Effect of Alkali Metal Atoms Doping on Structural and Nonlinear Optical Properties of the Gold-Germanium Bimetallic Clusters

The Key Laboratory for Surface Engineering and Remanufacturing in Shaanxi Province, School of Chemical Engineering, Xi’an University, Xi’an 710065, China
*
Author to whom correspondence should be addressed.
Nanomaterials 2017, 7(7), 184; https://doi.org/10.3390/nano7070184
Submission received: 16 June 2017
/
Revised: 8 July 2017
/
Accepted: 10 July 2017
/
Published: 17 July 2017
(This article belongs to the Special Issue Semiconductor Core/Shell Nanocrystals for Optoelectronic Applications)
Abstract
:A new series of alkali-based complexes, AM@GenAu (AM = Li, Na, and K), have been theoretically designed and investigated by means of the density functional theory calculations. The geometric structures and electronic properties of the species are systematically analyzed. The adsorption of alkali metals maintains the structural framework of the gold-germanium bimetallic clusters, and the alkali metals prefer energetically to be attached on clusters’ surfaces or edges. The high chemical stability of Li@Ge12Au is revealed by the spherical aromaticity, the hybridization between the Ge atoms and Au-4d states, and delocalized multi-center bonds, as well as large binding energies. The static first hyperpolarizability (βtot) is related to the cluster size and geometric structure, and the AM@GenAu (AM = Na and K) clusters exhibit the much larger βtot values up to 13050 a.u., which are considerable to establish their strong nonlinear optical (NLO) behaviors. We hope that this study will promote further application of alkali metals-adsorbed germanium-based semiconductor materials, serving for the design of remarkable and tunable NLO materials.
1. Introduction
Alkalis metals have attracted much attention because they serve as building blocks for novel materials with tunable properties [1], and a great deal of new alkali-based complexes have been recently reported [2,3,4]. In particular, a series of alkalides, proposed as the candidates of nonlinear optical (NLO) materials, exhibit exceptionally large NLO responses and are promising for their spectacular semiconductor potentials in photoelectricity devices and optical communications, e.g., M@C60 (M = Li, Na, Cs) [5], Li@C60-BX4 (X = F, Cl, Br) [6], LinF (n = 2–5) [7], Li2@BN nanotubes [8], OLi3-M-Li3O (M = Li, Na, K) [9], Li3+(calyx[4]pyrrole)M− [10], Li(NH3)4M (M = Li, Na, K) [11], and Li(CH3NH2)nNa [12], etc., in which both theoretical and experimental studies play a crucial role in finding ways to enhance the NLO behaviors. On the other hand, the different NLO materials can be expressed by the largely high second-order NLO response at the molecular level [13,14], and the first hyperpolarizability can be quantitatively used to evaluate the potential NLO materials of alkali-based complexes.
Meanwhile, owing to the unique electronic properties of alkalides, their derivatives have been extensively reported with the extension to silicon-based clusters, e.g., Si10(Li, Na, K)n (n = 1, 2) [15], (Li, Na, K)@SinNb (n = 1–12) [16], Si10Li8 [17], and SinFe (n = 1–14) [18], which ascribe to loosely bound electrons in alkali-based complexes, resulting in the large NLO responses. It is also noteworthy that although the analogous germanium and silicon species are isovalent, their structures and physicochemical properties are quite different [19,20,21,22,23,24,25]. For example, the static hyperpolarizabilities of pure SimGen (m + n = 7, n = 0–7) clusters were studied by using the density functional theory (DFT) and MP2 ab initio methods [26], and revealed that the enhancement of the hyperpolarizabilities arises from more polarizable character on the germanium atoms, and thus alkali metals-adsorbed germanium-based semiconductor clusters may have the unusual features in high-performance optoelectronic devices for the potential applications. In addition, Knoppe and Ozga et al. [27,28,29] found that the gold-containing clusters appear to have strong NLO responses, and the Au atom plays an important role in the optoelectronic application. Obviously, the knowledge of geometries, electronic structures, chemical bonding, and nonlinear optical properties of the species is very important for understanding these applications, but these are rarely reported. With this motivation, we explored the stability and electronic structures of alkali metals-adsorbed GenAu semiconductor clusters for the first time, labeled as AM@GenAu (AM = Li, Na, and K; n = 2–13), and investigated the effect of alkali metals on the dipole moment, polarizability, and first hyperpolarizability (βtot). The results suggest that the AM@GenAu (AM = Na and K) clusters may be proposed as novel potential high-performance NLO materials, especially for the Na@Ge7Au cluster with a large βtot value.
2. Computational Details
The geometrical optimizations of the AM@GenAu (AM = Li, Na, and K; n = 2–13) clusters were carried out by using the hybrid DFT-B3LYP functional [30,31], implemented in the Gaussian 09 suite of programs [32]. This method provides a good prediction on energy evaluation [33,34,35], in conjunction with the Karlsruhe split-valence basis set augmented with polarization functions (def-SVP) for all alkali metal atoms and the double-ζ LanL2DZ [36,37,38] with effective core potentials (ECPs) for the Ge and Au atoms, and then the low-lying isomers are further reoptimized at the B3LYP/def-TZVP level of theory. In the calculations, the singlet and triplet spin states were examined for each initial structure, and the zero-point vibrational correction was included into the relative energies. According to the previous works on the neutral [24] and cationic [39] GenAu clusters, we searched a large number of initial isomers for the alkali metals-adsorbed gold-doped germanium clusters from the following steps. One is that the alkali metal atoms are attached to different germanium positions on the surface, edge or apex of the lowest-energy structures of the GenAu clusters. The second is the attaching structure with gold positions adsorbed by alkali metals. The remaining structures were constructed by us. Vibrational frequency computations were conducted to ensure that these low-lying structures are local minima on its potential energy surface.
The energy of a molecular system in the presence of a homogeneous electric field can be written as the following equation [40,41,42]:
Here, E0 is the total energy of the molecule without electric field present, Fi is the electric field component in the α direction; the μi, αij, βijk terms are the dipole, the polarizability, and the first hyperpolarizability, respectively. The static dipole moment (μ0) and polarizability (αiso) are defined as follows:
μ0 = (μx2 + μy2 + μz2)1/2
The static first hyperpolarizability (βtot) is obtained as follows:
where .
βtot = (βx2 + βy2 + βz2)1/2
The density-of-states (DOS) spectra were convoluted utilizing the GaussSum 2.2 program [43] with the full-width at half maximum (FWHM) of 0.3 eV. Chemical bonding analyses were performed using the adaptive natural density partitioning (AdNDP) method proposed by Zubarev and Boldyrev [44]. The molecular orbitals were plotted with the isodensity surfaces (0.02 e1/2/(bohr)3/2), and the molecular graphs were visualized using the VMD program [45]. The long-range corrected functional CAM-B3LYP [46] was utilized to calculate the excited energies within the framework of the time-dependent density functional theory (TDDFT) [47], as well as for the evaluation of linear and nonlinear (L&NLO) optical properties, e.g., dipole moments (μ0), isotropic polarizability (αiso), and first hyperpolarizability (βtot) of the AM@GenAu clusters.
3. Results and Discussion
3.1. Geometric Structures
The low-lying structures of the AM@GenAu (n = 2–13) clusters are displayed in Figure 1. The relative energies of these low-lying isomers, obtained by using the two basis sets as mentioned above, are listed in Table S1 of Supplementary Materials, in which all the calculations indicate that the global minimum for each cluster is in singlet spin states. Meanwhile, one can see from Figure 1 that adsorption of the AM atoms does not largely change the structural framework of the Au-doped germanium clusters, as reported previously by Li et al. [24], and the alkali metals prefer to be attached on clusters’ surface or edge with the multi-adsorbed bonds (multi-bonds) rather than the AM–Ge or AM–Au single-adsorbed bond in the stable structures.
(Li, Na, K)@Ge2Au. The lowest-energy structure (2A) of AM@Ge2Au (AM = Li, Na, and K) takes the tetrahedron structure with Cs symmetry, generated from the AM atoms being capped on the clusters’ surface of the triangular Ge–Ge–Au base. The corresponding triplet states are less stable in energy than those with singlet spin states by 1.03–1.43 eV high at the B3LYP/def-TZVP level of theory (see Table S1 of Supplementary Materials). According to our calculations, the equilibrium Ge–Ge bond lengths are predicted to be 2.399–2.601 Å in the AM@Ge2Au clusters.
(Li, Na, K)@Ge3Au. 3A is a planar structure with Cs symmetry, which can be regarded as the alkali metals being bonded on the edge (Au–Ge) of the rhombic Ge3Au cluster, whereas the structure adsorbed on another edge (Ge–Ge) by alkali metals can be found to be unstable. The 3A geometry can be considered as the global minimum of the AM@Ge3Au (AM = Li, Na, and K) cluster. In Li@Ge3Au, the equilibrium bond lengths are evaluated to be 2.597 Å for the Li–Au bond, 2.515 Å for the Li–Ge bond, 2.558–2.722 Å for two Au-Ge bonds, and 2.353–2.876 Å for three Ge–Ge bonds.
(Li, Na, K)@Ge4Au. As previously reported [24], the ground-state structure of the Ge4Au cluster is a pyramid-distorted isomer. When the AM atoms are attached on the side face (Ge–Ge–Au) of Ge4Au, the most stable 4A structure is formed, in which the equilibrium Li–Ge and Li–Au bond lengths are predicted to be 2.597, 2.615, and 2.771 Å at the B3LYP/def-TZVP level of theory, respectively.
(Li, Na, K)@Ge5Au. In the search for the low-lying configurations of AM@Ge5Au (AM = Li, Na, and K), there are two stable structures (5A and 5B) with close energies (0.13–0.16 eV in singlet spin states) to be obtained while their triplet spin states have the higher relative energies (at least 0.78 eV) at the B3LYP/def-TZVP level of theory. The geometric differences between the two isomers are that the AM atoms are adsorbed on the side face (Ge–Ge–Ge–Au) or side edge (Ge–Ge) of the distorted triangular prism. Obviously, the global minimum of AM@Ge5Au (AM = Li, Na, and K) is found to be the 5A structure. It is noteworthily that for small clusters with n ≤ 5, the lowest-energy structures take the AM-adsorbed on Au-adjacent edge or surface of cluster base, to form the stable AM–Au chemical bonds.
(Li, Na, K)@Ge6Au. Similar to the n = 5 cluster as discussed above, we have manually designed a great number of initial geometries for the n = 6 cluster size, e.g., adsorbing the AM atoms to different position sites and substituting the apical Ge atom by the AM atoms in the Ge7Au cluster, and so on, but the 6A cagelike with Cs symmetry is the lowest-energy structure for the AM@Ge6Au (AM = Li, Na, and K). The 6B isomer is a distorted quadrangular prism, which is less stable than 6A by at least 0.13 eV higher in energy. Meanwhile, it is found that the two different basis sets used herein provide the same energetic ordering for these small clusters (see Table S1 of Supplementary Materials).
(Li, Na, K)@Ge7Au. As mentioned above, the AM atoms prefer energetically to be adsorbed on the Au-adjacent surface (Ge–Ge–Ge–Au) or edge (Ge–Au) of the quadrangular prism (QP), thus the two stable structures (7A and 7B) can be obtained. According to our calculation, the 7A isomer is predicted as a putative global minimum for the (Li, K)@Ge7Au clusters, whereas the 7B isomer is the most stable structure for Na@Ge7Au.
(Li, Na, K)@Ge8Au. The 8A and 8B isomers are one bicapped square prism, which can be viewed as the alkali metal and Au atom capped on different faces of the lowest-energy Ge7Au cluster (called as square prism). Of which 8A is the lowest-energy structure for the Li@Ge8Au cluster at the B3LYP/LanL2DZ(Ge,Au)/def-SVP(AM) level of theory, while the most stable structure for (Na, K)@Ge8Au is the 8B isomer lying only 0.02–0.04 eV under the 8A isomer. However, the energetic ordering of 8A and 8B for (Na, K)@Ge8Au can be reversed at the B3LYP/Def-TZVP level of theory. Thus, the 8A geometry can be found to be the global minimum for the AM@Ge8Au (AM = Li, Na, and K) cluster at the B3LYP/Def-TZVP level of theory.
(Li, Na, K)@Ge9Au. Based on the lowest-energy Ge9Au cluster [24], the alkali metal-adsorbed complexes can be generated from the AM atoms attaching on the above Au-Ge layer. It is obvious that the Au-adjacent isomer (9A) is more stable than the Au-outlying one (9B) by ~0.04–0.06 eV in singlet spin states, being consistent with the small clusters (n ≤ 5).
(Li, Na, K)@Ge10Au. The Ge10Au cluster is a C2v-symmetrical pentagonal prism with the Au-encapsulated into the central position of structure [24]. When the alkali metal atoms are directly face-capped on side quadrangle of Ge10Au, a new equilibrium 10A structure, considered as global minimum, is yielded using the DFT-B3LYP functional. Of which the theoretical bond lengths are predicted to be 2.769 Å for the four equivalent Li−Ge bonds, and 2.715, 2.733, and 2.742 Å for three kinds of the equivalent Ge–Au bonds.
(Li, Na, K)@Ge11Au. The 11A structure can be regarded as the Ge atom being directly capped on the side edge of the above pentagon of 10A or alkali metal atoms being directly adsorbed on the face-sided pentagon of the lowest-energy Ge11Au cluster, as published previously [24]. Similarly, the low-lying 11B isomer can be formed by capping the alkali metal atoms on the face-sided quadrangle of the Ge11Au cluster, which is less stable than 11A by at least 0.07 eV high in energy (see Table S1 of Supplementary Materials ).
(Li, Na, K)@Ge12Au. Similar to the n = 11 cluster as discussed above, the 12A and 12B structures can be generated from the different position sites adsorbed by alkali metal atoms on the lowest-energy Ge12Au cluster. Interestingly, the 12B isomer has a highly coordinated surface adsorbed by the Li atom to eliminate the number of dangling bonds on the clusters’ surface, regarded as the global minimum of Li@Ge12Au. Contrastingly, the adsorption of Na/K in lower coordinated edge position (12A) will form the lowest-energy structure for (Na, K)@Ge12Au cluster.
(Li, Na, K)@Ge13Au. Referring to the equilibrium geometries of the low-lying Ge13Au cluster reported previously [24], a great number of initial structures of alkali metal-adsorbed complexes were extensively constructed and optimized in order to locate the global minimum of the cluster size. For comparison, we only show two first stable 13A and 13B isomers for (Li, Na, K)@Ge13Au (see Figure 1), which shows the close energy difference between 0.03 eV and 0.22 eV at the B3LYP/Def-TZVP level of theory (see Table S1 of Supplementary Materials). Obviously, the lowest-energy 13A structure undergoes the structural relaxation, whereas the low-lying 13B structure keeps the structural framework of the lowest-energy Ge13Au cluster.
3.2. Chemical Stability and Electronic Structures
In order to explore the size selectivity and the electronic properties of the AM@GenAu (n = 2–13) clusters, we plotted the average binding energy (Eb), the dissociation energy (De, AM@GenAu → GenAu + AM), the HOMO-LUMO gaps (GAPs), and the vertical ionization potentials (VIP) and vertical electron affinities (VEA) in Figure 2 and Figure 3, respectively, which are defined by:
where the E term represents the total energy with zero-point vibrational corrections.
Eb(n) = [E(AM) + nE(Ge) + E(Au) − E(AM@GenAu)]/(n + 2)
De(n) = E(GenAu) + E(AM) − E(AM@GenAu)
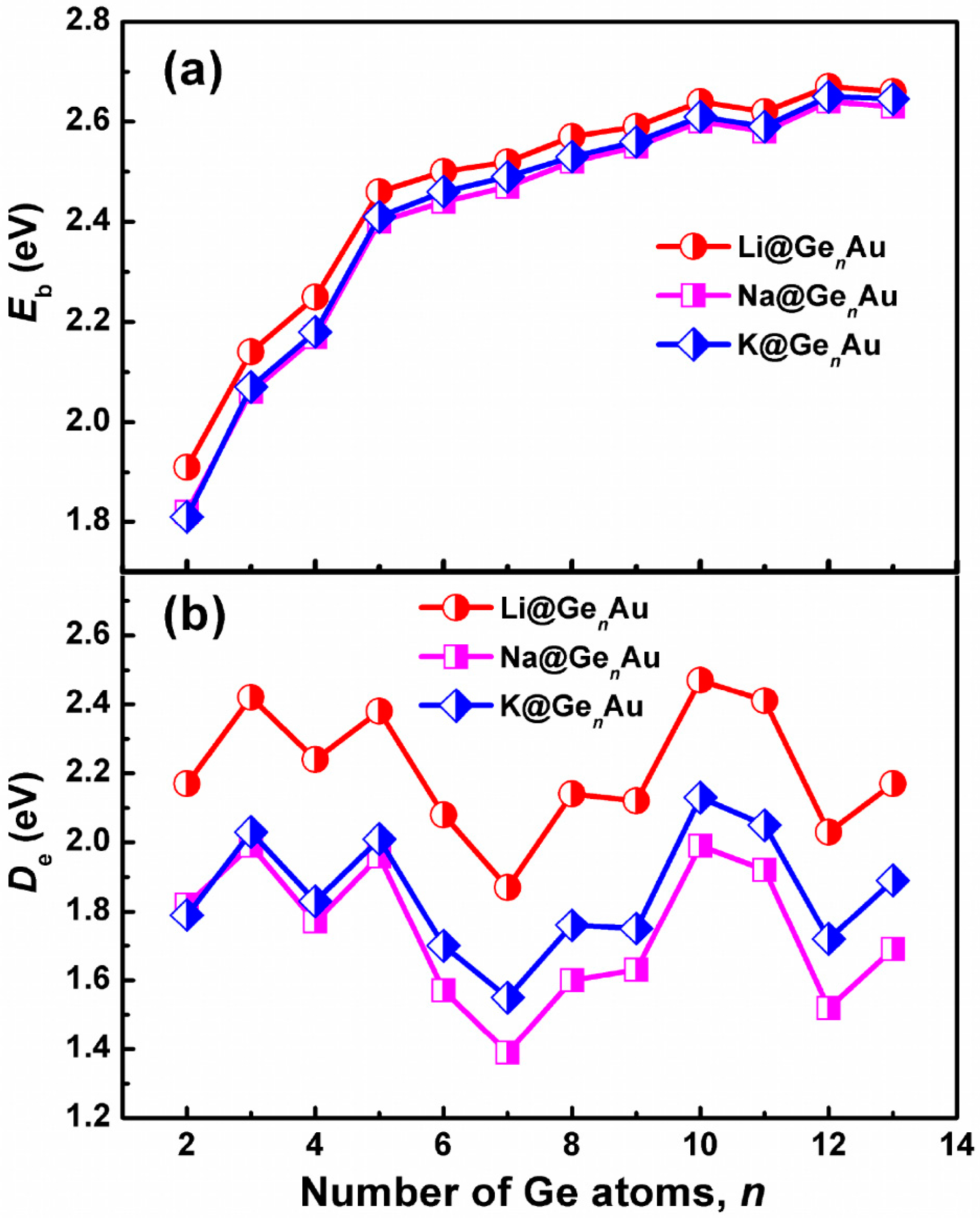
It is well known that a larger Eb value indicates a higher chemical stability of cluster. We can see from Figure 2a that the average binding energies of AM@GenAu (AM = Na and K) have almost similar values for each cluster size, which are slightly lower than that of Li@GenAu by ~0.03–0.10 eV. However, all of the AM@GenAu (AM = Li, Na, and K) clusters show the same increased trends. For example, their average binding energies dramatically increase up to the size of n = 5 and smoothly increase with the size n = 6–13, indicating that the large-sized doped clusters are more stable than the small-sized ones, especially for the n = 10 and 12 clusters. The dissociation energy (De) with the removal of the AM atoms is another useful physical quantity that can also reflect the relative stability of AM@GenAu. It is apparent from Figure 2b that the n = 3, 5, and 10 clusters are the local maximum De peaks, which are more stable than their corresponding neighbors. Meanwhile, the De energetic ordering by alkali metals is Li > K > Na.
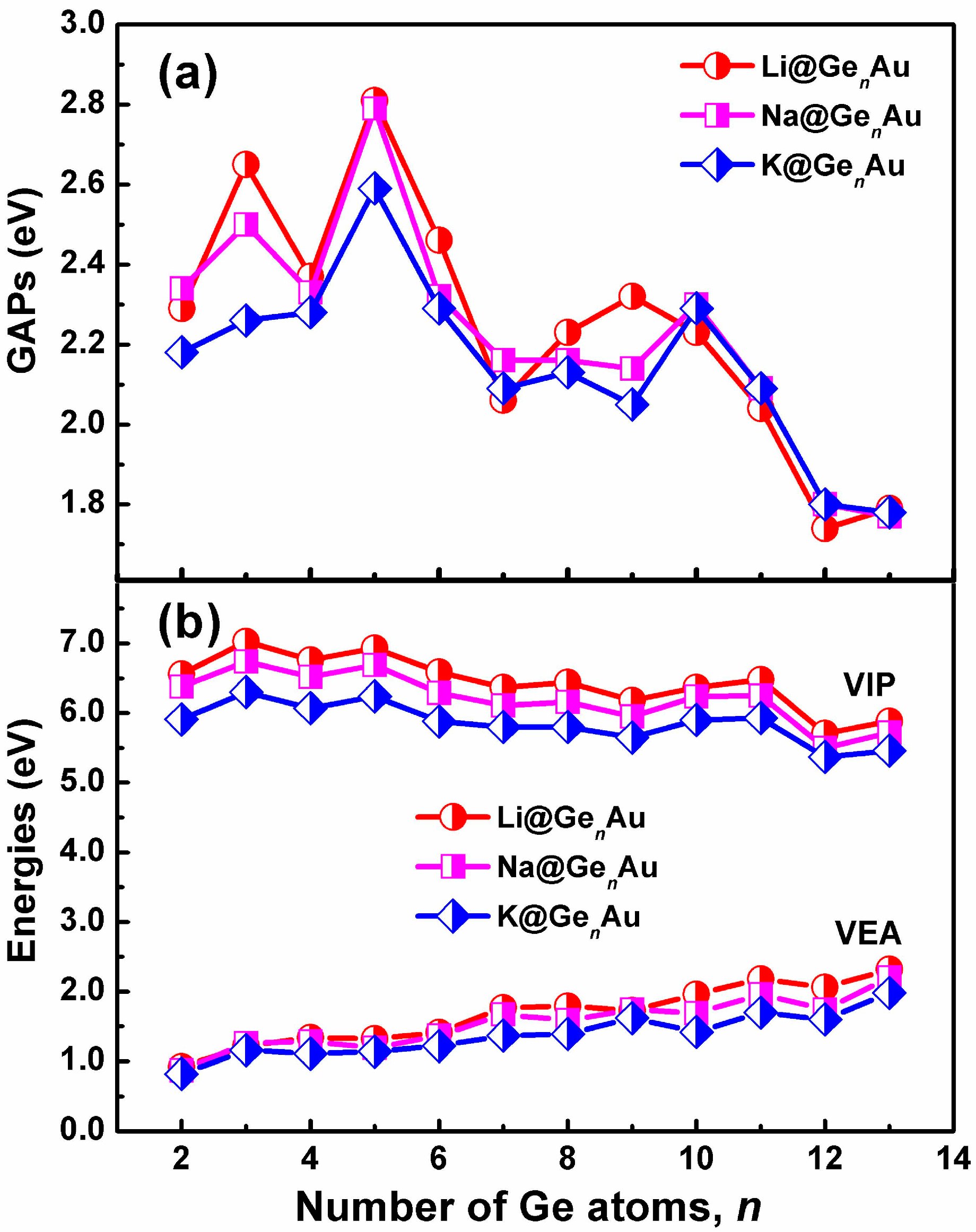
The HOMO-LUMO gaps (GAPs) of the AM@GenAu clusters are listed in Figure 3a. One can find that the gaps (2.04–2.81 eV) in the small-sized clusters with the size n ≤ 11 are larger than those, 1.74–1.80 eV, in the large-sized clusters. Clearly, the HOMO-LUMO gap values with n = 12 and 13 lie in the typically optical region (e.g., less than 2 eV), making these clusters attractive for the cluster-assembled optoelectronic materials, which are consistent with previous reports[48], e.g., Zn@Ge12 and Cd@Sn12.
The vertical ionization potentials (VIPs) and vertical electron affinities (VEAs) of AM@GenAu (n = 2–13) are considered to explore the dependence of electronic structures on the cluster size. The VIP can be evaluated by the energy difference between the optimized neutral species and single-point cationic species at the optimized neutral geometry. The VEA can be computed by adding one electron to the neutral species in its equilibrium geometry and taking their energy differences. The calculated VIPs and VEAs of the most stable clusters are plotted in Figure 3b. As shown in Figure 3b, the curve reveals that there is a gradually decreased behavior for VIPs along with increasing number of Ge atom, and the cluster with small VIPs (e.g., n = 12) will be more close to a metallic species [49]; reversely, there is a gradually increased trend for VEAs. Similar to the Eb and De values, the Li@GenAu clusters give the larger VIPs and VEAs than the Na- and K-based complexes. It is notable that the VIPs of AM@Ge10Au (AM = Na and K) are 5.48 eV and 5.32 eV, respectively, smaller than that of Li (5.62 eV) and Na (5.40 eV), respectively, indicating these clusters should have an electronic structure reminiscent of an alkali atom.
3.3. Density of States
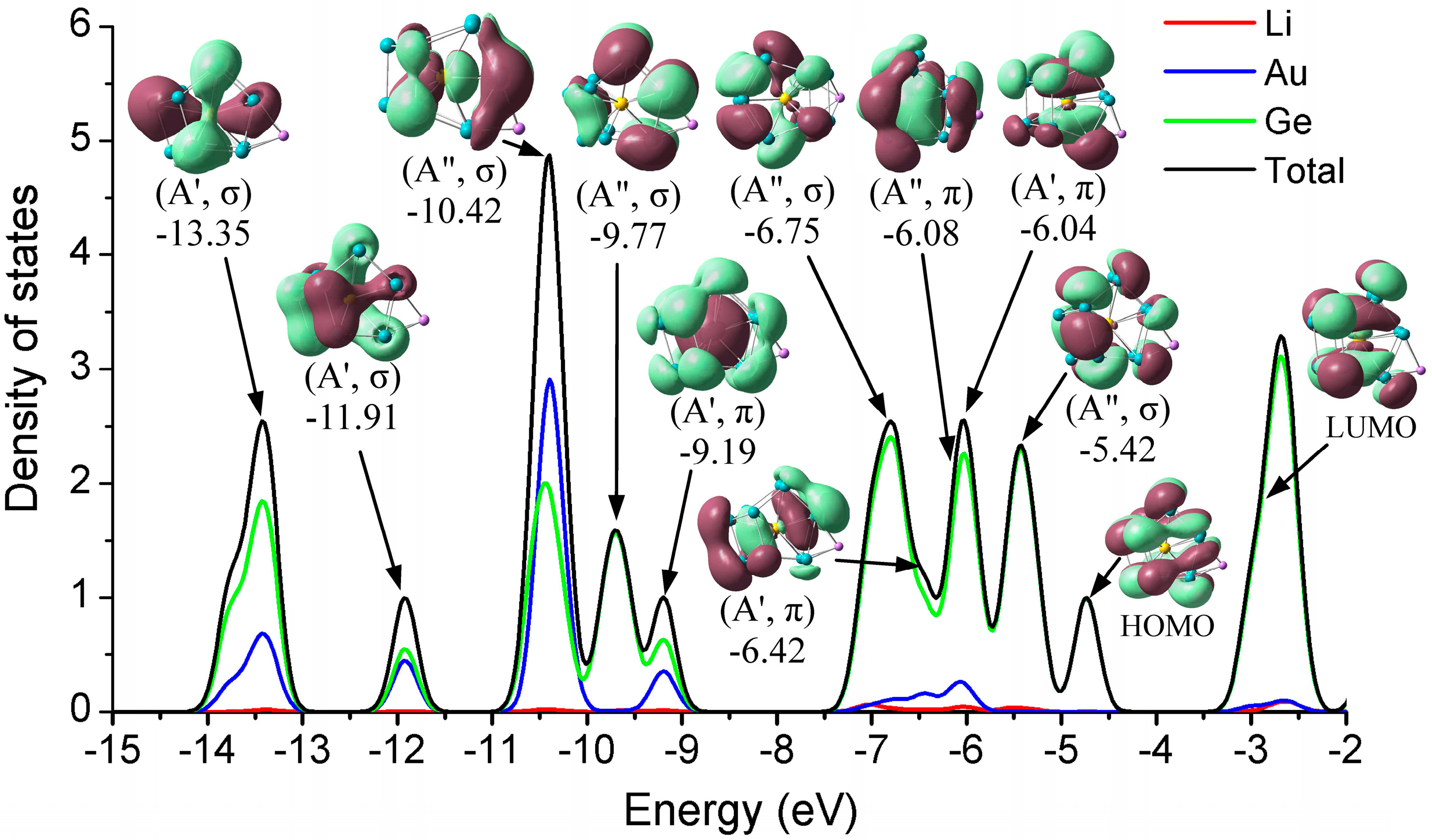
To further investigate the electronic effect of the alkali metals adsorption on the clusters’ surface, we explored the total (TDOS) and partial (PDOS) density of states, taking the stable Li@Ge12Au (12B) cluster as a typical case, shown in Figure 4, which includes all atomic contributions (Li, Ge, and Au) of the clusters to PDOS, while the contributions of different atomic shells (s, p, d) to the molecular orbitals (MOs) are also discussed.
As for this case, each Ge atom is expected to contribute its four valence electrons (4s24p2) to the shell configuration while the Li and Au atoms can contribute one (2s1) and eleven (5d106s1) valence electrons, respectively. Thus, the Li@Ge12Au cluster with Cs symmetry contains 60 valence electrons, which are distributed in the following orbital configuration: (1a′)2(2a′)2(1a″)2(3a′)2(2a″)2 (3a″)2(4a′)2(5a′)2(6a′)2(4a″)2(5a″)2(7a′)2(8a′)2(6a″)2(9a′)2(7a″)2(10a′)2(11a′)2(12a′)2(13a′)2(8a″)2(14a′)2(15a′)2 (9a″)2(16a′)2(10a″)2(17a′)2(11a″)2(18a′)2(12a″)2. The main valence molecular orbitals (0.02 e/a.u.3) of the cluster are depicted in Figure 4. We can clearly see that, for the stable Li@Ge12Au (12B) clusters, the σ-type HOMO has an interesting double pumpkin shape with the electron cloud delocalized on the two sides of the Ge–Ge–Au cross section, mostly involving the five-membered Ge5 rings, due to the hybridization of 12.38% Ge-4s and 85.51% Ge-4p states, whereas the σ-type LUMO originates mainly from the dominant interactions between Au-5dxz state and 4s4p hybridized orbitals of the Ge12 cage. The Au-5dxz orbital has only 3.37% contribution to the LUMO, while the 4s4p hybridized orbitals of 12 Ge atoms have 93.70% contribution to the LUMO. Obviously, the small HOMO-LUMO gap (1.74 eV) of the cluster should be largely related to the electron distributions of the frontier molecular orbitals, and the hybridization of Ge atoms with metal dopants can increase the HOMO-LUMO gap, and enhance the chemical stability of the cluster [22]. The strongest DOS band at around −10.42 eV mainly ascribes to the σ-type HOMO-19 orbital, which comes mostly from different atomic shells, e.g., 66.77% Au-dyz, 23.19% Ge-4s, and 2.92% Ge-4py states. In addition, the Au-4d states have strong interactions (28.86%, 46.57%, and 14.26%) with Ge atoms for HOMO-22 (−13.35 eV), HOMO-21 (−11.91 eV), and HOMO-12 (−9.19 eV) orbitals, respectively, indicating that the 4d states of Au atom in these orbitals are involved in the chemical bonding. One can see that the valence electron orbitals of Li@Ge12Au are divided into two different subsets occupied by σ and π electrons, and the doped cluster contains eight valence π-electrons in four molecular orbitals, e.g., −9.19(A′, π), −6.42(A′, π), −6.08(A″, π), and −6.04(A′, π) orbitals, which belong to one 1S- and three 1P-subshells of the doped cluster, according to the electron shell model [22,50,51].
3.4. Chemical Bonding Analysis
In order to further explore the bonding properties of metal dopant and Ge atoms, we performed the adaptive natural density partitioning (AdNDP) analysis proposed by Zubarev and Boldyrev [44], taking the most stable Li@Ge12Au (12B) cluster as an example. The AdNDP method is based on the concept of electron pairs as the main elements of chemical bonding, and represents the electronic structure in terms of n-center two-electron (nc-2e) bonds, in which the n values go from one (lone-pair) to the maximum number of clusters. This method has been successfully applied to gain insight into the bonding characters not only for fullerene derivatives [41], but also for boron [52,53,54,55] and transition-metal doped Si/Ge clusters [22].
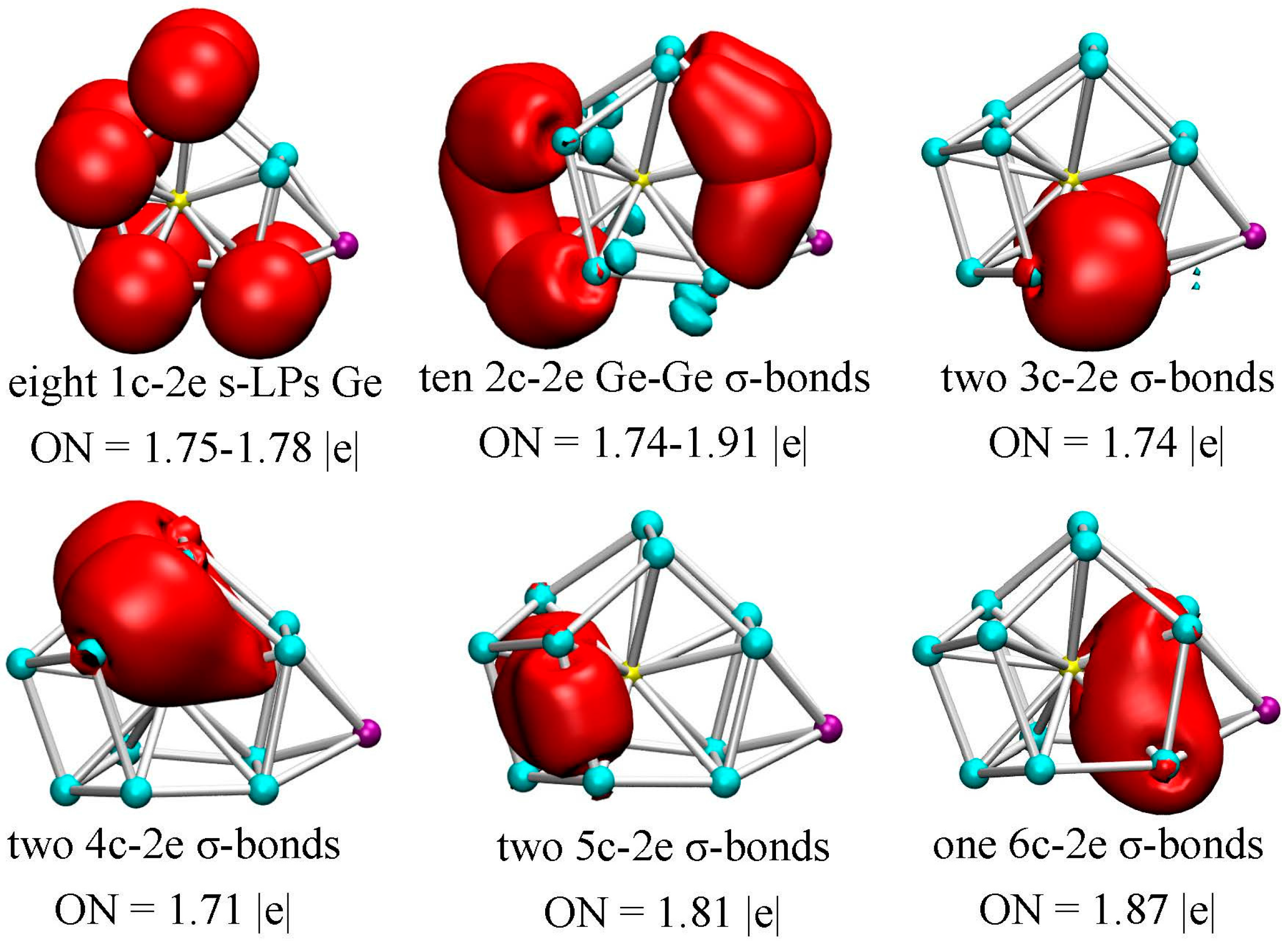
In the method, the occupation numbers (ONs) indicate the number of electrons per bond, and the ONs exceed the established threshold value and are close to the ideal limit of 2.00 |e|. As mentioned above, the Li@Ge12Au (12B) cluster has 60 valence electrons and thus 30 chemical bonds with each bond occupied by two electrons. According to the AdNDP results, there are five typical d-lone pairs (LPs) on Au with ON = 1.93–1.98 |e|, and eight s-lone pairs on the Ge atoms with ON = 1.75–1.78 |e| (see Figure 5), which reveals that electrons on the Ge atoms are not completely localized into lone pairs, but partially participate into the localized or delocalized bondings. As can be seen from Figure 5, ten two-center two-electron (2c-2e) σ-bonds are localized on clusters’ surface with ON = 1.74–1.91 |e|. Meanwhile, a total of seven delocalized σ-bonds can be readily identified: two 3c-2e σ-bonds (ON = 1.74 |e|) on the Ge-Au-Ge triangles under the cage structure, two 4c-2e σ-bonds (ON = 1.71 |e|) on the top structure, two 5c-2e σ-bonds (ON = 1.81 |e|) on the tetragonal pyramid of left structure and one 6c-2e σ-bonds (ON = 1.87 |e|) on the tetragonal bipyramid of right structure with two vertices occupied by Au and Li atoms. Obviously, the localized 2c-2e σ-bonds are mainly located on the clusters’ surface, whereas all of the delocalized σ-bonds are always involved in the endohedral Au dopant. This indicates that the delocalized Au–Ge interactions will be responsible for the structural stabilization of the lowest-energy Li@Ge12Au (12B) cluster.
3.5. Spherical Aromaticity
It is well known that aromaticity is one of important measures of many compounds, and analogous aromatic compounds commonly take on high chemical stability relative to non-aromatic ones. For planar structures, the aromatic characters are identified by using the 4N + 2 Hückel rule [56]. However, In 2000, Hirsch et al. [57] proposed another electron counting rule for three-dimensional (3D) structures, namely 2(N + 1)2 rule, which has been proven as an effective aromaticity criterion, with the extension to inorganic clusters [22,50,58]. In the proposal, the π-electron system of the spherical species can be approximately regarded as a spherical electron gas, which surrounds the spherical surface [59]. According to the Pauli principle, if the number of π electrons in a spherical species satisfies the 2(N + 1)2 counting rule, and then the 3D structures can be considered to be aromatic.
Figure 4 shows the molecular orbitals of the stable Li@Ge12Au (12B) cluster. One can note that the valence electron orbitals are divided into two different orbital sets occupied by σ or π electrons. In particular, the Li@Ge12Au (12B) cluster contains eight π-electrons in four molecular orbitals as mentioned above, and these π-electrons fully satisfy the 2(Nπ + 1)2[Nπ = 1] counting rule. As a result, the π-electron species makes Li@Ge12Au spherically aromatic, and the aromatic character can be regarded as one of the main reasons in the structural stabilization of endohedrally cluster. However, the 2(N + 1)2 electron counting rule cannot be solely applied to explore the aromaticity of compounds, e.g., the bianionic Si122− cluster contains eight π-electrons, but is predicted to be antiaromatic [60]. Therefore, the aromaticity of the Li@Ge12Au (12B) cluster has to be further confirmed by the nucleus-independent chemical shifts (NICS) calculations proposed by Chen and co-workers [61], on the basis of magnetic shieldings with GIAO approximation. Aromaticity is expected to be estimated by a negative NICS value, whereas antiaromaticity is expected to be estimated by a positive NICS value. In the study, a ghost atom is placed at the center of spherical geometry to compute the NICS value. At the B3LYP/def-TZVP level of theory, it is found that the NICS(0) value is −295.7 ppm for the Li@Ge12Au (12B) cluster. Thus, the aromatic character of the cluster, identified by the 2(Nπ + 1)2[Nπ = 1] counting rule, can be confirmed by the largely negative NICS values, and the large diatropic NICS(0) value can contribute to the high chemical stability of the geometry.
3.6. Linear and Nonlinear Optical Properties
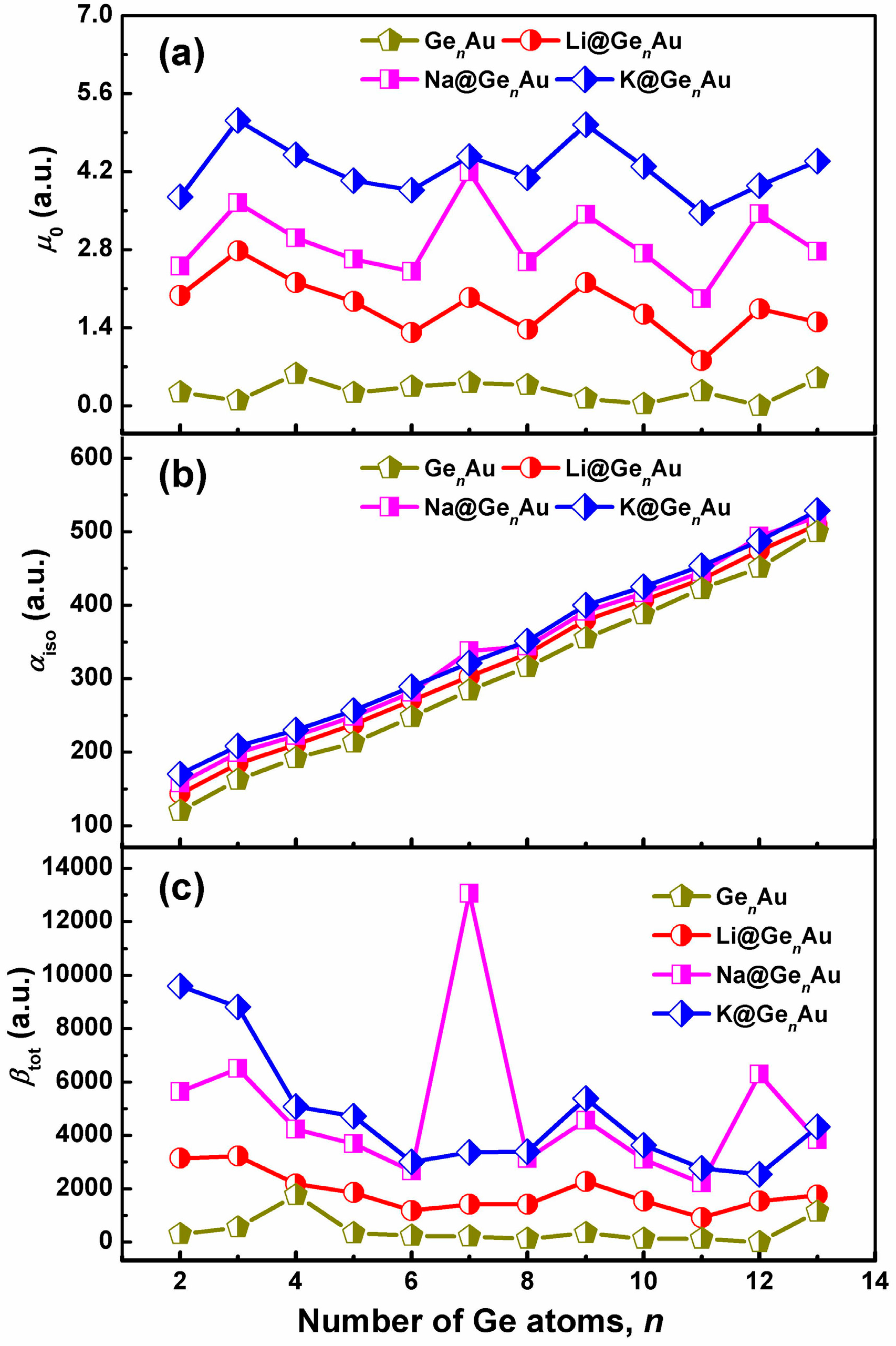
In order to explore the L&NLO behavior of the alkali metals-adsorbed gold-doped germanium clusters, we have computed the static dipole moments (μ0), isotropic polarizabilities (αiso), and static first hyperpolarizability (βtot) using the long-range corrected CAM-B3LYP functional in conjunction with the def2-TZVPD basis sets, shown in Figure 6. According to the results, it shown in Figure 6 that the doping of alkali metals (Li, Na and K) on the clusters’ surface largely enhances the electric properties of the considered systems.
It is evident that the dipole moments of AM@GenAu take on the fluctuating behaviors with the increasing number of Ge atom, and the local maximum peaks are found at n = 3, 7, and 9 for the alkali-based complexes, which are much larger than those of bare GenAu clusters, see Figure 6a. However, what is different is that the isotropic polarizability of all these complexes increases linearly with increasing cluster size, as shown in Figure 6b, similar to the Li2-doped boron nitride clusters (n = 4–8) [8]. In particular, the doping of the K atom can be predicted to improve the αiso values by ~7–41%, indicating that the alkali metal atoms provide the possibility for inducing the isotropic polarizability. Additionally, according to the hard soft acids bases (HSAB) principle [62], the species with small HOMO-LUMO gaps are less hard and more polarizable, and this reveals that the large polarizabilities of the studied species have close relations with their small energy gaps.
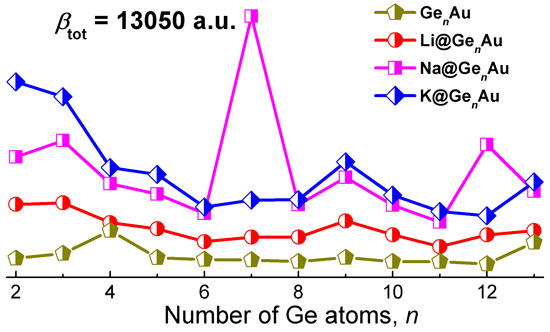
As shown in Figure 6c, the variation of the first hyperpolarizabilities (βtot) is interesting and has become the focus of our attention. One can see that the βtot values of all the complexes distinctly decrease up to the size of n = 6, and are slightly fluctuating for larger sizes, with the exception of local maximum peaks, e.g., n = 7, 9, and 12. Compared with the bare GenAu clusters, it is found that the alkali metals can dramatically enhance βtot, but there are strong dependencies on the cluster size, and they are more sensitive to the geometric structures. As shown in Figure 6c, the ordering of the enhanced βtot values by alkali metals is nearly K > Na > Li, with exception of the Na@Ge7Au and Na@Ge12Au clusters. Clearly, the first hyperpolarizabilities of AM@GenAu (AM = Na and K) are large enough to establish their strong nonlinear optical response, due to increased βtot values (~140–6111%) induced by the two alkali metal atoms. Especially, the Na@GenAu (n = 7 and 12) and K@GenAu (n = 2 and 3) clusters possess remarkable NLO responses, with the βtot values of 13,050, 6288, 9602, and 8812 a.u., respectively. Furthermore, the largest βtot value (13,050 a.u.) of Na@Ge7Au is comparable to those of Li2F (12,347 a.u.) [7] and Li2@BN-clusters(8,0) (12,282 a.u.) [8]. On the other hand, the largest βtot values is also about 1.87 times larger than that of the Na@Si9Nb+ cluster (6987 a.u.), which has the largest βtot value among the AM@SinNb+ clusters reported previously [16], and it shows that the germanium-based clusters doped by alkali metals provide the greater βtot than the silicon-based clusters. This result suggests that the germanium-based clusters with the large βtot, served as building blocks with tunable properties, may be promising for the design of novel macroscopic NLO materials.
To further understand the NLO behavior, we have performed the TDDFT calculations on the clusters with large βtot values at the CAM-B3LYP/def2-TZVPD level of theory. The most widely common two-level model is considered and it gains more insight into the NLO response [40,63]. The static first hyperpolarizability can be expressed by the following equation:
where ∆E, f0, and ∆μ are the transition energy, the oscillator strength, and the difference in dipole moment between the ground state and the crucial excited state, respectively. Of which the crucial excited state is specified by the largest f0 value, and the third-power of transition energy is inversely proportional to the βtot value. The calculated ∆E, f0, and ∆μ values by TDDFT for the crucial excited states are listed in Table 1. One can see that the ∆E values for K@Ge2Au, K@Ge3Au, and Na@Ge7Au are 6.056 eV, 5.702 eV and 3.798 eV, respectively. The smallest ∆E corresponds to the first excited state of the Na@Ge7Au cluster, which is in accordance with its largest βtot value. Thus, the two-level approximation can be used for qualitative description of the polarization mechanism, and the low transition energy is the most significant factor in designing the nonlinear optical materials.
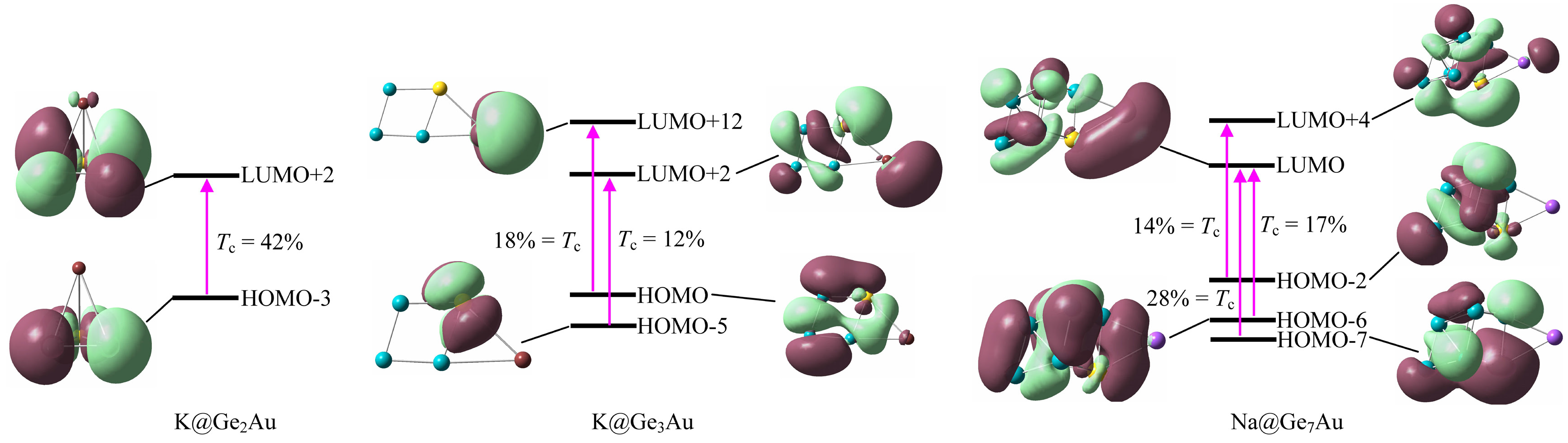
In order to explore the origin of the second-order NLO response, the molecular orbitals (MOs) features involving in the crucial transitions are presented in Figure 7. One can note that the crucial excitation of K@Ge2Au originates from a HOMO-3 → LUMO+2 (42%) transition at ~205 nm, and the two MOs are mainly localized on the two Ge atoms with s-lone pairs, and the Au atom with the dxy- and py-lone pairs, respectively. For K@Ge3Au, however, the large absorption band at ~217 nm with f0 = 0.297 ascribes to two mainly electron transitions, listed in Table 1, being the HOMO → LUMO+12 (18%) and HOMO-5 → LUMO+2 (12%) transitions, respectively. Of which the HOMO is mostly delocalized on the clusters surface (rhombic moiety) via the σ chemical bonds (e.g., Ge–Ge, Au–Ge, etc.), whereas the LUMO+12 is intensely localized on the K atom with the px-lone pair (see Figure 7). It is noteworthy that the strong transitions can be designated as intramolecular charge transfer, and this should be related to the large βtot value. Moreover, the difference of charge transfer can give rise to various electronic effects, but all of other MOs contribute little to electronic transitions of the cluster (≤8%). Table 1 lists the major contributions of electronic transition of the Na@Ge7Au cluster in crucial excited states. Its strongest transitions can be assigned to be composed by three mainly mixed excitations of HOMO-7 → LUMO (28%), HOMO-6 → LUMO (17%), and HOMO-2 → LUMO+4 (14%). From Figure 7, it is evident that HOMO-7 is delocalized on QP’s surface via σ bonds, and HOMO-2 and HOMO-6 can be formed by two π bonds, located at the inner and outer QP, whereas the electron distributions of LUMO are mainly associated with the Na–Au bond instead of other atoms, and LUMO+4 is mostly delocalized on the QP’s surface with additional distribution on Au atom. Therefore, the situation of intramolecular charge transfer of cluster directly influences the transition energy, which is a decisive factor that leads to a considerably large first hyperpolarizability.
4. Conclusions
In the present work, we have systematically investigated the structures, chemical stabilities and nonlinear optical properties as well as the chemical bonding and electronic structures of a series of alkali metals-adsorbed gold-germanium bimetallic clusters using the hybrid DFT-B3LYP method. Structurally, it has been determined that the adsorption of alkali metal atoms does not largely affect the structural framework of the gold-germanium clusters, and the alkali metals prefer energetically to be attached on clusters’ surface or edge. The Li-adsorbed bicapped pentagonal prism of the Li@Ge12Au cluster is electronically stable due to obeying the spherical aromaticity counting rule. Meanwhile, the molecular orbitals analysis reveals that the Au-4d state has strong interactions with the germanium atoms, and the hybridization between the Ge and Au atoms can enhance the chemical stability of the bimetallic cluster. This AdNDP analysis indicates that the localized 2c-2e σ-bonds are located on the clusters’ surface, while all the delocalized σ-bonds are involved in the endohedral Au dopant, which are responsible for structural stabilization of Li@Ge12Au. The static first hyperpolarizabilities are strongly related to the cluster size and geometric structure, and the AM@GenAu (AM = Na and K) clusters display the large βtot values, which are enough to establish their strong nonlinear optical behaviors, especially for Na@GenAu (n = 7 and 12) and K@GenAu (n = 2 and 3). The present results will inevitably stimulate future experimental and theoretical studies of germanium-based semiconductor clusters doped by alkali metals for the design of novel nonlinear optical materials.
Supplementary Materials
The following are available online at https://www.mdpi.com/2079-4991/7/7/184/s1. Table S1: Comparison of relative energies for the low-lying neutral AM@GenAu (AM = Li, Na, and K; n = 2–13) clusters, calculated by using the hybrid DFT-B3LYP functionals together with two different basis sets.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21603173, 21643014, and 21303135), the Natural Science Foundation of Shaanxi Province (Nos. 2016JQ5110, 2016JQ2030), the Special Natural Science Foundation of Science and Technology Bureau of Xi’an City (Nos. 2016CXWL16, 2016CXWL02, and 2016CXWL18).
Author Contributions
Xiaojun Li conceived and designed the project, performed the chemical bonding analyses and the linear and nonlinear optical (L&NLO) properties with all of the members, and wrote the manuscript; Shuna Li carried out the structural optimizations and contributed to the density of states; Hongjiang Ren analyzed the chemical stabilities of clusters and electronic structures; Juxiang Yang performed the TDDFT calculations and analyzed the results; Yongqiang Tang analyzed the spherical aromaticity. All authors contributed to the interpretation and discussion of the data at all stages.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khanna, S.N.; Jena, P. Atomic clusters: Building blocks for a class of solids. Phys. Rev. B 1995, 51, 13705. [Google Scholar] [CrossRef]
- Hou, N.; Wu, D.; Li, Y.; Li, Z.-R. Lower the electron affinity by halogenation: An unusual strategy to design superalkali cations. J. Am. Chem. Soc. 2014, 136, 2921–2927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-C.; Xu, H.-L.; Hu, Y.-Y.; Sun, S.-L.; Su, Z.-M. Quantum chemical research on structures, linear and nonlinear optical properties of the Li@n-acenes salt (n = 1, 2, 3, and 4). J. Phys. Chem. A 2011, 115, 2035–2040. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-M.; Wu, D.; Li, Y.; Li, Z.-R. Theoretical study on superalkali (Li3) in ammonia: Novel alkalides with considerably large first hyperpolarizabilities. Dalton Trans. 2014, 43, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Yaghobi, M.; Rafie, R.; Koohi, A. Optical and structural properties of the endohedral complexes M@C60 (M = Cs, Li, and Na). J. Mol. Struct. THEOCHEM 2009, 905, 48–50. [Google Scholar] [CrossRef]
- Wang, S.-J.; Li, Y.; Wang, Y.-F.; Wu, D.; Li, Z.-R. Structures and nonlinear optical properties of the endohedral metallofullerene-superhalogen compounds Li@C60–BX4 (X = F, Cl, Br). Phys. Chem. Chem. Phys. 2013, 15, 12903–12910. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Misra, N. Nonlinear optical behavior of LinF (n = 2–5) superalkali clusters. J. Mol. Model. 2015, 21, 305. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Zhou, Z.-J.; Liu, Y.-T. Li2 trapped inside tubiform [n] boron nitride clusters (n = 4–8): Structures and first hyperpolarizability. ChemPhysChem 2012, 13, 1307–1312. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Misra, N. Competition between alkalide characteristics and nonlinear optical properties in OLi3-M-Li3O (M = Li, Na, and K) complexes. Int. J. Quantum Chem. 2017, 117, 208–212. [Google Scholar] [CrossRef]
- Sun, W.-M.; Fan, L.-T.; Li, Y.; Liu, J.-Y.; Wu, D.; Li, Z.-R. On the potential application of superalkali clusters in designing novel alkalides with large nonlinear optical properties. Inorg. Chem. 2014, 53, 6170–6178. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.-Q.; Li, Z.-R.; Wu, D.; Li, Y.; Wang, B.-Q.; Gu, F.L.; Aoki, Y. Effect of the complexant shape on the large first hyperpolarizability of alkalides Li+(NH3)4M−. ChemPhysChem 2006, 7, 1759–1763. [Google Scholar] [CrossRef] [PubMed]
- DeBacker, M.G.; Mkadmi, E.B.; Sauvage, F.X.; Lelieur, J.-P.; Wagner, M.J.; Concepcion, R.; Kim, J.; McMills, L.E.H.; Dye, J.L. The lithium-sodium-methylamine system: Does a low-melting sodide become a liquid metal? J. Am. Chem. Soc. 1996, 118, 1997–2003. [Google Scholar] [CrossRef]
- Muhammad, S.; Xu, H.; Liao, Y.; Kan, Y.; Su, Z. Quantum mechanical design and structure of the Li@B10H14 basket with a remarkably enhanced electro-optical response. J. Am. Chem. Soc. 2009, 131, 11833–11840. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-L.; Li, Z.-R.; Su, Z.-M.; Muhammad, S.; Gu, F.L.; Harigaya, K. Knot-isomers of möbius cyclacene: How does the number of knots influence the structure and first hyperpolarizability? J. Phys. Chem. C 2009, 113, 15380–15383. [Google Scholar] [CrossRef]
- Karamanis, P.; Marchal, R.; Carbonniére, P.; Pouchan, C. Doping-enhanced hyperpolarizabilities of silicon clusters: A global ab initio and density functional theory study of Si10 (Li, Na, K)n (n = 1, 2) clusters. J. Chem. Phys. 2011, 135, 044511. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Han, Q.; Yang, X.; Song, R.; Song, L. Modification of alkali metals on silicon-based nanoclusters: An enhanced nonlinear optical response. Chem. Phys. Lett. 2016, 659, 93–99. [Google Scholar] [CrossRef]
- Donoso-Tauda, O.; Yepes, D.; Jaque, P.; Santos, J.C. Stability analysis of lithio-silicon si10li8 clusters: Planar bicyclic ring vs. Three-dimensional structures. Chem. Phys. Lett. 2014, 604, 72–76. [Google Scholar] [CrossRef]
- Li, X.; Su, K.; Yang, X.; Song, L.; Yang, L. Size-selective effects of geometry and electronic property on bimetallic au-ge nanoclusters. Comput. Theor. Chem. 2013, 1010, 32–37. [Google Scholar] [CrossRef]
- Deng, X.-J.; Kong, X.-Y.; Xu, X.-L.; Xu, H.-G.; Zheng, W.-J. Structural and magnetic properties of CoGen− (n = 2–11) clusters: Photoelectron spectroscopy and density functional calculations. ChemPhysChem 2014, 15, 3987–3993. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Claes, P.; Haertelt, M.; Lievens, P.; Janssens, E.; Fielicke, A. Structural determination of niobium-doped silicon clusters by far-infrared spectroscopy and theory. Phys. Chem. Chem. Phys. 2016, 18, 6291–6300. [Google Scholar] [CrossRef] [PubMed]
- Ngan, V.T.; Haeck, J.D.; Le, H.T.; Gopakumar, G.; Lievens, P.; Nguyen, M.T. Experimental detection and theoretical characterization of germanium-doped lithium clusters LinGe (n = 1–7). J. Phys. Chem. A 2009, 113, 9080–9091. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, Z.; Li, S. The nature of structure and bonding between transition metal and mixed si-ge tetramers: A 20-electron superatom system. J. Comput. Chem. 2016, 37, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-J.; Xu, X.-L.; Feng, G.; Xu, H.-G.; Zheng, W.-J. Structural and electronic properties of AuSin− (n = 4–12) clusters: Photoelectron spectroscopy and ab initio calculations. J. Phys. Chem. C 2016, 120, 25628–25637. [Google Scholar] [CrossRef]
- Li, X.; Su, K. Structure, stability and electronic property of the gold-doped germanium clusters: AuGen (n = 2–13). Theor. Chem. Acc. 2009, 124, 345–354. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Yang, J.; Jalbout, A.F. The structures, thermochemistry, and electron affinities of hydrogenated silicon clusters Si6Hn/Si6Hn− (n = 3–14). Int. J. Quantum Chem. 2009, 109, 1283–1301. [Google Scholar] [CrossRef]
- Xenides, D.; Karamanis, P.; Pouchan, C. A critical analysis of the performance of new generation functionals on the calculation of the (hyper) polarizabilities of clusters of varying stoichiometry: Test case the SimGen (m + n = 7, n = 0–7) clusters. Chem. Phys. Lett. 2010, 498, 134–139. [Google Scholar] [CrossRef]
- Knoppe, S.; Hakkinen, H.; Verbiest, T. Nonlinear optical properties of thiolate-protected gold clusters: A theoretical survey of the first hyperpolarizabilities. J. Phys. Chem. C 2015, 119, 27676–27682. [Google Scholar] [CrossRef]
- Knoppe, S.; Vanbel, M.; van Cleuvenbergen, S.; Vanpraet, L.; Buergi, T.; Verbiest, T. Nonlinear optical properties of thiolate-protected gold clusters. J. Phys. Chem. C 2015, 119, 6221–6226. [Google Scholar] [CrossRef]
- Ozga, K.; Kawaharamura, T.; Umar, A.A.; Oyama, M.; Nouneh, K.; Slezak, A.; Fujita, S.; Piasecki, M.; Reshak, A.H.; Kityk, I.V. Second order optical effects in Au nanoparticle-deposited ZnO nanocrystallite films. Nanotechnology 2008, 19, 185709. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ngan, V.T.; Nguyen, M.T. The aromatic 8-electron cubic silicon clusters Be@Si8, B@Si8+, and C@Si82+. J. Phys. Chem. A 2010, 114, 7609–7615. [Google Scholar] [CrossRef] [PubMed]
- Bauschlicher, C.W. A comparison of the accuracy of different functionals. Chem. Phys. Lett. 1995, 246, 40–44. [Google Scholar] [CrossRef]
- Garoufalis, C.S.; Zdetsis, A.D.; Grimme, S. High level ab initio calculations of the optical gap of small silicon quantum dots. Phys. Rev. Lett. 2001, 87, 276402. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Lu, S.-J.; Hu, L.-R.; Xu, X.-L.; Xu, H.-G.; Chen, H.; Zheng, W.-J. Transition from exohedral to endohedral structures of AuGen− (n = 2–12) clusters: Photoelectron spectroscopy and ab initio calculations. Phys. Chem. Chem. Phys. 2016, 18, 20321–20329. [Google Scholar] [CrossRef] [PubMed]
- Kanis, D.R.; Ratner, M.A.; Marks, T.J. Design and construction of molecular assemblies with large second-order optical nonlinearities. Quantum chemical aspects. Chem. Rev. 1994, 94, 195–242. [Google Scholar] [CrossRef]
- Li, X.; Ren, H.; Yang, X.; Song, J. Exploring the chemical bonding, infrared and UV–vis absorption spectra of oh radicals adsorption on the smallest fullerene. Spectrochim. Acta Part A 2015, 144, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.D.; Roothaan, C.C.J. Electric dipole polarizability of atoms by the hartree-fock method. I. Theory for closed-shell systems. J. Chem. Phys. 1965, 43, S34–S39. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. Cclib: A library for packageindependent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Yanaia, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange—Correlation functional using the coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Li, X.; Hopmann, K.H.; Hudecová, J.; Isaksson, J.; Novotná, J.; Stensen, W.; Andrushchenko, V.; Urbanová, M.; Svendsen, J.-S.; Bouř, P.; et al. Determination of absolute configuration and conformation of a cyclic dipeptide by nmr and chiral spectroscopic methods. J. Phys. Chem. A 2013, 117, 1721–1736. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kawazoe, Y. Metal-encapsulated icosahedral superatoms of germanium and tin with large gaps: Zn@Ge12 and Cd@Sn12. Appl. Phys. Lett. 2002, 80, 859–861. [Google Scholar] [CrossRef]
- Mahtout, S.; Tariket, Y. Electronic and magnetic properties of CrGen (15 ≤ n ≤ 29) clusters: A DFT study. Chem. Phys. 2016, 472, 270–277. [Google Scholar] [CrossRef]
- Tai, T.B.; Nguyen, M.T. A stochastic search for the structures of small germanium clusters and their anions: Enhanced stability by spherical aromaticity of the Ge10 and Ge122− systems. J. Chem. Theory Comput. 2011, 7, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Brack, M. The physics of simple metal clusters: Self-consistent jellium model and semiclassical approaches. Rev. Mod. Phys. 1993, 65, 677–732. [Google Scholar] [CrossRef]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.A.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.A.; Li, W.-L.; Piazza, Z.A.; Boldyrev, A.I.; Wang, L.-S. Complexes between planar boron clusters and transition metals: A photoelectron spectroscopy and ab initio study of CoB12− and RhB12−. J. Phys. Chem. A 2014, 118, 8098–8105. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.; Wang, L.-S. Beyond organic chemistry: Aromaticity in atomic clusters. Phys. Chem. Chem. Phys. 2016, 18, 11589–11605. [Google Scholar] [CrossRef] [PubMed]
- Popov, I.A.; Jian, T.; Lopez, G.V.; Boldyrev, A.I.; Wang, L.-S. Cobalt-centred boron molecular drums with the highest coordination number in the cob16− cluster. Nat. Commun. 2015, 6, 8654. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.I.; Wang, L.-S. All-metal aromaticity and antiaromaticity. Chem. Rev. 2005, 105, 3716–3757. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.; Chen, Z.; Jiao, H. Spherical aromaticity in ih symmetrical fullerenes: The 2(n + 1)2 rule. Angew. Chem. Int. Ed. 2000, 39, 3915–3917. [Google Scholar] [CrossRef]
- Hirsch, A.; Chen, Z.; Jiao, H. Spherical aromaticity of inorganic cage molecules. Angew. Chem. Int. Ed. 2001, 40, 2834–2838. [Google Scholar] [CrossRef]
- Tai, T.B.; Nguyen, M.T. Lithium atom can be doped at the center of a germanium cage: The stable icosahedral ge12li− cluster and derivatives. Chem. Phys. Lett. 2010, 492, 290–296. [Google Scholar] [CrossRef]
- King, R.B.; Heine, T.; Corminboeuf, C.; Schleyer, P.v.R. Antiaromaticity in bare deltahedral silicon clusters satisfying wade’s and hirsch’s rules: An apparent correlation of antiaromaticity with high symmetry. J. Am. Chem. Soc. 2004, 126, 430–431. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and soft acids and bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Oudar, J.L. Optical nonlinearities of conjugated molecules. Stilbene derivatives and highly polar aromatic compounds. J. Chem. Phys. 1977, 67, 446–457. [Google Scholar] [CrossRef]
Figure 1.
Low-lying structures of the AM@GenAu (n = 2–13) clusters, obtained by using the hybrid B3LYP functional. The blue, yellow and purple balls represent the Ge, Au and alkali metal (AM) atoms, respectively.
Figure 1.
Low-lying structures of the AM@GenAu (n = 2–13) clusters, obtained by using the hybrid B3LYP functional. The blue, yellow and purple balls represent the Ge, Au and alkali metal (AM) atoms, respectively.
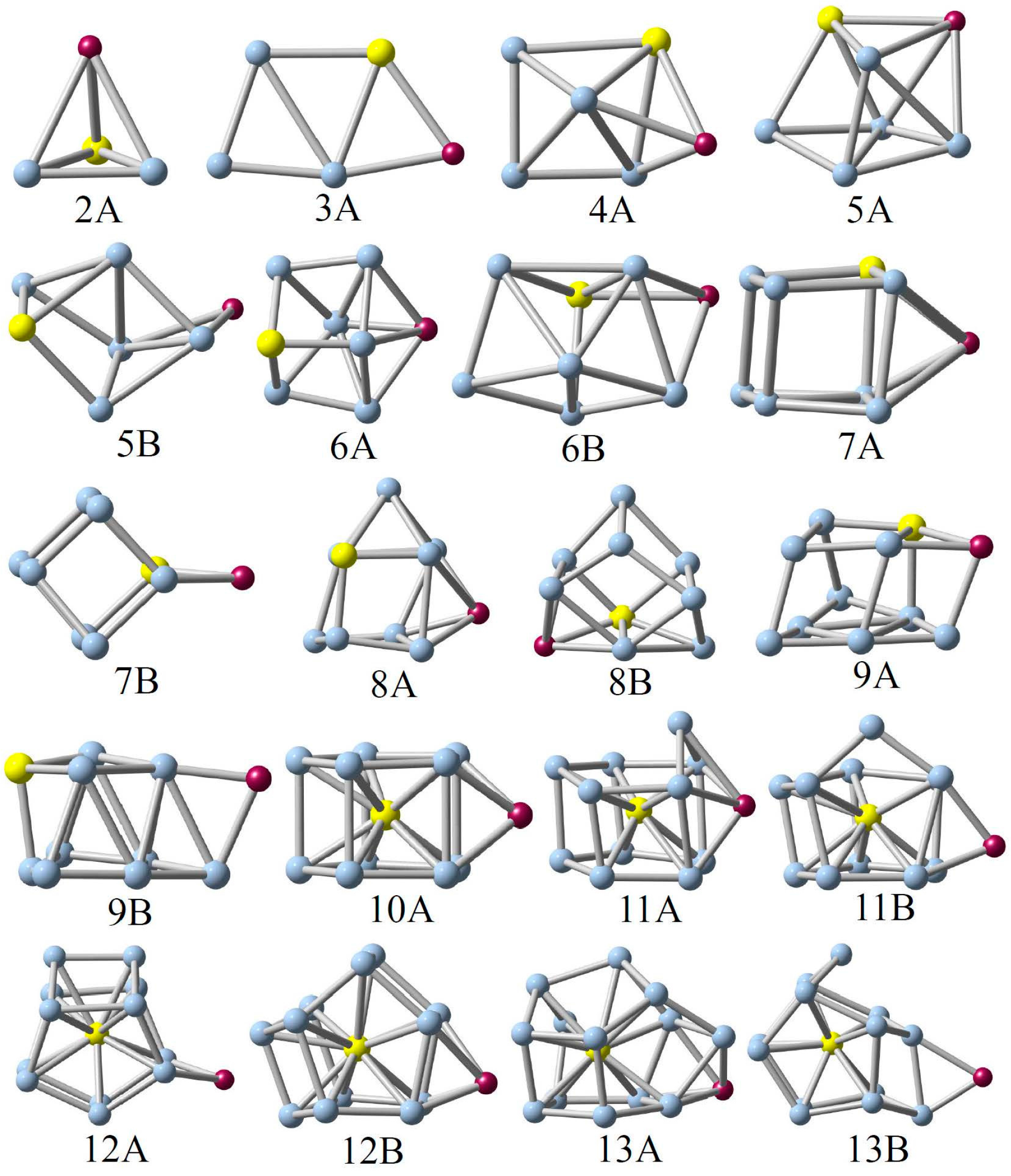
Figure 2.
Size dependences of (a) the average binding energies (Eb) and (b) dissociation energies (De) for the AM@GenAu (n = 2–13) clusters, obtained by using the hybrid B3LYP functional, in conjunction with the Def-TZVP basis set.
Figure 2.
Size dependences of (a) the average binding energies (Eb) and (b) dissociation energies (De) for the AM@GenAu (n = 2–13) clusters, obtained by using the hybrid B3LYP functional, in conjunction with the Def-TZVP basis set.
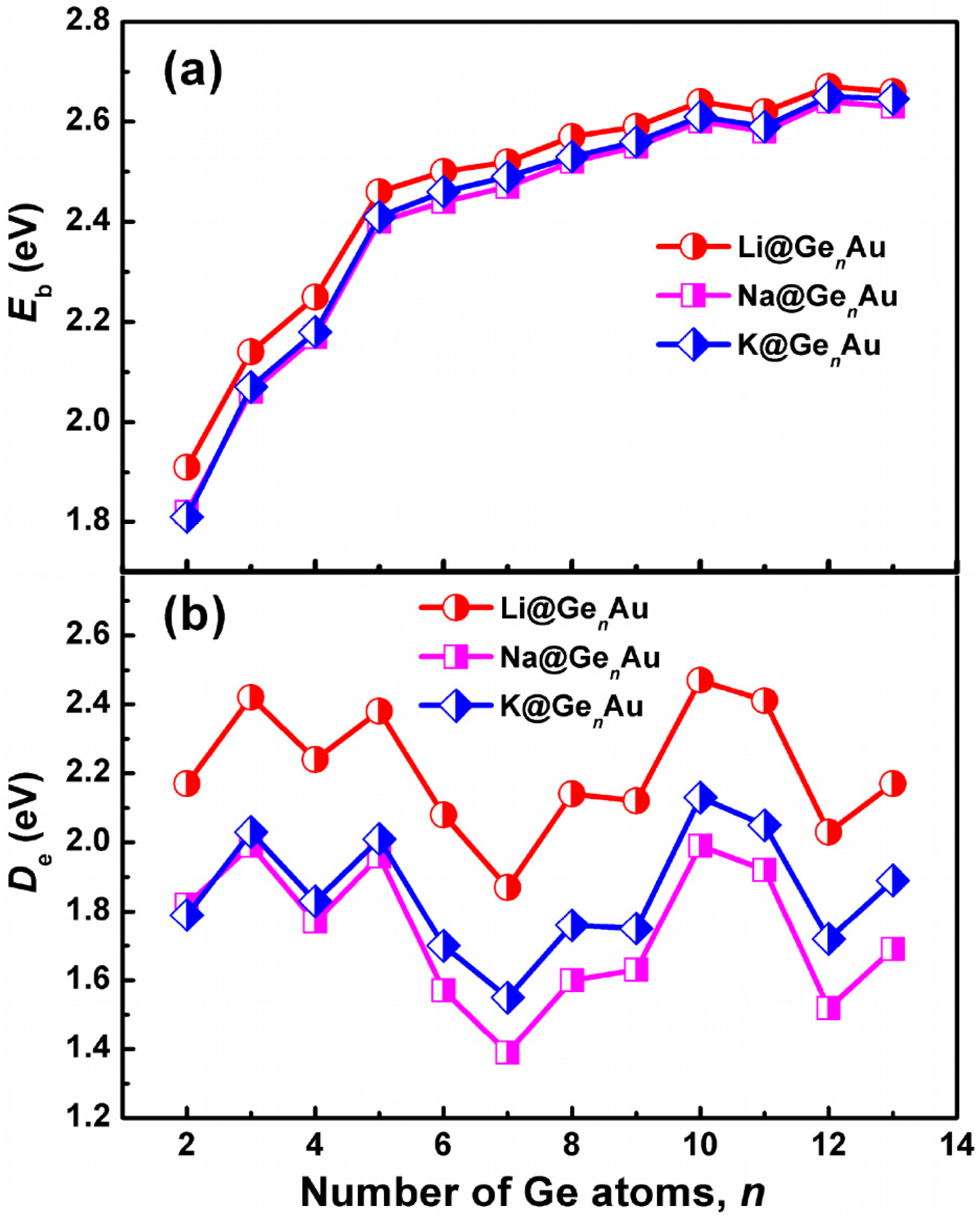
Figure 3.
Size dependences of (a) HOMO-LUMO gaps (GAPs), and (b) vertical ionization potentials (VIP) and vertical electron affinities (VEA) for the AM@GenAu (n = 2–13) clusters, obtained by using the hybrid B3LYP functional, in conjunction with the Def-TZVP basis set.
Figure 3.
Size dependences of (a) HOMO-LUMO gaps (GAPs), and (b) vertical ionization potentials (VIP) and vertical electron affinities (VEA) for the AM@GenAu (n = 2–13) clusters, obtained by using the hybrid B3LYP functional, in conjunction with the Def-TZVP basis set.
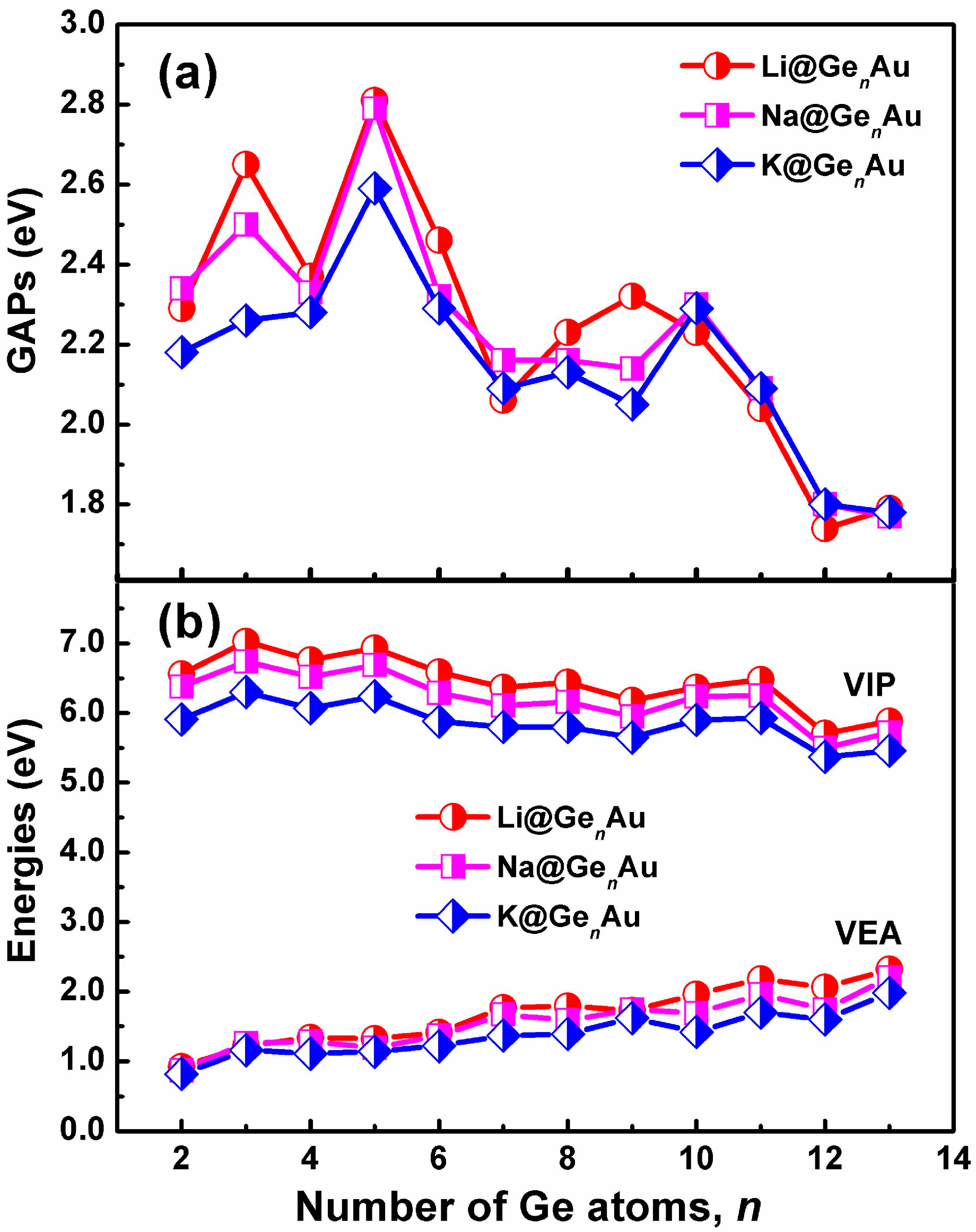
Figure 4.
Total (DOS) and partial (PDOS) density of states of the most stable Li@Ge12Au (12B) cluster, and the valence orbitals are obtained at the B3LYP/def-TZVP level of theory. The atomic contributions (Li, Au, and Ge) of the cluster to the DOS spectrum are labeled.
Figure 4.
Total (DOS) and partial (PDOS) density of states of the most stable Li@Ge12Au (12B) cluster, and the valence orbitals are obtained at the B3LYP/def-TZVP level of theory. The atomic contributions (Li, Au, and Ge) of the cluster to the DOS spectrum are labeled.
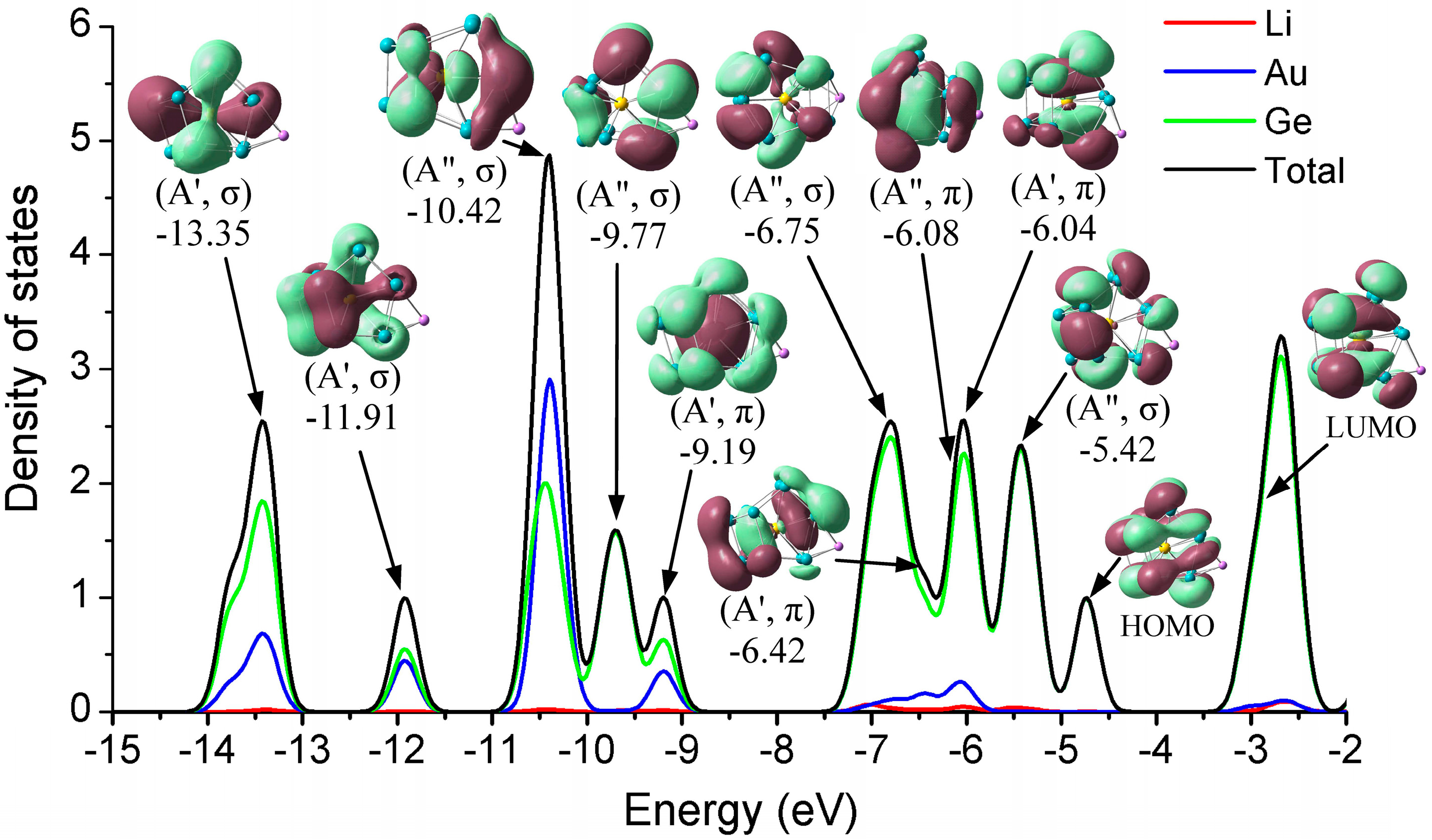
Figure 5.
AdNDP chemical bonding analyses of the Li@Ge12Au (12B) cluster. ON denotes the electron occupation number and is close to the ideal population of 2.00 |e|.
Figure 5.
AdNDP chemical bonding analyses of the Li@Ge12Au (12B) cluster. ON denotes the electron occupation number and is close to the ideal population of 2.00 |e|.
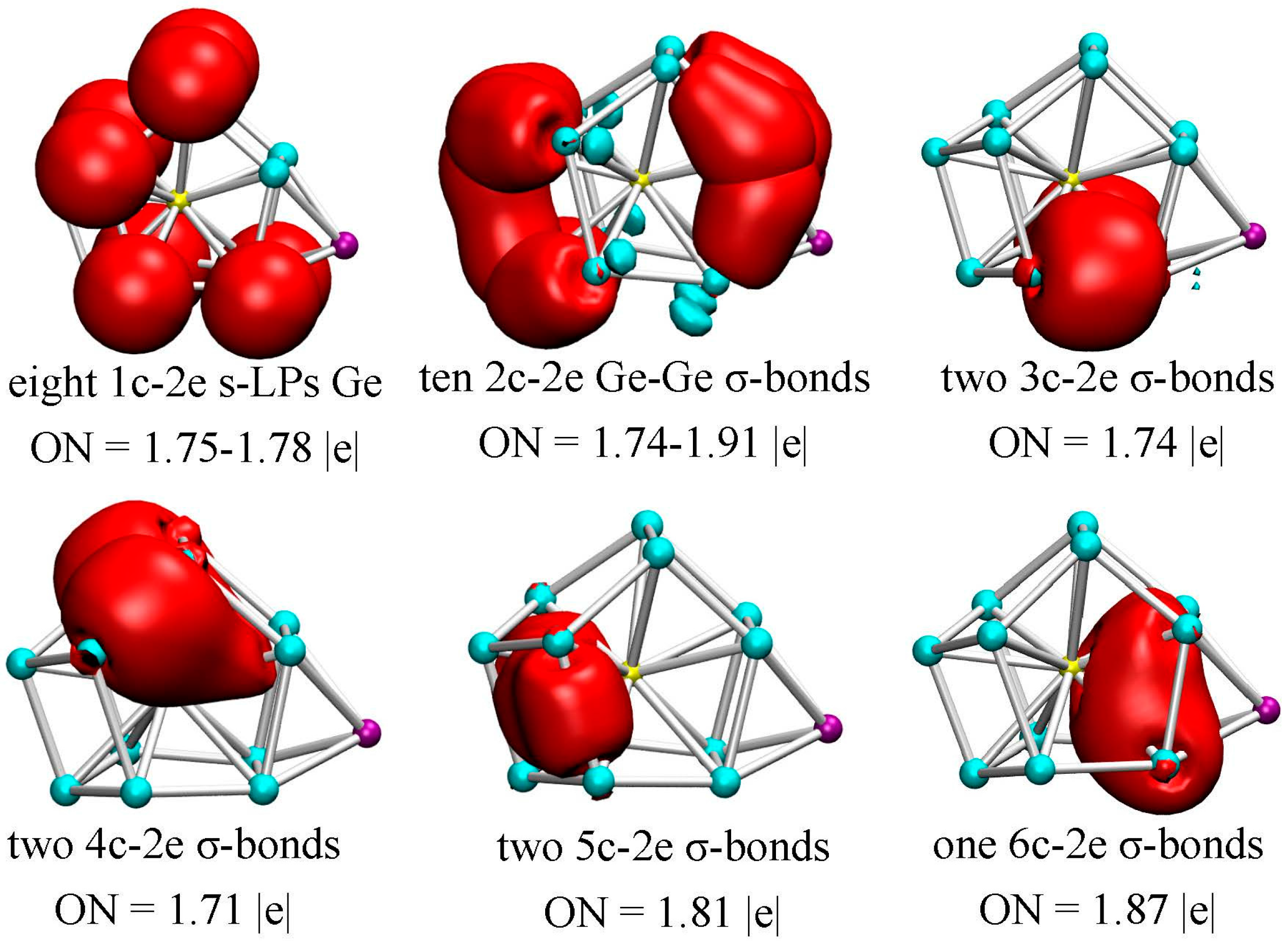
Figure 6.
Comparison of evaluated (a) dipole moments (μ0), (b) isotropic polarizabilities (αiso), and (c) static first hyperpolarizability (βtot) of AM@GenAu (AM = Li, Na, and K; n = 2–13) with GenAu cluster, calculated at the CAM-B3LYP/def2-TZVPD level of theory.
Figure 6.
Comparison of evaluated (a) dipole moments (μ0), (b) isotropic polarizabilities (αiso), and (c) static first hyperpolarizability (βtot) of AM@GenAu (AM = Li, Na, and K; n = 2–13) with GenAu cluster, calculated at the CAM-B3LYP/def2-TZVPD level of theory.
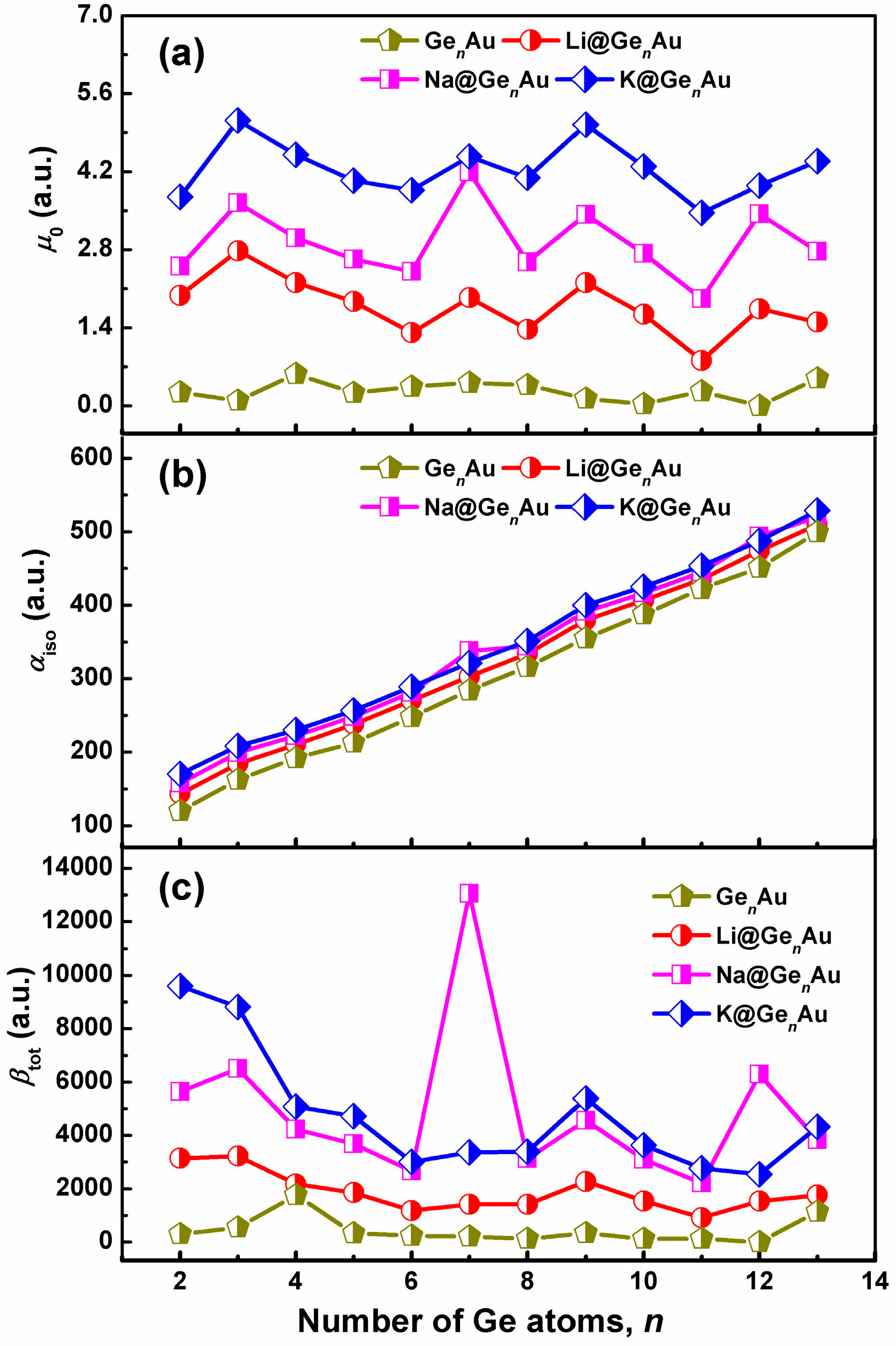
Figure 7.
Frontier molecular orbitals involving in the crucial transitions (Tc) for the K@Ge2Au, K@Ge3Au, and Na@Ge7Au clusters, computed at the CAM-B3LYP/def2-TZVPD level of theory.
Figure 7.
Frontier molecular orbitals involving in the crucial transitions (Tc) for the K@Ge2Au, K@Ge3Au, and Na@Ge7Au clusters, computed at the CAM-B3LYP/def2-TZVPD level of theory.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The isotropic polarizabilities (αiso, in a.u.), static first hyperpolarizability (βtot, in a.u.), transition energy (∆E, in eV), maximum oscillator strength (f0, in a.u.), and the change in dipole moment (∆μ, in Debye) for crucial excited states of the following clusters.
Table 1.
The isotropic polarizabilities (αiso, in a.u.), static first hyperpolarizability (βtot, in a.u.), transition energy (∆E, in eV), maximum oscillator strength (f0, in a.u.), and the change in dipole moment (∆μ, in Debye) for crucial excited states of the following clusters.
| Clusters | αiso | βtot | ∆E | f0 | ∆μ | Crucial Transitions * (%) |
|---|---|---|---|---|---|---|
| K@Ge2Au | 170 | 9602 | 6.056 | 0.358 | 0.371 | H-3 → L+2 (42%) |
| K@Ge3Au | 209 | 8812 | 5.702 | 0.297 | 0.623 | H → L+12 (18%), H-5 → L+2 (12%) |
| Na@Ge7Au | 338 | 13050 | 3.798 | 0.128 | 3.245 | H-7 → L (28%), H-6 → L (17%), H-2 → L+4 (14%) |
* H = HOMO, L = LUMO.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, X.; Li, S.; Ren, H.; Yang, J.; Tang, Y. Effect of Alkali Metal Atoms Doping on Structural and Nonlinear Optical Properties of the Gold-Germanium Bimetallic Clusters. Nanomaterials 2017, 7, 184. https://doi.org/10.3390/nano7070184
AMA Style
Li X, Li S, Ren H, Yang J, Tang Y. Effect of Alkali Metal Atoms Doping on Structural and Nonlinear Optical Properties of the Gold-Germanium Bimetallic Clusters. Nanomaterials. 2017; 7(7):184. https://doi.org/10.3390/nano7070184
Chicago/Turabian StyleLi, Xiaojun, Shuna Li, Hongjiang Ren, Juxiang Yang, and Yongqiang Tang. 2017. "Effect of Alkali Metal Atoms Doping on Structural and Nonlinear Optical Properties of the Gold-Germanium Bimetallic Clusters" Nanomaterials 7, no. 7: 184. https://doi.org/10.3390/nano7070184
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.