to Human Serum Samples

4. Conclusions
Acknowledgements
References
- Rothenberg, M.L. Irinotecan (CPT-11): Recent developments and future directions-colorectal cancer and beyond. Oncologist 2001, 6, 66–80. [Google Scholar] [CrossRef]
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991, 51, 4187–4191. [Google Scholar]
- Hageman, M.J.; Morozowich, W. Case study: Irinotecan (CPT-11), a water-soluble prodrug of SN-38. In Biotechnology: Pharmaceutical Aspects; Stella, V.J., Borchardt, R.T., Hageman, M.J., Oliyai, R., Maag, H., Tilley, J.W., Eds.; Springer: New-York, NY, USA, 2007; Volume 5, pp. 569–579. [Google Scholar]
- Rivory, L.P.; Bowles, M.R.; Robert, J.; Pond, S.M. Conversion of irinotecan (CPT-11) to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), by human liver carboxylesterase. Biochem. Pharmacol. 1996, 52, 1103–1111. [Google Scholar] [CrossRef]
- Khanna, R.; Morton, C.L.; Danks, M.K.; Potter, P.M. Proficient metabolism of irinotecan by a human intestinal carboxylesterase. Cancer Res. 2000, 60, 4725–4728. [Google Scholar]
- Mathijssen, R.H.; van Alphen, R.J.; Verweij, J.; Loos, W.J.; Nooter, K.; Stoter, G.; Sparreboom, A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin. Cancer Res. 2001, 7, 2182–2194. [Google Scholar]
- Gupta, E.; Lestingi, T.M.; Mick, R.; Ramirez, J.; Vokes, E.E.; Ratain, M.J. Metabolic fate of irinotecan in humans: Correlation of glucuronidation with diarrhea. Cancer Res. 1994, 54, 3723–3725. [Google Scholar]
- Lyer, L.; King, C.D.; Whitington, P.F.; Green, M.D.; Roy, S.K.; Tephly, T.R.; Coffman, B.L.; Ratain, M.J. Genetic predisposition to the metabolism of irinotecan (CPT-11). J. Clin. Invest. 1998, 101, 847–854. [Google Scholar] [CrossRef]
- Fassberg, J.; Stella, V.J. A kinetic and mechanistic study of the hydrolysis of camptothecin and some analogues. J. Pharm. Sci. 1992, 81, 676–684. [Google Scholar] [CrossRef]
- Sano, K.; Yoshikawa, M.; Hayasaka, S.; Satake, K.; Ikegami, Y.; Yoshida, H.; Ishikawa, T.; Sawada, S.; Tanabe, S. Simple non-ion-paired high-performance liquid chromatographic method for simultaneous quantitation of carboxylate and lactone forms of 14 new camptothecin derivatives. J. Chromatogr. B 2003, 795, 25–34. [Google Scholar] [CrossRef]
- Slichenmeyer, W.J.; Rowinsky, E.K.; Donehower, R.C.; Kaufmann, S.H. The current status of camptothecin analogues as antitumor agents. J. Natl. Cancer Inst. 1993, 85, 271–291. [Google Scholar] [CrossRef]
- Mullangi, R.; Ahlawat, P.; Srinivas, N.R. Irinotecan and its active metabolite, SN-38: Review of bioanalytical methods and recent update from clinical pharmacology perspective. Biomed. Chromatogr. 2010, 24, 104–123. [Google Scholar] [CrossRef]
- Rivory, L.P.; Chatelut, E.; Canal, P.; Mathieu-Boué, A.; Robert, J. Kinetics of the in vivo interconversion of the carboxylate and lactone forms of irinotecan (CPT-11) and of its metabolite SN-38 in patients. Cancer Res. 1994, 54, 6330–6333. [Google Scholar]
- Barilero, I.; Gandia, D.; Armand, J.P.; Mathieu-Boué, A.; Ré, M.; Gouyette, A.; Chabot, G.G. Simultaneous determination of the camptothecin analogue CPT-11 and its active metabolite SN-38 by high-performance liquid chromatography: application to plasma pharmacokinetic studies in cancer patients. J. Chromatogr. B 1992, 575, 275–280. [Google Scholar] [CrossRef]
- Boyd, G.; Smyth, J.F.; Jodrell, D.I.; Cummings, J. High-performance liquid chromatographic technique for the simultaneous determination of lactone and hydroxy acid forms of camptothecin and SN-38 in tissue culture media and cancer cells. Anal. Biochem. 2001, 297, 15–24. [Google Scholar] [CrossRef]
- D'Esposito, F.; Tattam, B.N.; Ramzan, I.; Murray, M. A liquid chromatography/electrospray ionization mass spectrometry (LC-MS/MS) assay for the determination of irinotecan (CPT-11) and its two major metabolites in human liver microsomal incubations and human plasma samples. J. Chromatogr. B 2008, 875, 522–530. [Google Scholar] [CrossRef]
- Andersson, L.I. Molecular imprinting for drug bioanalysis: A review on the application of imprinted polymers to solid-phase extraction and binding assay. J. Chromatogr. B 2000, 739, 163–173. [Google Scholar] [CrossRef]
- Ge, Y.; Turner, A.P.F. Molecularly imprinted sorbent assays: Recent developments and applications. Chem. Eur. J. 2009, 15, 8100–8107. [Google Scholar] [CrossRef]
- Pichon, V. Selective sample treatment using molecularly imprinted polymers. J. Chromatogr. A 2007, 1152, 41–53. [Google Scholar]
- Vo Duy, S.; Lefebvre-Tournier, I.; Pichon, V.; Hugon-Chapuis, F.; Puy, J.-Y.; Périgaud, C. Molecularly imprinted polymer for analysis of zidovudine and stavudine in human serum by liquid chromatography-mass spectrometry. J. Chromatogr. B 2009, 877, 1101–1108. [Google Scholar] [CrossRef]
- Bansal, T.; Awasthi, A.; Jaggi, M.; Khar, R.K.; Talegaonkar, S. Development and validation of reversed phase liquid chromatographic method utilizing ultraviolet detection for quantification of irinotecan (CPT-11) and its active metabolite, SN-38, in rat plasma and bile samples: Application to pharmacokinetic studies. Talanta 2008, 76, 1015–1021. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Roy, B.; Vo Duy, S.; Puy, J.-Y.; Martin, C.; Guitton, J.; Dumontet, C.; Périgaud, C.; Lefebvre-Tournier, I. Synthesis and Evaluation of a Molecularly Imprinted Polymer for Selective Solid-Phase Extraction of Irinotecan from Human Serum Samples. J. Funct. Biomater. 2012, 3, 131-142. https://doi.org/10.3390/jfb3010131
Roy B, Vo Duy S, Puy J-Y, Martin C, Guitton J, Dumontet C, Périgaud C, Lefebvre-Tournier I. Synthesis and Evaluation of a Molecularly Imprinted Polymer for Selective Solid-Phase Extraction of Irinotecan from Human Serum Samples. Journal of Functional Biomaterials. 2012; 3(1):131-142. https://doi.org/10.3390/jfb3010131
Chicago/Turabian StyleRoy, Béatrice, Sung Vo Duy, Jean-Yves Puy, Charlotte Martin, Jérome Guitton, Charles Dumontet, Christian Périgaud, and Isabelle Lefebvre-Tournier. 2012. "Synthesis and Evaluation of a Molecularly Imprinted Polymer for Selective Solid-Phase Extraction of Irinotecan from Human Serum Samples" Journal of Functional Biomaterials 3, no. 1: 131-142. https://doi.org/10.3390/jfb3010131