First-Principles Study of Structural, Electronic and Magnetic Properties of Metal-Centered Tetrahexahedral V15+ Cluster


The Key Laboratory for Surface Engineering and Remanufacturing in Shaanxi Province, School of Chemical Engineering, Xi’an University, Xi’an 710065, China
*
Author to whom correspondence should be addressed.
Nanomaterials 2017, 7(7), 164; https://doi.org/10.3390/nano7070164
Submission received: 30 May 2017
/
Revised: 22 June 2017
/
Accepted: 27 June 2017
/
Published: 30 June 2017
(This article belongs to the Special Issue Noble Metal Nanoparticles in Catalysis)
Abstract
:The V-centered bicapped hexagonal antiprism structure (A), as the most stable geometry of the cationic V15+ cluster, is determined by using infrared multiple photo dissociation (IR-MPD) in combination with density functional theory computations. It is found that the A structure can be stabilized by 18 delocalized 3c-2e σ-bonds on outer V3 triangles of the bicapped hexagonal antiprism surface and 12 delocalized 4c-2e σ-bonds on inner trigonal pyramidal V4 moiety, and the features are related to the strong p-d hybridization of the cluster. The total magnetic moments on the cluster are predicted to be 2.0 µB, which come mainly from the central vanadium atom.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Transition metal clusters have attracted a lot of attention because of their especially important roles in nanostructured material science [1,2,3,4,5,6,7,8,9], which are quite different from those of individual atoms or bulk phase [10,11], and present a strong size dependence on their interesting physicochemical properties, e.g., optical [12,13,14], magnetic [15,16] and catalytic [17,18,19] properties, serving in various industrial application [20]. In particular, vanadium clusters are of special interest, due to their magnetic properties [21,22,23,24] and novel catalysis [25,26], which are closely related to their electronic structures. It is well-known that one of the major challenges for understanding the fascinating electronic properties is to determine the cluster structure, which can be solved well by using a combination of state-of-the-art experimental and theoretical studies. For example, Fielicke et al. [27,28] explored the geometric structures of vanadium clusters using infrared multiple photon dissociation (IR-MPD) spectroscopy and density functional theory (DFT) computations, and identified the ground-state structures of some vanadium clusters. Also, the stable structures of large vanadium clusters have been theoretically determined by classical potentials, such as tight-binding potential [23,29]. In 2001, Taneda et al. [30] performed molecular-dynamics (MD) simulations on the disordered structures of the Vn clusters (n = 2–17, 55, 147 and 309), and found that the globally stable structures of small clusters are in agreement with first-principle calculations. Meanwhile, vanadium clusters have been experimentally measured using several experimental techniques, e.g., using the IR-MPD for Vn+ (n = 6–23) [31], photoelectron spectroscopy (PES) for Vn− (n = 3–65) [16,32], and collision-induced dissociation (CID) of Vn+ (n = 2–20) with Xe by guided ion beam mass spectrometry [33].
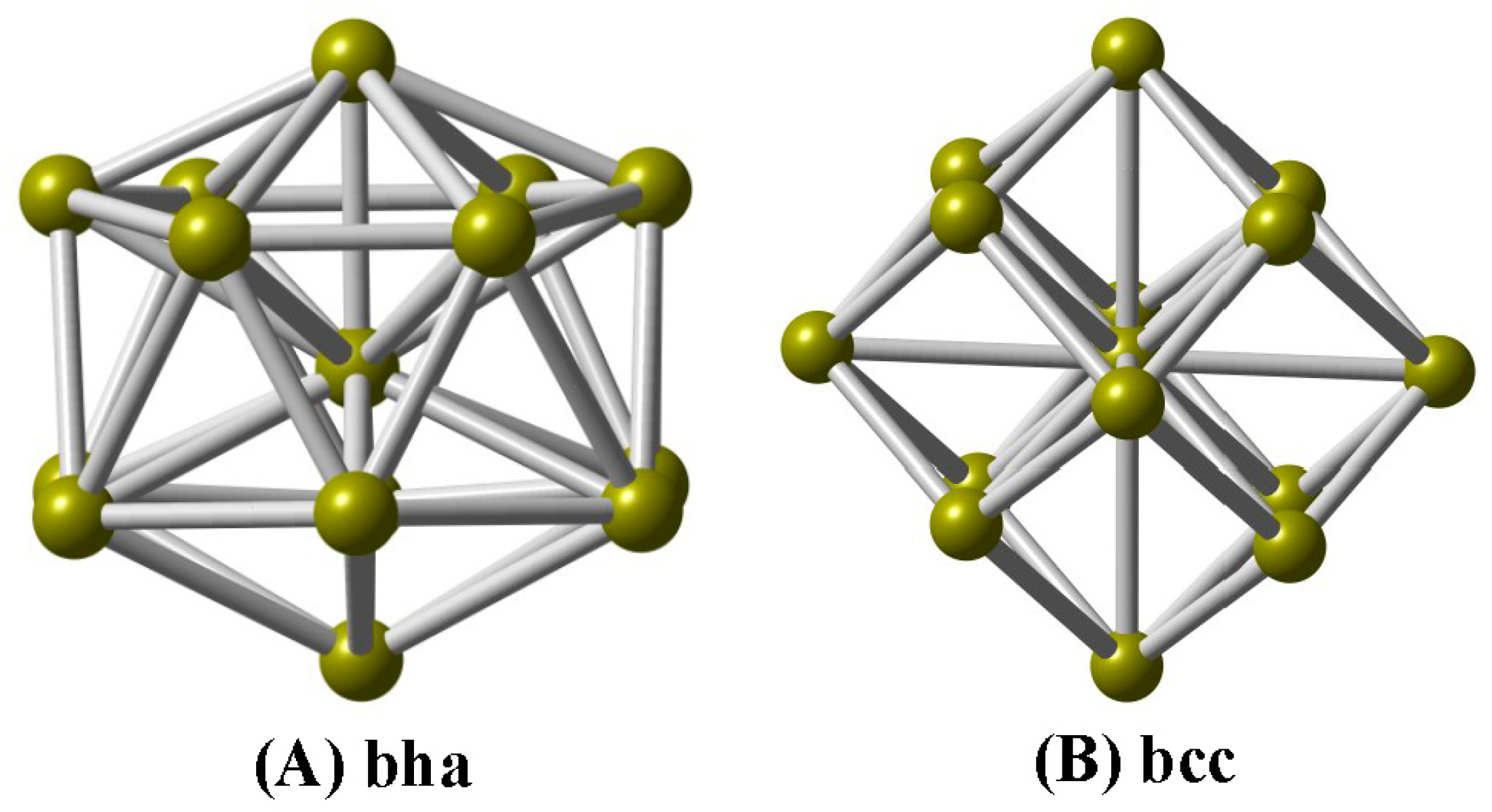
In this paper, we explore the geometric structures of V-centered bicapped hexagonal antiprisms (A bha, Figure 1) and body-centered cubes (B bcc) for the cationic V15+ cluster using a combination of DFT computations and IR-MPD spectroscopy, and determine the ground-state structure to be a V-centered tetrahexahedral geometry (A), which reproduces experimentally observed spectra well. Based on the assigned structure, the electronic structures, chemical bondings, and magnetic properties are investigated.
2. Materials and Methods
Cluster structure computations were performed on the Gaussian 09 suite of programs [34] using the following four density functionals: (a) PBE [35,36]; (b) BLYP [37,38]; (c) BPW91 [37,39]; and (d) BP86 [37,40,41]. The Karlsruhe split-valence (def-SVP) basis set, augmented with polarization functions and the all-electron 6-311+G(d) basis set, was applied for the two vanadium clusters. In the computation, the singlet and triplet spin electronic states (spin multiplicities 2S + 1 = 1, 3) were taken into account for the energy evaluation of the low-lying isomers. The two low-lying structures were optimized without any symmetry constraints. Harmonic vibrational frequencies were employed to confirm that the two structures correspond to real local minima, and zero-point vibrational corrections were included in the relative energies.
To aid the comparison with the experimental IR-MPD spectrum, the calculated infrared stick spectra were convoluted with a Gaussian function with a full width at half-maximum (FWHM) of 6 cm−1. In order to fit well with the experimental data, a frequency scaling factor of 0.87 was applied, similar to previous studies [28,31]. The density-of-states (DOS) spectra were convoluted utilizing the GaussSum 2.2 program [42] with a full-width at half maximum (FWHM) of 0.3 eV. The molecular orbitals were plotted with the isodensity surfaces (0.02 e1/2/(Bohr)3/2), and the molecular graphs were visualized using the VMD program [43]. Chemical bonding analyses were performed using the adaptive natural density partitioning (AdNDP) method proposed by Zubarev and Boldyrev [44].
3. Results and Discussion
3.1. Structural Determination
Structurally, we can see that the A isomer of the V15+ cluster is a V-centered tetrahexahedral structure (Figure 1A), consisting of a bicapped hexagonal antiprism (bha), in which the hexagonal faces are capped by a V atom above and below. Interestingly, the A isomer can be regarded as a wheel structure, and the V–V–V central axle can be surrounded by the V12 hexagonal antiprism. The A structure is found to be consistent with Fe@Si14 [45] and Fe@Ge14 [46] clusters. At the PBE/def-SVP level of theory, the average V–V bond lengths for the A isomer are predicted to be 2.506 Å, while a large average coordination number (6.67) is obtained on the vanadium atoms, which are close to previous calculations for V15 [23], i.e., 2.503 Å and 6.67 for V–V bond lengths and coordination number, respectively. The B isomer is a stuffed fullerene-like structure (Figure 1B), consisting of a body-centered cube (bcc, 8 + 1 atoms) framework with six tetrahedral pyramids on each face. One can find that the average V–V bond lengths for the B isomer are deemed to be 2.511 Å, while the average coordination number is also 6.67; there are no other experimental or theoretical data for comparison. The same structure was predicted for the isovalent Ta15+ cluster [6].
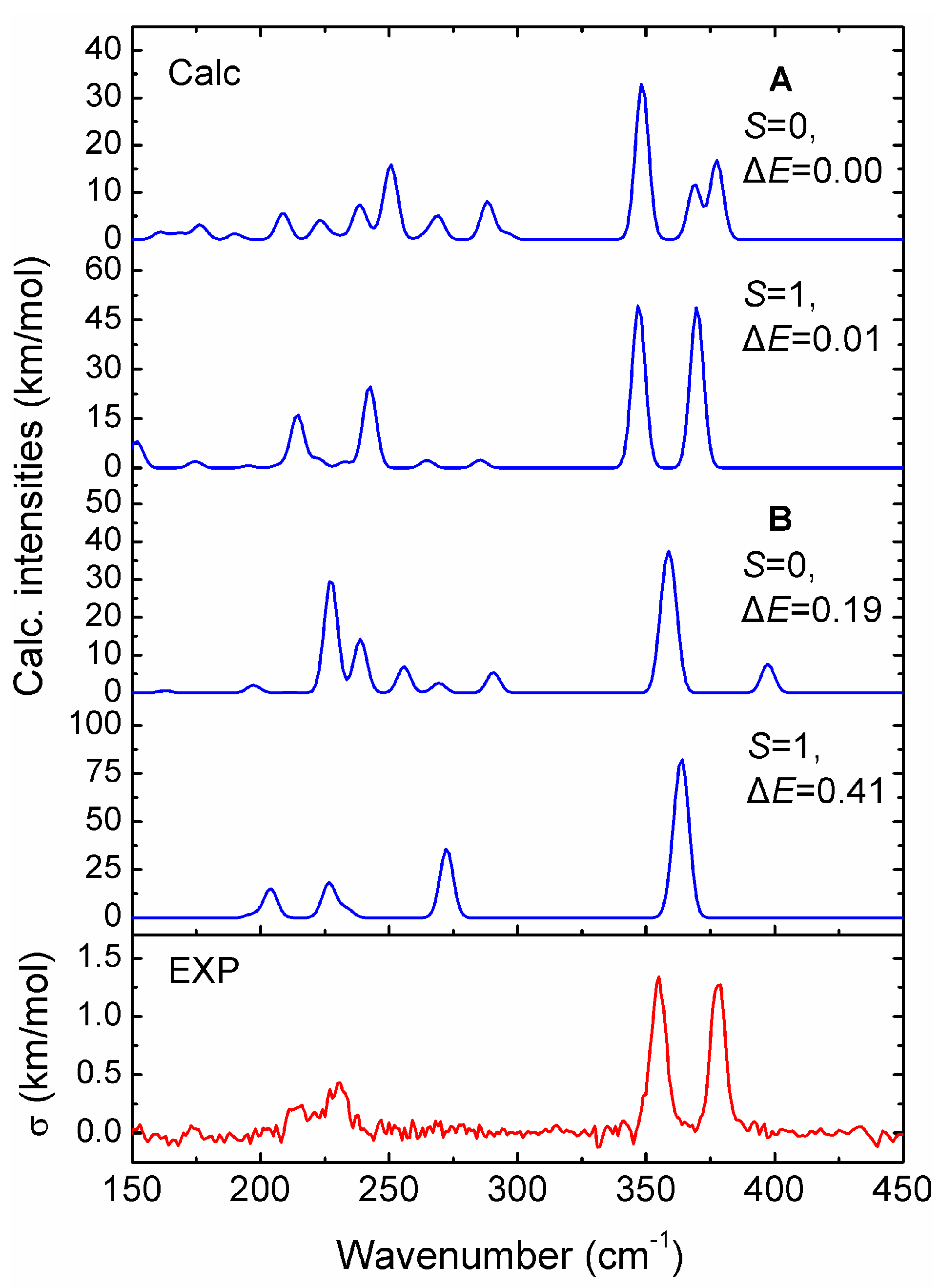
Figure 2 compares the relative energies of two low-lying isomers (A and B) for the V15+ cluster in singlet (S = 0) and triplet (S = 1) spin states, which are calculated from the five different theoretical methods. The relative energy can be evaluated by the energy differences of the two low-lying isomers in different spin states relative to the lowest-energy states (A, S = 0). According to the calculated results, it is found that the PBE, BPW91 and BP86 functionals provide similar energy orderings, whereas the BLYP functional predicts a B isomer with S = 0 to be more stable than A with S = 1 by 0.02 eV. At the same time, it clearly shows that the A geometry with two spin states is more stable than the B geometry, which is different from the results reported in [28], which predicted that the B structure is more stable in energy than the A geometry. Moreover, it is seen from Figure 2 that the A isomer with spin singlet (S = 0) is energetically preferred, and its spin triplet (S = 1) is less stable than the singlet state, having a higher energy by only 0.01–0.06 eV. Conversely, the B structure has larger relative energies by up to 0.46 eV among these theoretical methods. In addition, we have also performed global optimizations for the cluster using the semi-empirical Gupta potential with the basin-hopping method, and found that the A structure is more stable than the B structure by 0.39 eV. Thus, one can see from Figure 2 that the two spin states of the A isomer display a very small energy difference, but we cannot predict with certainty which of two spin states will be the true ground-state.
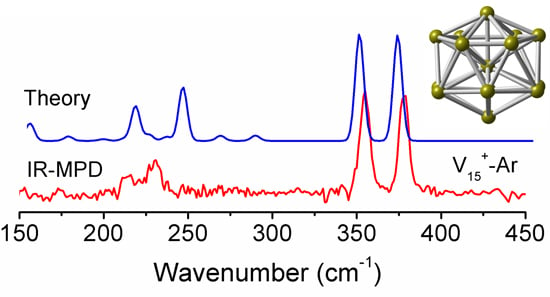
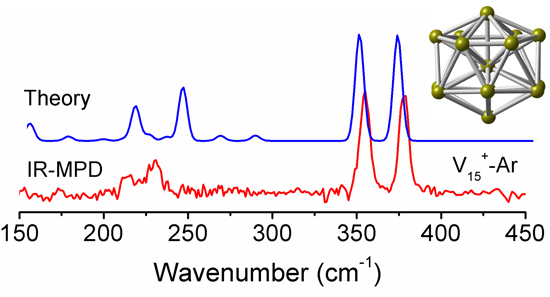
In order to test the reliability of the prediction for the V15+ cluster based on the stable structures, we compare the calculated infrared spectra of the two low-lying isomers (A and B) in different spin states obtained at the PBE/def-SVP level of theory, with the experimental IR-MPD spectrum from previous work [31], to correctly obtain structural assignments for the cluster. As shown in Figure 3, the experimental IR-MPD spectrum of V15+-Ar shows four characteristic absorptions with two intense bands at around 355 and 378 cm−1, and two weak bands at around 214 and 232 cm−1. It is apparent that the experimental spectroscopic bands are in excellent agreement with the theoretical prediction for the low-lying A isomer with spin triplet (S = 1). By comparison, the former two main experimental bands correspond to calculated bands centered on 347 and 370 cm−1, respectively, in which the first intense peak is primarily caused by the V–V stretching vibrations along the central V–V–V axis, while another intense peak is largely attributed to the horizontal wagging vibrations on the central V atom of the V–V–V axis. Other weak experimental bands at 214 and 232 cm−1 are reproduced well by those predicted at 214 and 242 cm−1, respectively. Thus, the A structure with spin triplet (S = 1) should be present in the molecular beam, and should contribute significantly to the experimental IR spectrum, which updates the previous report for the cluster [28].
3.2. Electronic Structures
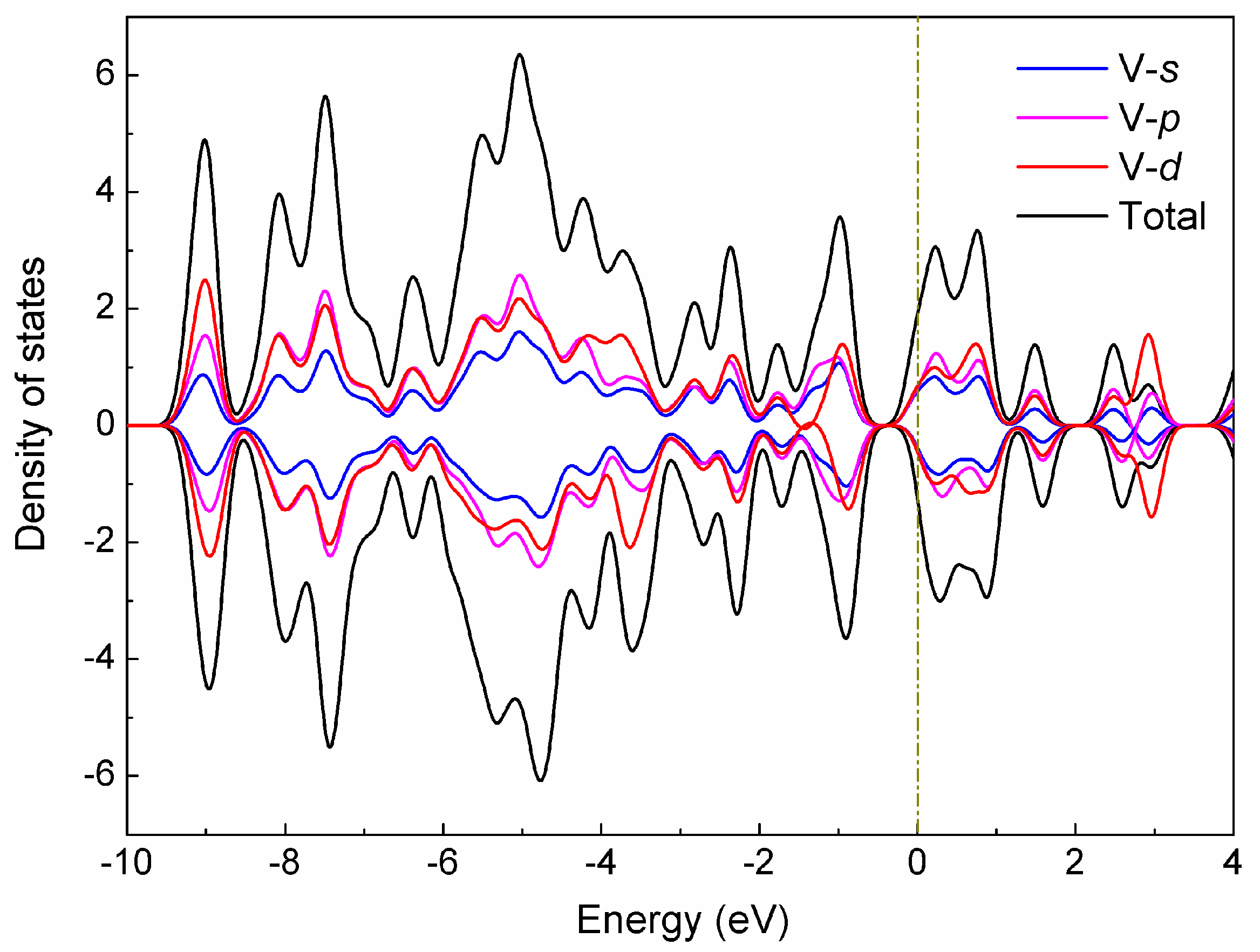
To systematically investigate the electronic structures of the V15+ cluster, we calculated the total (TDOS) and partial (PDOS) density of states for the low-lying A structure in terms of Mulliken population analysis [47], which includes the orbital contributions (V-s, V-p, and V-d) of the vanadium atom in the cluster to the TDOS, as depicted in Figure 4. As evidenced by the diagram, a strong hybridization occurred between the s, p, and d states, especially for p-d hybridization, and these hybridizations are mainly responsible for the structural stabilization of the cluster. It can also be observed from Figure 4 that the electronic features largely originate from the d states near the Fermi level, while the s and p states produce nearly non-negligible contributions. Meanwhile, the up-spin and down-spin bands of these different states are not substantially split for the A structure with spin triplet (S = 1), which may result in a smaller magnetic moment.
To further understand the electronic properties of the low-lying A structure, we also explore the up-spin orbital contributions in the DOS spectra (see Figure S1), involving the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). We can clearly see that there are three strong DOS peaks, located at −9.02, −7.49, and −5.03 eV, respectively. More impressively, the HOMO-32 orbital at −9.02 eV has a p-type MO pattern, which is contributed to significantly by the s-p hybridization between the interior V atom and surrounding V atoms, with the 26.44% V-4s and 32.16% V-4py states. The strongest occupied orbital peak (HOMO-12), located at −7.49 eV, is mainly attributable to the shell contributions (24.33% V-3dxy, 21.05% V-3dz2 states) of the completely delocalized σ-type MO on the hexagonal antiprism V atoms, with small components from 14.79% V-4pz state of the vanadium atoms. Obviously, the HOMO and LUMO orbitals have weak DOS peaks, where the HOMO orbital originates largely from the shell contributions of 20.24% V-3dxy and 31.93% V-3dx2y2 states, while the LUMO significantly involves the 24.77% V-4s and 35.34% V-3dz2 states, which results in the formation of strong localized atomic orbitals on each vanadium atom, especially for two capped V atoms (Figure S1). One can see that the HOMO-LUMO gap for the A isomer is predicted to be 0.38 eV, and the energy gap is mostly associated with the electron distributions of the HOMO and LUMO orbitals. It is expected that these theoretical results will be helpful to understand the electronic properties of the V15+ cluster, and gain insight into the origins of its stable geometry.
3.3. Chemical Bonding Analyses
In order to gain insight into the chemical bonding of the V15+ cluster, the n-center two-electron (nc-2e, n going from one (lone-pair) to the maximum number of clusters) bonds were, for the first time, explored using the adaptive natural density partitioning (AdNDP) method proposed by Zubarev and Boldyrev [44], which has been successfully used to reveal bonding characteristics not only for organic aromatic molecules [48,49], but also for boron [50,51] and transition-metal doping clusters [52,53,54].
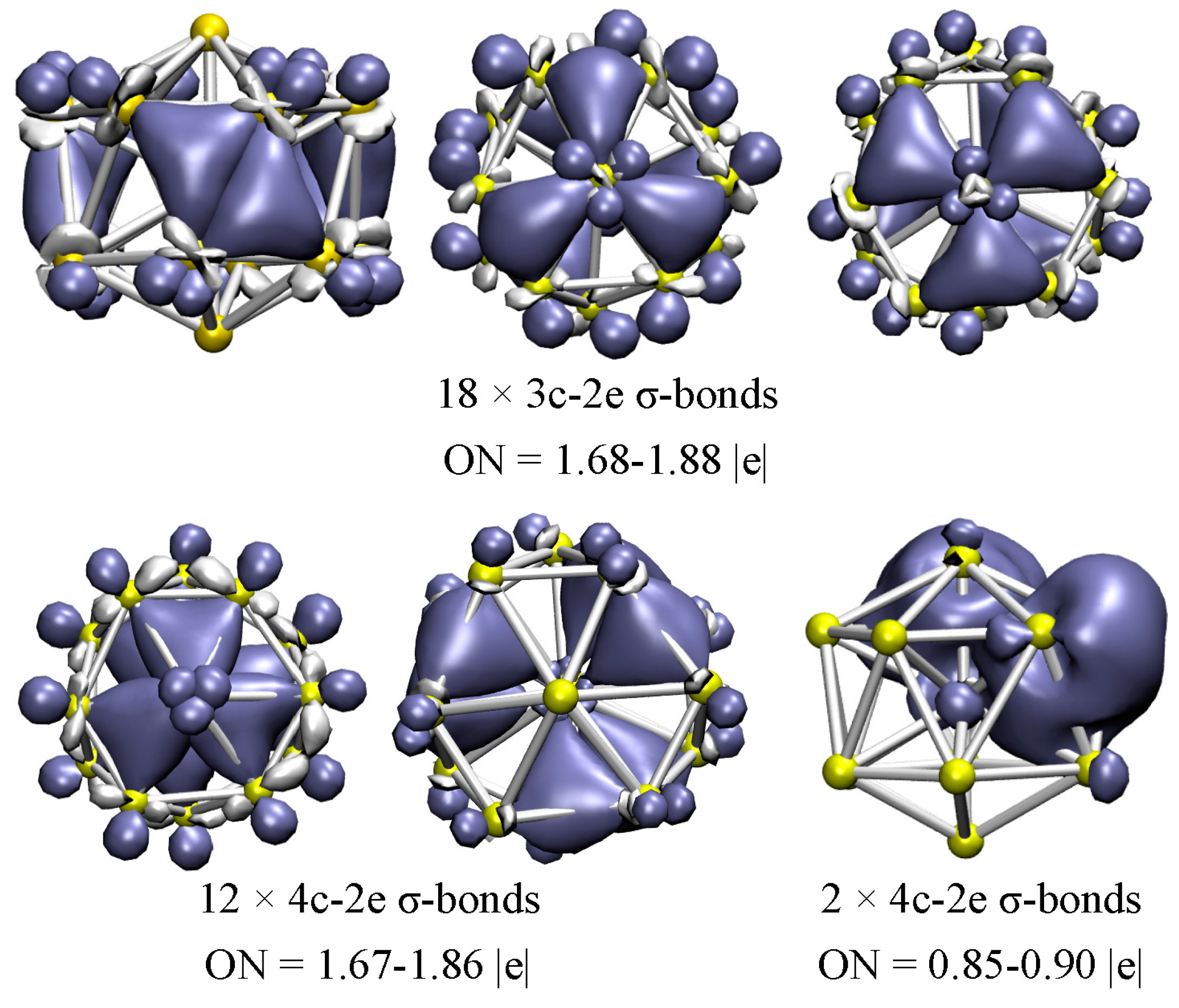
As mentioned above, the low-lying A structure is a bicapped hexagonal antiprism (bha) with an endohedral V atom. According to the AdNDP results (Figure 5), a total of 18 delocalized 3c-2e σ-bonds can be readily identified: six 3c-2e σ-bonds (ON = 1.84–1.88 |e|) on the outer V3 triangles of the hexagonal antiprism surface, and twelve 3c-2e σ-bonds (ON = 1.68–1.81 |e|) above and under the V3 triangles of the bicapped hexagonal faces. On the other hand, 12 delocalized 4c-2e σ-bonds are identified, with ON = 1.67–1.86 |e|, which are located on the inner trigonal pyramidal V4 moiety, involving the 12-atom outer ring and endohedral V atom, while none of the valence electrons in this structure are delocalized π-bonds or localized 2c-2e bonds. Accordingly, it is interesting that the low-lying A structure can be stabilized by the 3c-2e σ-bonds on the outer shell, and the 4c-2e σ-bonds on the inner shell. The two extra electrons in the open-shell A structure are clearly delocalized over the two V4 trigonal pyramid, and the ONs of the two σ-bonds are 0.85–0.90 |e|. In addition, the remaining twelve valence electrons can be characterized as six totally delocalized molecular orbitals (MOs) with ON = 2.00 |e|, distributed over the cage surface of the structure (see Figure S2).
3.4. Magnetic Moments and Charge Transfers
Local magnetic moments and charge transfers were performed to understand the magnetic properties of the cationic V15+ cluster, see Table S1. One can see that the average magnetic moments for the low-lying A and B structures in spin triplet (S = 1) are predicted to be 0.13 µB per atom, and are primarily produced by the central vanadium atom, e.g., 0.27 and 0.22 µB for A and B, respectively. Meanwhile, the average local moments for the first shell atoms are calculated to be 0.18 and 0.16 µB for A and B, respectively, obtained by using the PBE/def-SVP level of theory. The charge transfer can be used to reflect the interaction between the central vanadium atom and the cluster surface. Obviously, the charge largely transfers the 0.95 electron from the cluster surface to the central vanadium atom, which governs the structural stability of the endohedral cluster. It is worth mentioning that the strong interactions between the central vanadium atom and cluster surface are consistent with the chemical bonding analysis as discussed above, i.e., 12 delocalizedly inner 4c-2e σ-bonds. The electronic dipole moments are closely related to the structural features and electronic properties of the clusters. At the PBE/def-SVP level of theory, the theoretical dipole moments of the low-lying A structure in spin singlet and triplet are predicted to be 4.26 and 4.62 D, respectively, with the cluster’s symmetries being largely responsible for the moderate dipole moments.
4. Conclusions
In summary, the geometric structures of V-centered bicapped hexagonal antiprisms (A) and body-centered cubes (B) for cationic V15+ cluster are identified by using infrared multiple photo dissociation in combination with density functional theory computations. It is found that the A isomer with spin triplet reproduces well the experimentally observed spectrum, and the characteristic peaks are properly assigned according to theory. Based on the AdNDP analysis, we can see that the A isomer can be stabilized by 18 delocalized 3c-2e σ-bonds on the outer V3 triangles of the bicapped hexagonal antiprism surface, and 12 delocalized 4c-2e σ-bonds on the inner trigonal pyramidal V4 moiety. Meanwhile, there is strong hybridization between the s, p, and d states, especially for p-d hybridization, which could further explain the electronic structures of the cluster, with the local magnetic moments of the cluster originating mainly from the central vanadium atom. Thereby, our results will inevitably stimulate future theoretical and experimental studies for the exploration of novel vanadium-based catalytic materials.
Supplementary Materials
The following are available online at http://www.mdpi.com/2079-4991/7/7/164/s1. Figure S1: Up-spin DOS and MOs of the low-lying A structure for V15+, Figure S2: Six totally delocalized bonding patterns for V15+ revealed by AdNDP analysis, Table S1: Magnetic moments of two low-lying isomers (A and B) for V15+, Table S2: Optimized atomic coordinates of the lowest-energy A isomer for V15+.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21603173 and 21643014), the Natural Science Foundation of Shaanxi Province (No. 2016JQ5110), the Natural Science Foundation of Science and Technology Bureau of Xi’an City (Nos. 2016CXWL16, 2016CXWL02, CXY1443WL13, and CXY1531WL03), and the Natural Science Foundation of Education Department of Shaanxi Province (No. 15JK2145). The authors gratefully acknowledge André Fielicke for offering the original IR-MPD vibrational data of the vanadium cluster.
Author Contributions
Xiaojun Li designed the project, performed the chemical bonding analyses, analyzed the calculated results with all of the members, and wrote the manuscript. Hongjiang Ren carried out the calculations of density of states, and analyzed the electronic structure. Xinwei Huang performed the optimization calculations by several functionals, and determined the global structure. Shuna Li calculated the magnetic moments and charge transfers. All authors contributed to the interpretation and discussion of the data at all stages.
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Michaelian, K.; Rendón, N.; Garzón, I.L. Structure and energetics of Ni, Ag, and Au nanoclusters. Phys. Rev. B 1999, 60, 2000. [Google Scholar] [CrossRef]
- Schaefer, B.; Pal, R.; Khetrapal, N.S.; Amsler, M.; Sadeghi, A.; Zeng, V.B.X.C.; Goedecker, S.; Wang, L.-S. Isomerism and structural fluxionality in the Au26 and Au26− nanoclusters. ACS Nano 2014, 8, 7413–7422. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, X.; Zhai, H.J.; Wang, L.S. Au20: A tetrahedral cluster. Science 2003, 299, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Bowlan, J.; Harding, D.J.; Jalink, J.; Kirilyuk, A.; Meijer, G.; Fielicke, A. Communication: Structure of magnetic lanthanide clusters from far-ir spectroscopy:Tbn+ (n = 5–9). J. Chem. Phys. 2013, 138, 031102. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Huang, W.; Mei, W.-N.; Wang, L.-S.; Wu, Q.; Zeng, X.-C. Structural evolution of medium-sized gold clusters Aun− (n = 36, 37, 38): Appearance of bulk-like face centered cubic fragment. J. Phys. Chem. C 2014, 118, 6887–6892. [Google Scholar] [CrossRef]
- Du, J.; Sun, X.; Jiang, G. A theoretical study on Tan+ cluster cations: Structural assignments, stability, and electronic properties. J. Chem. Phys. 2012, 136, 094311. [Google Scholar] [CrossRef] [PubMed]
- Gruene, P.; Rayner, D.M.; Redlich, B.; van der Meer, A.F.G.; Lyon, J.T.; Meijer, G.; Fielicke, A. Structures of neutral Au7, Au19, and Au20 clusters in the gas phase. Science 2008, 321, 674–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tian, H.; Hao, C.; Zhao, J.; Wang, Q.; Liu, E.; Dong, C. First-principles calculations of elastic moduli of Ti-Mo-Nb alloys using a cluster-plus-glue-atom model for stable solid solutions. J. Mater. Sci. 2013, 48, 3138–3146. [Google Scholar] [CrossRef]
- Bhattacharjee, D.; Mishra, B.K.; Deka, R.C. Effect of double aluminium doping on the structure, stability and electronic properties of small gold clusters. J. Mater. Sci. 2015, 50, 4586–4599. [Google Scholar] [CrossRef]
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From theory to applications of alloy clusters and nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.A.; Shibuta, Y.; Wales, D.J. Global minima of transition metal clusters described by finnis–sinclair potentials: A comparison with semiempirical molecular orbital theory. Philos. Mag. 2009, 89, 3311–3332. [Google Scholar] [CrossRef]
- Lozano, X.L.; Mottet, C.; Weissker, H.-C. Effect of alloying on the optical properties of Ag–Au nanoparticles. J. Phys. Chem. C 2013, 117, 3062–3068. [Google Scholar] [CrossRef]
- Kreibig, U.; Vollmer, M. Optical Properties of Metal Clusters; Springer: Berlin, Germany, 1995. [Google Scholar]
- Koppen, J.V.; Hapka, M.; Szczęśniak, M.M.; Chałasiński, G. Optical absorption spectra of gold clusters Aun (n = 4, 6, 8,12, 20) from long-range corrected functionals with optimal tuning. J. Chem. Phys. 2012, 137, 114302. [Google Scholar] [CrossRef] [PubMed]
- Billas, I.M.L.; Becker, J.A.; Châtelain, A.; de Heer, W.A. Magnetic moments of iron clusters with 25 to 700 atoms and their dependence on temperature. Phys. Rev. Lett. 1993, 71, 4067. [Google Scholar] [CrossRef] [PubMed]
- Iseda, M.; Nishio, T.; Han, S.Y.; Yoshida, H.; Terasaki, A.; Kondow, T. Electronic structure of vanadium cluster anions as studied by photoelectron spectroscopy. J. Chem. Phys. 1997, 106, 2182. [Google Scholar] [CrossRef]
- Bell, A.T. The impact of nanoscience on heterogeneous catalysis. Science 2003, 299, 1688–1691. [Google Scholar] [CrossRef] [PubMed]
- Valden, M.; Lai, X.; Goodman, D.W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties. Science 1998, 281, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, X.; Zhang, H. Probing the activity of Ni13, Cu13, and Ni12Cu clusters towards the ammonia decomposition reaction by density functional theory. J. Mater. Sci. 2017, 52, 3162–3168. [Google Scholar] [CrossRef]
- Mariscal, M.M.; Oviedo, O.A.; Leiva, E.P.M. Metal Clusters and Nanoalloys: From Modeling to Applications; Springer: New York, NY, USA, 2013. [Google Scholar]
- Lee, K.; Callaway, J. Electronic structure and magnetism of small V and Cr clusters. Phys. Rev. B 1993, 48, 15358. [Google Scholar] [CrossRef]
- Douglass, D.C.; Bucher, J.P.; Bloomfield, L.A. Magnetic studies of free nonferromagnetic clusters. Phys. Rev. B 1992, 45, 6341. [Google Scholar] [CrossRef]
- Sun, H.; Luo, Y.-H.; Zhao, J.; Wang, G. Structural, electronic, and magnetic properties of small vanadium clusters. Phys. Status Solid B 1999, 215, 1127–1135. [Google Scholar] [CrossRef]
- Xu, J.; Rodgers, M.T.; Griffin, J.B.; Armentrout1, P.B. Guided ion beam studies of the reactions of Vn+ (n = 2–17) with O2: Bond energies and dissociation pathways. J. Chem. Phys. 1998, 108, 9339. [Google Scholar] [CrossRef]
- Jaeger, T.D.; Fielicke, A.; von Helden, G.; Meijer, G.; Duncan, M.A. Infrared spectroscopy of water adsorption on vanadium cluster cations (Vx+; x = 3–18). Chem. Phys. Lett. 2004, 392, 409–414. [Google Scholar] [CrossRef]
- Zakin, M.R.; Cox, D.M.; Whetten, R.L.; Trevor, D.J.; Kaldor, A. Effect of hydrogen chemisorption on the photoionization threshold of isolated transition metal clusters. Chem. Phys. Lett. 1987, 135, 223–228. [Google Scholar] [CrossRef]
- Fielicke, A.; von Helden, G.; Meijer, G. Far-infrared spectroscopy of isolated transition metal clusters. Eur. Phys. J. D 2005, 34, 83–88. [Google Scholar] [CrossRef]
- Ratsch, C.; Fielicke, A.; Kirilyuk, A.; Behler, J.; von Helden, G.; Meijer, G.; Scheffler, M. Structure determination of small vanadium clusters by density-functional theory in comparison with experimental far-infrared spectra. J. Chem. Phys. 2005, 122, 124302. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Z.; Sun, Q.; Liu, F.; Wang, G.; Lain, K.D. Tight-binding study of the structural and magnetic properties of vanadium clusters. Phys. B Condens. Matter 1995, 215, 377–382. [Google Scholar]
- Taneda, A.; Shimizu, T.; Kawazoe, Y. Stable disordered structures of vanadium clusters. J. Phys. Condens. Matter 2001, 13, L305–L312. [Google Scholar] [CrossRef]
- Fielicke, A.; Kirilyuk, A.; Ratsch, C.; Behler, J.; Scheffler, M.; von Helden, G.; Meijer, G. Structure determination of isolated metal clusters via far-infrared spectroscopy. Phys. Rev. Lett. 2004, 93, 023401. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Desai, S.R.; Wang, L.-S. Evolution of the electronic structure of small vanadium clusters from molecular to bulklike. Phys. Rev. Lett. 1996, 77, 2436. [Google Scholar] [CrossRef] [PubMed]
- Su, C.X.; Hales, D.A.; Armentrout, P.B. Collision-induced dissociation of Vn+ (n = 2–20) with Xe: Bond energies, dissociation pathways, and structures. J. Chem. Phys. 1993, 99, 6613. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Erratum: Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822. [Google Scholar] [CrossRef]
- Perdew, J.P. Erratum: Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 34, 7406. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for packageindependent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhao, J.; Wang, J.; Wang, B.; Lu, Q.; Wang, G. Growth behavior and magnetic properties of SinFe (n = 2–14) clusters. Phys. Rev. B 2006, 73, 125439. [Google Scholar] [CrossRef]
- Zhao, W.-J.; Wang, Y.-X. Geometries, stabilities, and electronic properties of FeGen (n = 9–16) clusters: Density-functional theory investigations. Chem. Phys. 2008, 352, 291–296. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1838. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Revealing intuitively assessable chemical bonding patterns in organic aromatic molecules via adaptive natural density partitioning. J. Org. Chem. 2008, 73, 9251–9258. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ren, H.; Song, X.Y.J. Exploring the chemical bonding, infrared and UV–vis absorption spectra of OH radicals adsorption on the smallest fullerene. Spectrochim. Acta Part A 2015, 144, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.A.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.A.; Li, W.-L.; Piazza, Z.A.; Boldyrev, A.I.; Wang, L.-S. Complexes between planar boron clusters and transition metals: A photoelectron spectroscopy and ab initio study of CoB12− and RhB12−. J. Phys. Chem. A 2014, 118, 8098–8105. [Google Scholar] [CrossRef] [PubMed]
- Galeev, T.R.; Boldyrev, A.I. Recent advances in aromaticity and antiaromaticity in transition-metal systems. Annu. Rep. Prog. Chem. Sect. C 2011, 107, 124–147. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Deciphering chemical bonding in golden cages. J. Phys. Chem. A 2009, 113, 866–868. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, Z.; Li, S. The nature of structure and bonding between transition metal and mixed Si-Ge tetramers: A 20-electron superatom system. J. Comput. Chem. 2016, 37, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Two low-lying structures of the cationic V15+ cluster. (A) bicapped hexagonal antiprism (bha); (B) body-centered cube (bcc).
Figure 1.
Two low-lying structures of the cationic V15+ cluster. (A) bicapped hexagonal antiprism (bha); (B) body-centered cube (bcc).
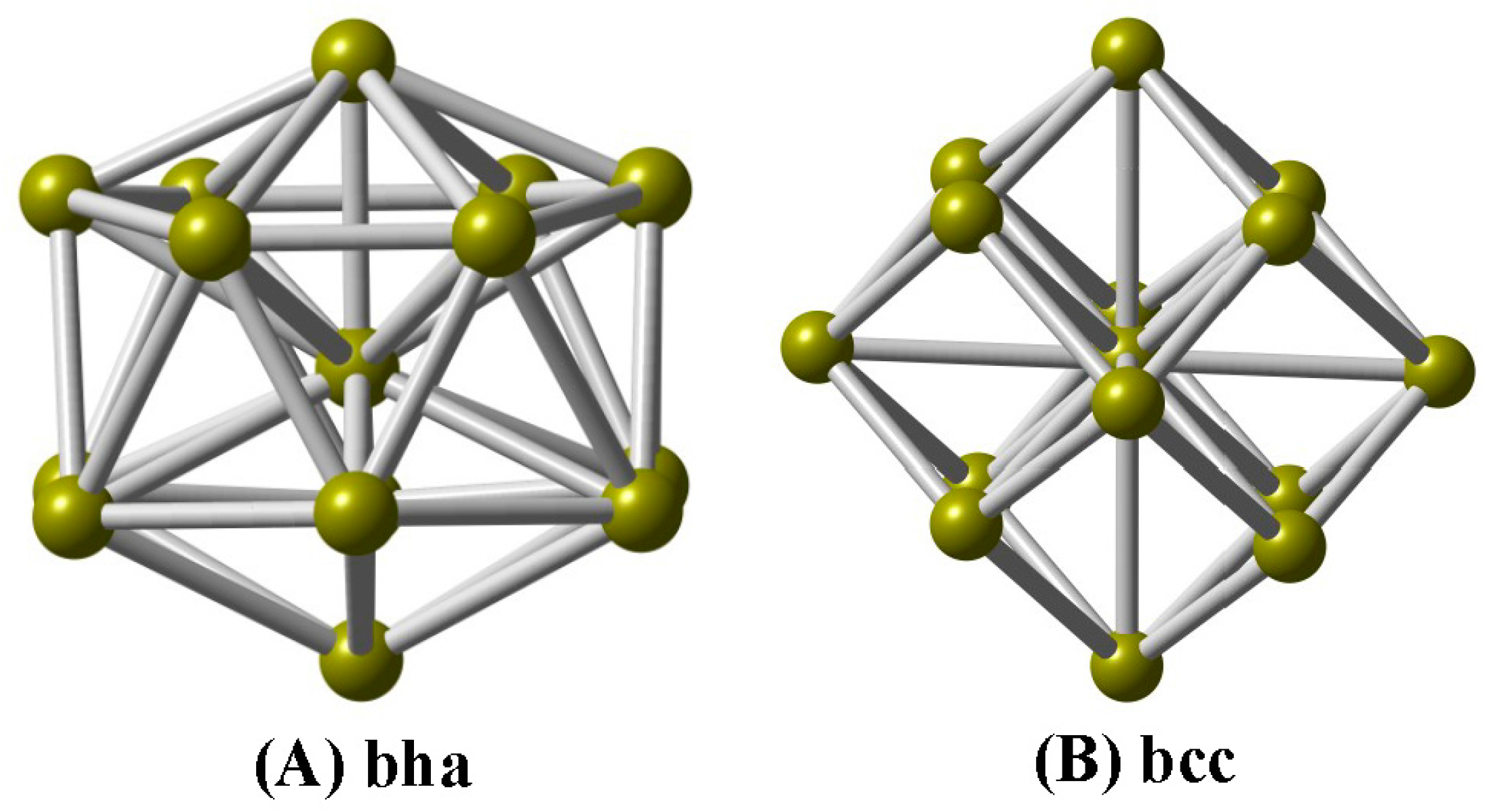
Figure 2.
Comparison of relative energies of the two low-lying isomers (A and B) for the cationic V15+ cluster in different spin states, obtained by using the five different methods. All the calculations are based on the zero-point corrected total energies upon full optimizations.
Figure 2.
Comparison of relative energies of the two low-lying isomers (A and B) for the cationic V15+ cluster in different spin states, obtained by using the five different methods. All the calculations are based on the zero-point corrected total energies upon full optimizations.
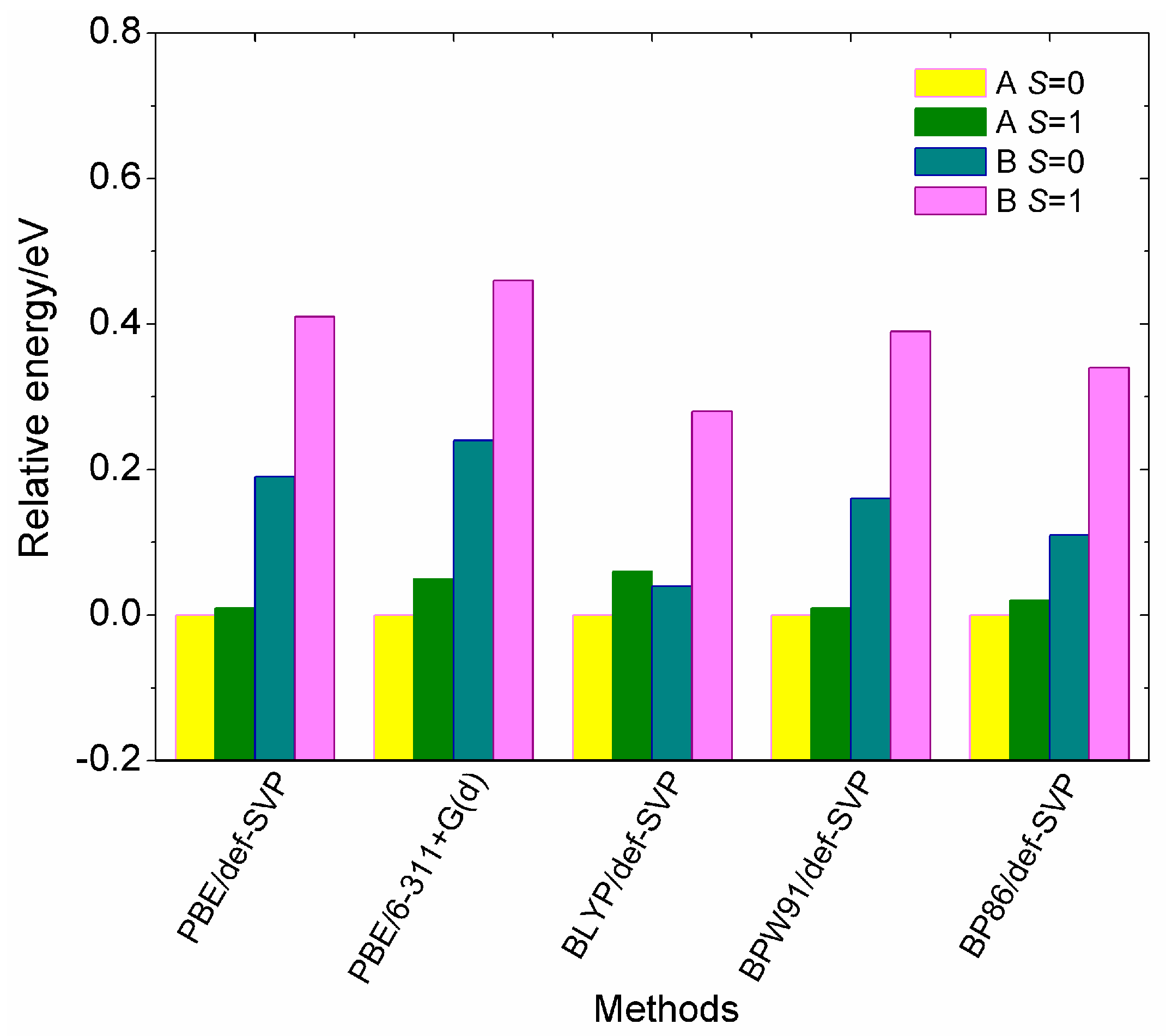
Figure 3.
Comparison of experimental IR-MPD spectrum (lower panel) of V15+-Ar cluster with the calculated IR spectra (upper panels) of the two low-lying structures (A and B) in different spin states (S = 0 and 1), obtained from the PBE/def-SVP level of theory. The σ symbol represents the experimental IR cross section. A Gaussian band with the full width at half-maximum (FWHM) of 6 cm−1 was used. A scaling factor of 0.87 was applied to correct all the calculated vibrational frequencies.
Figure 3.
Comparison of experimental IR-MPD spectrum (lower panel) of V15+-Ar cluster with the calculated IR spectra (upper panels) of the two low-lying structures (A and B) in different spin states (S = 0 and 1), obtained from the PBE/def-SVP level of theory. The σ symbol represents the experimental IR cross section. A Gaussian band with the full width at half-maximum (FWHM) of 6 cm−1 was used. A scaling factor of 0.87 was applied to correct all the calculated vibrational frequencies.

Figure 4.
Density of states (DOS) of the low-lying A structure for the V15+ cluster. The orbital contributions (V-s, V-p, and V-d) of the cluster to the TDOS spectrum are labeled. The Fermi level is shifted to zero.
Figure 4.
Density of states (DOS) of the low-lying A structure for the V15+ cluster. The orbital contributions (V-s, V-p, and V-d) of the cluster to the TDOS spectrum are labeled. The Fermi level is shifted to zero.
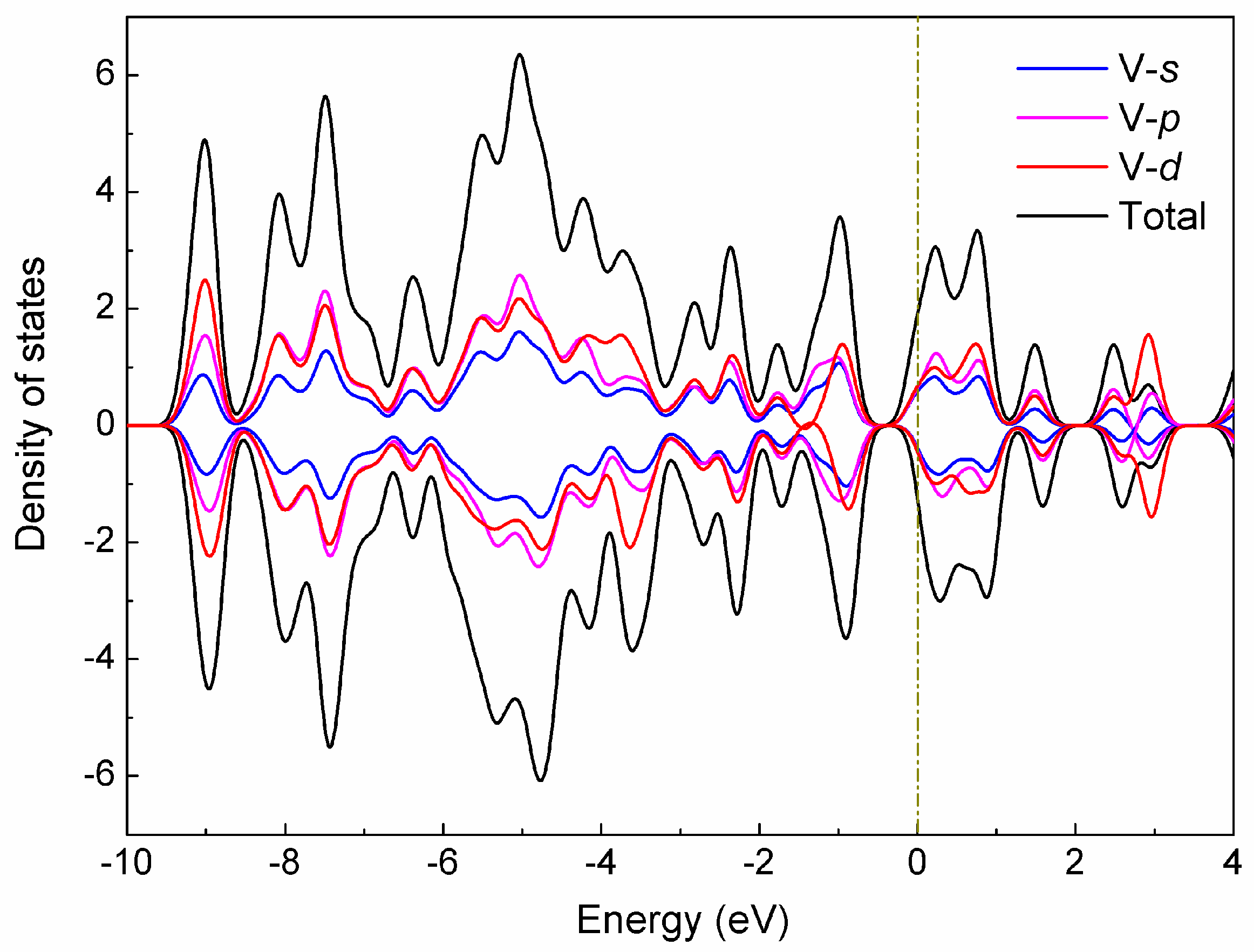
Figure 5.
AdNDP chemical bonding analyses of the low-lying A structure for the V15+ cluster. ON denotes the electron occupation number and is close to the ideal population of 2.00 |e|.
Figure 5.
AdNDP chemical bonding analyses of the low-lying A structure for the V15+ cluster. ON denotes the electron occupation number and is close to the ideal population of 2.00 |e|.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, X.; Ren, H.; Huang, X.; Li, S. First-Principles Study of Structural, Electronic and Magnetic Properties of Metal-Centered Tetrahexahedral V15+ Cluster. Nanomaterials 2017, 7, 164. https://doi.org/10.3390/nano7070164
AMA Style
Li X, Ren H, Huang X, Li S. First-Principles Study of Structural, Electronic and Magnetic Properties of Metal-Centered Tetrahexahedral V15+ Cluster. Nanomaterials. 2017; 7(7):164. https://doi.org/10.3390/nano7070164
Chicago/Turabian StyleLi, Xiaojun, Hongjiang Ren, Xinwei Huang, and Shuna Li. 2017. "First-Principles Study of Structural, Electronic and Magnetic Properties of Metal-Centered Tetrahexahedral V15+ Cluster" Nanomaterials 7, no. 7: 164. https://doi.org/10.3390/nano7070164
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.