Biological Fate of Fe3O4 Core-Shell Mesoporous Silica Nanoparticles Depending on Particle Surface Chemistry

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Synthesis and Characterization of Fe3O4@MSN
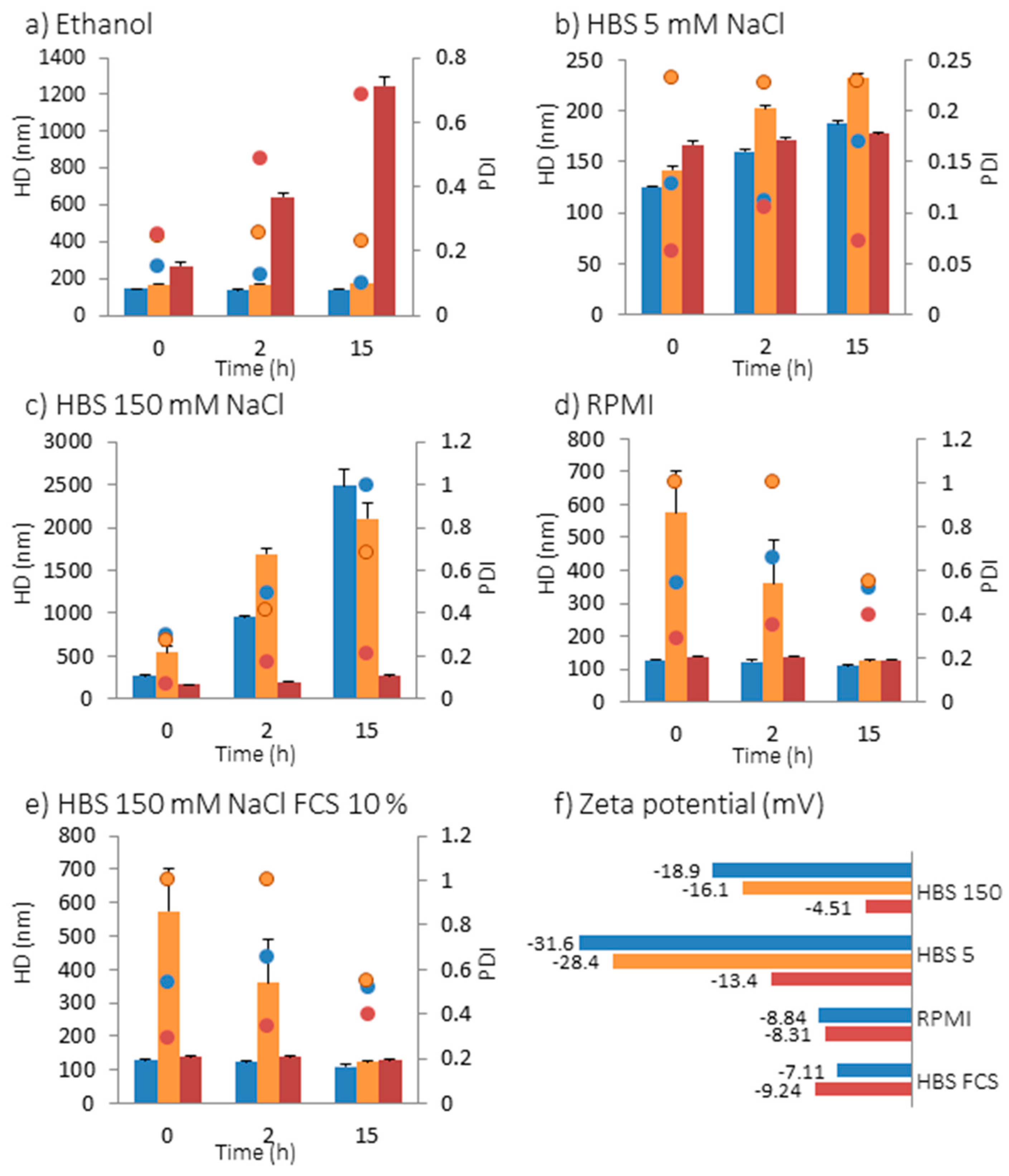
2.2. Interaction of NPs with Model Membranes Depending of the Surface Coating
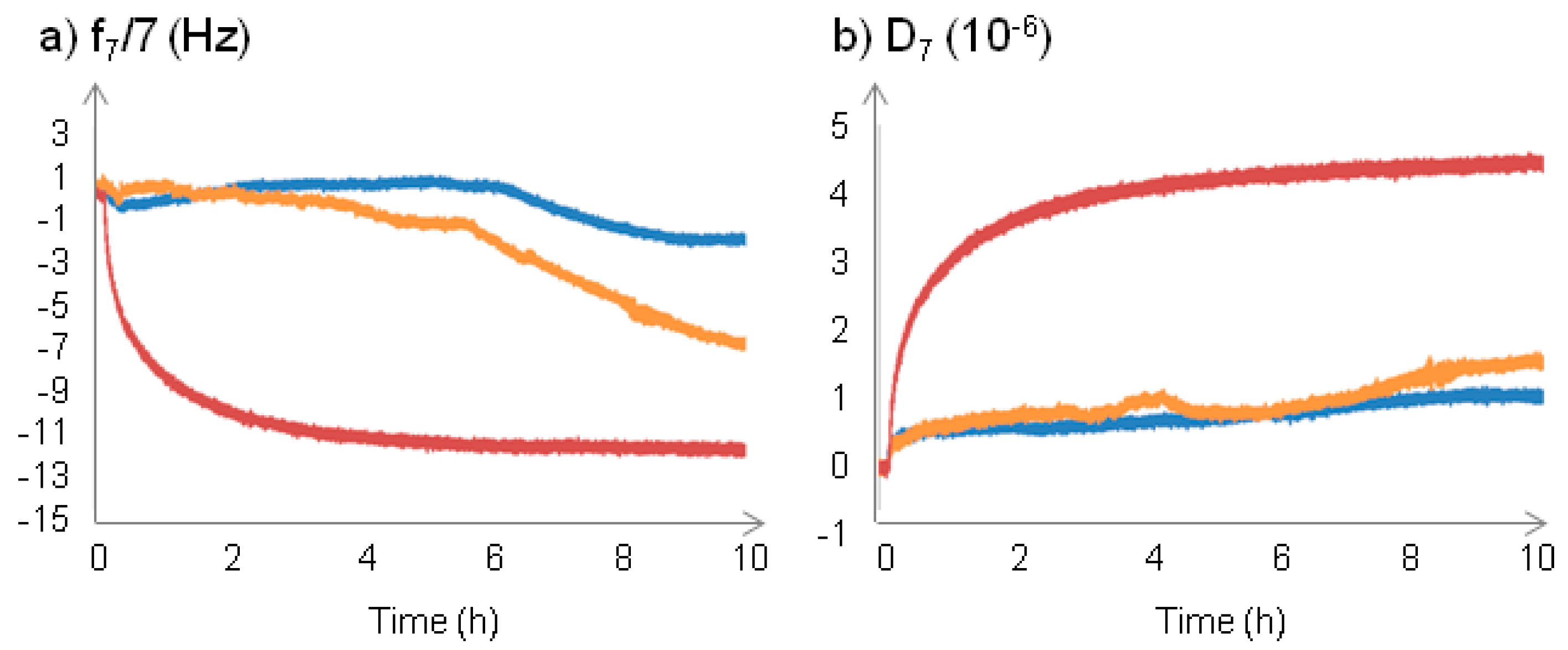
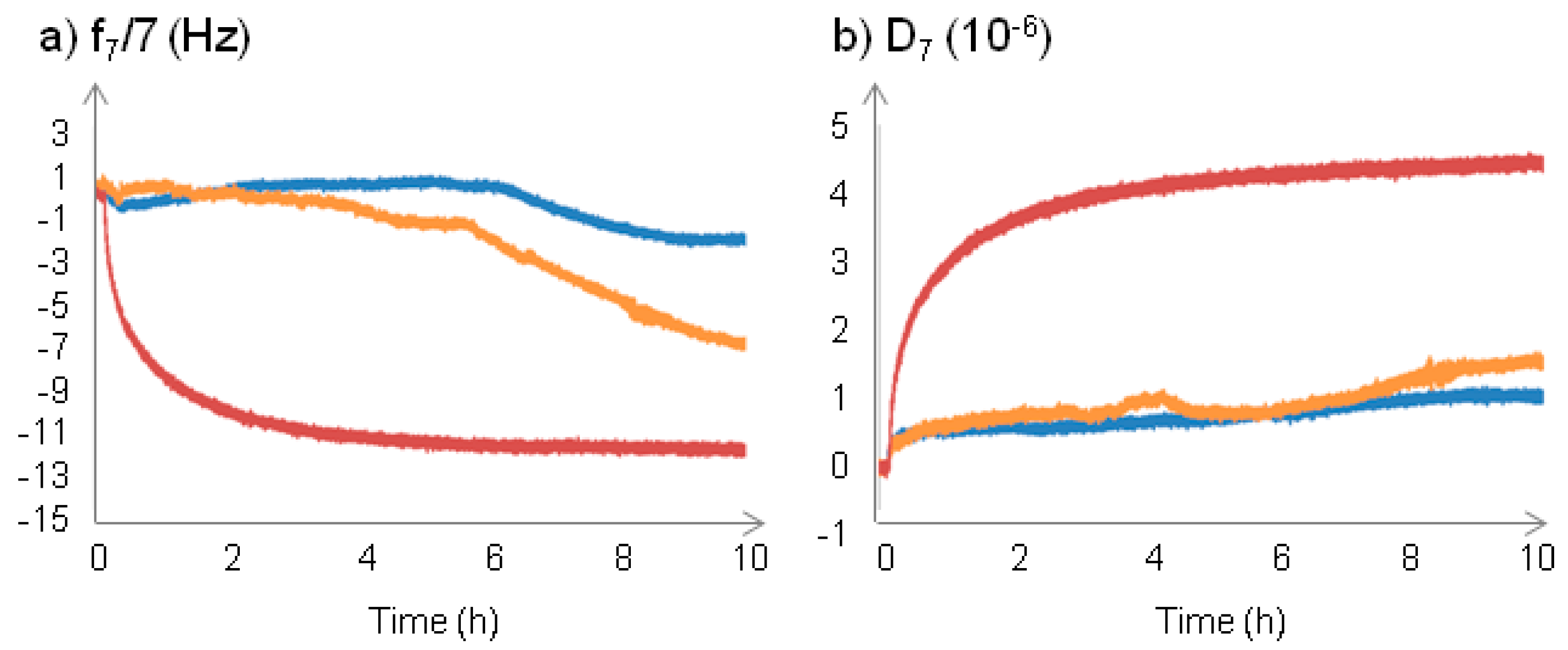
2.2.1. Changes at the Interface between NPs and a Supported Lipid Bilayer
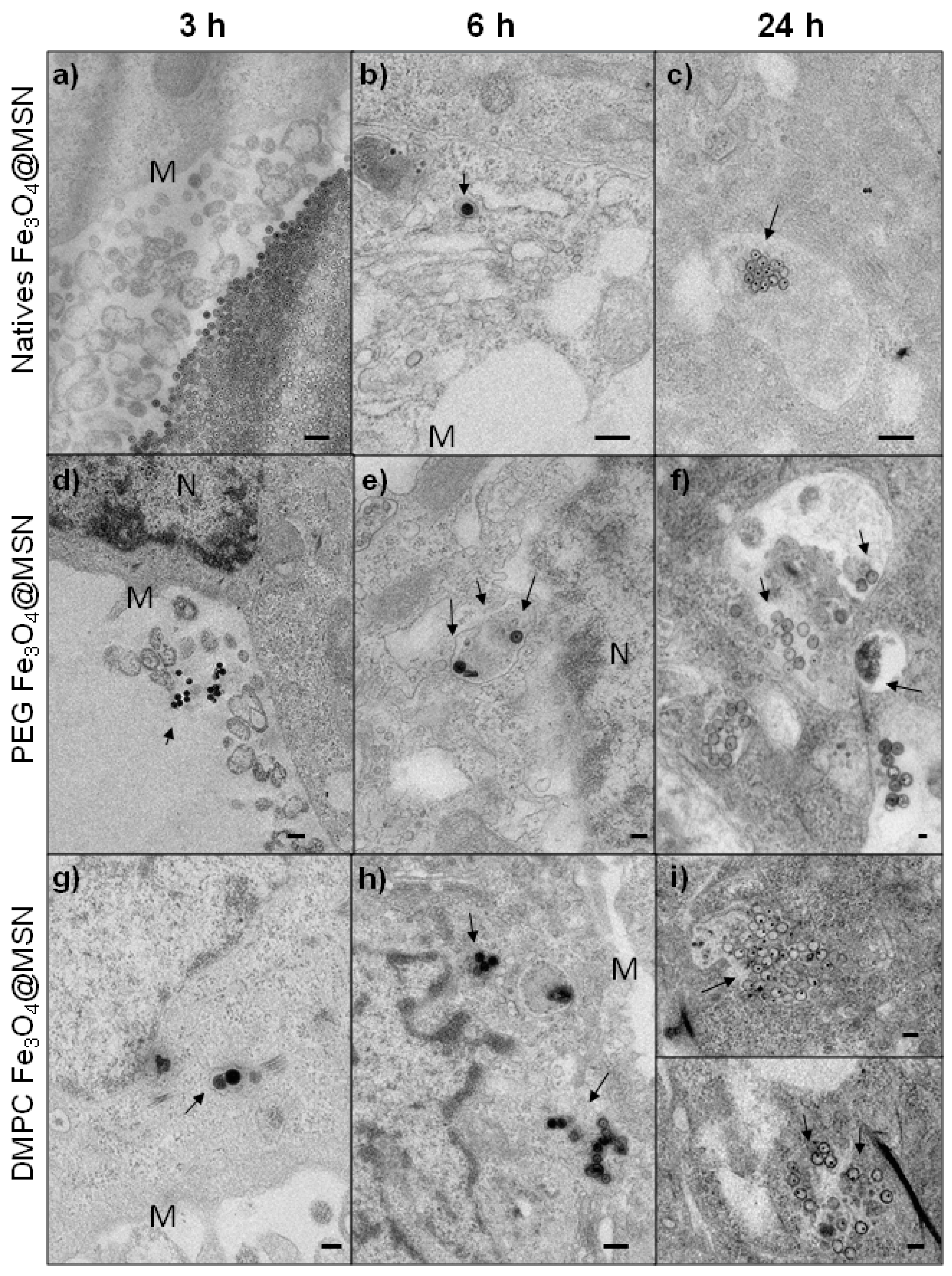
2.2.2. Time-Dependent Internalization of NPs in Hep-G2 cells
2.3. In Vivo Experiments
2.3.1. In Vivo Biocompatibility
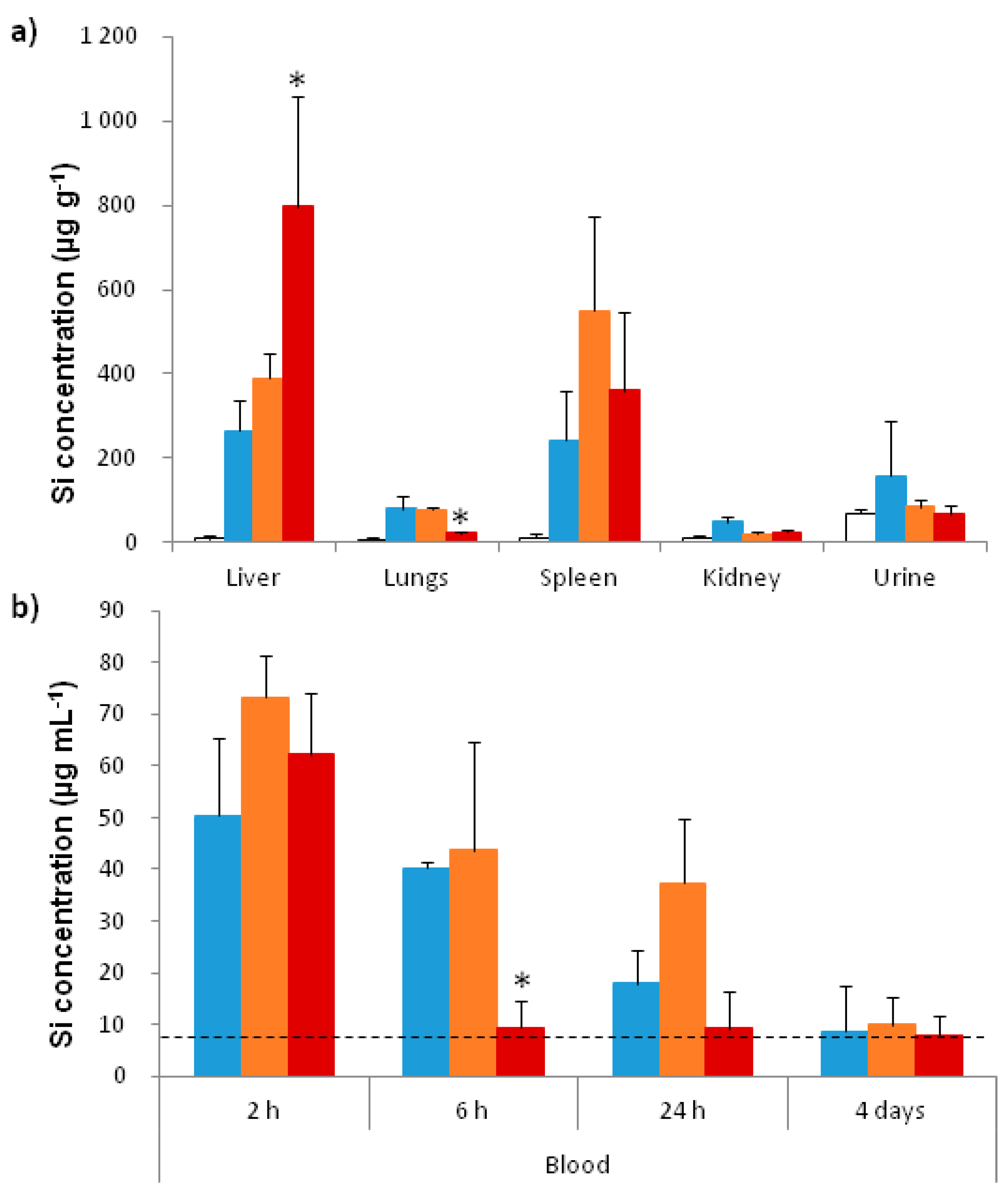
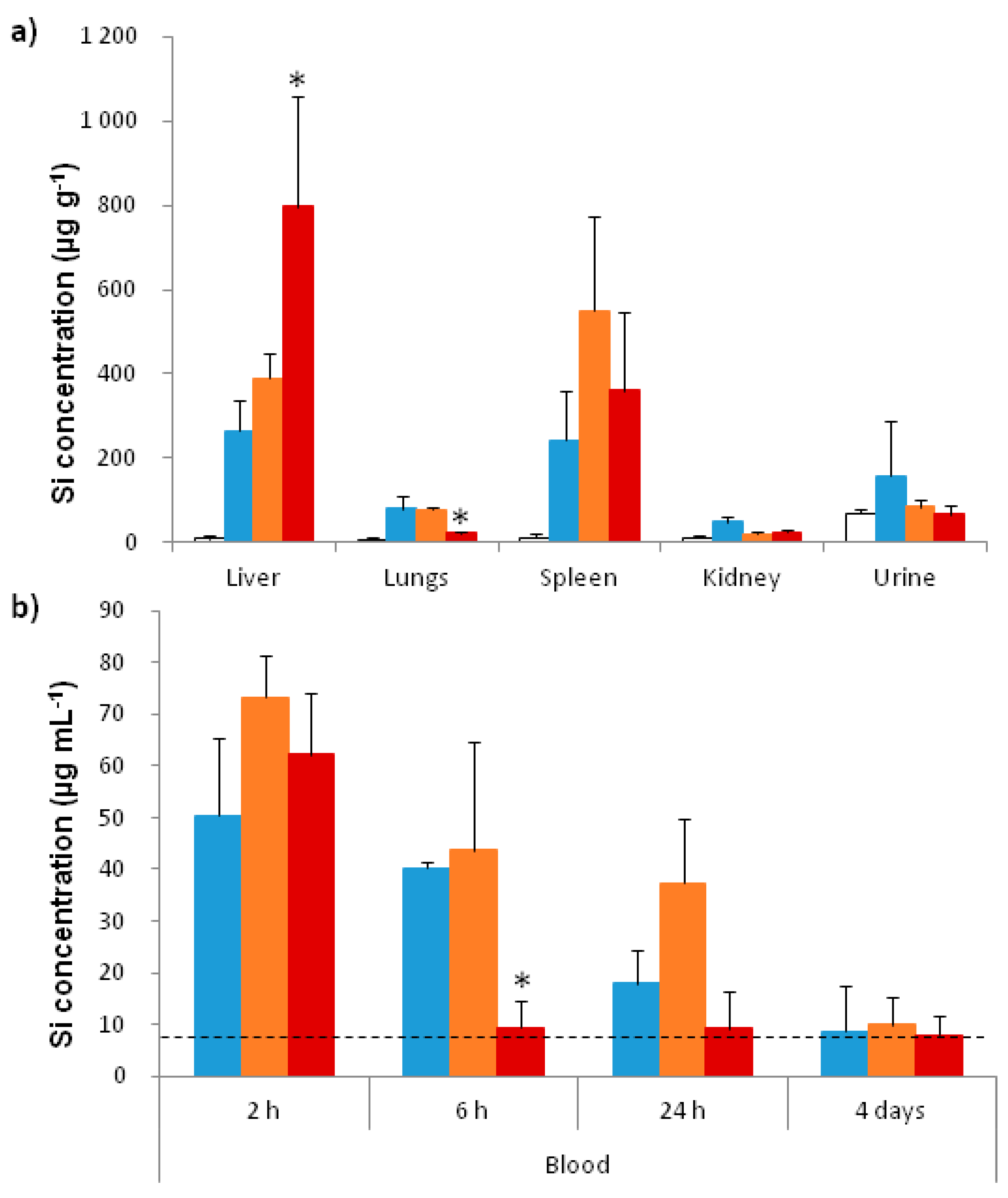
2.3.2. In Vivo Biodistribution
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Synthesis and Characterization of NPs
4.3. Interaction of NPs with Membrane Models
4.4. Cellular Assays
4.4.1. Cell Culture
4.4.2. Internalization Assay
4.5. Biodistribution in Mice
4.5.1. Animals
4.5.2. Preliminary Toxicological Assessment
4.5.3. In Vivo Distribution and Degradation of MSN
4.6. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tarn, D.; Ashley, C.E.; Xue, M.; Carnes, E.C.; Zink, J.I.; Brinker, C.J. Mesoporous silica nanoparticle nanocarriers: Biofunctionality and biocompatibility. Acc. Chem. Res. 2012, 46, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Lazarovits, J.; Chen, Y.Y.; Sykes, E.A.; Chan, W.C. Nanoparticle-blood interactions: The implications on solid tumour targeting. Chem. Commun. 2015, 51, 2756–2767. [Google Scholar] [CrossRef] [PubMed]
- Monopoli, M.P.; Walczyk, D.; Campbell, A.; Elia, G.; Lynch, I.; Bombelli, F.B.; Dawson, K.A. Physical-chemical aspects of protein corona: Relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Westmeier, D.; Stauber, R.H.; Docter, D. The concept of bio-corona in modulating the toxicity of engineered nanomaterials (ENM). Toxicol. Appl. Pharmacol. 2015, 299, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Tada, D.B.; Suraniti, E.; Rossi, L.M.; Leite, C.A.; Oliveira, C.S.; Tumolo, T.C.; Calemczuk, R.; Livache, T.; Baptista, M.S. Effect of lipid coating on the interaction between silica nanoparticles and membranes. J. Biomed. Nanotechnol. 2014, 10, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Caracciolo, G.; Capriotti, A.L.; Cavaliere, C.; La Barbera, G.; Anchordoquy, T.J.; Lagana, A. Surface chemistry and serum type both determine the nanoparticle-protein corona. J. Proteom. 2015, 119, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Hadjidemetriou, M.; Al-Ahmady, Z.; Mazza, M.; Collins, R.F.; Dawson, K.; Kostarelos, K. In vivo biomolecule corona around blood-circulating, clinically used and antibody-targeted lipid bilayer nanoscale vesicles. ACS Nano 2015, 9, 8142–8156. [Google Scholar] [CrossRef] [PubMed]
- Schottler, S.; Becker, G.; Winzen, S.; Steinbach, T.; Mohr, K.; Landfester, K.; Mailander, V.; Wurm, F.R. Protein adsorption is required for stealth effect of poly(ethylene glycol)—And poly(phosphoester)—Coated nanocarriers. Nat. Nanotechnol. 2016, 11, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Neun, B.W.; Man, S.; Ye, X.; Hansen, M.; Patri, A.K.; Crist, R.M.; McNeil, S.E. Protein corona composition does not accurately predict hematocompatibility of colloidal gold nanoparticles. Nanomedicine 2014, 10, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Hodges, C.; Ding, Y.; Biggs, S.; Brooker, A.; York, D. Adsorption kinetics of laponite and ludox silica nanoparticles onto a deposited poly(diallyldimethylammonium chloride) layer measured by a quartz crystal microbalance and optical reflectometry. Langmuir 2010, 26, 18105–18112. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Reiser, M.; Yu, X.; Gummuluru, S.; Wetzler, L.; Reinhard, B.M. Lipid-mediated targeting with membrane-wrapped nanoparticles in the presence of corona formation. ACS Nano 2016, 10, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Pombo Garcia, K.; Zarschler, K.; Barbaro, L.; Barreto, J.A.; O’Malley, W.; Spiccia, L.; Stephan, H.; Graham, B. Zwitterionic-coated “stealth” nanoparticles for biomedical applications: Recent advances in countering biomolecular corona formation and uptake by the mononuclear phagocyte system. Small 2014, 10, 2516–2529. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Ipe, B.I.; Misra, P.; Lee, J.H.; Bawendi, M.G.; Frangioni, J.V. Tissue- and organ-selective biodistribution of nir fluorescent quantum dots. Nano Lett. 2009, 9, 2354–2359. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Situ, A.; Kang, Y.; Villabroza, K.R.; Liao, Y.; Chang, C.H.; Donahue, T.; Nel, A.E.; Meng, H. Irinotecan delivery by lipid-coated mesoporous silica nanoparticles shows improved efficacy and safety over liposomes for pancreatic cancer. ACS Nano 2016, 10, 2702–2715. [Google Scholar] [CrossRef] [PubMed]
- Warther, D.; Jimenez, C.M.; Raehm, L.; Gerardin, C.; Durand, J.-O.; Morere, A.; El Cheikh, K.; Gallud, A.; Gary-Bobo, M.; Maynadier, M.; et al. Small sized mesoporous silica nanoparticles functionalized with mannose for retinoblastoma cell imaging. RSC Adv. 2014, 4, 37171–37179. [Google Scholar] [CrossRef]
- Mashaghi, S.; Jadidi, T.; Koenderink, G.; Mashaghi, A. Lipid nanotechnology. Int. J. Mol. Sci. 2013, 14, 4242–4282. [Google Scholar] [CrossRef] [PubMed]
- Bringas, E.; Koysuren, O.; Quach, D.V.; Mahmoudi, M.; Aznar, E.; Roehling, J.D.; Marcos, M.D.; Martinez-Manez, R.; Stroeve, P. Triggered release in lipid bilayer-capped mesoporous silica nanoparticles containing spion using an alternating magnetic field. Chem. Commun. 2012, 48, 5647–5649. [Google Scholar] [CrossRef] [PubMed]
- Van Schooneveld, M.M.; Vucic, E.; Koole, R.; Zhou, Y.; Stocks, J.; Cormode, D.P.; Tang, C.Y.; Gordon, R.E.; Nicolay, K.; Meijerink, A.; et al. Improved biocompatibility and pharmacokinetics of silica nanoparticles by means of a lipid coating: A multimodality investigation. Nano Lett. 2008, 8, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qiao, S.Z.; Hu, Q.H.; Lu, G.Q. Magnetic nanocomposites with mesoporous structures: Synthesis and applications. Small 2011, 7, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Mintri, S.; Menon, A.V.; Lee, H.Y.; Choi, H.S.; Kim, J. Pharmacokinetics, pharmacodynamics and toxicology of theranostic nanoparticles. Nanoscale 2015, 7, 18848–18862. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Detrembleur, C.; Pauw-Gillet, D.; Mornet, S.; Vander Elst, L.; Laurent, S.; Jérôme, C.; Duguet, E. Heat-triggered drug release systems based on mesoporous silica nanoparticles filled with a maghemite core and phase-change molecules as gatekeepers. J. Mater. Chem. B 2014, 2, 59–70. [Google Scholar] [CrossRef]
- Nyalosaso, J.L.; Rascol, E.; Pisania, C.; Dorandeu, C.; Dumail, X.; Maynadier, M.; Gary-Bobo, M.; Lai Kee Him, J.; Bron, P.; Garcia, M.; et al. Synthesis, decoration, and cellular effects of magnetic mesoporous silica nanoparticles. RSC Adv. 2016, 6, 57275–57283. [Google Scholar] [CrossRef]
- Moore, T.L.; Rodriguez-Lorenzo, L.; Hirsch, V.; Balog, S.; Urban, D.; Jud, C.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle colloidal stability in cell culture media and impact on cellular interactions. Chem. Soc. Rev. 2015, 44, 6287–6305. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Luyts, K.; Napierska, D.; Nemery, B.; Hoet, P.H. How physico-chemical characteristics of nanoparticles cause their toxicity: Complex and unresolved interrelations. Environ. Sci. Process. Impacts 2013, 15, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.L.; Bothun, G.D. Nanoparticles meet cell membranes: Probing nonspecific interactions using model membranes. Environ. Sci. Technol. 2013, 48, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Rascol, E.; Devoisselle, J.M.; Chopineau, J. The relevance of membrane models to understand nanoparticles-cell membrane interactions. Nanoscale 2016, 8, 4780–4798. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.A.; Kasemo, B. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys. J. 1998, 75, 1397–1402. [Google Scholar] [CrossRef]
- Richter, R.P.; Brisson, A.R. Following the formation of supported lipid bilayers on mica: A study combining AFM, QCM-D, and ellipsometry. Biophys. J. 2005, 88, 3422–3433. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, D.; Wu, J.; Kim, C.; Fortner, J.D. Aqueous aggregation and surface deposition processes of engineered superparamagnetic iron oxide nanoparticles for environmental applications. Environ. Sci. Technol. 2014, 48, 11892–11900. [Google Scholar] [CrossRef] [PubMed]
- Quevedo, I.R.; Olsson, A.L.; Tufenkji, N. Deposition kinetics of quantum dots and polystyrene latex nanoparticles onto alumina: Role of water chemistry and particle coating. Environ. Sci. Technol. 2013, 47, 2212–2220. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.M.; Kamaloo, E.; Waterman, K.L.; Wang, K.F.; Nagarajan, R.; Camesano, T.A. Size dependence of gold nanoparticle interactions with a supported lipid bilayer: A QCM-D study. Biophys. Chem. 2015, 203, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, K.L. Interactions of graphene oxide with model cell membranes: Probing nanoparticle attachment and lipid bilayer disruption. Langmuir 2015, 31, 12076–12086. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Chen, K.L. Interaction of multiwalled carbon nanotubes with supported lipid bilayers and vesicles as model biological membranes. Environ. Sci. Technol. 2013, 47, 5711–5719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lim, M.; Liu, X.; Wang, Z.; Chen, K.L. Influence of solution chemistry and soft protein coronas on the interactions of silver nanoparticles with model biological membranes. Environ. Sci. Technol. 2016, 50, 2301–2309. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Liu, T.; Li, L.; Liu, H.; Chen, D.; Tang, F. The absorption, distribution, excretion and toxicity of mesoporous silica nanoparticles in mice following different exposure routes. Biomaterials 2013, 34, 2565–2575. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Gao, Q.; Schutz, I.; Noufele, C.N.; Ruan, W.; Posselt, U.; Korotianskiy, E.; Nordmeyer, D.; Rancan, F.; Hadam, S.; et al. Surface functionalization of silica nanoparticles supports colloidal stability in physiological media and facilitates internalization in cells. Langmuir 2012, 28, 7598–7613. [Google Scholar] [CrossRef] [PubMed]
- Heuberger, M.; Drobek, T.; Voros, J. About the role of water in surface-grafted poly(ethylene glycol) layers. Langmuir 2004, 20, 9445–9448. [Google Scholar] [CrossRef] [PubMed]
- Pasche, S.; Voros, J.; Griesser, H.J.; Spencer, N.D.; Textor, M. Effects of ionic strength and surface charge on protein adsorption at pegylated surfaces. J. Phys. Chem. B 2005, 109, 17545–17552. [Google Scholar] [CrossRef] [PubMed]
- Fatisson, J.; Quevedo, I.R.; Wilkinson, K.J.; Tufenkji, N. Physicochemical characterization of engineered nanoparticles under physiological conditions: Effect of culture media components and particle surface coating. Colloids Surf. B Biointerfaces 2012, 91, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Pelaz, B.; del Pino, P.; Maffre, P.; Hartmann, R.; Gallego, M.; Rivera-Fernandez, S.; de la Fuente, J.M.; Nienhaus, G.U.; Parak, W.J. Surface functionalization of nanoparticles with polyethylene glycol: Effects on protein adsorption and cellular uptake. ACS Nano 2015, 9, 6996–7008. [Google Scholar] [CrossRef] [PubMed]
- Bagwe, R.P.; Hilliard, L.R.; Tan, W. Surface modification of silica nanoparticles to reduce aggregation and nonspecific binding. Langmuir 2006, 22, 4357–4362. [Google Scholar] [CrossRef] [PubMed]
- Card, J.W.; Zeldin, D.C.; Bonner, J.C.; Nestmann, E.R. Pulmonary applications and toxicity of engineered nanoparticles. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 295, L400–L411. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rascol, E.; Daurat, M.; Da Silva, A.; Maynadier, M.; Dorandeu, C.; Charnay, C.; Garcia, M.; Lai-Kee-Him, J.; Bron, P.; Auffan, M.; et al. Biological Fate of Fe3O4 Core-Shell Mesoporous Silica Nanoparticles Depending on Particle Surface Chemistry. Nanomaterials 2017, 7, 162. https://doi.org/10.3390/nano7070162
Rascol E, Daurat M, Da Silva A, Maynadier M, Dorandeu C, Charnay C, Garcia M, Lai-Kee-Him J, Bron P, Auffan M, et al. Biological Fate of Fe3O4 Core-Shell Mesoporous Silica Nanoparticles Depending on Particle Surface Chemistry. Nanomaterials. 2017; 7(7):162. https://doi.org/10.3390/nano7070162
Chicago/Turabian StyleRascol, Estelle, Morgane Daurat, Afitz Da Silva, Marie Maynadier, Christophe Dorandeu, Clarence Charnay, Marcel Garcia, Joséphine Lai-Kee-Him, Patrick Bron, Mélanie Auffan, and et al. 2017. "Biological Fate of Fe3O4 Core-Shell Mesoporous Silica Nanoparticles Depending on Particle Surface Chemistry" Nanomaterials 7, no. 7: 162. https://doi.org/10.3390/nano7070162