Biocompatibility of Chitosan Carriers with Application in Drug Delivery


Abstract
:1. Introduction
2. Chitosan Application in Drug Delivery
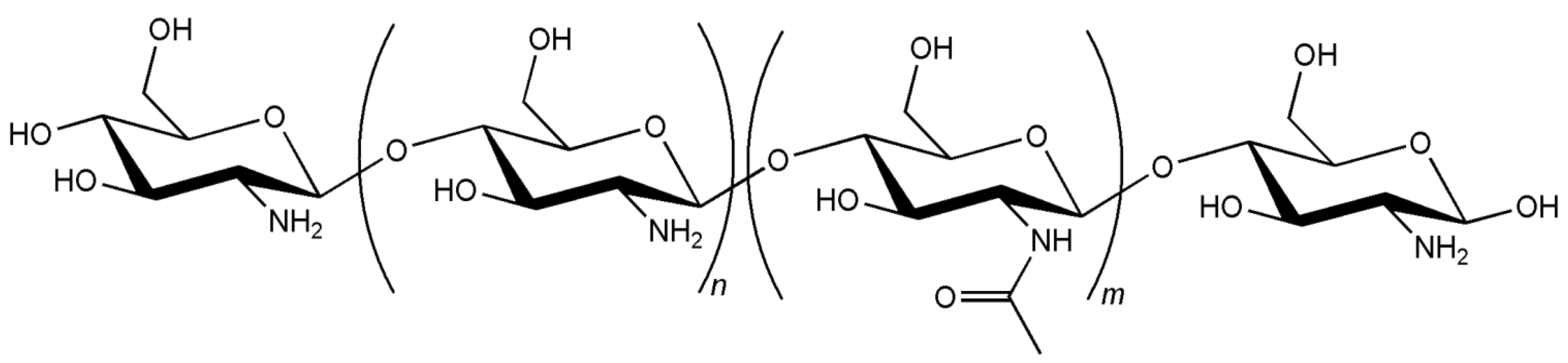
3. Biocompatibility of Chitosan Carriers
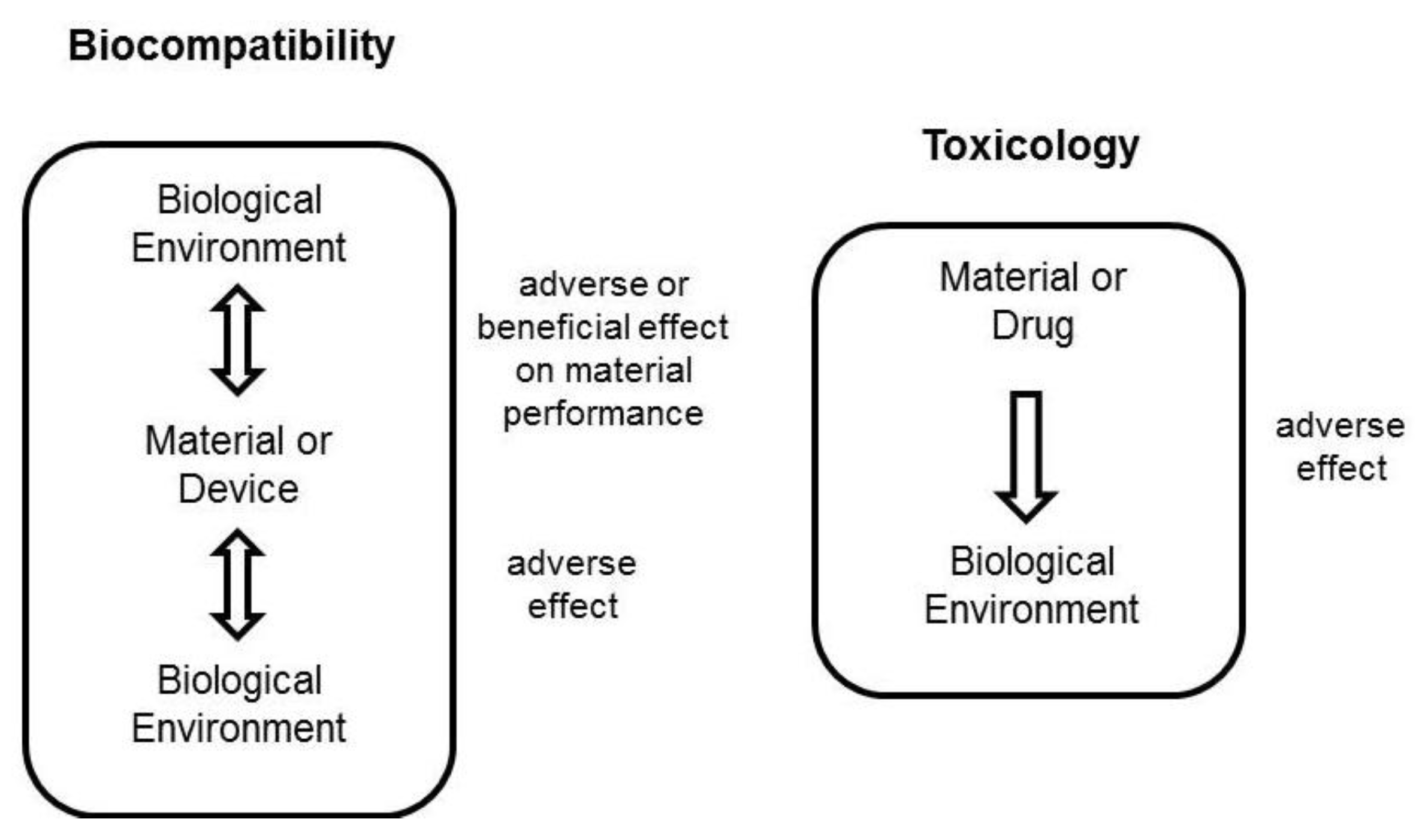
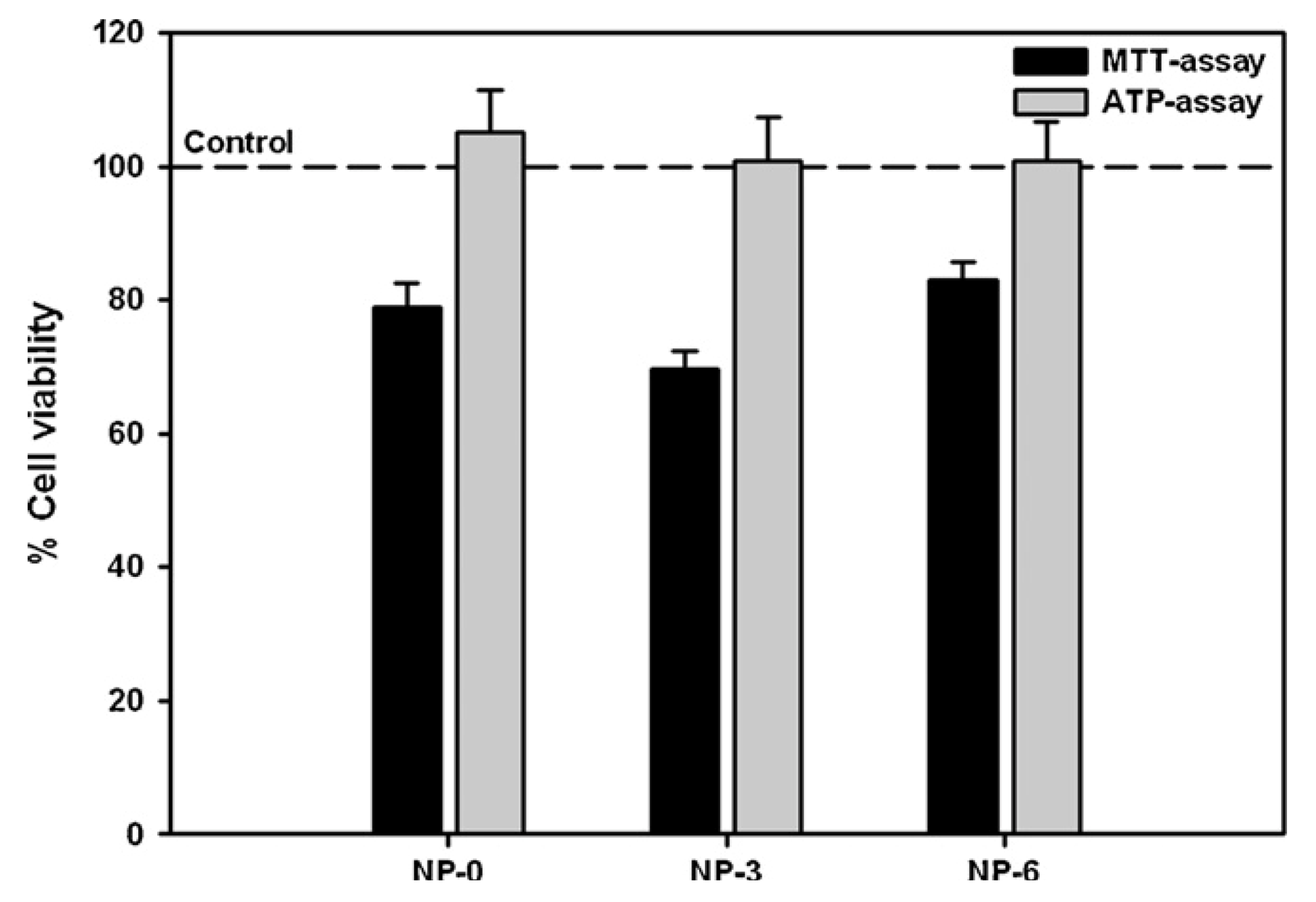
3.1. In Vitro Cell Toxicity

{kind=link}
{kind=link}
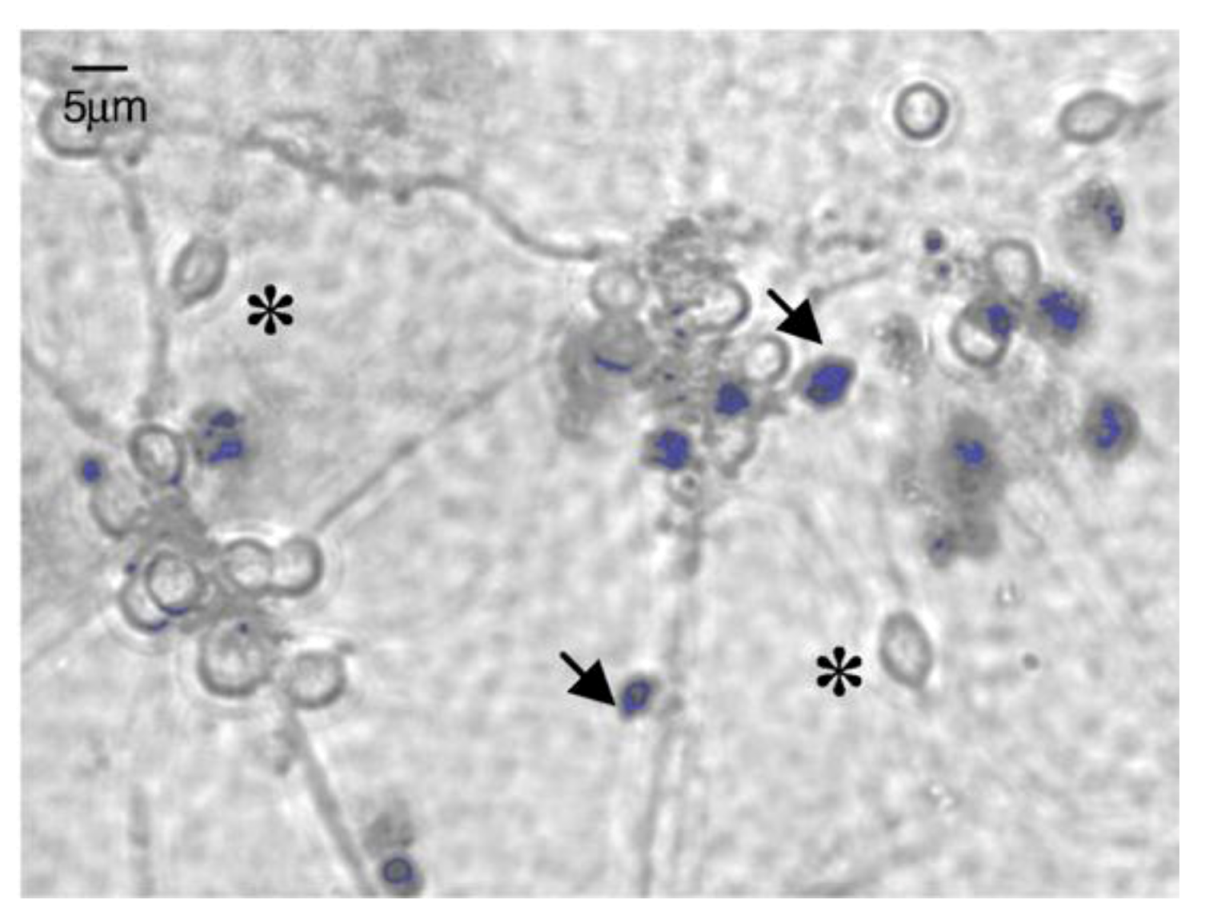{kind=link}
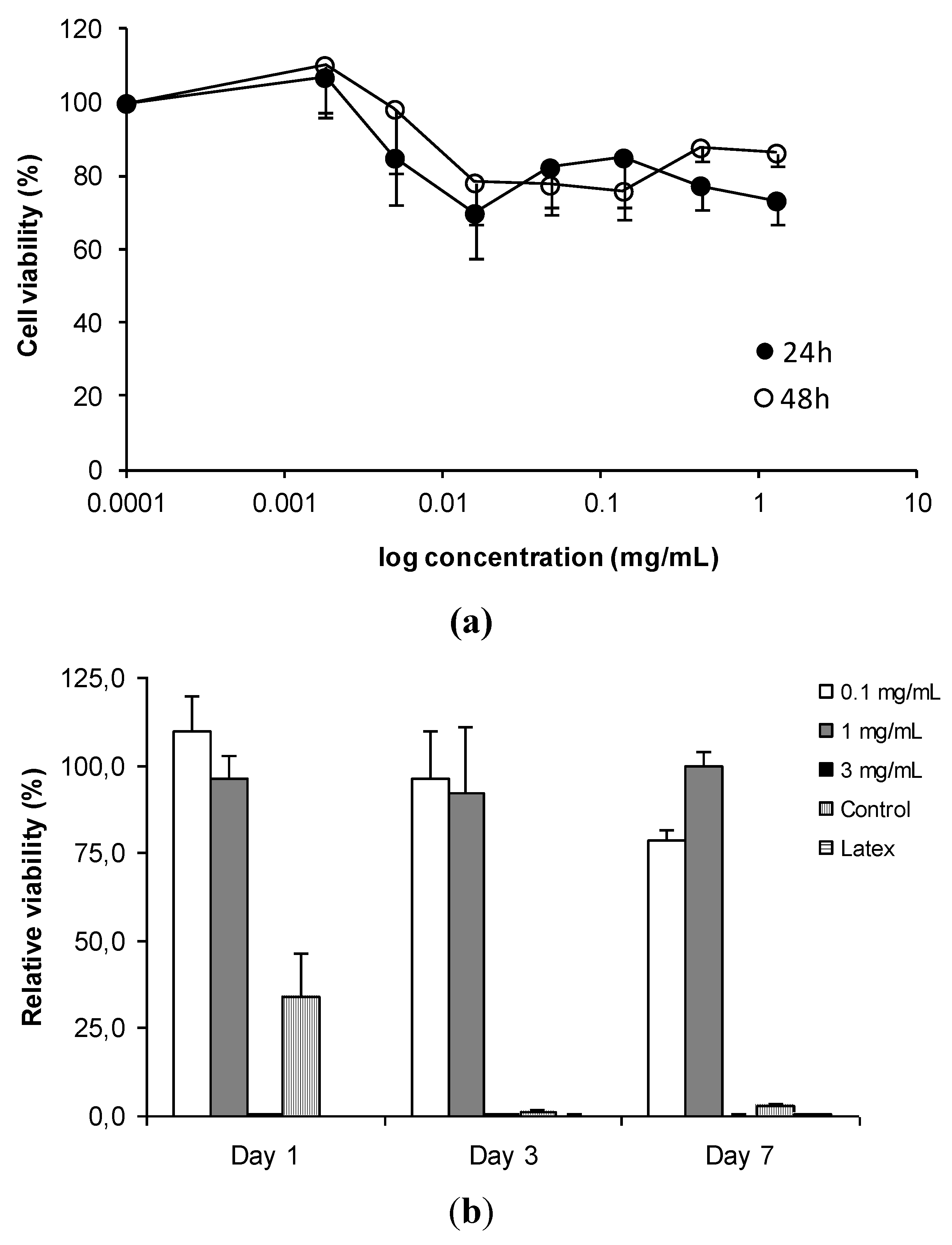{kind=link}
{kind=link}
{kind=link}
| Assay | Theoretical principle | Evaluated cellular function |
|---|---|---|
| Tryplan blue | Blue dye is excluded by viable cells | Cell membrane integrity |
| Propidium iodide | Red dye enters damaged cells and intercalates DNA, enhancing dye fluorescence | Cell membrane integrity |
| Lactate dehydrogenase | LDH leaks from damaged cell membrane. Enzyme transforms NADH + pyruvate into NAD+ + lactate: | Cell membrane integrity |
| Direct quantification of NADH at 340 nm | ||
| Tetrazolium reduction to formazan | ||
| Neutral red | Lysosomal uptake of red dye in live cells | Lysosomal membrane integrity |
| MTT, MTS, XTT | Tetrazolium reduction to blue formazan in metabolically active cells | Mitochondrial metabolism |
| Alamar blue | Resazurin reduction to pink resorufin by metabolically active cells | Mitochondrial metabolism |
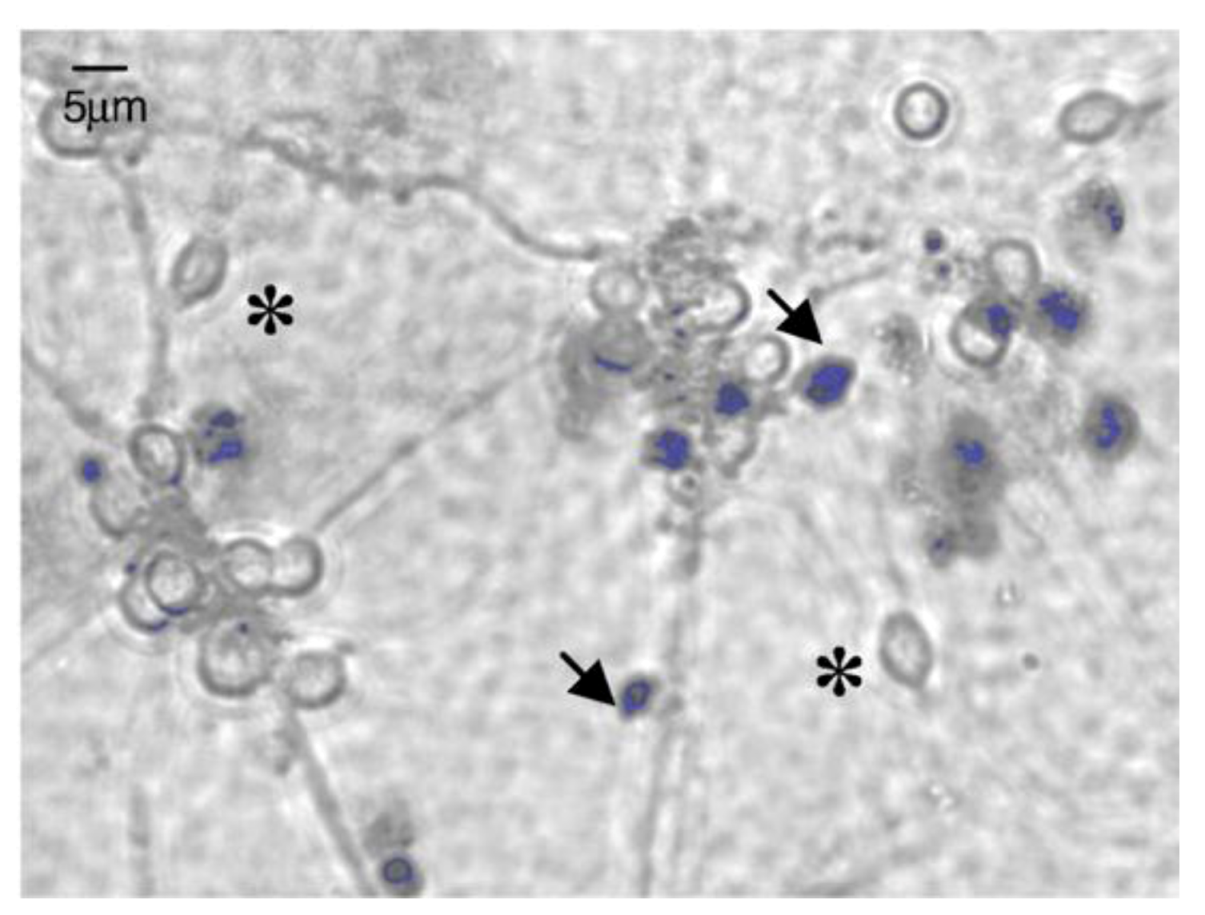
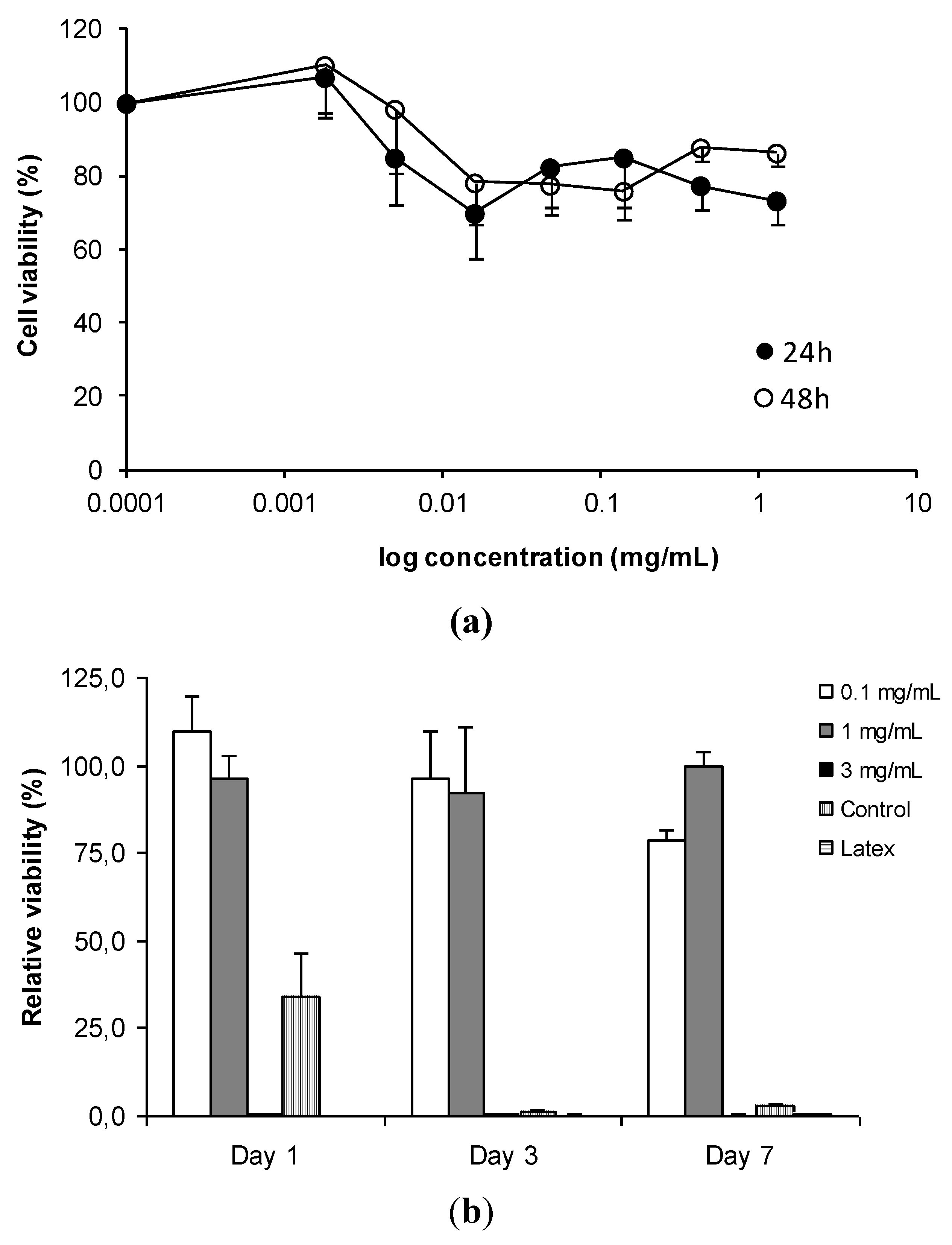
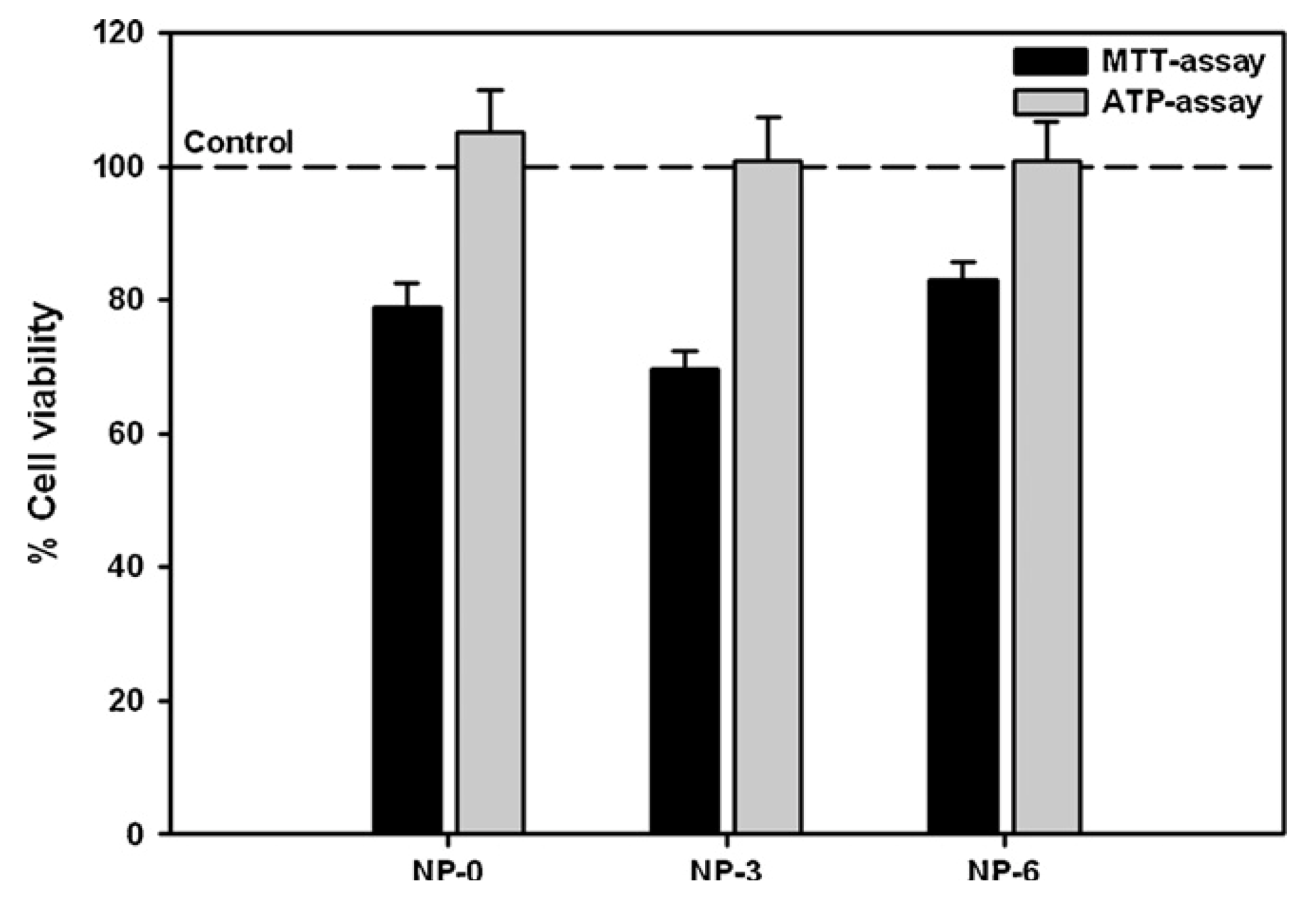
3.2. In Vitro Detection of Impaired Cell or Epithelial Function
| Assay | Theoretical principle | Evaluated cellular function |
|---|---|---|
| ATP | Reduction in ATP cytoplasmic level indicates cell injury | Cell functional integrity |
| Luciferase catalyses light formation from ATP and luciferin. Luminescence observation | ||
| TER | Cellular damage or stress induces TER decrease | Cell barrier integrity |
| Comet | Electrophoresis separation of broken DNA strands which form the tail of the comet. DNA staining with dye and observation by fluorescence microscopy | DNA damage |
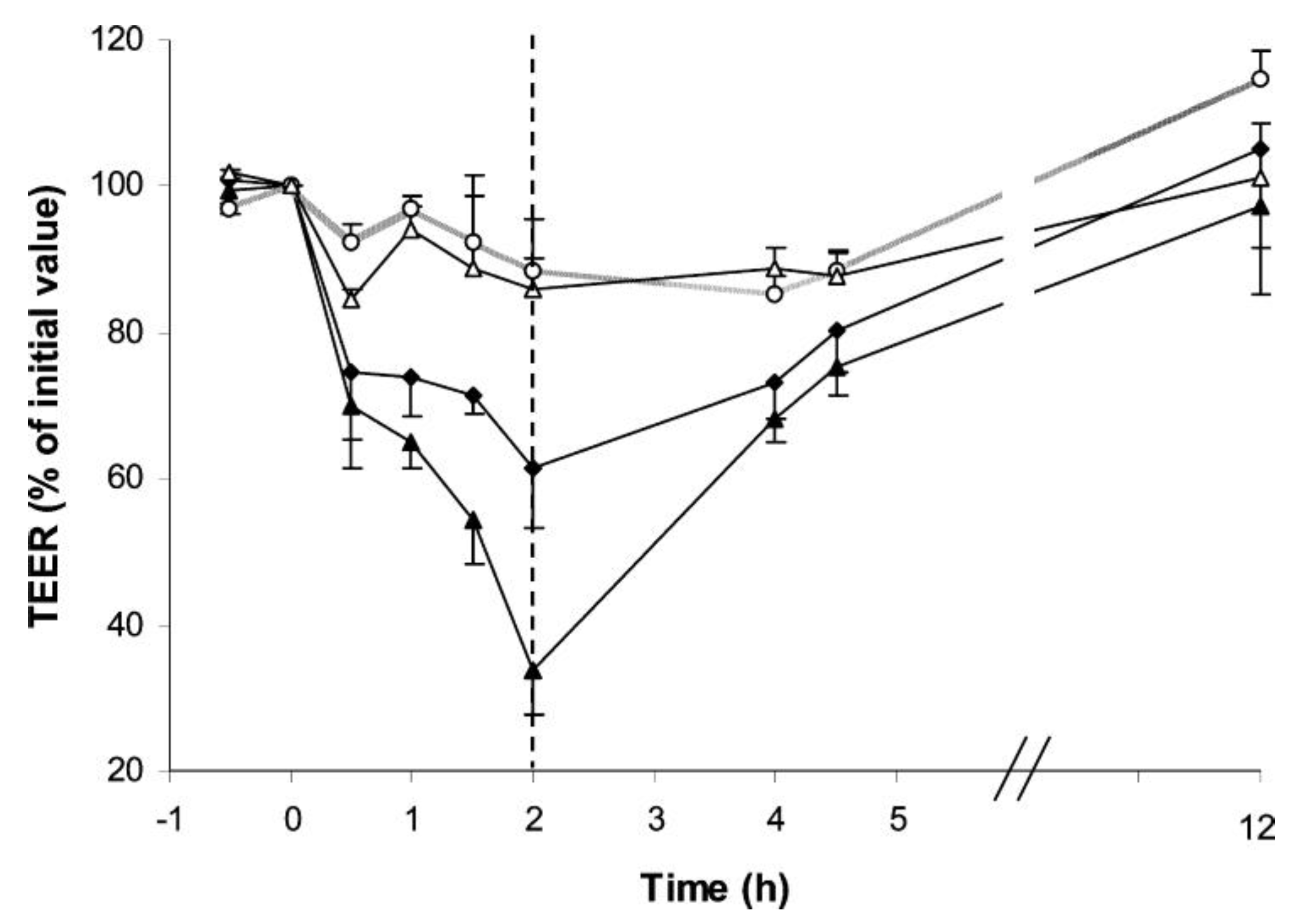
3.3. In Vitro Genotoxicity
3.4. In Vitro Monitorisation of Inflammatory Response
3.5. In Vivo Studies
4. Conclusions and Expert Opinion
Acknowledgments
References
- Antosova, Z.; Mackova, M.; Kral, V.; Macek, T. Therapeutic application of peptides and proteins: Parenteral forever? Trends Biotechnol. 2009, 27, 628–635. [Google Scholar] [CrossRef]
- Grenha, A.; Carrión-Recio, D.; Teijeiro-Osorio, D.; Seijo, B.; Remuñán-López, C. Nano- and micro-particulate carriers for pulmonary drug delivery. In Handbook of Particulate Drug Delivery; Ravi Kumar, M.N.V., Ed.; American Scientific Publishers: Valencia, CA, USA, 2008; Volume 2, pp. 165–192. [Google Scholar]
- Peppas, N.A.; Thomas, J.B.; McGinity, J. Molecular aspects of mucoadhesive carrier development for drug delivery and improved absorption. J. Biomater. Sci. Polym. Ed. 2009, 20, 1–20. [Google Scholar] [CrossRef]
- Amidi, M.; Mastrobattista, E.; Jiskoot, W.; Hennink, W.E. Chitosan-based delivery systems for protein therapeutics and antigens. Adv. Drug Deliv. Rev. 2010, 62, 59–82. [Google Scholar] [CrossRef]
- Grenha, A.; Al-Qadi, S.; Seijo, B.; Remuñán-López, C. The potential of chitosan for pulmonary drug delivery. J. Drug Deliv. Sci. Technol. 2010, 20, 33–43. [Google Scholar]
- Lehr, C.-M.; Bouwstra, J.A.; Schacht, E.H.; Junginger, H.E. In vitro evaluation of mucoadhesive properties of chitosan and some other natural polymers. Int. J. Pharm. 1992, 78, 43–48. [Google Scholar] [CrossRef]
- Deacon, M.P.; McGurk, S.; Roberts, C.J.; Williams, P.M.; Tendler, S.J.; Davies, M.C.; Davis, S.S.; Harding, S.E. Atomic force microscopy of gastric mucin and chitosan mucoadhesive systems. Biochem. J. 2000, 348, 557–563. [Google Scholar] [CrossRef]
- Shruti, C.; Saiqa, M.; Jasjeet, K.; Zeemat, I.; Sushma, T. Advances and potential applications of chitosan derivatives as mucoadhesive biomaterials in modern drug delivery. J. Pharm. Pharm. 2006, 58, 1021–1032. [Google Scholar]
- Kumar, M.N.; Muzzarelli, R.A.; Muzzarelli, C.; Sashiwa, H.; Domb, A.J. Chitosan chemistry and pharmaceutical perspectives. Chem. Rev. 2004, 104, 6017–6084. [Google Scholar]
- Baldrick, P. The safety of chitosan as a pharmaceutical excipient. Regul. Toxicol. Pharm. 2010, 56, 290–299. [Google Scholar] [CrossRef]
- Braz, L.; Rodrigues, S.; Fonte, P.; Grenha, A.; Sarmento, B. Mechanisms of chemical and enzymatic chitosan biodegradability and its application on drug delivery. In Biodegradable Polymers: Processing, Degradation and Applications; Felton, G., Ed.; Nova Science Publisher: New York, NY, USA, 2011. [Google Scholar]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Rinaudo, M. Main properties and current applications of some polysaccharides as biomaterials. Polym. Int. 2008, 57, 397–430. [Google Scholar] [CrossRef]
- Rodrigues, S.; Rosa da Costa, A.; Grenha, A. Chitosan/carrageenan nanoparticles: Effect of cross-linking with tripolyphosphate and charge ratios. Carbohydr. Polym. 2012, 89, 282–289. [Google Scholar] [CrossRef]
- Kurita, K. Chitin and chitosan: Functional biopolymers from marine crustaceans. Mar. Biotechnol. 2006, 8, 203–226. [Google Scholar] [CrossRef]
- Kean, T.; Thanou, M. Biodegradation, biodistribution and toxicity of chitosan. Adv. Drug Deliv. Rev. 2010, 62, 3–11. [Google Scholar] [CrossRef]
- Singh, D.; Ray, A. Biomedical applications of chitin, chitosan and their derivatives. Rev. Macromol. Chem. Phys. 2000, C40, 69–83. [Google Scholar] [CrossRef]
- Cho, Y.; Jang, J.; Park, C.; Ko, S. Preparation and solubility in acid and water of partially deacetylated chitins. Biomacromolecules 2000, 1, 609–614. [Google Scholar] [CrossRef]
- Prego, C.; Torres, D.; Alonso, M.J. The potential of chitosan for the oral administration of peptides. Expert Opin. Drug Deliv. 2005, 2, 843–854. [Google Scholar] [CrossRef]
- Şenel, S. Potential applications of chitosan in oral mucosal delivery. J. Drug Deliv. Sci. Technol. 2010, 20, 23–32. [Google Scholar]
- Wang, J.J.; Zeng, Z.W.; Xiao, R.Z.; Xie, T.A.; Zhou, G.L.; Zhan, X.R.; Wang, S.L. Recent advances of chitosan nanoparticles as drug carriers. Int. J. Nanomed. 2011, 6, 765–774. [Google Scholar]
- Andrade, F.; Antunes, F.; Nascimento, A.; da Silva, S.; das Neves, J.; Ferreira, D.; Sarmento, B. Chitosan formulations as carriers for therapeutic proteins. Curr. Drug Discov. Technol. 2011, 8, 157–172. [Google Scholar] [CrossRef]
- Park, J.H.; Saravanakumar, G.; Kim, K.; Kwon, I.C. Targeted delivery of low molecular drugs using chitosan and its derivatives. Adv. Drug Deliv. Rev. 2010, 62, 28–41. [Google Scholar] [CrossRef]
- Chiellini, F.; Piras, A.M.; Errico, C.; Chiellini, E. Micro/nanostructured polymeric systems for biomedical and pharmaceutical applications. Nanomedicine 2008, 3, 367–393. [Google Scholar] [CrossRef]
- Csaba, N.; Garcia-Fuentes, M.; Alonso, M.J. The performance of nanocarriers for transmucosal drug delivery. Expert Opin. Drug Deliv. 2006, 3, 463–478. [Google Scholar] [CrossRef]
- Fonte, P.; Andrade, F.; Araújo, F.; Andrade, C.; Neves, Jd.; Sarmento, B. Chitosan-coated solid lipid nanoparticles for insulin delivery. Methods Enzymol. 2012, 508, 295–314. [Google Scholar]
- Grenha, A. Chitosan nanoparticles: a survey of preparation methods. J. Drug Target. 2012, 20, 291–300. [Google Scholar] [CrossRef]
- El-Sherbiny, I.M.; Smyth, H.D.C. Controlled release pulmonary administration of curcumin using swellable biocompatible microparticles. Mol. Pharm. 2011, 9, 269–280. [Google Scholar] [CrossRef]
- Venishetty, V.K.; Chede, R.; Komuravelli, R.; Adepu, L.; Sistla, R.; Diwan, P.V. Design and evaluation of polymer coated carvedilol loaded solid lipid nanoparticles to improve the oral bioavailability: A novel strategy to avoid intraduodenal administration. Colloids Surf. B Biointerfaces 2012, 95, 1–9. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, M.; Zheng, A.; Cao, D.; Bi, Y.; Sun, J. Preparation and characterization of insulin-loaded bioadhesive PLGA nanoparticles for oral administration. Eur. J. Pharm.Sci. 2012, 45, 632–638. [Google Scholar]
- Wang, G.; Pan, L.; Zhang, Y.; Wang, Y.; Zhang, Z.; Lü, J.; Zhou, P.; Fang, Y.; Jiang, S. Intranasal delivery of cationic PLGA nano/microparticles-loaded FMDV DNA vaccine encoding IL-6 elicited protective immunity against FMDV challenge. PLoS One 2011, 6, e27605. [Google Scholar]
- Pawar, D.; Goyal, A.; Mangal, S.; Mishra, N.; Vaidya, B.; Tiwari, S.; Jain, A.; Vyas, S. Evaluation of mucoadhesive PLGA microparticles for nasal immunization. AAPS J. 2010, 12, 130–137. [Google Scholar] [CrossRef]
- Qiang, F.; Shin, H.-J.; Lee, B.-J.; Han, H.-K. Enhanced systemic exposure of fexofenadine via the intranasal administration of chitosan-coated liposome. Int. J. Pharm. 2012, 430, 161–166. [Google Scholar] [CrossRef]
- Sugihara, H.; Yamamoto, H.; Kawashima, Y.; Takeuchi, H. Effectiveness of submicronized chitosan-coated liposomes in oral absorption of indomethacin. J. Liposome Res. 2012, 22, 72–79. [Google Scholar] [CrossRef]
- Fernández-Urrusuno, R.; Calvo, P.; Remuñán-López, C.; Vila-Jato, J.L.; José Alonso, M. Enhancement of nasal absorption of insulin using chitosan nanoparticles. Pharm. Res. 1999, 16, 1576–1581. [Google Scholar] [CrossRef]
- Vila, A.; Sánchez, A.; Janes, K.; Behrens, I.; Kissel, T.; Jato, J.L.V.; Alonso, M.J. Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice. Eur. J. Pharm. Biopharm. 2004, 57, 123–131. [Google Scholar] [CrossRef]
- Artursson, P.; Lindmark, T.; Davis, S.S.; Illum, L. Effect of chitosan on the permeability of monolayers of intestinal epithelial cells (Caco-2). Pharm. Res. 1994, 11, 1358–1361. [Google Scholar] [CrossRef]
- Borchard, G.; Lueβen, H.L.; de Boer, A.G.; Verhoef, J.C.; Lehr, C.-M.; Junginger, H.E. The potential of mucoadhesive polymers in enhancing intestinal peptide drug absorption. III: Effects of chitosan-glutamate and carbomer on epithelial tight junctions in vitro. J. Control. Release 1996, 39, 131–138. [Google Scholar]
- Prego, C.; García, M.; Torres, D.; Alonso, M.J. Transmucosal macromolecular drug delivery. J. Control. Release 2005, 101, 151–162. [Google Scholar]
- De Campos, A.M.; Sánchez, A.; Alonso, M.J. Chitosan nanoparticles: A new vehicle for the improvement of the delivery of drugs to the ocular surface. Application to cyclosporin A. Int. J. Pharm. 2001, 224, 159–168. [Google Scholar]
- Portero, A.; Remuñán-López, C.; Nielsen, H.M. The potential of chitosan in enhancing peptide and protein absorption across the TR146 cell culture model—An in vitro model of the buccal epithelium. Pharm. Res. 2002, 19, 169–174. [Google Scholar] [CrossRef]
- Al-Qadi, S.; Grenha, A.; Carrión-Recio, D.; Seijo, B.; Remuñán-López, C. Microencapsulated chitosan nanoparticles for pulmonary protein delivery: In vivo evaluation of insulin-loaded formulations. J. Control. Release 2012, 157, 383–390. [Google Scholar] [CrossRef]
- Arca, H. C.; Gunbeyaz, M.; Senel, S. Chitosan-based systems for the delivery of vaccine antigens. Expert Rev. Vaccines 2009, 8, 937–953. [Google Scholar] [CrossRef]
- Jarmila, V.; Vavríková, E. Chitosan derivatives with antimicrobial, antitumour and antioxidant activities—A review. Curr. Pharm.Design 2011, 17, 3596–3607. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, W.; Liu, P.; Cheng, Q.; Tahirou, T.; Gu, W.; Li, B. Chitosan modification and pharmaceutical/biomedical applications. Mar. Drugs 2010, 8, 1962–1987. [Google Scholar] [CrossRef]
- Ren, D.; Yi, H.; Wang, W.; Ma, X. The enzymatic degradation and swelling properties of chitosan matrices with different degrees of N-acetylation. Carbohydr. Res. 2005, 340, 2403–2410. [Google Scholar] [CrossRef]
- Aillon, K.L.; Xie, Y.; El-Gendy, N.; Berkland, C.J.; Forrest, M.L. Effects of nanomaterial physicochemical properties on in vivo toxicity. Adv. Drug Deliv. Rev. 2009, 61, 457–466. [Google Scholar] [CrossRef]
- Hirano, S.; Tsuchida, H.; Nagao, N. N-acetylation in chitosan and the rate of its enzymic hydrolysis. Biomaterials 1989, 10, 574–576. [Google Scholar] [CrossRef]
- Aiba, S. Studies on chitosan: 4. Lysozymic hydrolysis of partially N-acetylated chitosans. Int. J. Biol. Macromol. 1992, 14, 225–228. [Google Scholar] [CrossRef]
- Pangburn, S.; Trescony, P.; Heller, J. Lysozyme degradation of partially deacetylated chitin, its films and hydrogels. Biomaterials 1982, 3, 105–108. [Google Scholar]
- Kofuji, K.; Qian, C.; Nishimura, M.; Sugiyama, I.; Murata, Y.; Kawashima, S. Relationship between physicochemical characteristics and functional properties of chitosan. Eur. Polym. J. 2005, 41, 2784–2791. [Google Scholar]
- Schipper, N.G.M.; Vårum, K.M.; Artursson, P. Chitosans as absorption enhancers for poorly absorbable drugs. 1: Influence of molecular weight and degree of acetylation on drug transport across human intestinal epithelial (Caco-2) cells. Pharm. Res. 1996, 13, 1686–1692. [Google Scholar] [CrossRef]
- Gaspar, R.; Duncan, R. Polymeric carriers: Preclinical safety and the regulatory implications for design and development of polymer therapeutics. Adv. Drug Deliv. Rev. 2009, 61, 1220–1231. [Google Scholar] [CrossRef]
- Williams, D. Definitions in Biomaterials; Elsevier: Amsterdam, The Netherlands, 1987. [Google Scholar]
- Williams, D.F. On the mechanisms of biocompatibility. Biomaterials 2008, 29, 2941–2953. [Google Scholar] [CrossRef]
- Power, K.A.; Fitzgerald, K.T.; Gallagher, W.M. Examination of cell–host–biomaterial interactions via high-throughput technologies: A re-appraisal. Biomaterials 2010, 31, 6667–6674. [Google Scholar] [CrossRef]
- Kohane, D.; Langer, R. Biocompatibility and drug delivery systems. Chem. Sci. 2010, 1, 441–446. [Google Scholar] [CrossRef]
- Guyton, A.; Hall, J. Textbook of Medical Physiology, 12th ed; Elsevier: Philadelphia, PA, USA, 2011. [Google Scholar]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef]
- Huang, M.; Khor, E.; Lim, L.-Y. Uptake and cytotoxicity of chitosan molecules and nanoparticles: Effects of molecular weight and degree of deacetylation. Pharm. Res. 2004, 21, 344–353. [Google Scholar]
- ISO, Biological Evaluation of Medical Devices Part 5: Tests for in vitro Cytotoxicity, 10993-5: 2009; International Organization for Standardization: Geneva, Switzerland, 2009.
- ISO, Biological Evaluation of Medical Devices Part 1: Evaluation and Testing, 10993-1: 2009; International Organization for Standardization: Geneva, Switzerland, 2009.
- ISO, Biological Evaluation of Medical Devices Part 3: Tests for Genotoxicity, Carcinogenicity, and Reproductive Toxicity, 10993-3: 2003; International Organization for Standardization: Geneva, Switzerland, 2003.
- Lewinski, N.; Colvin, V.; Drezek, R. Cytotoxicity of nanoparticles. Small 2008, 4, 26–49. [Google Scholar] [CrossRef]
- Keong, L.; Halim, A. In vitro models in biocompatibility assessment for biomedical-grade chitosan derivatives in wound management. Int. J. Mol. Sci. 2009, 10, 1300–1313. [Google Scholar] [CrossRef]
- ISO, Biological Evaluation of Medical Devices Part 2: Animal Welfare Requirements, 10993-2: 2006; International Organization for Standardization: Geneva, Switzerland, 2006.
- Scherließ, R. The MTT assay as tool to evaluate and compare excipient toxicity in vitro on respiratory epithelial cells. Int. J. Pharm. 2011, 411, 98–105. [Google Scholar] [CrossRef]
- Oh, H.; Livingston, R.; Smith, K.; Abrishamian-Garcia, L. Comparative study of the time dependency of cell death assays. MIT Undergrad. Res. J. 2004, 11, 53–62. [Google Scholar]
- Altman, S.A.; Randers, L.; Rao, G. Comparison of trypan blue dye exclusion and fluorometric assays for mammalian cell viability determinations. Biotechnol. Prog. 1993, 9, 671–674. [Google Scholar] [CrossRef]
- Soenen, S.; de Cuyper, M. Assessing cytotoxicity of (iron oxide-based) nanoparticles: An overview of different methods exemplified with cationic magnetoliposomes. Contrast Media Mol. Imaging 2009, 4, 207–219. [Google Scholar] [CrossRef]
- Monteiro-Riviere, N.A.; Inman, A.O.; Zhang, L.W. Limitations and relative utility of screening assays to assess engineered nanoparticle toxicity in a human cell line. Toxicol. Appl. Pharm. 2009, 234, 222–235. [Google Scholar] [CrossRef]
- Stoddart, M.J. Cell viability assays: Introduction. Methods Mol. Biol. 2011, 740, 1–6. [Google Scholar] [CrossRef]
- King, M.A. Detection of dead cells and measurement of cell killing by flow cytometry. J. Immunol. Methods 2000, 243, 155–166. [Google Scholar] [CrossRef]
- Aden, P.; Goverud, I.; Liestøl, K.; Løberg, E.M.; Paulsen, R.E.; Mæhlen, J.; Lømo, J. Low-potency glucocorticoid hydrocortisone has similar neurotoxic effects as high-potency glucocorticoid dexamethasone on neurons in the immature chicken cerebellum. Brain Res. 2008, 1236, 39–48. [Google Scholar] [CrossRef]
- Han, X.; Gelein, R.; Corson, N.; Wade-Mercer, P.; Jiang, J.; Biswas, P.; Finkelstein, J.N.; Elder, A.; Oberdörster, G. Validation of an LDH assay for assessing nanoparticle toxicity. Toxicology 2011, 287, 99–104. [Google Scholar] [CrossRef]
- Fotakis, G.; Timbrell, J.A. In vitro cytotoxicity assays: Comparison of LDH, neutral red, MTT and protein assay in hepatoma cell lines following exposure to cadmium chloride. Toxicol. Lett. 2006, 160, 171–177. [Google Scholar] [CrossRef]
- Racher, A.J.; Looby, D.; Griffiths, J.B. Use of lactate dehydrogenase release to assess changes in culture viability. Cytotechnology 1990, 3, 301–307. [Google Scholar] [CrossRef]
- Zhao, C.; Li, X.; Luo, S.; Chang, Y. Assessments of lysosomal membrane responses to stresses with neutral red retention assay and its potential application in the improvement of bivalve aquaculture. Afr. J. Biotechnol. 2011, 10, 13968–13973. [Google Scholar]
- Hamid, R.; Rotshteyn, Y.; Rabadi, L.; Parikh, R.; Bullock, P. Comparison of alamar blue and MTT assays for high through-put screening. Toxicol. in Vitro 2004, 18, 703–710. [Google Scholar] [CrossRef]
- Gonzalez, R.J.; Tarloff, J.B. Evaluation of hepatic subcellular fractions for Alamar blue and MTT reductase activity. Toxicol. in Vitro 2001, 15, 257–259. [Google Scholar] [CrossRef]
- Bernas, T.; Dobrucki, J. Mitochondrial and nonmitochondrial reduction of MTT: Interaction of MTT with TMRE, JC-1, and NAO mitochondrial fluorescent probes. Cytometry 2002, 47, 236–242. [Google Scholar] [CrossRef]
- Jena, P.; Mohanty, S.; Mallick, R.; Jacob, B.; Sonawane, A. Toxicity and antibacterial assessment of chitosancoated silver nanoparticles on human pathogens and macrophage cells. Int. J. Nanomed. 2012, 7, 1805–1818. [Google Scholar]
- Xu, J.; Ma, L.; Liu, Y.; Xu, F.; Nie, J.; Ma, G. Design and characterization of antitumor drug paclitaxel-loaded chitosan nanoparticles by W/O emulsions. Int. J. Biol. Macromol. 2012, 50, 438–443. [Google Scholar]
- Grenha, A.; Gomes, M.E.; Rodrigues, M.; Santo, V.E.; Mano, J.F.; Neves, N.M.; Reis, R.L. Development of new chitosan/carrageenan nanoparticles for drug delivery applications. J. Biomed. Mater. Res. Part A 2010, 92A, 1265–1272. [Google Scholar]
- Jeong, Y.-I.; Jin, S.-G.; Kim, I.-Y.; Pei, J.; Wen, M.; Jung, T.-Y.; Moon, K.-S.; Jung, S. Doxorubicin-incorporated nanoparticles composed of poly(ethylene glycol)-grafted carboxymethyl chitosan and antitumor activity against glioma cells in vitro. Colloids Surf. B Biointerf. 2010, 79, 149–155. [Google Scholar] [CrossRef]
- Anitha, A.; Chennazhi, K.; Nair, S.; Jayakumar, R. 5-flourouracil loaded N,O-carboxymethyl chitosan nanoparticles as an anticancer nanomedicine for breast cancer. J. Biomed. Nanotechnol. 2012, 8, 29–42. [Google Scholar] [CrossRef]
- Silva, C.M.; Veiga, F.; Ribeiro, A.J.; Zerrouk, N.; Arnaud, P. Effect of chitosan-coated alginate microspheres on the permeability of Caco-2 cell monolayers. Drug Dev. Ind. Pharm. 2006, 32, 1079–1088. [Google Scholar] [CrossRef]
- Huang, R.; Mendis, E.; Rajapakse, N.; Kim, S.-K. Strong electronic charge as an important factor for anticancer activity of chitooligosaccharides (COS). Life Sci. 2006, 78, 2399–2408. [Google Scholar]
- Kowapradit, J.; Opanasopit, P.; Ngawhirunpat, T.; Apirakaramwong, A.; Rojanarata, T.; Ruktanonchai, U.; Sajomsang, W. In vitro permeability enhancement in intestinal epithelial cells (Caco-2) monolayer of water soluble quaternary ammonium chitosan derivatives. AAPS PharmSciTech 2010, 11, 497–508. [Google Scholar] [CrossRef]
- Loh, J.W.; Yeoh, G.; Saunders, M.; Lim, L.-Y. Uptake and cytotoxicity of chitosan nanoparticles in human liver cells. Toxicol. Appl. Pharm. 2010, 249, 148–157. [Google Scholar] [CrossRef]
- Grenha, A.; Grainger, C.I.; Dailey, L.A.; Seijo, B.; Martin, G.P.; Remuñán-López, C.; Forbes, B. Chitosan nanoparticles are compatible with respiratory epithelial cells in vitro. Eur. J. Pharm. Sci. 2007, 31, 73–84. [Google Scholar] [CrossRef]
- Lozano, M.V.; Torrecilla, D.; Torres, D.; Vidal, A.; Dominguez, F.; Alonso, M.J. Highly efficient system to deliver taxanes into tumor cells: Docetaxel-loaded chitosan oligomer colloidal carriers. Biomacromolecules 2008, 9, 2186–2193. [Google Scholar]
- Zaki, N.; Hafez, M. Enhanced antibacterial effect of ceftriaxone sodium-loaded chitosan nanoparticles against intracellular Salmonella typhimurium. AAPS PharmSciTech 2012, 13, 411–421. [Google Scholar] [CrossRef]
- Wang, Z.H.; Wang, Z.Y.; Sun, C.S.; Wang, C.Y.; Jiang, T.Y.; Wang, S.L. Trimethylated chitosan-conjugated PLGA nanoparticles for the delivery of drugs to the brain. Biomaterials 2010, 31, 908–915. [Google Scholar] [CrossRef]
- Guerra, G.D.; Cerrai, P.; Tricoli, M.; Maltinti, S.; Guerra, R.S.D. In vitro cytotoxicity testing of chitosan-containing polyelectrolyte complexes. J. Mater. Sci. Mater. Med. 1998, 9, 73–76. [Google Scholar] [CrossRef]
- Mura, S.; Hillaireau, H.; Nicolas, J.; Le Droumaguet, B.; Gueutin, C.; Zanna, S.; Tsapis, N.; Fattal, E. Influence of surface charge on the potential toxicity of PLGA nanoparticles towards Calu-3 cells. Int. J. Nanomed. 2011, 6, 2591–2605. [Google Scholar]
- Chang, S.; Kang, B.; Dai, Y.; Zhang, H.; Chen, D. One-step fabrication of biocompatible chitosan-coated ZnS and ZnS:Mn2+ quantum dots via a γ-radiation route. Nanoscale Res. Lett. 2011, 6, 591–597. [Google Scholar]
- Tan, W.B.; Zhang, Y. Surface modification of gold and quantum dot nanoparticles with chitosan for bioapplications. J. Biomed. Mater. Res. Part A 2005, 75A, 56–62. [Google Scholar] [CrossRef]
- Woitiski, C.B.; Sarmento, B.; Carvalho, R.A.; Neufeld, R.J.; Veiga, F. Facilitated nanoscale delivery of insulin across intestinal membrane models. Int. J. Pharm. 2011, 412, 123–131. [Google Scholar] [CrossRef]
- Shu, S.; Zhang, X.; Teng, D.; Wang, Z.; Li, C. Polyelectrolyte nanoparticles based on water-soluble chitosan–poly(l-aspartic acid)–polyethylene glycol for controlled protein release. Carbohyd. Res. 2009, 344, 1197–1204. [Google Scholar] [CrossRef]
- Nafee, N.; Schneider, M.; Schaefer, U.F.; Lehr, C.-M. Relevance of the colloidal stability of chitosan/PLGA nanoparticles on their cytotoxicity profile. Int. J. Pharma. 2009, 381, 130–139. [Google Scholar] [CrossRef]
- Weyermann, J.; Lochmann, D.; Zimmer, A. A practical note on the use of cytotoxicity assays. Int. J. Pharm. 2005, 288, 369–376. [Google Scholar] [CrossRef]
- Smith, J.; Wood, E.; Dornish, M. Effect of chitosan on epithelial cell tight junctions. Pharm. Res. 2004, 21, 43–49. [Google Scholar]
- Ulluwishewa, D.; Anderson, R.C.; McNabb, W.C.; Moughan, P.J.; Wells, J.M.; Roy, N.C. Regulation of tight junction permeability by intestinal bacteria and dietary components. J. Nut. 2011, 141, 769–776. [Google Scholar]
- Johnson, P.H.; Frank, D.; Costantino, H.R. Discovery of tight junction modulators: Significance for drug development and delivery. Drug Discov. Today 2008, 13, 261–267. [Google Scholar] [CrossRef]
- Salama, N.N.; Eddington, N.D.; Fasano, A. Tight junction modulation and its relationship to drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 15–28. [Google Scholar] [CrossRef]
- Pasternak, A.S.; Miller, W.M. Measurement of trans-epithelial electrical resistance in perfusion: Potential application for in vitro ocular toxicity testing. Biotechnol. Bioeng. 1996, 50, 568–579. [Google Scholar] [CrossRef]
- Schneeberger, E.E.; Lynch, R.D. The tight junction: a multifunctional complex. Am. J. Physiol. Cell Physiol. 2004, 286, C1213–C1228. [Google Scholar] [CrossRef]
- Stewart, C.; Torr, E.; Jamili, N.; Bosquillon, C.; Sayers, I. Evaluation of differentiated human bronchial epithelial cell culture systems for asthma research. J. Allergy 2012, 2012, 1–11. [Google Scholar]
- Forbes, B.; Hashmi, N.; Martin, G.; Lansley, A. Formulation of inhaled medicines: Effect of delivery vehicle on immortalized epithelial cells. J. Aerosol Med. 2000, 13, 281–288. [Google Scholar] [CrossRef]
- Van der Lubben, I.M.; Verhoef, J.C.; Borchard, G.; Junginger, H.E. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur. J. Pharm. Sci. 2001, 14, 201–207. [Google Scholar] [CrossRef]
- González-Mariscal, L.; Nava, P.; Hernández, S. Critical role of tight junctions in drug delivery across epithelial and endothelial cell layers. J. Membr. Biol. 2005, 207, 55–68. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, L.; Hernández, S.; Vega, J. Inventions designed to enhance drug delivery across epithelial and endothelial cells through the paracellular pathway. Recent Pat. Drug Deliv. Formul. 2008, 2, 145–176. [Google Scholar] [CrossRef]
- Vllasaliu, D.; Exposito-Harris, R.; Heras, A.; Casettari, L.; Garnett, M.; Illum, L.; Stolnik, S. Tight junction modulation by chitosan nanoparticles: Comparison with chitosan solution. Int. J. Pharm. 2010, 400, 183–193. [Google Scholar] [CrossRef]
- Teijeiro-Osorio, D.; Remuñán-López, C.; Alonso, M.J. New generation of hybrid poly/oligosaccharide nanoparticles as carriers for the nasal delivery of macromolecules. Biomacromolecules 2009, 10, 243–249. [Google Scholar]
- Loh, J.W.; Saunders, M.; Lim, L.-Y. Cytotoxicity of monodispersed chitosan nanoparticles against the Caco-2 cells. Toxicol. Appl. Pharm. 2012, 262, 273–282. [Google Scholar] [CrossRef]
- Sadeghi, A.M.M.; Dorkoosh, F.A.; Avadi, M.R.; Weinhold, M.; Bayat, A.; Delie, F.; Gurny, R.; Larijani, B.; Rafiee-Tehrani, M.; Junginger, H.E. Permeation enhancer effect of chitosan and chitosan derivatives: Comparison of formulations as soluble polymers and nanoparticulate systems on insulin absorption in Caco-2 cells. Eur. J. Pharm. Biopharm. 2008, 70, 270–278. [Google Scholar] [CrossRef]
- Ma, Z.; Lim, L.-Y. Uptake of chitosan and associated insulin in Caco-2 cell monolayers: A comparison between chitosan molecules and chitosan nanoparticles. Pharma. Res. 2003, 20, 1812–1819. [Google Scholar] [CrossRef]
- Olive, P.L. The comet assay: An overview of techniques. In In Situ Detection of DNA Damage: Methods and Protocols; Didenko, V.V., Ed.; Humana Press: Totowa, NJ, USA, 2002; Volume 203, pp. 179–194. [Google Scholar]
- Fernandes, J.C.; Borges, M.; Nascimento, H.; Bronze-da-Rocha, E.; Ramos, O.S.; Pintado, M.E.; Malcata, F.X.; Santos-Silva, A. Cytotoxicity and genotoxicity of chitooligosaccharides upon lymphocytes. Int. J. Biol. Macromol. 2011, 49, 433–438. [Google Scholar]
- Lewis, C.; McGee, J.D. The Macrophage; IRL Press: Oxford, UK, 1992; p. 423. [Google Scholar]
- Liu, L.; Song, C.; Song, L.; Zhang, H.; Dong, X.; Leng, X. Effects of alkylated-chitosan-DNA nanoparticles on the function of macrophages. J. Mater. Sci. Mater. Med. 2009, 20, 943–948. [Google Scholar] [CrossRef]
- Zhang, C.; Qu, G.; Sun, Y.; Wu, X.; Yao, Z.; Guo, Q.; Ding, Q.; Yuan, S.; Shen, Z.; Ping, Q.; Zhou, H. Pharmacokinetics, biodistribution, efficacy and safety of N-octyl-O-sulfate chitosan micelles loaded with paclitaxel. Biomaterials 2008, 29, 1233–1241. [Google Scholar]
- Jiang, H.-L.; Lim, H.-T.; Kim, Y.-K.; Arote, R.; Shin, J.-Y.; Kwon, J.-T.; Kim, J.-E.; Kim, J.-H.; Kim, D.; Chae, C.; Nah, J.-W.; Choi, Y.-J.; Cho, C.-S.; Cho, M.-H. Chitosan-graft-spermine as a gene carrier in vitro and in vivo. Eur. J. Pharm. Biopharm. 2011, 77, 36–42. [Google Scholar] [CrossRef]
- Choi, M.; Cho, M.; Han, B.S.; Hong, J.; Jeong, J.; Park, S.; Cho, M.-H.; Kim, K.; Cho, W.-S. Chitosan nanoparticles show rapid extrapulmonary tissue distribution and excretion with mild pulmonary inflammation to mice. Toxicol. Lett. 2010, 199, 144–152. [Google Scholar] [CrossRef]
- Huang, Y.C.; Vieira, A.; Huang, K.L.; Yeh, M.K.; Chiang, C.H. Pulmonary inflammation caused by chitosan microparticles. J. Biomed. Mater. Res. Part A 2005, 75, 283–287. [Google Scholar]
- Pandey, R.; Khuller, G.K. Chemotherapeutic potential of alginate-chitosan microspheres as anti-tubercular drug carriers. J. Antimicrob. Chemother. 2004, 53, 635–640. [Google Scholar] [CrossRef]
- Sonaje, K.; Lin, Y.H.; Juang, J.H.; Wey, S.P.; Chen, C.T.; Sung, H.W. In vivo evaluation of safety and efficacy of self-assembled nanoparticles for oral insulin delivery. Biomaterials 2009, 30, 2329–2339. [Google Scholar] [CrossRef]
- Sonaje, K.; Lin, K.-J.; Tseng, M.T.; Wey, S.-P.; Su, F.-Y.; Chuang, E.-Y.; Hsu, C.-W.; Chen, C.-T.; Sung, H.-W. Effects of chitosan-nanoparticle-mediated tight junction opening on the oral absorption of endotoxins. Biomaterials 2011, 32, 8712–8721. [Google Scholar]
- Zheng, F.; Shi, X.-W.; Yang, G.-F.; Gong, L.-L.; Yuan, H.-Y.; Cui, Y.-J.; Wang, Y.; Du, Y.-M.; Li, Y. Chitosan nanoparticle as gene therapy vector via gastrointestinal mucosa administration: Results of an in vitro and in vivo study. Life Sci. 2007, 80, 388–396. [Google Scholar] [CrossRef]
- Semete, B.; Booysen, L.I.J.; Kalombo, L.; Venter, J.D.; Katata, L.; Ramalapa, B.; Verschoor, J.A.; Swai, H. In vivo uptake and acute immune response to orally administered chitosan and PEG coated PLGA nanoparticles. Toxicol. Appl. Pharm. 2010, 249, 158–165. [Google Scholar] [CrossRef]
- Pokharkar, V.; Dhar, S.; Bhumkar, D.; Mali, V.; Bodhankar, S.; Prasad, B.L.V. Acute and subacute toxicity studies of chitosan reduced gold nanoparticles: A novel carrier for therapeutic agents. J. Biomed. Nanotechnol. 2009, 5, 233–239. [Google Scholar] [CrossRef]
- Kunzmann, A.; Andersson, B.; Thurnherr, T.; Krug, H.; Scheynius, A.; Fadeel, B. Toxicology of engineered nanomaterials: Focus on biocompatibility, biodistribution and biodegradation. Biochim. Biophys. Acta 2011, 1810, 361–373. [Google Scholar]
- Chen, X.-G.; Liu, C.-S.; Liu, C.-G.; Meng, X.-H.; Lee, C.M.; Park, H.-J. Preparation and biocompatibility of chitosan microcarriers as biomaterial. Biochem. Eng. J. 2006, 27, 269–274. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rodrigues, S.; Dionísio, M.; López, C.R.; Grenha, A. Biocompatibility of Chitosan Carriers with Application in Drug Delivery. J. Funct. Biomater. 2012, 3, 615-641. https://doi.org/10.3390/jfb3030615
Rodrigues S, Dionísio M, López CR, Grenha A. Biocompatibility of Chitosan Carriers with Application in Drug Delivery. Journal of Functional Biomaterials. 2012; 3(3):615-641. https://doi.org/10.3390/jfb3030615
Chicago/Turabian StyleRodrigues, Susana, Marita Dionísio, Carmen Remuñán López, and Ana Grenha. 2012. "Biocompatibility of Chitosan Carriers with Application in Drug Delivery" Journal of Functional Biomaterials 3, no. 3: 615-641. https://doi.org/10.3390/jfb3030615