
Modulation of the Tumor Microenvironment for Cancer Treatment: A Biomaterials Approach

Abstract
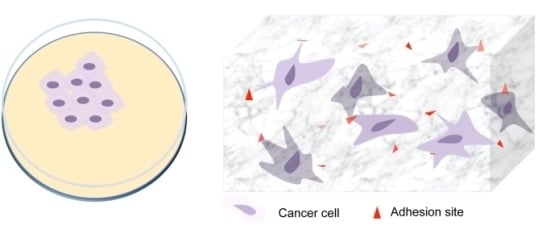
:
1. Introduction
2. Microenvironment in Cancer Initiation and Progression
2.1. Role of Different Cell Populations

{kind=link}
{kind=link}
{kind=link}
| Lineage | Role in tumorigenesis |
|---|---|
| Tumor-associated macrophages | Immunosuppression; produce cytokines and growth factors Tumor remodeling; secrete matrix metalloproteinases (MMPs) and urokinase-type plasminogen activator (uPA) |
| Neutrophils | Produce cytokines and reactive oxygen species |
| Treg cells | Immunosuppression; secrete TGF-β and IL-10 that inhibit the antitumor activity of cytotoxic T-cells and natural killer cells |
| Th cells | Production of cytokines that induce immunosuppression |
| B-cells | Production of cytokines and activation of mast cells |
| Mesenchymal stem cells | Produce cytokines that promote tumor invasiveness and metastasis; Replenish cancer cells |
| Tumor-associated fibroblasts | Secrete MMPs involved in tumor remodeling; Produce vascular endothelial growth factor (VEGF) that induce angiogenesis |
| Vascular endothelial cells | Form blood vessels that support tumor growth and metastasis |
2.1.1. Fibroblasts
2.1.2. Immune Cells
2.1.3. Stem Cells
2.1.4. Vascular Endothelial Cells
3. Modeling Cancer Progression Using Tissue Engineering Concepts

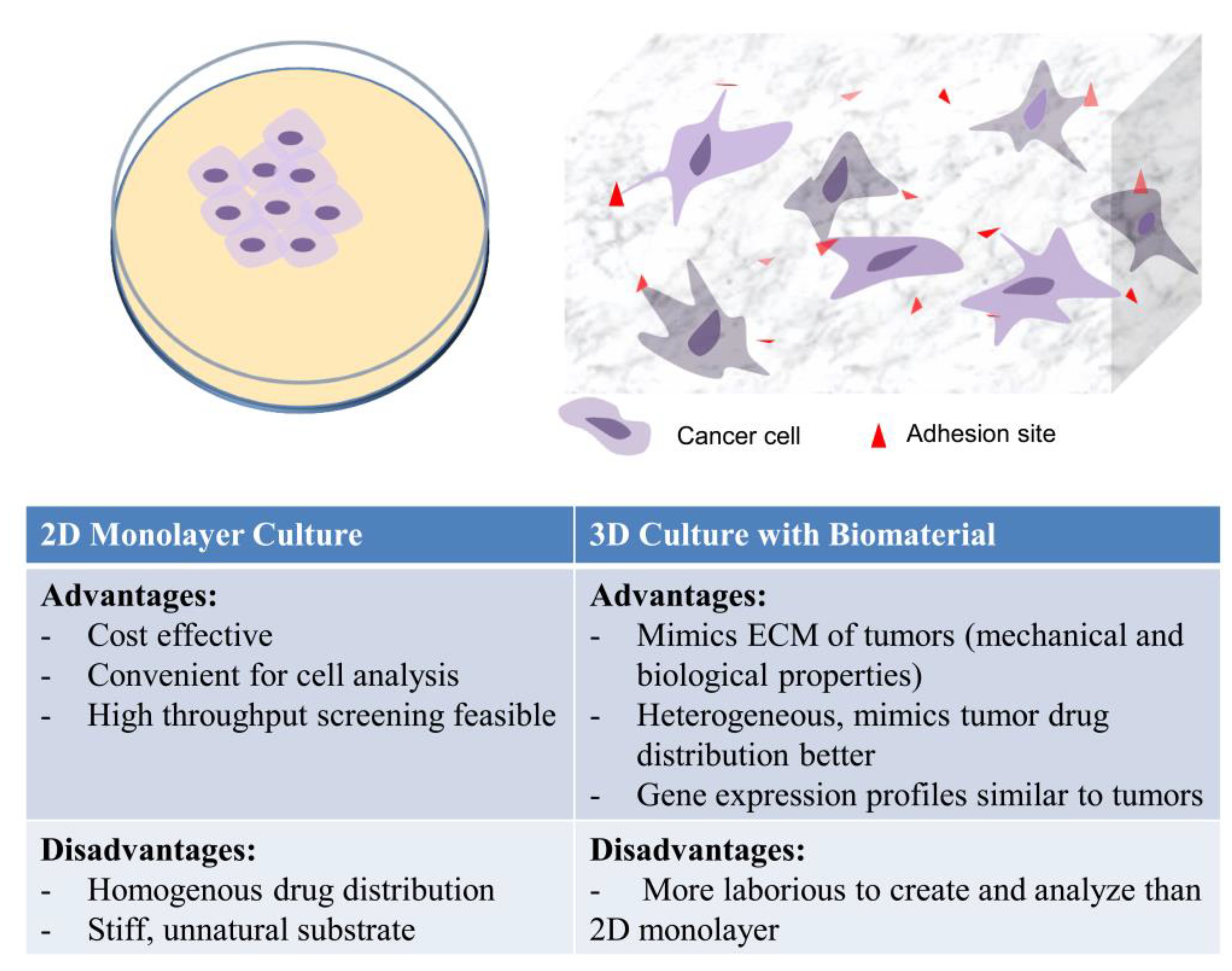
In Vitro 3D Models in Studying Cancer Biology
4. Regulating the Tumor Microenvironment with Biomaterials for Treatment
| Drug name | Nanomaterial | Therapeutic | State of development |
|---|---|---|---|
| Doxil | Liposome | Doxorubicin | Approved (US, 1995; EU, 1996) |
| DaunoXome® | Liposome | Daunorubicin citrate | Approved (US, 1996) |
| Feridex | Dextran coated superparamagnetic iron oxide nanoparticles (SPION) | – | Approved (US, 1996) |
| Myocet | Liposome | Doxorubicin | Approved (EU, 2000) |
| Abraxane | Albumin NPs | Paclitaxel | Approved (US, 2005; EU, 2006) |
| Genexol-PM | PEG-PLA Micelle NPs | Paclitaxel | Approved (South Korea, 2007) Phase III trials |
| Lipoplatin | Liposome | Cisplatin | Phase III trials |
| OPAXIO | Polymer-drug conjugate | Paclitaxel | Phase III trials |
| Clariscan | SPION | – | Phase III trials |
| ABI-008 | Albumin NPs | Docetaxel | Phase II trials |
| AP5250 | Polymer-drug conjugate | Carboplatine platinate | Phase II trials |
| CRLX101 | Polymeric NPs | Camptothecin | Phase II trials |
| MBP-426 | liposome | Oxaliplatin | Phase II trials |
| BIND-014 | Targeted polymeric NPs | Docetaxel | Phase I trials |
| MAG-CPT | Polymer-drug conjugate | Camptothecin | Phase I trial |

4.1. Drug Delivery to Tumor Stroma
4.2. Biomaterial-Mediated Modulation of Tumor Immune Components
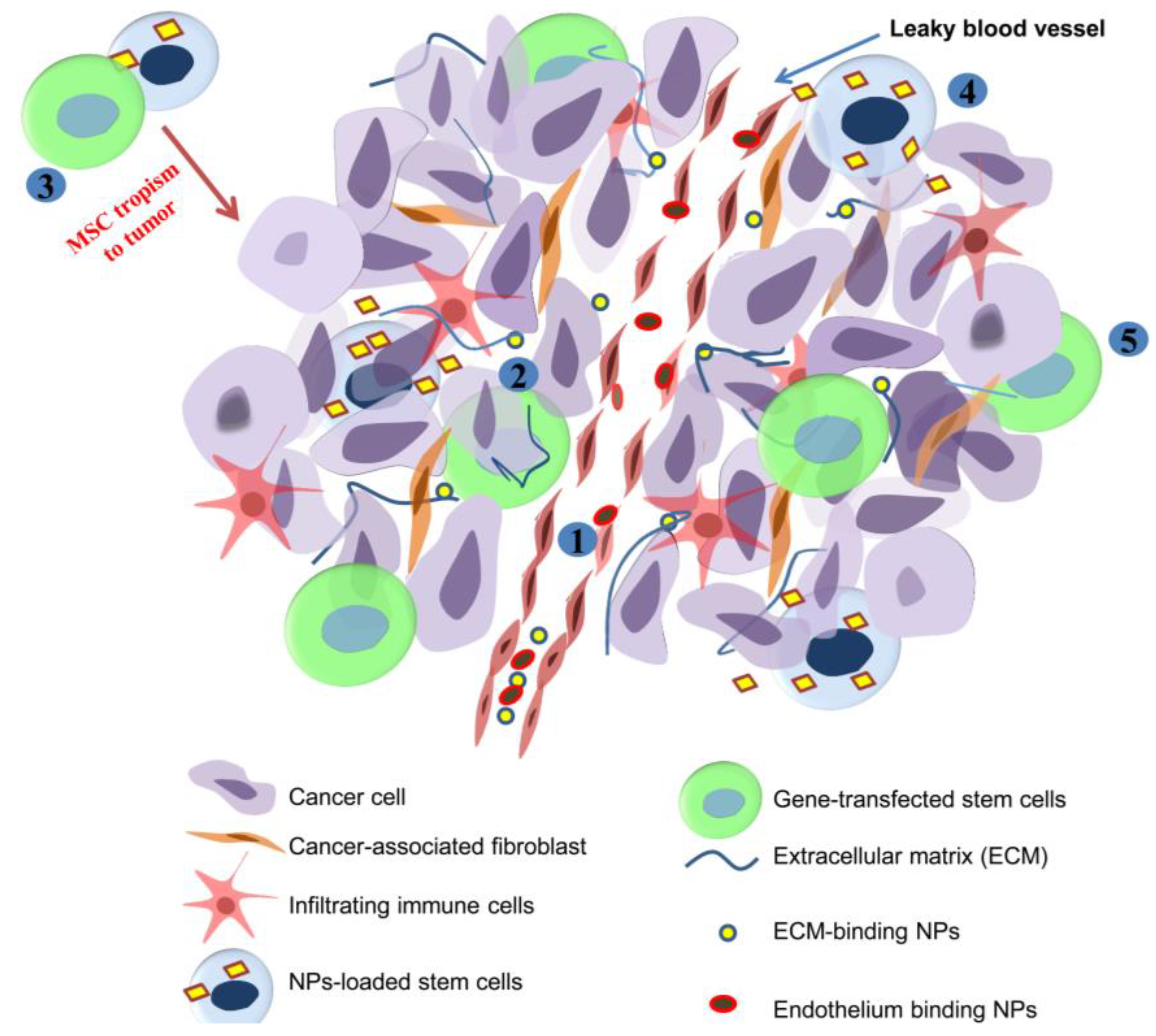
4.3. Modifying Stem Cells with Biomaterials to Control Tumor Growth
4.4. Identification and Regulation of Matrix Remodeling Enzymes with Biomaterials
5. Future Perspective
6. Conclusion
Author Contributions
Conflict of interest
References
- Nakasone, E.S.; Askautrud, H.A.; Kees, T.; Park, J.-H.; Plaks, V.; Ewald, A.J.; Fein, M.; Rasch, M.G.; Tan, Y.-X.; Qiu, J. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 2012, 21, 488–503. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited—The role of tumor‐stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Can cancer be reversed by engineering the tumor microenvironment? Semin. Cancer Biol. 2008, 18, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Thoma, C.R.; Zimmermann, M.; Agarkova, I.; Kelm, J.M.; Krek, W. 3D cell culture systems modeling tumor growth determinants in cancer target discovery. Adv. Drug Deliv. Rev. 2014, 69–70, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Gutschalk, C.M.; Hoffmann, C.; Yilmaz, D.; Mueller, M.M. Integrating macrophages into organotypic co-cultures: A 3D in vitro model to study tumor-associated macrophages. PloS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Ghajar, C.M.; Bissell, M.J. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv. Drug Deliv. Rev. 2014, 69–70, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 206–212. [Google Scholar] [CrossRef]
- Cho, N.-H.; Cheong, T.-C.; Min, J.H.; Wu, J.H.; Lee, S.J.; Kim, D.; Yang, J.-S.; Kim, S.; Kim, Y.K.; Seong, S.-Y. A multifunctional core-shell nanoparticle for dendritic cell-based cancer immunotherapy. Nat. Nanotechnol. 2011, 6, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Otranto, M.; Sarrazy, V.; Bonté, F.; Hinz, B.; Gabbiani, G.; Desmouliere, A. The role of the myofibroblast in tumor stroma remodeling. Cell Adhes. Migr. 2012, 6, 203–219. [Google Scholar] [CrossRef]
- Whiteside, T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; D’Asti, E.; Magnus, N.; Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles as mediators of intercellular communication in cancer—The emerging science of cellular ‘debris’. Semin. Immunopathol. 2011, 33, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Sahai, E. Cell communication networks in cancer invasion. Curr. Opin. Cell Biol. 2011, 23, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-κB-dependent manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Haviv, I.; Campbell, I.G. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009, 25, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, G.; Rüegg, C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem. Cell Biol. 2008, 130, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, S.P.; Restifo, N.P. Cellular constituents of immune escape within the tumor microenvironment. Cancer Res. 2012, 72, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Lichtenberger, B.M.; Hoste, E.; Kretzschmar, K.; Simons, B.D.; Charalambous, M.; Ferron, S.R.; Herault, Y.; Pavlovic, G.; Ferguson-Smith, A.C.; et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature 2013, 504, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Tassone, E.; Valacca, C.; Mignatti, P. Membrane‐type 1 matrix metalloproteinase downregulates fibroblast growth factor‐2 binding to the cell surface and intracellular signaling. J. Cell. Physiol. 2015, 230, 366–377. [Google Scholar] [CrossRef]
- Yun, Y.-R.; Won, J.E.; Jeon, E.; Lee, S.; Kang, W.; Jo, H.; Jang, J.-H.; Shin, U.S.; Kim, H.-W. Fibroblast growth factors: Biology, function, and application for tissue regeneration. J. Tissue Eng. 2010, 1. [Google Scholar] [CrossRef]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2012, 19, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Porsch, H.; Mehić, M.; Olofsson, B.; Heldin, P.; Heldin, C.-H. Platelet-derived growth factor β-receptor, transforming growth factor β type I receptor, and CD44 protein modulate each other’s signaling and stability. J. Biol. Chem. 2014, 289, 19747–19757. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.E.; Shaw, A.K.; Strand, D.W.; Hayward, S.W. Cancer associated fibroblasts in cancer pathogenesis. Semin. Cell Dev. Biol. 2010, 21, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Fullár, A.; Kovalszky, I.; Bitsche, M.; Romani, A.; Schartinger, V.H.; Sprinzl, G.M.; Riechelmann, H.; Dudás, J. Tumor cell and carcinoma-associated fibroblast interaction regulates matrix metalloproteinases and their inhibitors in oral squamous cell carcinoma. Exp. Cell Res. 2012, 318, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margadant, C.; Sonnenberg, A. Integrin–TGF-β crosstalk in fibrosis, cancer and wound healing. EMBO Rep. 2010, 11, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Mario, A.S.; Surabhi, D.-G.; Amanda, J.R.; Hidayatullah, G.M. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 2012, 441, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008, 6. [Google Scholar] [CrossRef]
- Feng, P.-H.; Yu, C.-T.; Wu, C.-Y.; Yen, T.-H.; Lee, K.-Y. The predictive role of tumor-associated macrophages in stage IIIA pN2 non-small cell lung cancer after neoadjuvant chemotherapy and surgery. Cancer Res. 2014, 74. [Google Scholar] [CrossRef] [PubMed]
- Caillou, B.; Talbot, M.; Weyemi, U.; Pioche-Durieu, C.; Al Ghuzlan, A.; Bidart, J.M.; Chouaib, S.; Schlumberger, M.; Dupuy, C. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PloS One 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Hammes, L.S.; Tekmal, R.R.; Naud, P.; Edelweiss, M.I.; Kirma, N.; Valente, P.T.; Syrjänen, K.J.; Cunha-Filho, J.S. Macrophages, inflammation and risk of cervical intraepithelial neoplasia (CIN) progression—Clinicopathological correlation. Gynecol. Oncol. 2007, 105, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, L.; Che, G.; Yu, N.; Dai, F.; You, Z. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Ohri, C.M.; Shikotra, A.; Green, R.H.; Waller, D.A.; Bradding, P. Macrophages within NSCLC tumour islets are predominantly of a cytotoxic M1 phenotype associated with extended survival. Eur. Respir. J. 2009, 33, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef]
- Mills, C.D.; Ley, K. M1 and M2 macrophages: The chicken and the egg of immunity. J. Innate Immune. 2014. [Google Scholar] [CrossRef]
- Varin, A.; Mukhopadhyay, S.; Herbein, G.; Gordon, S. Alternative activation of macrophages by IL-4 impairs phagocytosis of pathogens but potentiates microbial-induced signalling and cytokine secretion. Blood 2010, 115, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Robinson, S.C.; Schulz, M.; Trümper, L.; Balkwill, F.R.; Binder, C. Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-α dependent up-regulation of matrix metalloproteases. Carcinogenesis 2004, 25, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Tuszynski, G. The role of matrix metalloproteinases in tumor angiogenesis and tumor metastasis. Pathol. Oncol. Res. 2001, 7, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Germano, G.; Marchesi, F.; Locatelli, M.; Biswas, S.K. Cancer‐promoting tumor‐associated macrophages: New vistas and open questions. Eur. J. Immunol. 2011, 41, 2522–2525. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. What are regulatory T cells (Treg) regulating in cancer and why? Semin. Cancer Biol. 2012, 22, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Ugolini, S.; Blaise, D.; Chabannon, C.; Brossay, L. Targeting natural killer cells and natural killer T cells in cancer. Nat. Rev. Immunol. 2012, 12, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; de Sousa e Melo, F.; Richel, D.J.; Medema, J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet Oncol. 2012, 13, e83–e89. [Google Scholar] [CrossRef] [PubMed]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-R.; Yuan, Y.; Wang, X.-J.; Wei, L.-L.; Chen, Y.-N.; Cong, C.; Li, S.-F.; Long, D.; Tan, W.-D.; Mao, Y.-Q.; et al. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol. Ther. 2008, 7, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Bergfeld, S.A.; DeClerck, Y.A. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metastasis Rev. 2010, 29, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Khakoo, A.Y.; Pati, S.; Anderson, S.A.; Reid, W.; Elshal, M.F.; Rovira, I.I.; Nguyen, A.T.; Malide, D.; Combs, C.A.; Hall, G.; et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J. Exp. Med. 2006, 203, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Xu, Z.; Zhao, T.; Zhao, Z.; Shi, M.; Zhao, R.C.; Ye, L.; Zhang, X. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008, 18, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Klopp, A.H.; Gupta, A.; Spaeth, E.; Andreeff, M.; Marini, F. Concise review: Dissecting a discrepancy in the literature: Do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011, 29, 11–19. [Google Scholar] [CrossRef] [PubMed]
- D’souza, N.; Burns, J.S.; Grisendi, G.; Candini, O.; Veronesi, E.; Piccinno, S.; Horwitz, E.M.; Paolucci, P.; Conte, P.; Dominici, M. MSC and tumors: Homing, differentiation, and secretion influence therapeutic potential. In Mesenchymal Stem Cells—Basics and Clinical Application II; Weyand, B., Dominici, M., Hass, R., Jacobs, R., Kasper, K., Eds.; Springer Berlin Heidelberg: Berlin, Germany, 2012; Volume 130, pp. 209–266. [Google Scholar]
- Studeny, M.; Marini, F.C.; Dembinski, J.L.; Zompetta, C.; Cabreira-Hansen, M.; Bekele, B.N.; Champlin, R.E.; Andreeff, M. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 2004, 96, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Kanehira, M.; Xin, H.; Hoshino, K.; Maemondo, M.; Mizuguchi, H.; Hayakawa, T.; Matsumoto, K.; Nakamura, T.; Nukiwa, T.; Saijo, Y. Targeted delivery of NK4 to multiple lung tumors by bone marrow-derived mesenchymal stem cells. Cancer Gene Ther. 2007, 14, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of tumor endothelial cells: Comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harbor Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Yamasaki, T.; Kamba, T.; Kanno, T.; Inoue, T.; Shibasaki, N.; Arakaki, R.; Yamada, T.; Kondo, K.; Kamoto, T.; Nishiyama, H. Tumor microvasculature with endothelial fenestrations in VHL null clear cell renal cell carcinomas as a potent target of anti‐angiogenic therapy. Cancer Sci. 2012, 103, 2027–2037. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, H.; Kadowaki, K.; Matsusaki, M.; Cabral, H.; Nishihara, H.; Ijichi, H.; Koike, K.; Kataoka, K.; Miyazono, K.; Akashi, M. Engineering fibrotic tissue in pancreatic cancer: A novel three-dimensional model to investigate nanoparticle delivery. Biochem. Biophys. Res. Commun. 2012, 419, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell plasticity and heterogeneity in cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Ronnov-Jessen, L.; Petersen, O.W.; Bissell, M.J. Cellular changes involved in conversion of normal to malignant breast: Importance of the stromal reaction. Physiol. Rev. 1996, 76, 69–125. [Google Scholar] [PubMed]
- Zhang, S. Beyond the petri dish. Nat. Biotechnol. 2004, 22, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.S.; Kwak, B.; Han, B.; Park, K. Development of an in vitro 3D tumor model to study therapeutic efficiency of an anticancer drug. Mol. Pharm. 2013, 10, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Benton, G.; Arnaoutova, I.; George, J.; Kleinman, H.K.; Koblinski, J. Matrigel: From discovery and ECM mimicry to assays and models for cancer research. Adv. Drug Deliv. Rev. 2014, 79–80, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W.; Loessner, D.; Rizzi, S.; Kaplan, D.L.; Mooney, D.J.; Clements, J.A. Can tissue engineering concepts advance tumor biology research? Trends Biotechnol. 2010, 28, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Håkanson, M.; Cukierman, E.; Charnley, M. Miniaturized pre-clinical cancer models as research and diagnostic tools. Adv. Drug Deliv. Rev. 2014, 69–70, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Song, H.-H.G.; Park, K.M.; Gerecht, S. Hydrogels to model 3D in vitro microenvironment of tumor vascularization. Adv. Drug Deliv. Rev. 2014, 79–80, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Birgersdotter, A.; Sandberg, R.; Ernberg, I. Gene expression perturbation in vitro—A growing case for three-dimensional (3D) culture systems. Semin. Cancer Biol. 2005, 15, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Horning, J.L.; Sahoo, S.K.; Vijayaraghavalu, S.; Dimitrijevic, S.; Vasir, J.K.; Jain, T.K.; Panda, A.K.; Labhasetwar, V. 3-D tumor model for in vitro evaluation of anticancer drugs. Mol. Pharm. 2008, 5, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Miroshnikova, Y.; Jorgens, D.; Spirio, L.; Auer, M.; Sarang-Sieminski, A.; Weaver, V. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Phys. Biol. 2011, 8. [Google Scholar] [CrossRef]
- Plodinec, M.; Loparic, M.; Monnier, C.A.; Obermann, E.C.; Zanetti-Dallenbach, R.; Oertle, P.; Hyotyla, J.T.; Aebi, U.; Bentires-Alj, M.; LimRoderick, Y.H.; et al. The nanomechanical signature of breast cancer. Nat. Nanotech. 2012, 7, 757–765. [Google Scholar] [CrossRef]
- Hansen, T.D.; Koepsel, J.T.; Le, N.N.; Nguyen, E.H.; Zorn, S.; Parlato, M.; Loveland, S.G.; Schwartz, M.P.; Murphy, W.L. Biomaterial arrays with defined adhesion ligand densities and matrix stiffness identify distinct phenotypes for tumorigenic and non-tumorigenic human mesenchymal cell types. Biomater. Sci. 2014, 2, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.P.; Kraning-Rush, C.M.; Williams, R.M.; Reinhart-King, C.A. Biophysical control of invasive tumor cell behavior by extracellular matrix microarchitecture. Biomaterials 2012, 33, 4157–4165. [Google Scholar] [CrossRef] [PubMed]
- LaBarbera, D.V.; Reid, B.G.; Yoo, B.H. The multicellular tumor spheroid model for high-throughput cancer drug discovery. Expert Opin. Drug Discov. 2012, 7, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Haisler, W.L.; Timm, D.M.; Gage, J.A.; Tseng, H.; Killian, T.C.; Souza, G.R. Three-dimensional cell culturing by magnetic levitation. Nat. Protoc. 2013, 8, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, H.; Gage, J.; Leonard, F.; Srinivasan, S.; Souza, G.R.; Dave, B.; Godin, B. Three-dimensional in vitro co-culture model of breast tumor using magnetic levitation. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Horman, S.R.; To, J.; Orth, A.P.; Slawny, N.; Cuddihy, M.J.; Caracino, D. High-content analysis of three-dimensional tumor spheroids: Investigating signaling pathways using small hairpin RNA. Nat. Meth. 2013, 10, v–vi. [Google Scholar]
- Pickl, M.; Ries, C. Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene 2008, 28, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Szot, C.S.; Buchanan, C.F.; Freeman, J.W.; Rylander, M.N. 3D in vitro bioengineered tumors based on collagen I hydrogels. Biomaterials 2011, 32, 7905–7912. [Google Scholar] [CrossRef] [PubMed]
- Hanjaya-Putra, D.; Wong, K.T.; Hirotsu, K.; Khetan, S.; Burdick, J.A.; Gerecht, S. Spatial control of cell-mediated degradation to regulate vasculogenesis and angiogenesis in hyaluronan hydrogels. Biomaterials 2012, 33, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, C.; Kong, H.J.; Hsiong, S.X.; Evangelista, M.B.; Yuen, W.; Mooney, D.J. Cancer cell angiogenic capability is regulated by 3D culture and integrin engagement. Proc. Natl. Acad. Sci. 2009, 106, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, S.; Mandal, M.; Hutmacher, D.W.; Russell, P.J.; Soekmadji, C.; Kundu, S.C. Engineered silk fibroin protein 3D matrices for in vitro tumor model. Biomaterials 2011, 32, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Pathi, S.P.; Lin, D.D.W.; Dorvee, J.R.; Estroff, L.A.; Fischbach, C. Hydroxyapatite nanoparticle-containing scaffolds for the study of breast cancer bone metastasis. Biomaterials 2011, 32, 5112–5122. [Google Scholar] [CrossRef] [PubMed]
- Zervantonakis, I.K.; Hughes-Alford, S.K.; Charest, J.L.; Condeelis, J.S.; Gertler, F.B.; Kamm, R.D. Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. 2012, 109, 13515–13520. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, L.; Maffini, M.V.; Soto, A.; Sonnenschein, C.; Kaplan, D.L. A complex 3D human tissue culture system based on mammary stromal cells and silk scaffolds for modeling breast morphogenesis and function. Biomaterials 2010, 31, 3920–3929. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J. Clin. Oncol. 2013, 31, 2205–2218. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Jin, H.; Kim, H.W.; Cho, M.-H.; Cho, C.S. Mannosylated chitosan nanoparticle-based cytokine gene therapy suppressed cancer growth in BALB/c mice bearing CT-26 carcinoma cells. Mol. Cancer Ther. 2006, 5, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.Y.; Chung, H.; Min, K.H.; Yoon, H.Y.; Kim, K.; Park, J.H.; Kwon, I.C.; Jeong, S.Y. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials 2010, 31, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Fukuda, T.; Nagaoka, Y.; Hasumura, T.; Morimoto, H.; Yoshida, Y.; Maekawa, T.; Venugopal, K.; Kumar, D.S. Curcumin loaded-PLGA nanoparticles conjugated with Tet-1 peptide for potential use in alzheimer’s disease. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.K.; Sun, I.-C.; Ryu, J.H.; Koo, H.; Park, C.W.; Youn, I.-C.; Choi, K.; Kwon, I.C.; Kim, K.; Ahn, C.-H. Matrix metalloproteinase sensitive gold nanorod for simultaneous bioimaging and photothermal therapy of cancer. Bioconjugate Chem. 2010, 21, 2173–2177. [Google Scholar] [CrossRef]
- Yu, T.; Malugin, A.; Ghandehari, H. Impact of silica nanoparticle design on cellular toxicity and hemolytic activity. ACS Nano 2011, 5, 5717–5728. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lytton-Jean, A.K.; Chen, Y.; Love, K.T.; Park, A.I.; Karagiannis, E.D.; Sehgal, A.; Querbes, W.; Zurenko, C.S.; Jayaraman, M. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 2012, 7, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.J.; Hao, L.; Narayan, S.P.; Auyeung, E.; Mirkin, C.A. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc. Natl. Acad. Sci. 2013, 110, 7625–7630. [Google Scholar] [CrossRef] [PubMed]
- Saranya, N.; Moorthi, A.; Saravanan, S.; Devi, M.P.; Selvamurugan, N. Chitosan and its derivatives for gene delivery. Int. J. Biol. Macromol. 2011, 48, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Dimou, A.; Syrigos, K.N.; Saif, M.W. Overcoming the stromal barrier: Technologies to optimize drug delivery in pancreatic cancer. Ther. Adv. Med. Oncol. 2012. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Ruoslahti, E. Targeting tumor vasculature with homing peptides from phage display. Semin. Cancer Biol. 2000, 10, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Greenwald, D.R.; Ruoslahti, E. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Sci. 2010, 328, 1031–1035. [Google Scholar] [CrossRef]
- Guo, J.; Gao, X.; Su, L.; Xia, H.; Gu, G.; Pang, Z.; Jiang, X.; Yao, L.; Chen, J.; Chen, H. Aptamer-functionalized PEG–PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 2011, 32, 8010–8020. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Heldin, P.; Hascall, V.C.; Karamanos, N.K.; Skandalis, S.S.; Markwald, R.R.; Ghatak, S. Hyaluronan–CD44 interactions as potential targets for cancer therapy. FEBS J. 2011, 278, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Skandalis, S.; Gialeli, C.; Theocharis, A.; Karamanos, N. Advances and advantages of nanomedicine in the pharmacological targeting of hyaluronan-CD44 interactions and signaling in cancer. Adv. Cancer Res. 2013, 123, 277–317. [Google Scholar]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Elbayoumi, T.; Torchilin, V. Tumor-specific liposomal nanomedicines: Antitumor antibody-modified doxorubicin-loaded liposomes. Nanomed. Health Dis. 2011, 336–355. [Google Scholar]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Mejías, R.; Pérez-Yagüe, S.; Gutiérrez, L.; Cabrera, L.I.; Spada, R.; Acedo, P.; Serna, C.J.; Lázaro, F.J.; Villanueva, Á.; del Puerto Morales, M.; et al. Dimercaptosuccinic acid-coated magnetite nanoparticles for magnetically guided in vivo delivery of interferon gamma for cancer immunotherapy. Biomaterials 2011, 32, 2938–2952. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Cho, H.R.; Oh, M.H.; Lee, S.H.; Kim, K.; Kim, B.H.; Shin, K.; Ahn, T.-Y.; Choi, J.W.; Kim, Y.-W. Multifunctional Fe3O4/TaOx core/shell nanoparticles for simultaneous magnetic resonance imaging and X-ray computed tomography. J. Am. Chem. Soc. 2012, 134, 10309–10312. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-S.; Liu, S.-Q.; Ng, S.Y.; Froix, M.; Ohno, T.; Heller, J. Controlled release of interleukin-2 for tumour immunotherapy using alginate/chitosan porous microspheres. J. Control. Release 1997, 43, 65–74. [Google Scholar] [CrossRef]
- Linehan, S.A.; Martínez-Pomares, L.; Gordon, S. Macrophage lectins in host defence. Microbes Infect. 2000, 2, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Z.; Jiang, Y.; Zhang, D.; Chen, J.; Dong, L.; Zhang, J. Targeted delivery of oligonucleotides into tumor-associated macrophages for cancer immunotherapy. J. Control. Release 2012, 158, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.-W.; Wang, Y.-C.; Lin, P.-L.; Cheng, Y.-W.; Chen, C.-Y.; Wu, T.-C.; Lee, H. IL-10 promotes tumor aggressiveness via upregulation of CIP2A transcription in lung adenocarcinoma. Clin. Cancer Res. 2013, 19, 4092–4103. [Google Scholar] [CrossRef] [PubMed]
- Downey, C.M.; Aghaei, M.; Schwendener, R.A.; Jirik, F.R. DMXAA causes tumor site-specific vascular disruption in murine non-small cell lung cancer, and like the endogenous non-canonical cyclic dinucleotide sting agonist, 2′3′-cGAMP, induces M2 macrophage repolarization. PloS One 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, X.; Wang, S.; Deng, W.; Zan, L.; Yu, S. In vitro repolarized tumor macrophages inhibit gastric tumor growth. Oncol. Res. Featur. Preclinical Clin. Cancer Ther. 2013, 20, 275–280. [Google Scholar]
- Sheng, W.-Y.; Huang, L. Cancer immunotherapy and nanomedicine. Pharm. Res. 2011, 28, 200–214. [Google Scholar] [CrossRef] [PubMed]
- De Jong, S.; Chikh, G.; Sekirov, L.; Raney, S.; Semple, S.; Klimuk, S.; Yuan, N.; Hope, M.; Cullis, P.; Tam, Y. Encapsulation in liposomal nanoparticles enhances the immunostimulatory, adjuvant and anti-tumor activity of subcutaneously administered CpG ODN. Cancer Immunol. Immunother. 2007, 56, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Tongchusak, S.; Mizukami, Y.; Kang, Y.J.; Ioji, T.; Touma, M.; Reinhold, B.; Keskin, D.B.; Reinherz, E.L.; Sasada, T. Induction of anti-tumor cytotoxic t cell responses through PLGA-nanoparticle mediated antigen delivery. Biomaterials 2011, 32, 3666–3678. [Google Scholar] [CrossRef] [PubMed]
- Hamdy, S.; Molavi, O.; Ma, Z.; Haddadi, A.; Alshamsan, A.; Gobti, Z.; Elhasi, S.; Samuel, J.; Lavasanifar, A. Co-delivery of cancer-associated antigen and toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine 2008, 26, 5046–5057. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Litwin, T.; Nagaraja, A.R.; Kwong, B.; Katz, J.; Watson, N.; Irvine, D.J. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core–shell nanoparticles. Nano Lett. 2007, 7, 3056–3064. [Google Scholar] [CrossRef] [PubMed]
- Swartz, M.A.; Hirosue, S.; Hubbell, J.A. Engineering approaches to immunotherapy. Sci. Transl. Med. 2012, 4, 148–149. [Google Scholar]
- Reddy, S.T.; van der Vlies, A.J.; Simeoni, E.; Angeli, V.; Randolph, G.J.; O’Neil, C.P.; Lee, L.K.; Swartz, M.A.; Hubbell, J.A. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007, 25, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.T.; Rehor, A.; Schmoekel, H.G.; Hubbell, J.A.; Swartz, M.A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 2006, 112, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.N.; Vokali, E.; Lund, A.W.; Hubbell, J.A.; Swartz, M.A. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials 2014, 35, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.; Dembinski, J.; Sasser, A.K.; Studeny, M.; Andreeff, M.; Marini, F. Mesenchymal stem cells in cancer: Tumor-associated fibroblasts and cell-based delivery vehicles. Int. J. Hematol. 2007, 86, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Parekkadan, B.; Milwid, J.M. Mesenchymal stem cells as therapeutics. Annu. Rev. Biomed. Eng. 2010, 12, 87–117. [Google Scholar] [CrossRef] [PubMed]
- Loebinger, M.R.; Sage, E.K.; Davies, D.; Janes, S.M. Trail-expressing mesenchymal stem cells kill the putative cancer stem cell population. Br. J. Cancer 2010, 103, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Shah, K. Mesenchymal stem cells engineered for cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Peetla, C.; Adjei, I.M.; Labhasetwar, V. Selective biophysical interactions of surface modified nanoparticles with cancer cell lipids improve tumor targeting and gene therapy. Cancer Lett. 2013, 334, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Oliveira, H.; Pandita, D.; Rodrigues, J.; Pêgo, A.P.; Granja, P.L.; Tomás, H. Functionalization of poly(amidoamine) dendrimers with hydrophobic chains for improved gene delivery in mesenchymal stem cells. J. Control. Release 2010, 144, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Morshed, R.; Tobias, A.; Cheng, Y.; Ahmed, A.U.; Lesniak, M.S. Drug-loaded nanoparticle systems and adult stem cells: A potential marriage for the treatment of malignant glioma? Oncotarget 2013, 4, 378–396. [Google Scholar] [PubMed]
- Li, L.; Guan, Y.; Liu, H.; Hao, N.; Liu, T.; Meng, X.; Fu, C.; Li, Y.; Qu, Q.; Zhang, Y. Silica nanorattle–doxorubicin-anchored mesenchymal stem cells for tumor-tropic therapy. ACS Nano 2011, 5, 7462–7470. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.W.; Shah, K. Stem cell-based therapies for cancer treatment: Separating hope from hype. Nat. Rev. Cancer 2014, 14, 638–691. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer—Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Veiseh, O.; Gunn, J.W.; Kievit, F.M.; Sun, C.; Fang, C.; Lee, J.S.; Zhang, M. Inhibition of tumor‐cell invasion with chlorotoxin‐bound superparamagnetic nanoparticles. Small 2009, 5, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xing, G.; Blanco, E.; Song, Y.; Zhao, L.; Sun, B.; Li, X.; Wang, P.C.; Korotcov, A.; Li, W.; et al. Gadolinium metallofullerenol nanoparticles inhibit cancer metastasis through matrix metalloproteinase inhibition: Imprisoning instead of poisoning cancer cells. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 136–146. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adjei, I.M.; Blanka, S. Modulation of the Tumor Microenvironment for Cancer Treatment: A Biomaterials Approach. J. Funct. Biomater. 2015, 6, 81-103. https://doi.org/10.3390/jfb6010081
Adjei IM, Blanka S. Modulation of the Tumor Microenvironment for Cancer Treatment: A Biomaterials Approach. Journal of Functional Biomaterials. 2015; 6(1):81-103. https://doi.org/10.3390/jfb6010081
Chicago/Turabian StyleAdjei, Isaac M., and Sharma Blanka. 2015. "Modulation of the Tumor Microenvironment for Cancer Treatment: A Biomaterials Approach" Journal of Functional Biomaterials 6, no. 1: 81-103. https://doi.org/10.3390/jfb6010081