Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts

1
Fuels Research Center, Department of Chemical Technology, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand
2
Graduate School of Engineering & Green Mobility Collaborative Research Center, Nagoya University, Nagoya 464-8603, Japan
3
Center of Excellence on Petrochemical and Materials Technology (PETRO-MAT), Chulalongkorn University, Bangkok 10330, Thailand
4
Associate Fellow of Royal Society of Thailand (AFRST), Sanam Suea Pa, Dusit, Bangkok 10300, Thailand
*
Author to whom correspondence should be addressed.
Nanomaterials 2018, 8(4), 269; https://doi.org/10.3390/nano8040269
Submission received: 26 March 2018
/
Revised: 14 April 2018
/
Accepted: 19 April 2018
/
Published: 23 April 2018
(This article belongs to the Special Issue Nanostructure Metal Alloys for the Transformation of Biomass)
Abstract
:In this study, sol-immobilization was used to prepare gold nanoparticle (Au NP)-decorated titanium dioxide (TiO2) photocatalysts at different Au weight % (wt. %) loading (Aux/TiO2, where x is the Au wt. %) and Au–M NP-decorated TiO2 photocatalysts (Au3M3/TiO2), where M is bismuth (Bi), platinum (Pt) or palladium (Pd) at 3 wt. %. The Aux/TiO2 photocatalysts exhibited a stronger visible light absorption than the parent TiO2 due to the localized surface plasmon resonance effect. Increasing the Au content from 1 wt. % to 7 wt. % led to increased visible light absorption due to the increasing presence of defective structures that were capable of enhancing the photocatalytic activity of the as-prepared catalyst. The addition of Pt and Pd coupled with the Au3/TiO2 to form Au3M3/TiO2 improved the photocatalytic activity of the Au3/TiO2 photocatalyst by maximizing their light-absorption property. The Au3/TiO2, Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts promoted the formation of glyceraldehyde from glycerol as the principle product, while Au3Bi3/TiO2 facilitated glycolaldehyde formation as the major product. Among all the prepared photocatalysts, Au3Pd3/TiO2 exhibited the highest photocatalytic activity with a 98.75% glycerol conversion at 24 h of reaction time.
1. Introduction
Biodiesel has recently become a globally attractive renewable biomass energy source. The increasing production of biodiesel has led to glycerol, the inevitable by-product from the biodiesel production process, becoming oversupplied in the market. To capitalize on the economic aspect of biodiesel production, it is essential to contrive a process for using glycerol in more valuable products. Due to the highly functionalized chemical molecule of glycerol, it is a potential bio-platform molecule that can be converted into viable chemicals and intermediate products for the chemical industry through a range of catalytic routes, such as fermentation [1], dehydrogenation [2], dehydration [3], etherification [4], esterification [5] and oxidation [6]. Consequently, the valorization of biodiesel-derived glycerol has gained much attention for increasing the proficiency and economic viability of the use of biomass resources. Among a large number of effective processes for glycerol transformation, aerobic oxidation is one of the extensively studied routes, since it can promote the generation of high-value fine chemicals and useful intermediates in organic synthesis, such as glycolic acid (GCOA), glyceric acid (GCA), glyceraldehyde (GCD), and dihydroxyacetone (DHA) [6,7]. However, the major hindrance in the glycerol oxidation process at a commercial scale is its requirement for an alkaline condition, high operating temperature and pressure, and the deactivation of the utilized catalyst. Thus, there is an urgent need to find a new, green process to address the increasing demands of sustainable and clean-energy technologies. In the last two decades, photocatalytic technology has been regarded as one of the most attractive processes due to its superior features, such as being environmentally benign, high efficiency, and operation at room temperature and atmospheric pressure. Many researchers have focused on this process, with versatile applications ranging from the field of wastewater and air remediation [8,9], carbon dioxide (CO2) reduction [10], clean hydrogen (H2) production [11] and the synthesis of high-value organic compounds [12], towards the development of an efficient solar-to-energy conversion [13].
A wide spectrum of semiconductors, such as TiO2, bismuth(III) oxide (Bi2O3), zinc oxide (ZnO), silicon carbide (SiC), tungsten(VI) oxide (WO3) and carbon nitride (C3N4), have been explored for their use in photocatalytic processes. Among all the commercial photocatalysts, TiO2 has the greatest appeal as a benchmark material in the fields of environmental remediation and solar fuel production, and still remains the most efficient and suitable material for light-energy harvesting. This is due to its many positive features, such as excellent photo-stability, high conductivity, low toxicity, low price and corrosion resistance. Despite these advantages, TiO2 is only optically active in the ultraviolet (UV) light region (~5% of the total solar spectrum), and together with the rapid recombination rate of photo-induced charge carriers. These are the main barriers that make TiO2 industrially less attractive. Several approaches to improve the photocatalytic performance of TiO2 nanostructures have been explored. The loading of TiO2 with a co-catalyst, such as metal nanoparticles (NPs) or secondary narrowed-bandgap semiconductors, to act as an electron-sink in order to boost the quantum yield is a promising effective approach. In addition, the decoration of metal NPs can also provide active sites for the reaction and alter the adsorption behavior of molecules by changing their polarizability, among other properties, leading to the preferential generation of the desired value-added products [14]. Various plasmonic noble metal NP-decorated TiO2 nanostructures that enhance the generation and migration of hot electrons from the metal to TiO2 have been widely reported [15,16,17]. Among all the decorated metal co-catalysts, Au has been found to be the most active metal, which is attributed to its noble nature, i.e., visible light active arising from the collective coherent oscillation of surface electrons through localized surface plasmon resonance (LSPR) [18,19,20,21,22], its low susceptibility to oxygen, high chemical stability [23] and effective charge transfer from TiO2 to it and/or vice versa [24]. The synthesis of Au NPs has become of great interest in recent decades. The most impressive technique for AuNP synthesis is the two-phase reduction method developed by Brust and co-workers [25,26]. The breakthrough for using Au NPs as a heterogenous catalyst has occurred since Haruta and Hutching provided the significant observations that Au NPs was very active for CO oxidation and ethylene hydrochlorination [27,28]. Apart from that, to date many reports have pointed out that Au NPs are also active in a range of catalytic routes such as glycerol conversion [7,29,30], water splitting [20,31], H2 generation [21,32], the photo-oxidation of organic substrates [22,33,34,35] and the photo-reduction of organic compounds [36,37].
Besides the monometallic Au, the combination of Au NPs with other metals can enable an enhanced global reaction system and promote the desired reaction pathway depending on their structural and/or electronic properties at the nanoscale level. Bimetallic Au-based NPs have been studied in a variety of reactions, including solvent-free benzyl alcohol oxidation to benzaldehyde [38]; selective oxidation of methanol to methyl formate [39]; photocatalytic H2 production from ethanol reforming over core–shell Au-palladium (Pd) promoters [40] and over bimetallic Au-platinum (Pt) NPs [41]; selective oxidation of furfural to furoic acid [42]; selective oxidation of 1,2-propanediol to lactic acid [43]; selective oxidation of ethanol to acetaldehyde [44]; and the degradation of ciprofloxacin [45]. Apart from these reactions, glycerol oxidation in a liquid phase system has also been intensively investigated over a bimetallic Au-based system [46,47,48,49]. The synergism between Au and a coupled metal (e.g., Pt or Pd) can boost up the glycerol conversion and tune the selectivity of oxidation products. Moreover, the synthesis procedure and particle size of as-prepared bimetallic Au-based NPs also significantly affected the product distribution and route of glycerol transformation [47,50,51].
As far as we know, there have been only a few research reports on the photocatalytic oxidation of glycerol over metal-decorated TiO2. Panagiotopoulou et al. [52] prepared Pt impregnated on TiO2 and utilized the prepared Pt/TiO2 for the photo-reforming and photo-oxidation of glycerol. The Pt/TiO2 photocatalyst exhibited a more efficient reaction rate for glycerol oxidation than bare TiO2, which was mainly due to the increased separation of the charge carriers and the elevation of the rate-limiting cathodic half reaction (oxygen (O2) reduction) in the glycerol photo-oxidation. Likewise, the addition of any of Bi, Au, Pt or Pd on TiO2 was reported in our previous work to further enhance the rate of glycerol photo-oxidation, where Au-decorated TiO2 exhibited the best performance [53]. However, the photocatalytic conversion of glycerol over the surface of metals and metal oxides to the desired products still requires a thorough comprehension of the surface chemistry and reaction mechanism. The bias towards certain reaction pathways can be modulated by varying the reaction conditions (light, pressure, temperature, etc.), reaction media (water with O2 or hydrogen peroxide) and the nature of the catalyst surface (i.e., monometal, bimetal and/or metal oxide). Thus, in this work, we investigated the photocatalytic oxidation of glycerol over Au-based mono- and bimetallic NP-decorated TiO2. The aim was to explore the individual as well as the combined effects of Au, Au-Bi, Au-Pt and Au-Pd on the photocatalytic performance of TiO2 for the photocatalytic oxidation of glycerol. The Bi, Pt and Pd NPs were selected to combine with Au because Bi prefers to react with the secondary hydroxyl group of glycerol to form DHA while the Pt and Pd can promote the activity of glycerol oxidation [53,54,55,56].
2. Experiment
2.1. Preparation of the Au/TiO2 and AuM/TiO2 Photocatalysts
Both mono- and bimetallic NP-decorated TiO2 photocatalysts were prepared by sol-immobilization, which has been previously reported to be an outstanding preparation method [51,57]. Commercial TiO2 powder (Sigma Aldrich, St. Louis, MO, USA) containing ≥99% anatase phase structure was used as the support semiconductor material. The Au NP-decorated TiO2 (Au/TiO2) at different Au contents from 1 wt. % to 7 wt. % were prepared using sodium borohydride (NaBH4; Loba Chemie, Mumbai, India) as a reducing agent. In brief, for the 1 wt. % Au NP-decorated TiO2 (Au1/TiO2), 40.8 mg of HAuCl4·3H2O (Sigma Aldrich) was dissolved in deionized (DI) water. Then, 2 wt. % polyvinyl alcohol (PVA; [-CH2CHOH-]n, 99% hydrolyzed, Sigma Aldrich) was added to a 1:10 molar ratio of PVA: HAuCl4·3H2O with stirring at a constant rate of 300 rpm in order to stabilize the metal dispersion on the support and prevent agglomeration. Next, 1.98 g of TiO2 (Anatase, Sigma Aldrich) was added to the prepared solution and then excess NaBH4 was slowly added to the colloidal solution. The reaction was held for 24 h at atmospheric pressure and temperature to obtain the complete sol immobilization. The obtained mixture was filtered and washed thoroughly several times with DI water until no chloride ions remained, which was detected in the filtrate solution by the silver nitrate test. Finally, the obtained solid was dried at 110 °C in a hot air oven overnight and, subsequently, the organic scaffold residue was removed and the catalyst was activated by heat treatment under a N2 flow at 350 °C for 3 h followed by a H2 flow at 350 °C for 3 h, respectively, to yield the ready-to-use Au1/TiO2. A similar procedure was repeated for the 3, 5 and 7 wt. % Au NP-decorated TiO2, except using 122.45, 204.06 and 285.72 mg of HAuCl4·3H2O instead of 40.8 mg to yield the Au3/TiO2, Au5/TiO2 and Au7/TiO2 photocatalysts, respectively.
To prepare the Au-based bimetallic NP-decorated TiO2 photocatalysts with a 3 wt. % loading of both Au and the selected metal NPs (Au3M3/TiO2), three types of second metal (Bi, Pt, and Pd) were separately incorporated with the Au on TiO2 to yield the Au3Bi3/TiO2, Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts, respectively, via the same method. For the Au3Bi3/TiO2 photocatalyst, 122.45 mg of HAuCl4·3H2O was dissolved in 10 mL of DI water, while 90.9 mg of bismuth(III) chloride (BiCl3; Sigma Aldrich) was dissolved in 10 mL of 0.2 M of hydrochloric acid (HCl; 37% purity, Sigma Aldrich). Both solutions were mixed together and added to 10.5 mL of 2 wt. % PVA with stirring at constant rate of 300 rpm in order to stabilize the metal dispersion on the support and prevent agglomeration. Then, 1.88 g of TiO2 was added to the prepared solution and subsequently reduced by the slow addition of NaBH4 to get the colloid solution. The reaction was held for 24 h to obtain complete sol immobilization. Then, the obtained mixture was filtered and washed thoroughly with DI water until no chloride ions. Finally, the obtained solid was dried in an oven and subsequently removed the organic scaffold residue and activated by heat treatment under a N2 flow followed by a H2 flow as above to yield the ready-to-use Au3Bi3/TiO2. Similar procedures were repeated for the synthesis of the Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts except using 159.6 mg of chloroplatinic acid hexahydrate (H2PtCl6·6H2O; Sigma Aldrich) dissolved in 10 mL of DI water and 100 mg of palladium (II) chloride (PdCl2; Sigma Aldrich) solution dissolved in 10 mL of 0.2 M HCl instead of the BiCl3.
2.2. Characterization of the as-Prepared Aux/TiO2 and Au3M3/TiO2 Photocatalysts
The diffractogram peaks of all prepared photocatalysts were first characterized by X-ray diffraction (XRD) using a D8 Discover-Bruker AXS X-ray diffractometer (Billerica, MA, USA) equipped with Cu Kα in order to evaluate the crystal and phase structure. The X-ray diffractometer was operated at 40 mA and 40 kV with acquiring step of 0.02° between angles of 10–80 degrees. The diffractogram peaks were identified using the JCPDS database. The loading content of the decorated metal on the TiO2 surface was examined by scanning electron microscopy (SEM; JSM-6610LV) (Peabody, MA, USA) equipped with energy-dispersive X-ray spectrometry (EDS) to perform elemental analysis at the atomic resolution. The dispersion of decorated-metals and particle size distribution were observed by transmission electron microscopy (TEM; JEOL-2100Plus, Tokyo, Japan) at an electron acceleration of 200 kV. Elemental line scan and mapping of the decorated metal were also observed on a JEOL 2100 Plus equipped with a Bruker EDS using a nominal electron beam size of 1 nm. The average metal particle sizes and their distribution were calculated by averaging not less than 100 particles randomly distributed in the TEM images. The diffuse reflectance spectra were monitored via UV-visible near infrared spectrometry (UV-Vis-NIR; Perkin Elmer, Lambda 950, Waltham, MA, USA) over a wavelength of 320–820 nm. The elemental oxidation states of all the prepared photocatalysts were assessed by X-ray photoelectron spectroscopy (XPS; PHI 5000 VersaProbeII) (ULVAC-PHI, Inc., Kanagawa, Japan) with a monochromatized Al Kα source (hν = 1486.6 eV). Accurate binding energies (±0.1 eV) were established with respect to the position of the adventitious carbon C1s peak at 284.8 eV. The spectra deconvolution was performed using the XPSPEAK41 software package (Kratos Analytical Ltd., Manchester, UK).
2.3. Photocatalytic Activity Test
The photocatalytic activity of all the prepared photocatalysts was tested via the photocatalytic oxidation of glycerol at room temperature and ambient pressure using a 120 W high-pressure mercury lamp (RUV 533 BC, Holland, The Netherlands) at light intensity 4.7 mW/cm2. The photoreactor was placed in the middle of a UV-protected box (0.68 m × 0.68 m × 0.78 m) containing 100 mL of 0.3 M glycerol (QReC). The solution was agitated at 300 rpm to achieve complete mixing. Oxygen was supplied continuously into the reactor at a constant feed rate of 200 mL/min. Prior to light illumination, the selected photocatalyst was suspended in the glycerol solution in a dark room for 30 min to reach an adsorption equilibrium. As the experiment progressed, 2 mL of sample was collected and then centrifuged on a KUBOTA KC-25 digital laboratory centrifuge (KUBOTA, Tokyo, Japan) to separate the solid catalyst from the aqueous product. The variation in the glycerol concentration and the generation of all monitored compounds, including GCD, DHA, hydroxypyruvic acid (HPA), GCOA, formaldehyde (FMD) and glycolaldehyde (GCAD), were analyzed by high-performance liquid chromatography with a RID-10A refractive index detector (Shimadzu, LC-10 ADVP, Kyoto, Japan). The stationary phase was an Aminex HPX-87H ion-exclusion column (300 × 7.8 mm), and the mobile phase was a 70:30 (v/v) water: acetonitrile solution in 0.5 mM H2SO4 at a constant flow rate of 0.4 mL/min. Standard solutions of glycerol, GCD (Sigma Aldrich), DHA (Merck, Kenilworth, NJ, USA), HPA (Sigma Aldrich), GCA (Ajax Finechem, Taren Point, Australia), FMD (Merck), GCOA (Ajax Finechem) and GCAD (Sigma Aldrich) were run to identify the retention times and determine the relationships between the peak area and concentration. The conversion of glycerol (X) and yield (Y) of each selected product of the photocatalytic oxidation were calculated according to Equations (1) and (2), respectively:
3. Results and Discussion
3.1. Au/TiO2 Photocatalysts
3.1.1. Photocatalyst Morphology
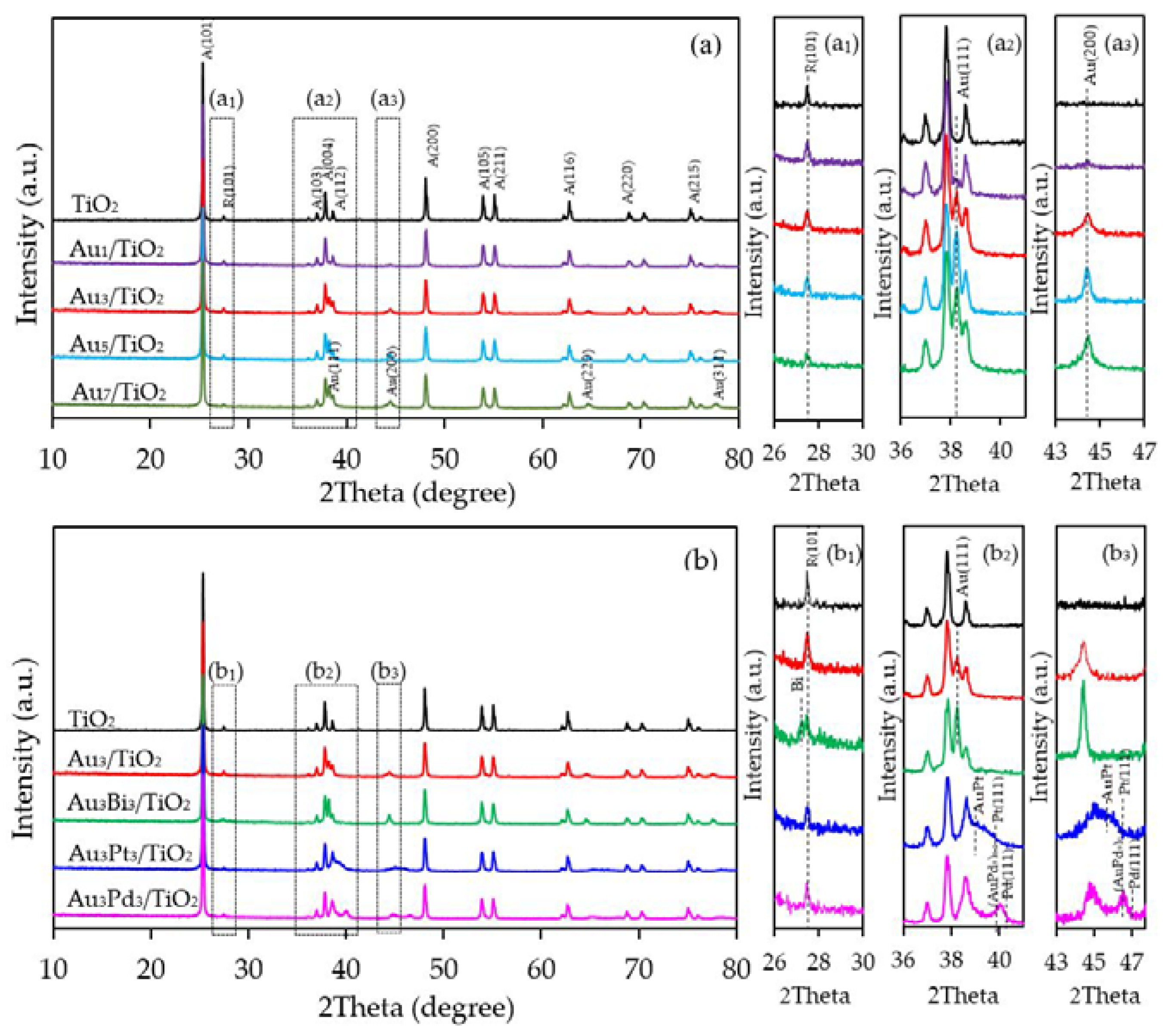
Representative XRD patterns of the as-synthesized Aux/TiO2 photocatalysts with different metal loading contents are displayed in Figure 1a. All photocatalysts exhibited the main diffraction peaks of TiO2 in the anatase phase at a 2θ of 25.3°, 37.0°, 37.9°, 38.6°, 48.1°, 53.9°, 55.1°, 62.7°, 68.8°, 70.3° and 75.1°, corresponding to the (101), (103), (004), (112), (200), (105), (221), (204), (116), (220) and (215) crystal planes, respectively (JCPDS No. 21-1272), with an additional rutile peak in trivial content at 27.5° corresponding to the (101) crystal plane (JCPDS No. 04-0802) (Figure 1a1). In addition, the diffraction peaks of Au NPs appeared at about 38.2°, 44.4°, 64.6° and 77.6°, assigning to the face-centered cubic structure of Au with (111), (200), (220) and (311) planes (JCPDS No. 002-1095), indicating the presence of metallic Au in the as-synthesized Aux/TiO2 photocatalysts. The intensity of the Au peaks increased as the content of Au decorated on the TiO2 surface increased, but without a shift in the peak position (Figure 1a2,a3).
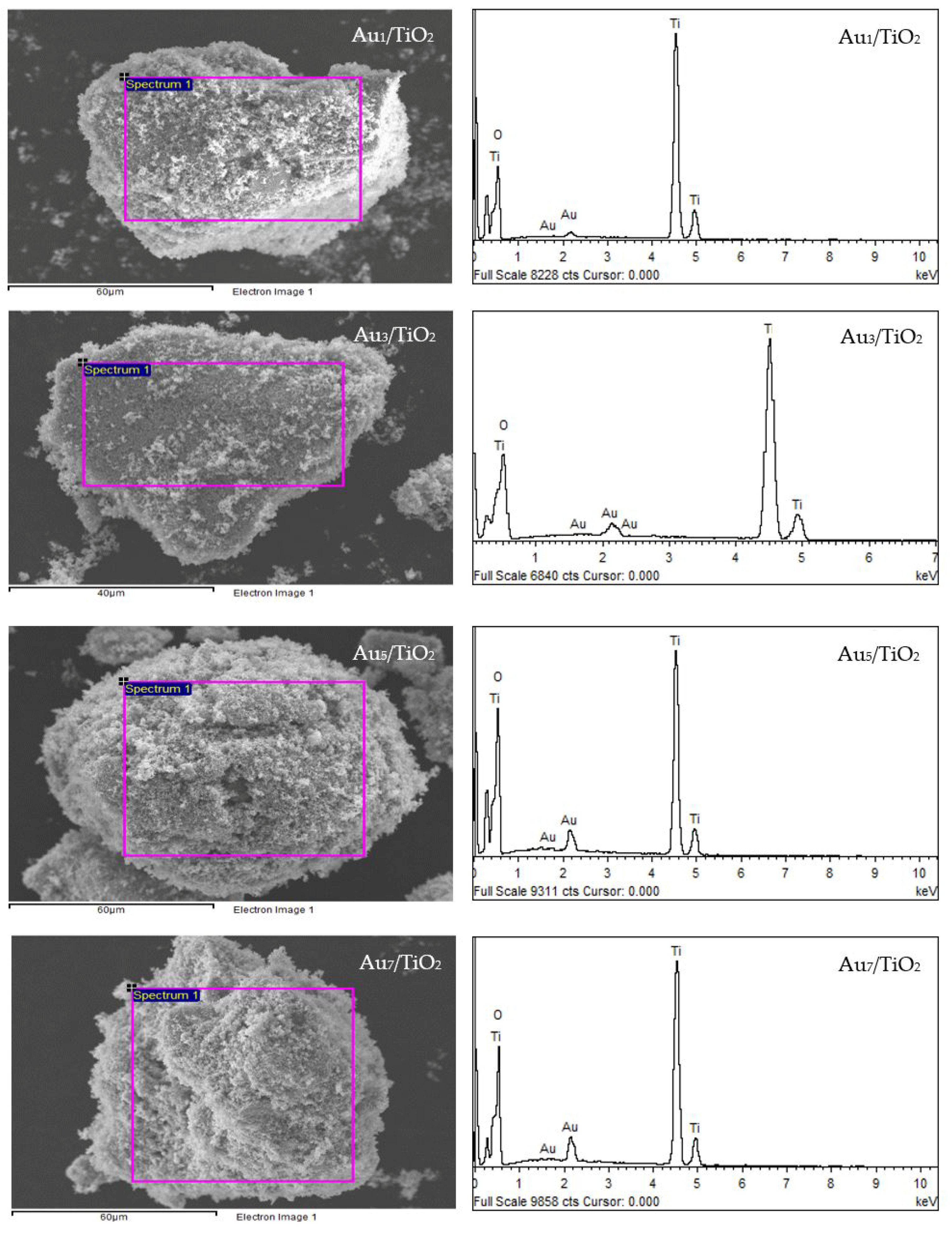
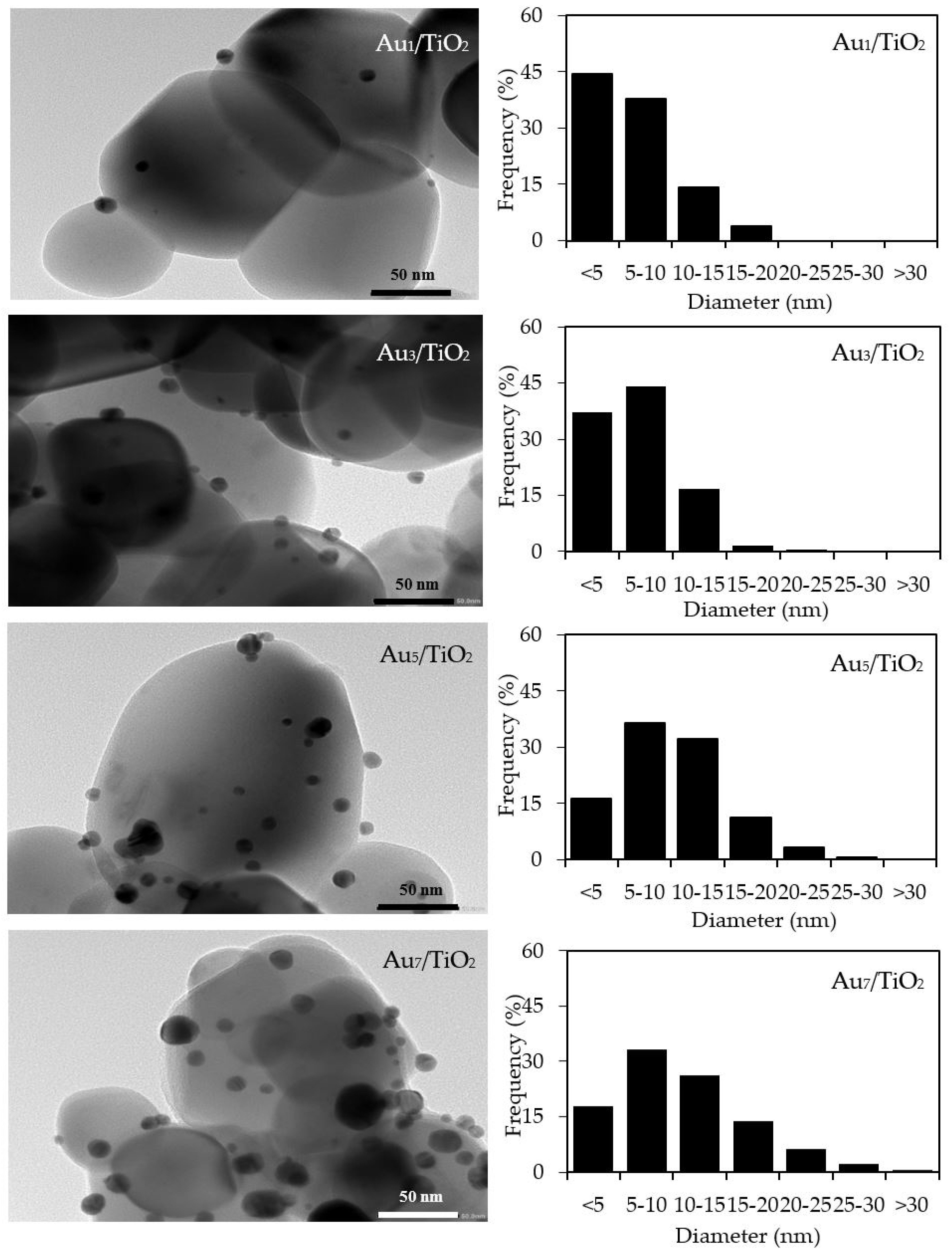
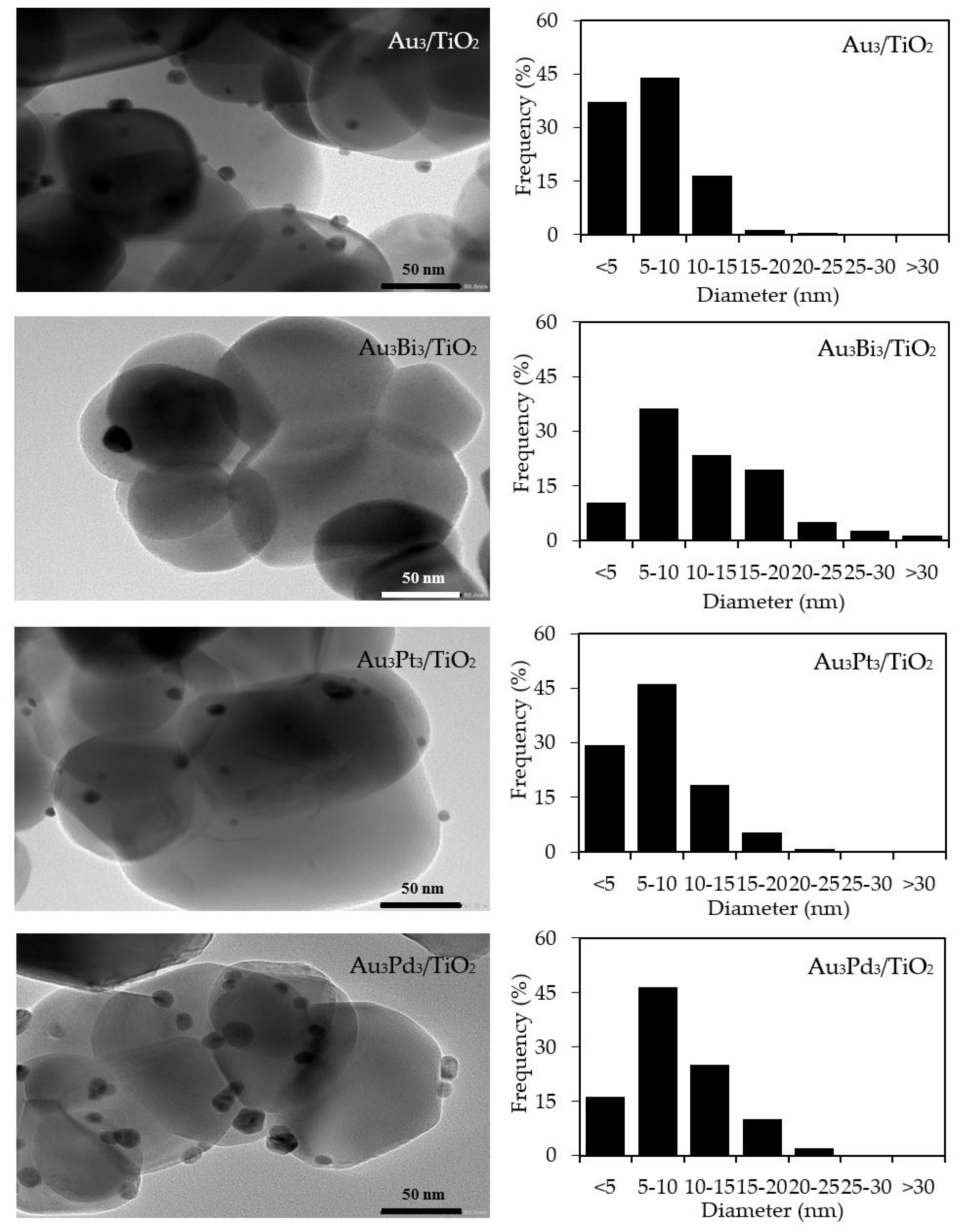
The qualitative presence of Au in all the Aux/TiO2 photocatalysts was also confirmed by SEM–EDS analysis (Figure 2). Quantitatively, as summarized in Table 1, the Au content in each Aux/TiO2 photocatalyst was close to the set value. The TEM images of the as-prepared photocatalysts with the derived Au NPs size distribution are displayed in Figure 3. They exhibited well-dispersed decorated Au NPs on the TiO2 surface with a narrow size distribution. The average size of Au NPs increased slightly as the Au content increased, suggesting an agglomeration of Au NPs in the presence of a high Au loading content.
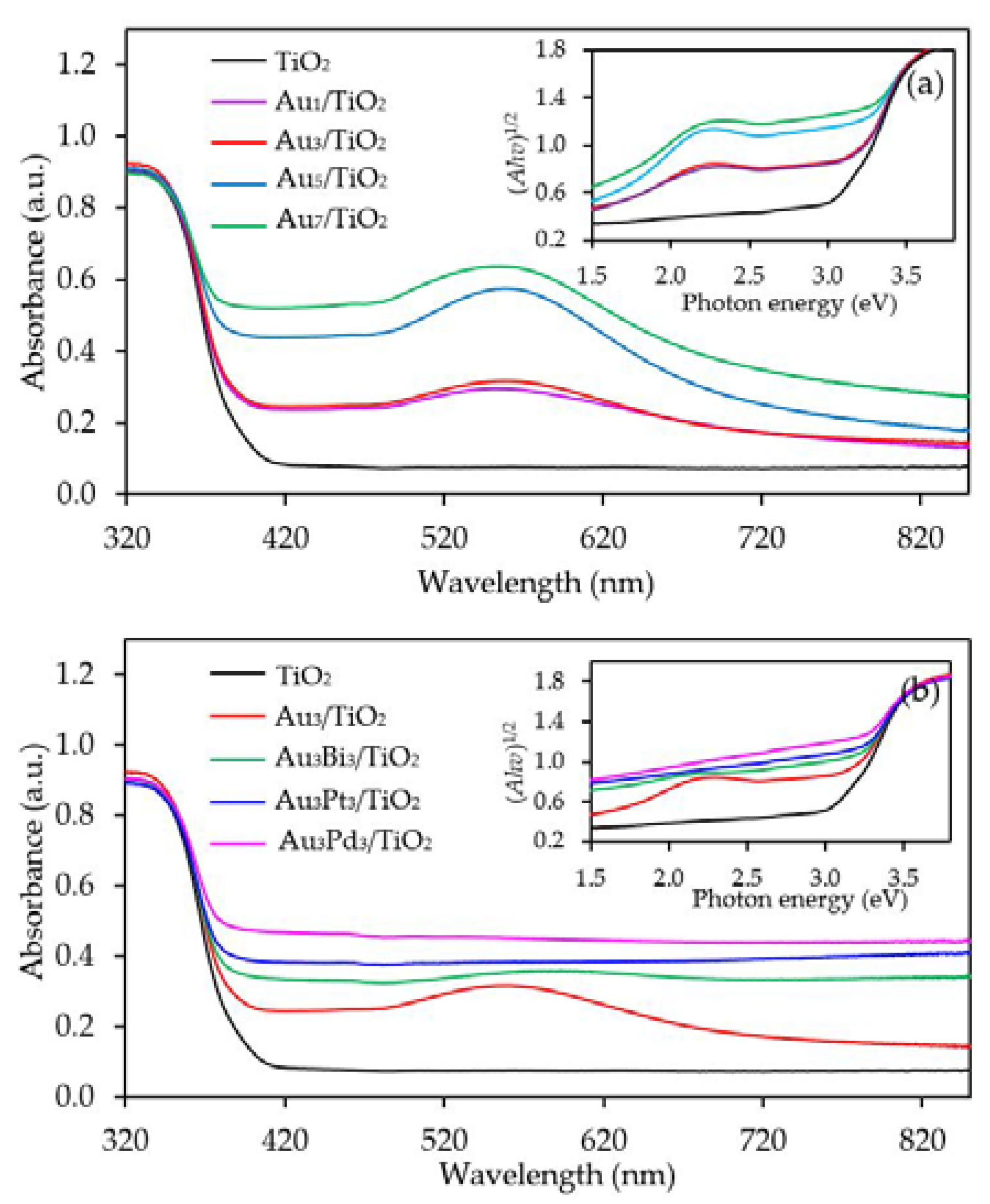
Figure 4a exhibits the UV-vis absorption spectra of the Aux/TiO2 photocatalysts with different Au contents. The parent TiO2 exhibited the main intense UV absorption band at a wavelength of less than 400 nm with no absorption band in the visible light region or at a wavelength of greater than 400 nm. This was mainly due to its large bandgap energy that enabled the electron transfer from the filled valence O 2p orbitals of the valence band to the vacant Ti 3d orbitals of the conduction band only by the excitation of the photon energy from the UV light region. However, all the Aux/TiO2 photocatalysts exhibited an enhanced visible light absorption with an intense broad absorption band centered at wavelength around 540 nm. The peak intensity increased as the Au content increased from 1.0 wt. % to 7.0 wt. %. The presence of the visible light absorption of all Aux/TiO2 photocatalysts might be caused by the LSPR effect of the decorated Au NPs that can absorb visible light through the polarization and oscillation of the conduction electrons in the metal structure [58]. However, as reported previously, the LSPR band of well-dispersed spherical Au nanocrystals is generally sharp and appears at a wavelength of 520 nm [59]. The absorption center of all the prepared Aux/TiO2 photocatalysts in this study deviated from the mentioned wavelength (520 nm) with a broad tail extending towards longer wavelengths, which might be attributed to the anisotropy of the trigonal/prismatic shape (non-spherical) of the Au nanocrystals [60]. The bandgap values of all samples were determined from a Tauc’s plot (inset of Figure 4a), and are listed in Table 1. The addition of Au NPs at 1 and 3 wt. % affected insignificantly the bandgap energy of the parent TiO2. In that, both contents decreased the bandgap value of TiO2 from 3.02 eV to 3.0 eV, probably because both low Au contents can induce only a small formation of Ti3+ defect structure which cannot intensely alter the major electronic state position of the O2p valence orbital as well as the localized band bending of the O2p valence band edge maximum. However, addition of Au NPs at loading of 5 and 7 wt. % importantly decreased the band-gap value of TiO2 to 2.94 and 2.80 eV, respectively. This indicated that the presence of a high defective TiO2 structure in the presence of high Au content (5 and 7 wt. %) can alter the major electronic state of the valence band and conduction band position.
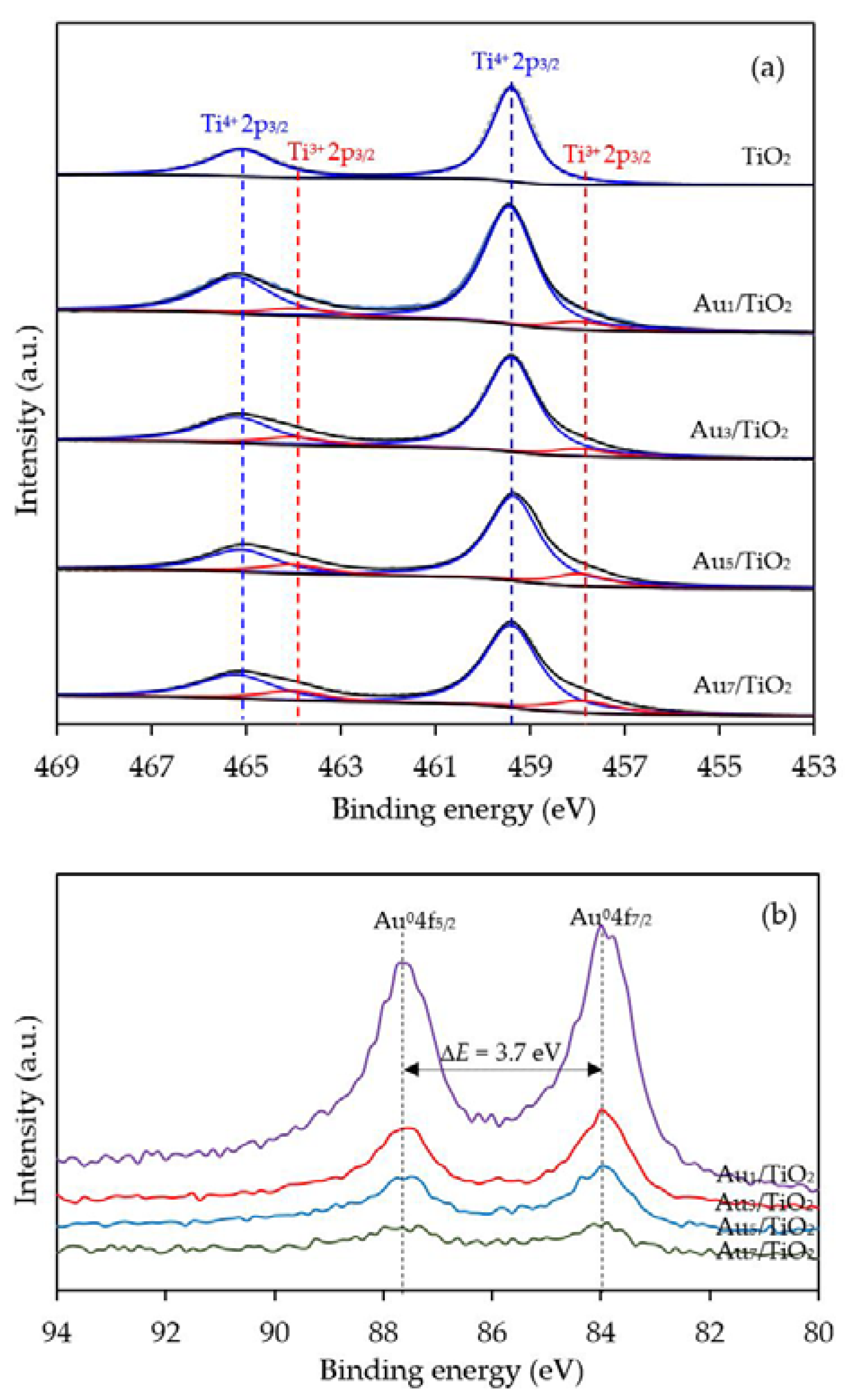
To confirm the formation of defective structure of TiO2 in the presence of Au NPs, XPS analysis with a high resolution (HR) Ti2p state was employed. As demonstrated in Figure 5a, two symmetrical peaks at 459.4 and 465.1 eV, assigned to Ti2p3/2 and Ti2p1/2, were exhibited in the Ti2p core-level spectra of all the prepared Aux/TiO2 photocatalysts. The different binding energy of the observed spin-orbit splitting between the Ti2p3/2 and Ti2p1/2 was around 5.7 eV, in accordance with the typical value of Ti4+ sites coordinated to oxygen atoms in TiO2 [61]. Interestingly, two additional tailored peaks were observed at a lower binding energy of around 457.9 and 464.0 eV, which can be ascribed to the existence of Ti3+ species. The generation of Ti3+ could have originated from the incorporation of the decorated-metal on the TiO2 under the H2 treatment [62,63]. This is because the decorated metal, Au in this case, can induce the generation of oxygen vacancies that are subsequently essential to form the Ti3+-O−-Ti4+ defect structures [64]. After deconvolution, the relative content of Ti3+/Ti4+ in all the Aux/TiO2 photocatalysts was obtained, and is summarized in Table 1. The Ti3+/Ti4+ ratio increased as the Au content increased, becoming almost two-fold higher in Au7/TiO2 than in Au1/TiO2, suggesting that the decoration of TiO2 with a high Au content induced a high content of Ti3+ defective structures in the TiO2.
With respect to the valence state of the decorated metal, the Au 4f level spectra were also examined. All the prepared photocatalysts displayed the main Au 4f7/2 and Au 4f5/2 peaks at a binding energy of about 84.0 and 87.7 eV with a spin-orbit splitting of 3.7 eV (Figure 5b), which are in excellent agreement with the values of the monometallic Au0 state [65]. Therefore, the Au NPs existed totally in metallic form on the surface of the as-prepared Aux/TiO2 photocatalysts. No shift in the Au 4f binding energy was observed, suggesting that the addition of Au within the investigated weight content (1–7 wt. %) did not alter the chemical state of the decorated Au NPs on the TiO2.
3.1.2. Photocatalytic Activity
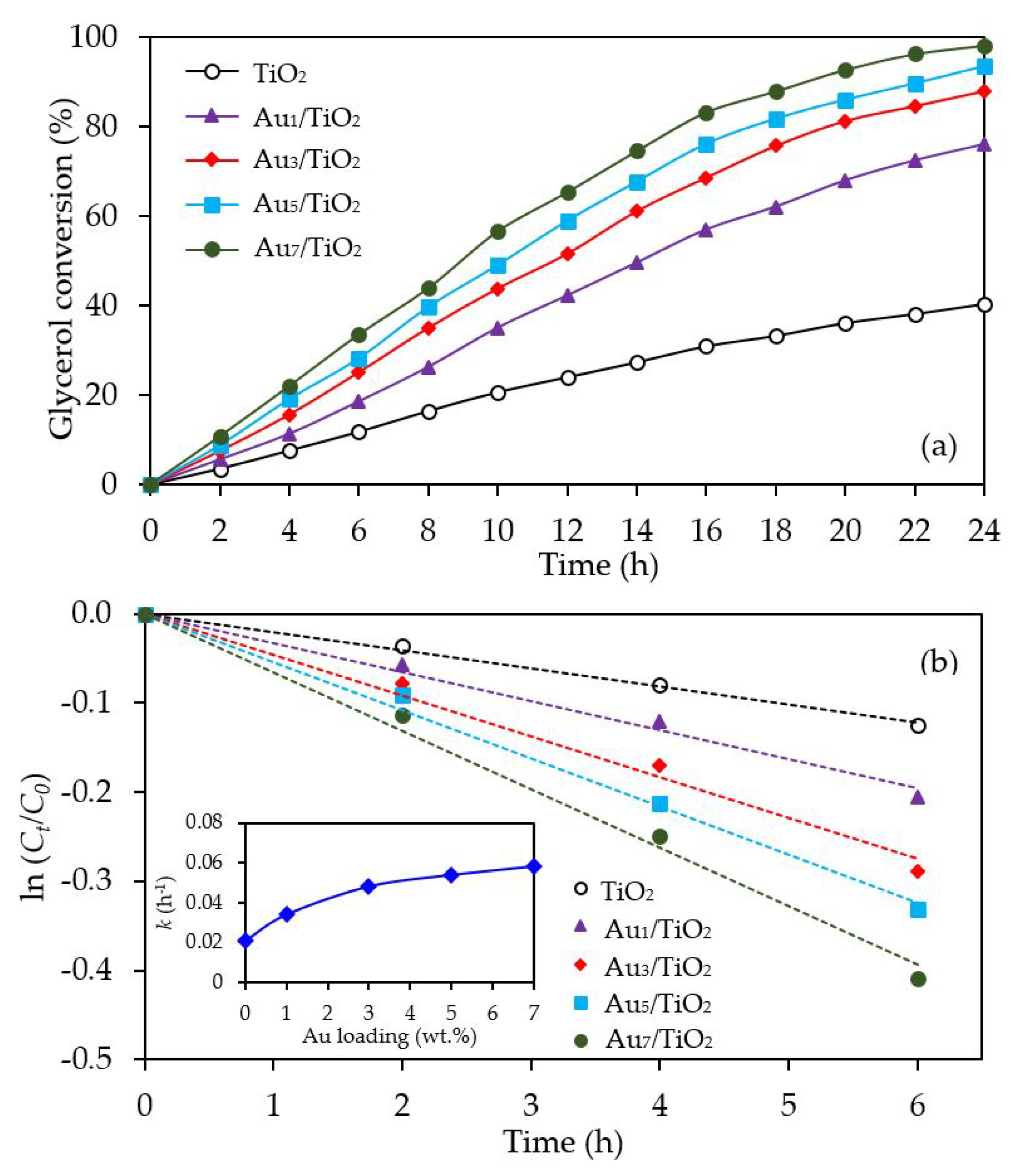
The glycerol conversion in the presence of the different Aux/TiO2 photocatalysts is summarized in Figure 6a. All the Aux/TiO2 photocatalysts significantly enhanced the glycerol conversion compared to the bare TiO2. The glycerol conversion increased as the Au content increased from 1 wt. % to 7 wt. %. Plotting the data with the pseudo first order reaction kinetic model (Figure 6b), where a plot of ln (Ct/C0) versus t provides a straight line and allows the determination of the kinetic rate constant (k), gave a good fit with a determination coefficient (R2) of greater than 0.9879. Accordingly, the kinetic rate constant of glycerol conversion over the Au1/TiO2, Au3/TiO2, Au5/TiO2 and Au7/TiO2 photocatalysts were deduced to be 0.0340, 0.0480, 0.0537 and 0.0606 h−1, respectively, compared to 0.021 h−1 for TiO2 (inset of Figure 6b). The enhanced glycerol conversion over Au-decorated TiO2 could have arisen from the plasmon induction effect of Au NPs. In addition, the presence of Au NPs was able to serve as electron sinks to facilitate interfacial electron transfer, which could effectively reduce the recombination rate of photogenerated holes (h+) and electrons (e−) [66].
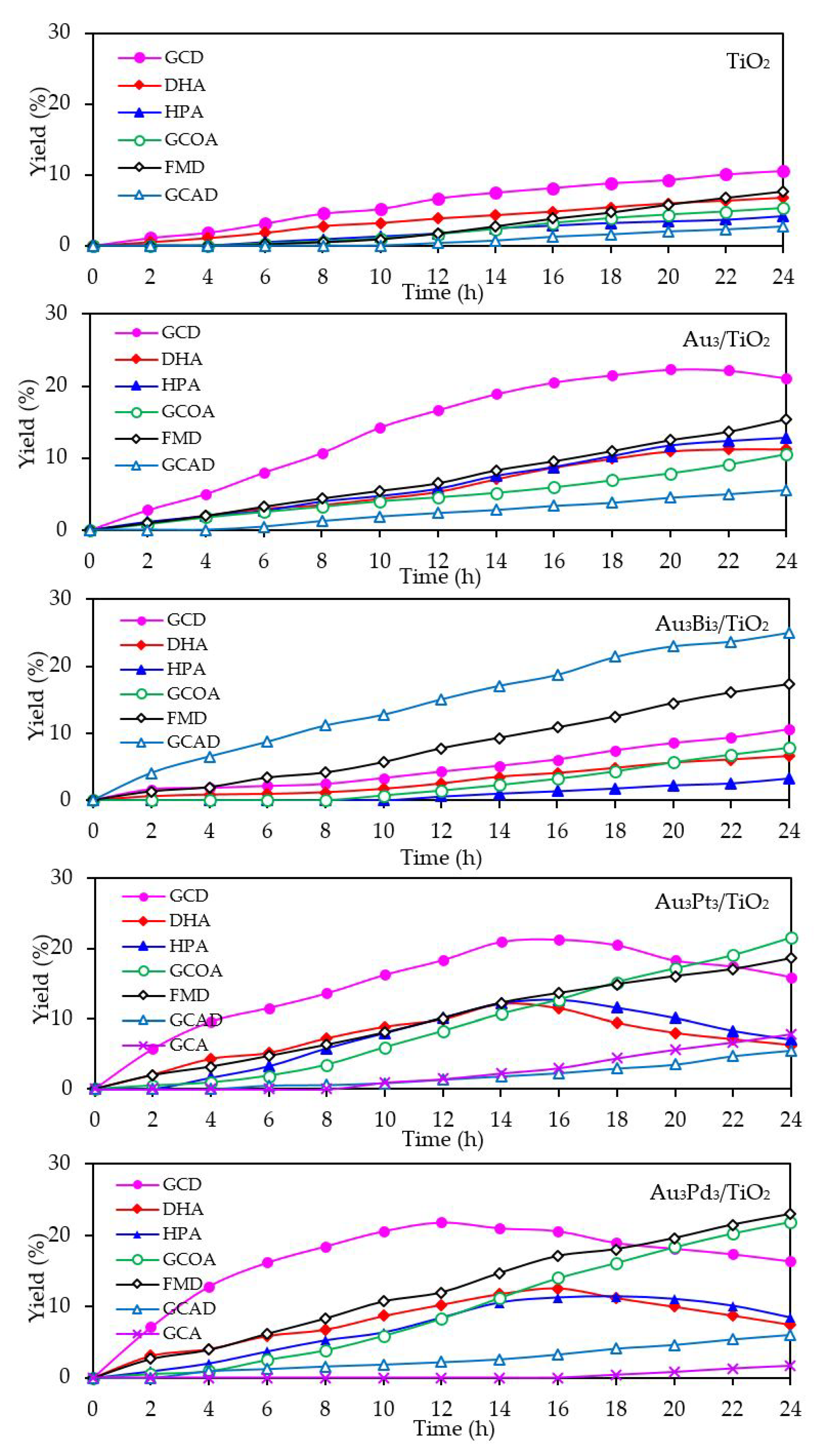
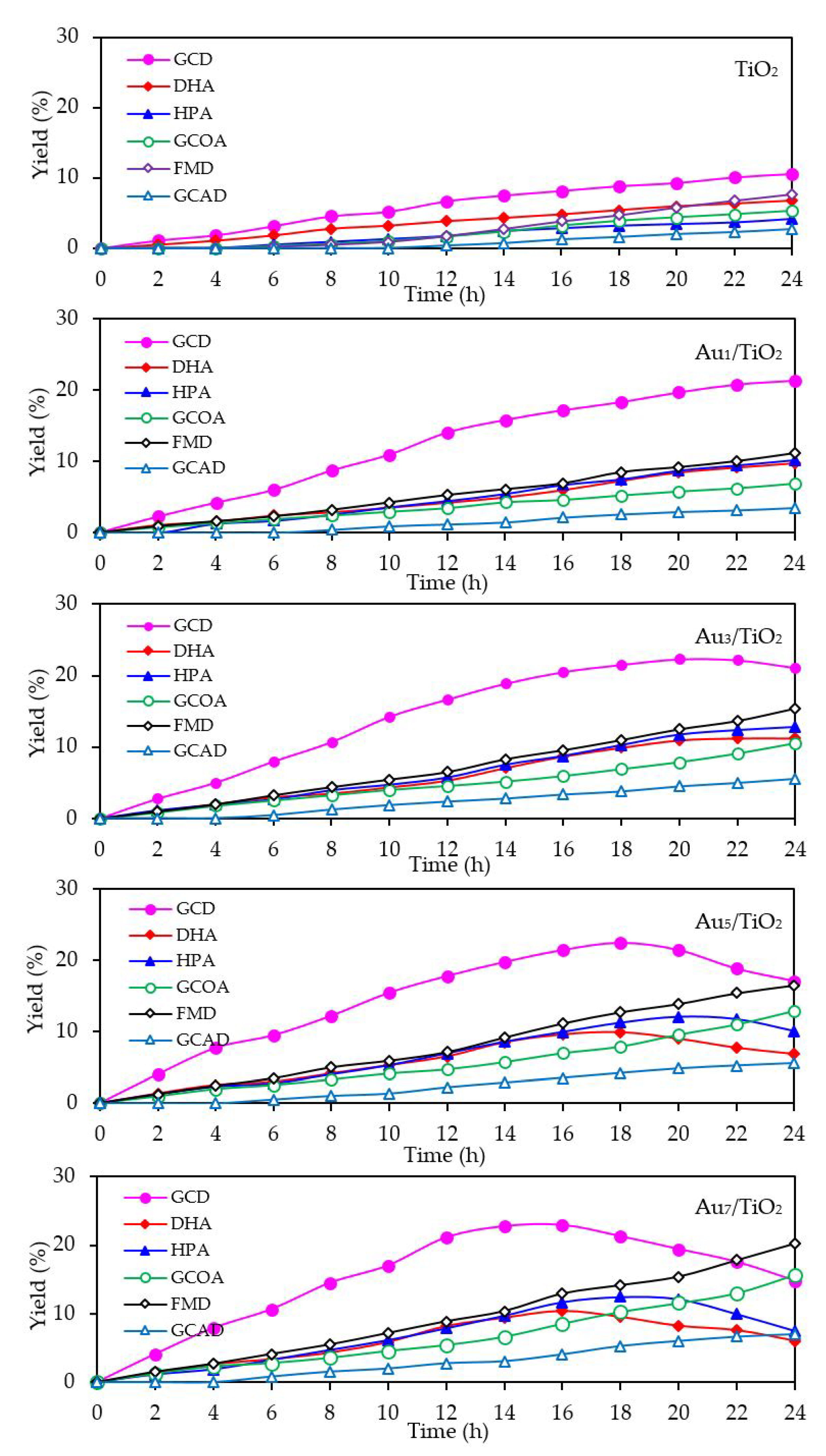
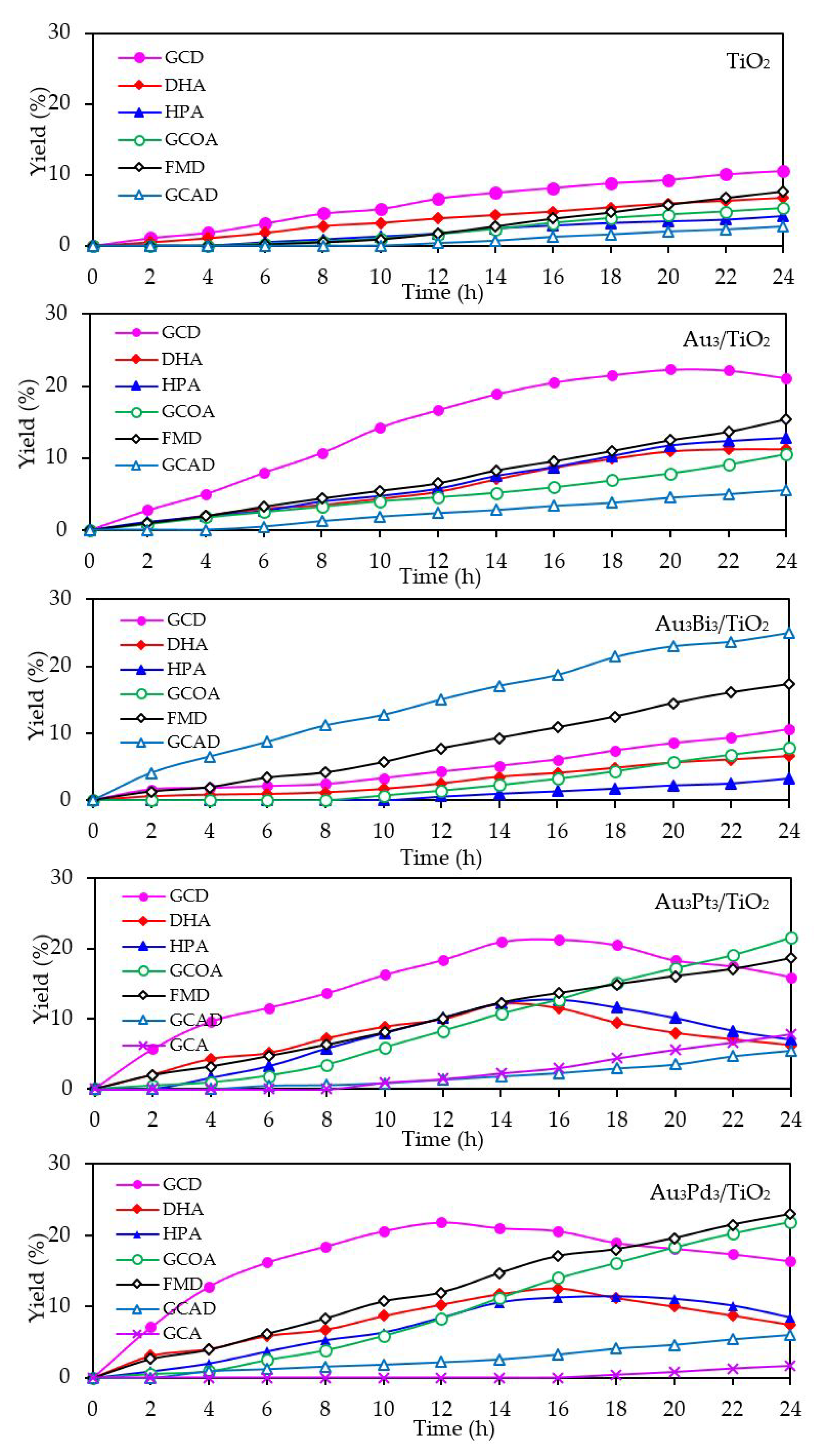
With respect to the chemical species generated from the glycerol conversion via the photocatalytic oxidation process in the presence of the Aux/TiO2 photocatalysts, similar chemical species, including GCD, DHA, HPA, GCOA, FMD and GCAD, were generated via photocatalytic oxidation with the Aux/TiO2 photocatalysts as well as with the parent TiO2 (Figure 7), where GCD was the main product. However, the GCD yield as a function of reaction time varied with the different Au NP contents on the Aux/TiO2 photocatalysts. The yield of GCD increased continuously in the presence of 1 wt. % Au1/TiO2 from the initial time to the end of reaction time, but in the presence of a high Au content (Au ≥ 3 wt. %), the yield of GCD increased continuously to reach a maximum value of around 22% and then dropped slightly. The Aux/TiO2 photocatalysts with a high Au NP content enhanced the fast generation of GCD to a maximum level and then accelerated the conversion of GCD to other chemical species, as seen by the decreased GCD yield in the later reaction times. This suggested that the Aux/TiO2 photocatalysts with a higher Au content had a higher photocatalytic activity than those with a lower Au content. This was presumably due to their higher content of defective structures, which could promote the visible light absorption due to the narrowing of the electronic bandgap via the continuum interstate between the Ti3d states of the conduction band and the O2p states of the valence band [67]. A high quantity of absorbed photons can achieve a high level of photogenerated h+ and e- on the TiO2 surface. Moreover, the function of the decorated Au NPs as an electron sink can also promote the lifetime of the photogenerated h+ and e−, and so enhanced the photocatalytic activity for glycerol conversion.
The mechanism of glycerol conversion to GCD, DHA, HPA, GCOA, FMD and GCAD has been proposed previously [12,68,69]. In brief, the photocatalytic oxidation of glycerol via TiO2 occurred mainly at the 1°-OH groups to form GCD as the principle product and partially occurred at the 2°-OH group to form DHA. The generated GCD can then be further oxidized with hydroxyl radicals (HO•) to form GCOA and FMD, while the DHA is then further oxidized with oxygen species (O2•−/1O2) to form HPA. The generation of a low GCAD content was probably caused by the adsorption of two molecules of generated FMD on two neighboring Ti3+ defective sites and their subsequent dimerization to form GCAD.
3.2. Au3M3/TiO2 Photocatalysts
According to the results obtained, to further explore the role of decorated bimetallic NPs and enhance the glycerol conversion efficiency and yield of product distribution, the Au3/TiO2 photocatalyst was selected to composite with other three metals (Bi, Pt and Pd) at the same loading of 3 wt. %.
3.2.1. Photocatalyst Morphology
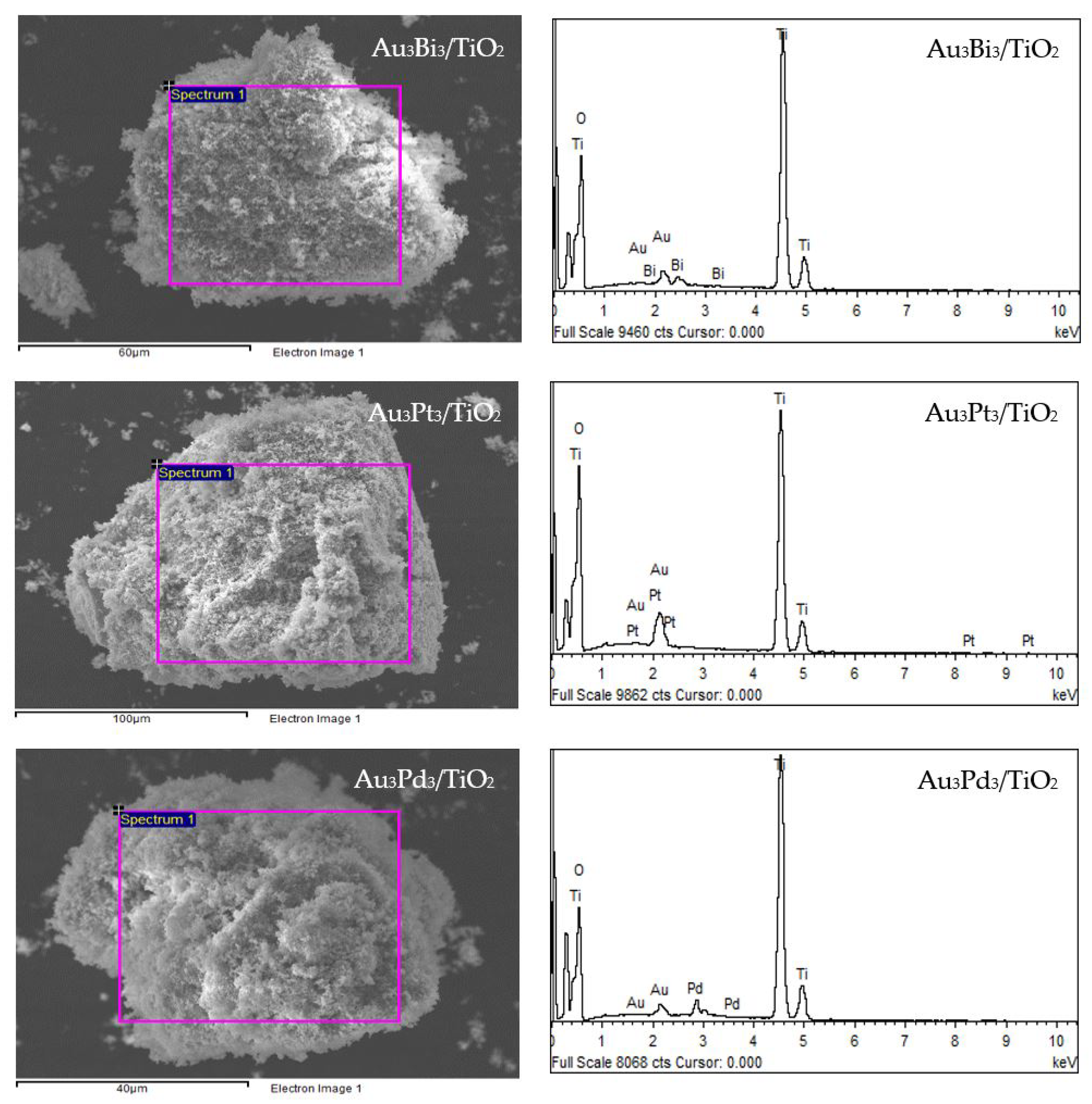
The XRD patterns of the prepared Au3M3/TiO2 photocatalysts are displayed in Figure 1b. All three exhibited the main diffraction peaks of TiO2 in the anatase phases, while the diffraction peak of Au NPs was still clearly observed in the XRD patterns of the Au3/TiO2 and Au3Bi3/TiO2 photocatalysts, but was not observed in the Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts (Figure 1b1). A clear diffraction peak of Bi in the rhombohedral lattice appeared separately at a 2θ of 27.0° (PDF No. 01-085-1329) (Figure 1b1), while no separate peak of Pt and Pd was clearly observed, probably due to their alloy formation with Au NPs. The track of AuPt and AuPd peaks was detected, as shown in Figure 1b2,b3. The presence of Au and the respective M in all three Au3M3/TiO2 photocatalysts was close to the set values (3 wt. % of both Au and M), as confirmed by the SEM–EDS analysis (Figure 8), and are listed in Table 1.
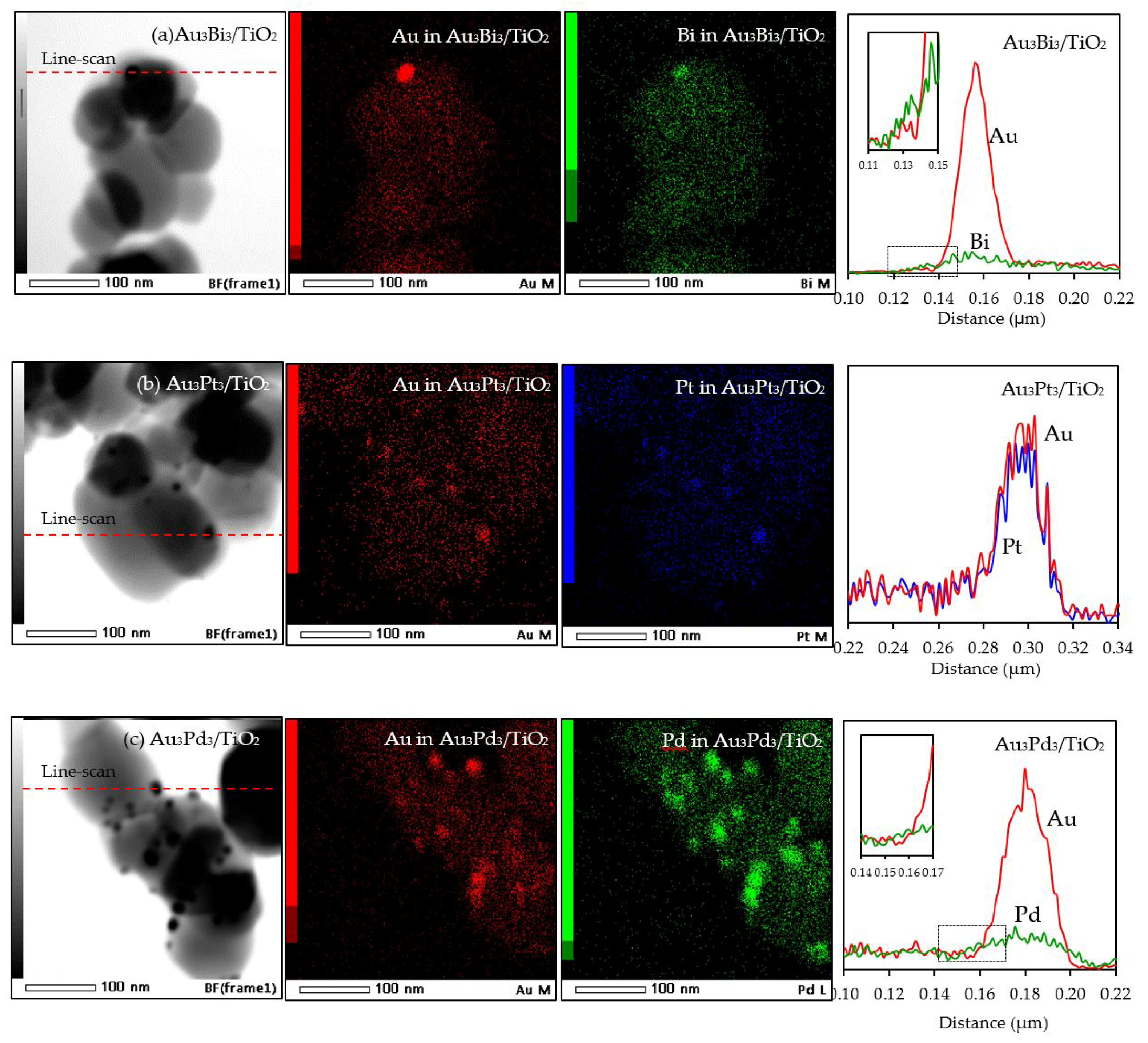
With respect to the particle size distribution, the TEM images (Figure 9) revealed a well-dispersed and narrow size distribution of the decorated AuM NPs on the TiO2 surface, with a slightly larger particle size than that in Au3/TiO2 (Table 1). This suggests that the addition of the second metal induced a slight agglomeration of the AuM NPs. To get insight into the morphology of the decorated metal NPs in the bimetallic catalysts, EDS line-scanning together with the elemental mapping analysis was performed. The compositional line scan of the Au3Bi3/TiO2 photocatalyst showed that the concentration of Au increased from the edge to the center of the metal particle, whereas the Bi distribution was almost constant from the edge to the center of the selected particle (Figure 10a), indicating a core–shell structure of the Bi–Au particle with Au as the core and Bi as the shell. The onset position of the Bi signal was around 0.13 μm, which was 0.008 μm lower than that of Au, indicating a 8 nm thickness of the dendritic Bi shell. A core–shell structure was also observed for the Au3Pd3/TiO2 photocatalyst, in which the high density of Au appeared at the center of the metal particle, while Pd was distributed uniformly from the edge to the center (Figure 10c), with the thickness of the dendritic Pd shell around the Au core being 7.5 nm. A different pattern of the line scan profile was observed for the Au3Pt3/TiO2 photocatalyst, in which the density of both Au and Pt NPs increased from the edge to the center of the selected particle (Figure 10b). This indicated that the AuPt ensembles existed as a solid solution of arbitrary composition or as an AuPt alloy form.
The capability of UV-vis absorption spectra of the Au3M3/TiO2 photocatalysts is shown in Figure 4b, where they all exhibited a high capability to absorb visible light at a wavelength of greater than 400 nm. The addition of Bi, Pt or Pd to the Au3/TiO2 attenuated an intense broad absorption band centered at around 540 nm of Au, probably due to the different dielectric function of the bimetals compared to the Au monometallic state [70]. Moreover, the strong interaction between the decorated Au–M can also influence the flattening of the Au LSPR band [71]. The bandgap values of the Au3Bi3/TiO2, Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts, as determined from a Tauc’s plot (inset of Figure 4b), were 2.85, 2.85 and 2.79 eV, respectively (Table 1), which suggested that the addition of the respective second metal caused the generation of the defective structure of TiO2. The formation of the Ti3+ defect structure likely induced an altered major electronic state of the valence band and conduction band, by which it formed a mid-gap state below the conduction band and also enabled the lengthening of the conduction band edge, resulting in the formation of the band tail structure and the narrowness of the bandgap energy [72].
To confirm the presence of the Ti3+ defective structures, Ti2p HR-XPS spectra of the Au3M3/TiO2 photocatalysts were investigated, and are shown in Figure 11a. The core-level Ti2p spectra still exhibited the two main symmetric peaks of Ti 2p3/2 and Ti 2p1/2 at a binding energy of 459.4 and 465.1 eV, respectively, with a difference of 5.7 eV, in accordance with the typical value of the Ti4+ state in TiO2. The shoulder peak of Ti2p at a low binding energy, assigned to the existence of the Ti3+ structure, was distinctly observed in all the Au3M3/TiO2 photocatalysts. The Ti3+/Ti4+ ratios, as obtained from the area under the deconvolution of both peaks, are listed in Table 1. The Au3M3/TiO2 photocatalysts all exhibited a higher Ti3+/Ti4+ ratio than the Au3/TiO2 and parent TiO2 photocatalysts, indicating that the addition of the bimetallic NPs (Au–Bi, Au–Pt and Au–Pd) induced a greater content of defective structures of TiO2 than the addition of the monometallic Au NPs. Among all the Au3M3/TiO2 photocatalysts, surprisingly, the Au3Pd3/TiO2 photocatalyst exhibited the highest Ti3+/Ti4+ ratio of 0.3561, which was 2.41-, 1.82- and 1.83-fold higher than that of the Au3/TiO2, Au3Bi3/TiO2 and Au3Pt3/TiO2 photocatalysts, respectively. The higher content of Ti3+ defect species in the bi- than mono-metallic system might be because the high content of decorated metal can incorporate into the TiO2 structure and induce formation of the Ti3+ defect site by transferring electrons between the decorated metal and oxygen vacancies of TiO2 in order to maintain the charge balance [73].
To distinguish the valence states of the decorated metal, the Au 4f level spectra of all Au3M3/TiO2 photocatalysts were analyzed, with respective examples shown in Figure 11b. The two main characteristic peaks of Au 4f7/2 and Au 4f5/2 were still observed with a spin-orbit splitting of 3.7 eV, indicating the presence of Au in the metallic state. A slight blue shift in the Au 4f binding energy was observed for the Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts in comparison with that of Au3/TiO2. Typically, the shift in the binding energy in each metal is attributed to various factors, such as a change in its chemical state or the charge compensation of added elements on the parent elements. However, in this case, the chemical state of Au in all the Au3M3/TiO2 photocatalysts was still in the metallic state (Figure 11b), and so this blue shift in the Au binding energy could be the result of charge compensation of the Pt or Pd elements on Au elements [74], by which Au accepted sp-electrons from Pt or Pd, but donated d-electrons to Pt or Pd, and possibly due to the partial formation of Au–M alloy structure, which are more negatively charged [75]. A low Au 4f intensity was observed in the case of the Au3Bi3/TiO2 photocatalyst compared with other two Au3M3/TiO2 photocatalysts, due to the presence of the Bi-wrapped Au NPs as a core–shell structure.
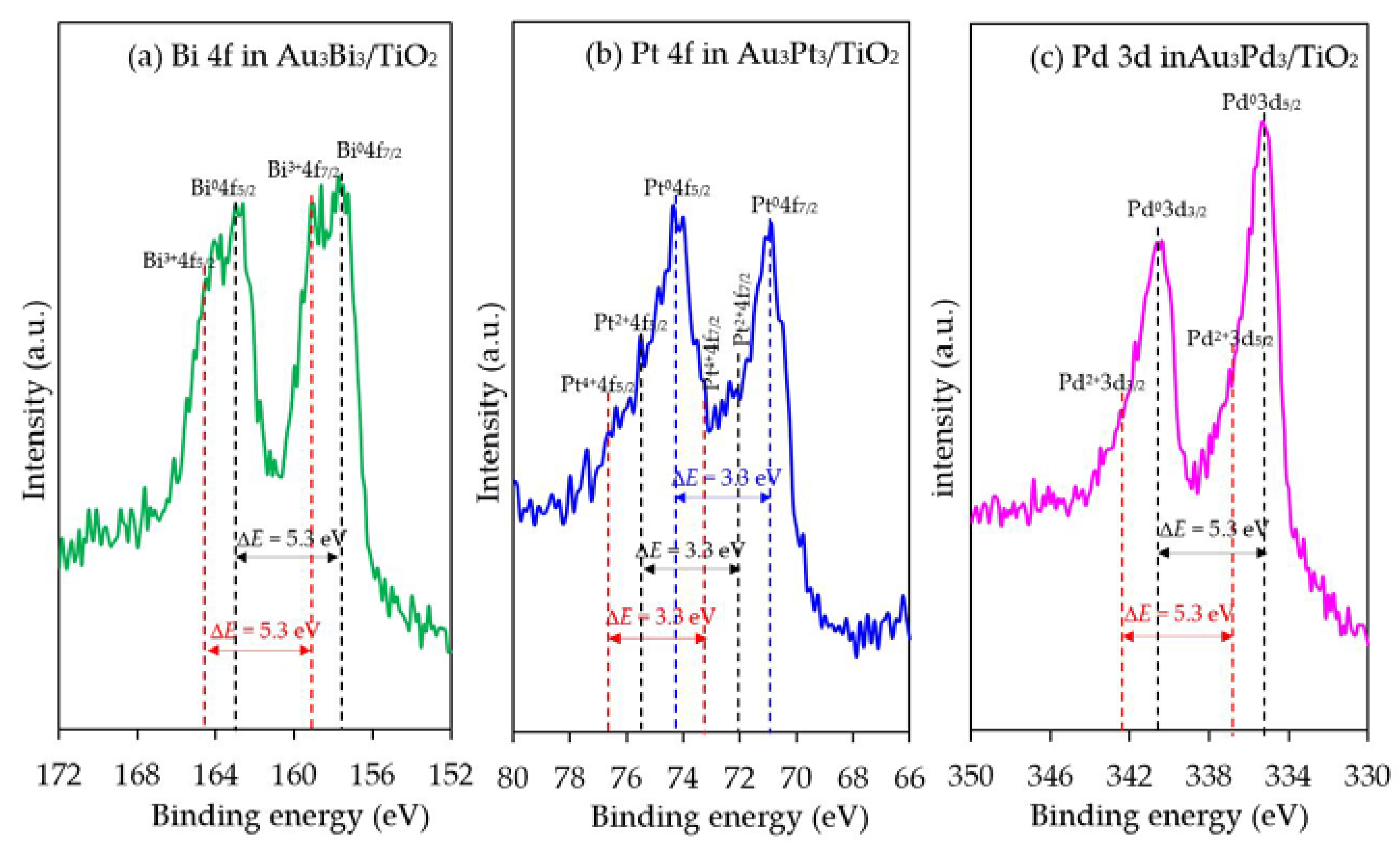
The chemical state of the second metal in all the photocatalysts was also analyzed by the HR-XPS. As demonstrated in Figure 12, all the second metals in the respective Au3M3/TiO2 photocatalysts were presented in both metallic and oxide forms. The peaks located at 162.9 eV and 157.6 eV for the Au3Bi3/TiO2 photocatalyst were assigned to Bi0 4f5/2 and Bi0 4f7/2, respectively, which is the characteristic value of the metallic Bi. A further two peaks at 164.5 eV and 159.2 eV were assigned to the Bi3+4f5/2 and Bi3+4f7/2 region, which are characteristic of Bi3+ in the as-prepared catalysts. These binding energy values of the Bi3+ ion-valent-form are too high to be assigned to the typical Bi3+ in bismuth oxide (Bi2O3), but rather were assigned to bismuth hydroxylated oxide (BiO(OH)) species on the photocatalyst surface [76]. For the Au3Pt3/TiO2 photocatalyst, the Pt 4f signals consisted of three pairs of doublets; the Pt0 4f5/2 and Pt0 4f7/2 peaks at binding energies of 74.2 eV and 70.9 eV, the Pt2+ 4f5/2 and Pt2+ 4f7/2 peaks at binding energies of 75.3 eV and 72.0 eV and the Pt4+ 4f5/2 and Pt4+ 4f7/2 peaks at binding energies of 76.5 eV and 73.2 eV, with an identical spin-orbit splitting of about 3.3 eV [77]. This suggests that the Pt NPs were present in both metallic zero-valent (Pt0) and metallic ion-valent (Pt2+ and Pt4+) forms. For the Au3Pd3/TiO2 photocatalyst, the most intensive doublet, at binding energies of 340.6 eV (Pd0 3d3/2) and 335.3 eV (Pd0 3d5/2), was attributed to metallic Pd. The shoulder peaks located at binding energies of around 342.2 eV and 336.9 eV were assigned to Pd2+ 3d3/2 and Pd2+ 3d5/2, respectively, which is characteristic of Pd2+ in PdO in the achieved catalyst. The spin-orbit splitting of about 5.3 eV in both the metallic Pd and Pd2+ was well in accordance with the reported value [14]. All the second metal NPs in the respective Au3M3/TiO2 catalysts existed predominantly in the metallic form, whereas the partial presence of the second metal in an oxide form might be attributed to the formation of a M–O bond driven by the oxygen chemisorption on the surface of the second metal nanostructure during the prepa ration process.
3.2.2. Photocatalytic Activity
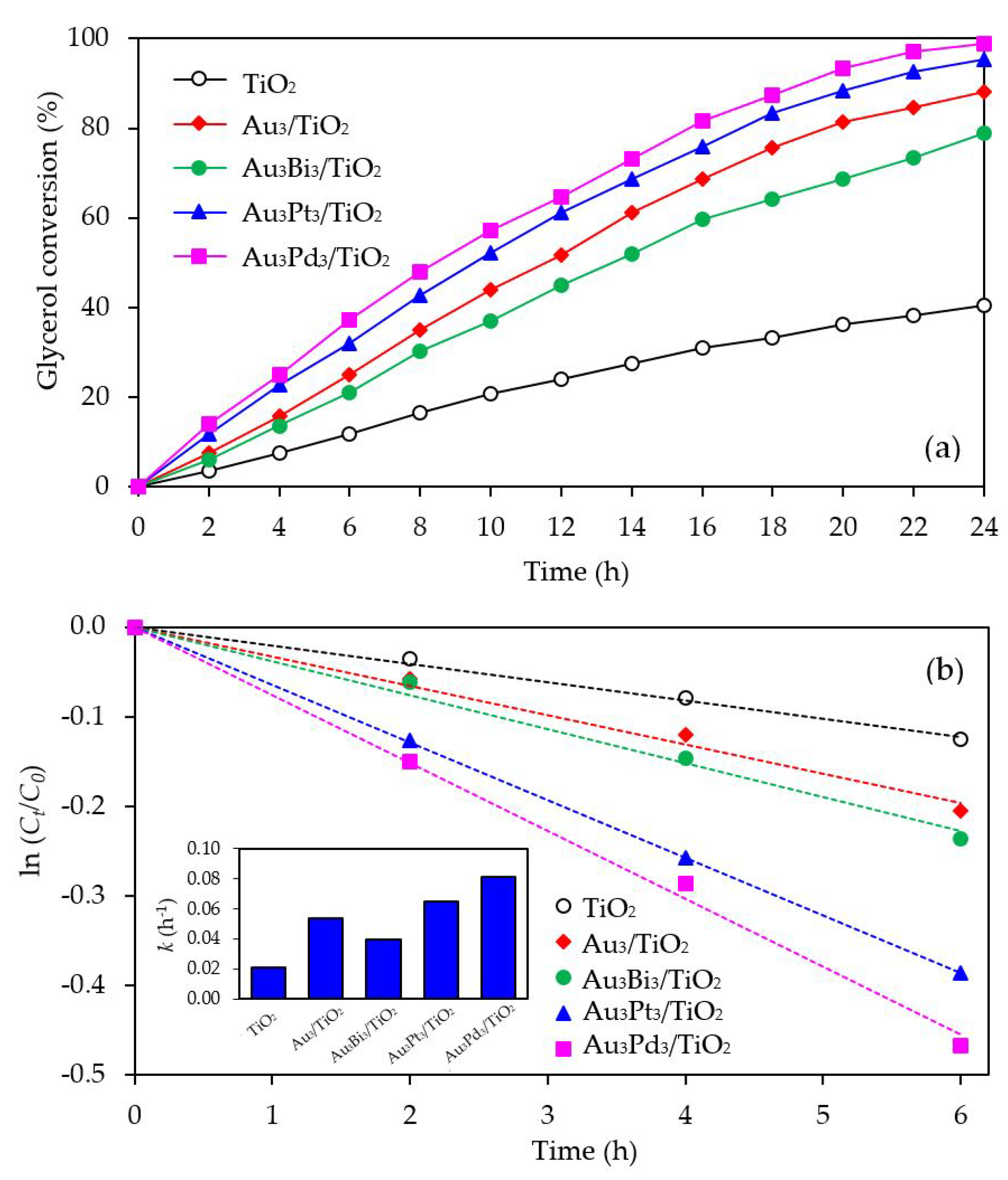
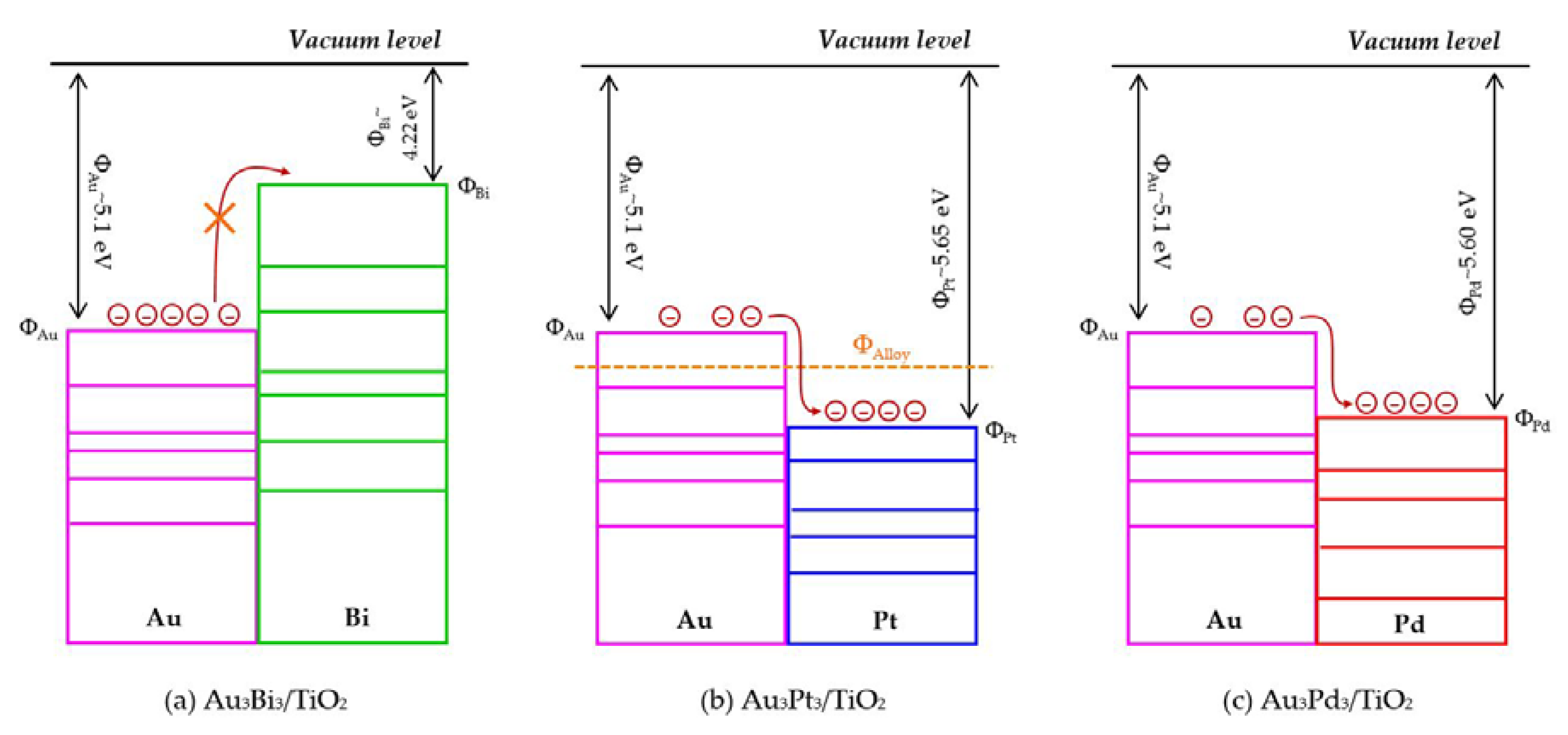
The photocatalytic activity of the Au3M3/TiO2 photocatalysts was tested for the photocatalytic oxidation of glycerol to selected value-added compounds. As demonstrated in Figure 13a, the addition of Pt or Pd NPs on the Au3/TiO2 photocatalyst to form Au3Pt3/TiO2 and Au3Pd3/TiO2, respectively, enhanced the glycerol conversion, while the addition of Bi NPs retarded the reaction. The first order kinetic rate constants of glycerol conversion over Au3Bi3/TiO2, Au3Pt3/TiO2 and Au3Pd3/TiO2 were 0.0396, 0.0644 and 0.0810 h−1, respectively (Figure 13b). The Au3Bi3/TiO2 photocatalyst exhibited a lower photocatalytic activity than the Au3/TiO2 photocatalyst despite having a lower bandgap energy and higher defective structure, probably due to the core–shell structure of the AuBi NPs. That is, the LSPR behavior of the Au NPs was diminished when they were wrapped by the Bi species. Moreover, the decorated Bi NPs cannot extract the excited e− from the Au or supported TiO2 due to its lower work function (4.22 eV) than that of Au (5.1 eV) and TiO2 (4.9 eV), resulting in unalleviated e−-h+ recombination (Figure 14a). The high photocatalytic activity of the Au3Bi3/TiO2 compared to the parent TiO2 was due to the presence of the valence band tail above the valence band, which was caused by the hybridization of the Bi 6s and O 2p orbital, and the existence of the Ti3+ state below the conduction band [53], which allowed an easy jump of photogenerated e− from either the valence band tail level to either a shallow trap (Ti3+ state) or conduction band. In addition, as previously mentioned, the Bi6s is widely dispersed in the hybrid orbital of Bi6s-O2p leading to an increased charge mobility and consequently an improved photocatalytic activity of the parent photocatalyst [78].
For the Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts, the enhanced glycerol conversion compared with the Au3/TiO2 photocatalyst was probably due to their lower bandgap energy, which allowed the electron excitation at a lower photon energy and the presence of LSPR behavior in the decorated noble metal structure, resulting in the transfer of e- from the metallic NPs to a shallow trap or conduction band of TiO2, or vice versa, under the light absorption. Moreover, in fact, the excited electron can be transferred by the driven work function. The difference in work function between Au and the second metal (Pt or Pd) can reduce the e−-h+ recombination rate by transferring the e- between the two metals after trapping the e- from the conduction band of TiO2 into equilibrium [79]. That is, the work function of Au is 5.1 eV and those of Pt and Pd are 5.65 eV and 5.22 eV, respectively. The decorated Au–Pt NPs formed an alloy structure in the Au3Pt3/TiO2 photocatalyst, which can initiate a new Fermi level of alloy NPs somewhere between the Fermi level of Au and Pt (Figure 14b) and so could prolong the e−-h+ lifetime compared with the Au–Bi NPs. This was shorter than the two-stage transferring of an excited electron between the two individual Fermi levels of each decorated metal in the core–shell structure generated in the Au3Pd3/TiO2 photocatalyst (Figure 14c). This is the reason why the Au3Pd3/TiO2 exhibited a higher photocatalytic activity than the Au3Pt3/TiO2 photocatalyst.
The core–shell structure of the Au3Pd3/TiO2 photocatalyst did not exhibit a negative effect on the LSPR behavior in this photocatalyst, probably because the plasmon metallic state of Pd can still occur, leading to an easy transfer of e- from the TiO2 conduction band to Pd NPs and/or Au NPs and vice versa.
With respect to the generated products, the same products (GCD, DHA, HPA, GCOA, FMD and GCAD) were generated via all the Au3M3/TiO2 photocatalysts and were similar to those generated in the presence of either the Au3/TiO2 or parent TiO2 photocatalyst (Figure 15). The principle product was GCD in the presence of all the evaluated photocatalysts except for Au3Bi3/TiO2, where GCAD was the principle product. As reported previously [53], monometallic Bi NPs decorated on TiO2 did not promote the formation of GCAD as a major product. Accordingly, it is suggested that the synergism between the bimetallic Au–Bi was a vital factor in the formation of GCAD from glycerol. Various published works on the aerobic oxidation of glycerol over bimetallic catalysts with Bi NPs have proposed that Bi played a major role as a geometric blocking site, which can facilitate the activation and transformation of the secondary hydroxyl group of glycerol [54,55,56]. In this present work, glycerol was mainly converted through the photocatalytic oxidation reaction over Au3Bi3/TiO2 to GCAD as the major product. It is hypothesized that the two terminal hydroxyl (-OH) groups of glycerol were bound on the surface of the Au–Bi NPs with Bi3+ species, whereas Bi adatoms function as blockers on the Au site, which is a high-energy site where the adsorption and reaction of the primary –OH group is preferred. This then serves to control the adsorbed orientation of glycerol molecules towards the preferential oxidation of the secondary –OH group to form DHA. In addition, due to the presence of various reactive oxygen species with a high oxidizing power in the photocatalytic system, the cleavage of the C–C bond of the generated DHA molecule to one molecule of GCAD and FMD is dramatically achieved. The generated FMD can eventually be oxidized to form CO2 as a gas-phase oxidation product. However, the synergetic role of Bi and the optimization of the Bi-promoted Au or other noble metals on TiO2 photocatalyst with the corresponding reaction conditions act to determine the highly selective oxidation of glycerol, which could also work to some extent that might be beneficial in academic research and the chemical industry.
4. Conclusions
Monometallic Au NPs with different metal contents and three types of Au-based bimetallic NPs (Au–Bi, Au–Pt and Au–Pd) with a nominal metal content of 3 wt. % were decorated on TiO2 by the sol-immobilization technique and then tested for photocatalytic oxidation of glycerol. The decoration of Au NPs in both mono- and bimetallic systems on TiO2 enhanced the photocatalytic glycerol conversion due to the narrowed bandgap energy and increased light harvesting capability of the as-prepared photocatalysts and the alleviation of e−-h+ recombination in the presence of the decorated metal. The Au3Pd3/TiO2 photocatalyst was found to be highly active and the most efficient tested photocatalyst for glycerol conversion, mainly because it had the lowest bandgap energy, the largest Ti3+ defect content and highest UV-Visible light absorption ability. The principle liquid phase product in the photocatalytic oxidation of glycerol over the Aux/TiO2, Au3Pt3/TiO2 and Au3Pd3/TiO2 photocatalysts was GCD. By contrast, Au3Bi3/TiO2 promoted the generation of GCAD as the major liquid phase product, which might be due to the promotional role of Bi species that act as a chelated site between the glycerol molecules and Au sites. This work paves a new way to undertake the rational design of mono- and bimetallic NPs decorated on TiO2 in order to enhance the performance of the photocatalytic process and promote the selective photocatalytic oxidation of glycerol.
Acknowledgments
The authors thank the Chulalongkorn University Dutsadi Phiphat Scholarship, the Ratchadapisek Sompoch Endowment Fund, Chulalongkorn University (Sci-Super II GF_58_08_23_01) and the Thailand Research Fund (IRG5780001) for financial support.
Author Contributions
T.J., M.H., and N.S. conceived and designed the experiments; T.J. performed the experiments; T.J., and M.H. analyzed the data; M.H., and N.S. contributed reagents/materials/analysis tools; and T.J., and M.H. wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Clomburg, J.M.; Gonzalez, R. Anaerobic fermentation of glycerol: A platform for renewable fuels and chemicals. Trends Biotechnol. 2013, 31, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotti, C.; Kašpar, J.; Farnetti, E. Dehydrogenation of glycerol to dihydroxyacetone catalyzed by iridium complexes with p–n ligands. Green Chem. 2010, 12, 1295–1300. [Google Scholar] [CrossRef]
- Zou, B.; Ren, S.; Ye, X.P. Glycerol dehydration to acrolein catalyzed by zsm-5 zeolite in supercritical carbon dioxide medium. ChemSusChem 2016, 9, 3268–3271. [Google Scholar] [CrossRef] [PubMed]
- Veiga, P.M.; Gomes, A.C.; Veloso, C.O.; Henriques, C.A. Acid zeolites for glycerol etherification with ethyl alcohol: Catalytic activity and catalyst properties. Appl. Catal. A Gen. 2017, 548, 2–15. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Y.; Huang, Y.; Wang, J.; Gao, J.; Xu, J. Selective esterification of glycerol with acetic acid to diacetin using antimony pentoxide as reusable catalyst. J. Energy Chem. 2015, 24, 632–636. [Google Scholar] [CrossRef]
- Katryniok, B.; Kimura, H.; Skrzyńska, E.; Girardon, J.-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Villa, A.; Dimitratos, N.; Chan-Thaw, C.E.; Hammond, C.; Prati, L.; Hutchings, G.J. Glycerol oxidation using gold-containing catalysts. Acc. Chem. Res. 2015, 48, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Puddu, V.; Choi, H.; Dionysiou, D.D.; Puma, G.L. TiO2 photocatalyst for indoor air remediation: Influence of crystallinity, crystal phase, and uv radiation intensity on trichloroethylene degradation. Appl. Catal. B Environ. 2010, 94, 211–218. [Google Scholar] [CrossRef]
- Pansa-Ngat, P.; Jedsukontorn, T.; Hunsom, M. Simultaneous H2 production and pollutant removal from biodiesel wastewater by photocatalytic oxidation with different crystal structure TiO2 photocatalysts. J. Taiwan Inst. Chem. Eng. 2017, 78, 386–394. [Google Scholar] [CrossRef]
- Low, J.; Cheng, B.; Yu, J. Surface modification and enhanced photocatalytic Co2 reduction performance of TiO2: A review. Appl. Surf. Sci. 2017, 392, 658–686. [Google Scholar] [CrossRef]
- Fujita, S.-I.; Kawamori, H.; Honda, D.; Yoshida, H.; Arai, M. Photocatalytic hydrogen production from aqueous glycerol solution using nio/tio2 catalysts: Effects of preparation and reaction conditions. Appl. Catal. B Environ. 2016, 181, 818–824. [Google Scholar] [CrossRef]
- Jedsukontorn, T.; Meeyoo, V.; Saito, N.; Hunsom, M. Route of glycerol conversion and product generation via TiO2-induced photocatalytic oxidation in the presence of H2O2. Chem. Eng. J. 2015, 281, 252–264. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Cao, S.-W.; Xue, C. Au/pt nanoparticle-decorated TiO2 nanofibers with plasmon-enhanced photocatalytic activities for solar-to-fuel conversion. J. Phys. Chem. C 2013, 117, 25939–25947. [Google Scholar] [CrossRef]
- Bashir, S.; Idriss, H. Mechanistic study of the role of au, pd and au-pd in the surface reactions of ethanol over TiO2 in the dark and under photo-excitation. Catal. Sci. Technol. 2017, 7, 5301–5320. [Google Scholar] [CrossRef]
- Sellappan, R.; Nielsen, M.G.; González-Posada, F.; Vesborg, P.C.; Chorkendorff, I.; Chakarov, D. Effects of plasmon excitation on photocatalytic activity of ag/TiO2 and au/TiO2 nanocomposites. J. Catal. 2013, 307, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Shuang, S.; Lv, R.; Xie, Z.; Zhang, Z. Surface plasmon enhanced photocatalysis of au/pt-decorated TiO2 nanopillar arrays. Sci. Rep. 2016, 6, 26670. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.H.; Chu, H.Y.; Ibrahim, S.; Saravanan, P. Palladium nanoparticles anchored to anatase TiO2 for enhanced surface plasmon resonance-stimulated, visible-light-driven photocatalytic activity. Beilstein J. Nanotechnol. 2015, 6, 428. [Google Scholar] [CrossRef] [PubMed]
- Linic, S.; Christopher, P.; Ingram, D.B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011, 10, 911. [Google Scholar] [CrossRef] [PubMed]
- Ide, Y.; Matsuoka, M.; Ogawa, M. Efficient visible-light-induced photocatalytic activity on gold-nanoparticle-supported layered titanate. J. Am. Chem. Soc. 2010, 132, 16762–16764. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Teramura, K.; Hosokawa, S.; Kominami, H.; Tanaka, T. Visible light-induced water splitting in an aqueous suspension of a plasmonic au/TiO2 photocatalyst with metal co-catalysts. Chem. Sci. 2017, 8, 2574–2580. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Photocatalytic reactions under irradiation of visible light over gold nanoparticles supported on titanium (iv) oxide powder prepared by using a multi-step photodeposition method. Catal. Sci. Technol. 2014, 4, 1931–1938. [Google Scholar] [CrossRef]
- Tanaka, A.; Hashimoto, K.; Ohtani, B.; Kominami, H. Non-linear photocatalytic reaction induced by visible-light surface-plasmon resonance absorption of gold nanoparticles loaded on titania particles. Chem. Commun. 2013, 49, 3419–3421. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; You, T.; Shi, W.; Li, J.; Guo, L. Au/TiO2/au as a plasmonic coupling photocatalyst. J. Phys. Chem. C 2012, 116, 6490–6494. [Google Scholar] [CrossRef]
- Hvolbæk, B.; Janssens, T.V.; Clausen, B.S.; Falsig, H.; Christensen, C.H.; Nørskov, J.K. Catalytic activity of au nanoparticles. Nano Today 2007, 2, 14–18. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase liquid–liquid system. J. Chem. Soc. Chem. Commun. 1994, 801–802. [Google Scholar] [CrossRef]
- Brust, M.; Fink, J.; Bethell, D.; Schiffrin, D.; Kiely, C. Synthesis and reactions of functionalised gold nanoparticles. J. Chem. Soc. Chem. Commun. 1995, 1655–1656. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 c. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Hutchings, G.J. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Wang, J.; Gu, H. Novel metal nanomaterials and their catalytic applications. Molecules 2015, 20, 17070–17092. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Ahmadpour, A.; Bamoharram, F.F.; Tanhaei, B.; Mänttäri, M.; Sillanpää, M. A review on catalytic applications of au/TiO2 nanoparticles in the removal of water pollutant. Chemosphere 2014, 107, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, R.; Bellardita, M.; D’Urso, L.; Compagnini, G.; Palmisano, L.; Scirè, S. Au/ TiO2-CeO2 catalysts for photocatalytic water splitting and vocs oxidation reactions. Catalysts 2016, 6, 121. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Preparation of au/TiO2 exhibiting strong surface plasmon resonance effective for photoinduced hydrogen formation from organic and inorganic compounds under irradiation of visible light. Catal. Sci. Technol. 2012, 2, 907–909. [Google Scholar] [CrossRef]
- Tanaka, A.; Hashimoto, K.; Kominami, H. A very simple method for the preparation of au/TiO2 plasmonic photocatalysts working under irradiation of visible light in the range of 600–700 nm. Chem. Commun. 2017, 53, 4759–4762. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hashimoto, K.; Kominami, H. Selective photocatalytic oxidation of aromatic alcohols to aldehydes in an aqueous suspension of gold nanoparticles supported on cerium (iv) oxide under irradiation of green light. Chem. Commun. 2011, 47, 10446–10448. [Google Scholar] [CrossRef] [PubMed]
- Kominami, H.; Tanaka, A.; Hashimoto, K. Mineralization of organic acids in aqueous suspensions of gold nanoparticles supported on cerium (iv) oxide powder under visible light irradiation. Chem. Commun. 2010, 46, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Tanaka, A.; Hashimoto, K.; Kominami, H. Photocatalytic hydrogenation of furan to tetrahydrofuran in alcoholic suspensions of metal-loaded titanium (iv) oxide without addition of hydrogen gas. Phys. Chem. Chem. Phys. 2017, 19, 20206–20212. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nishino, Y.; Sakaguchi, S.; Yoshikawa, T.; Imamura, K.; Hashimoto, K.; Kominami, H. Functionalization of a plasmonic au/TiO2 photocatalyst with an ag co-catalyst for quantitative reduction of nitrobenzene to aniline in 2-propanol suspensions under irradiation of visible light. Chem. Commun. 2013, 49, 2551–2553. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Han, Y.; Fu, H.; Qu, X.; Xu, Z.; Zheng, S. Au@ pd/TiO2 with atomically dispersed pd as highly active catalyst for solvent-free aerobic oxidation of benzyl alcohol. Chem. Eng. J. 2017, 313, 1–9. [Google Scholar] [CrossRef]
- Colmenares, J.C.; Lisowski, P.; Łomot, D.; Chernyayeva, O.; Lisovytskiy, D. Sonophotodeposition of bimetallic photocatalysts pd–au/TiO2: Application to selective oxidation of methanol to methyl formate. ChemSusChem 2015, 8, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.; Su, R.; Wells, P.P.; Shen, Y.; Dimitratos, N.; Bowker, M.; Morgan, D.; Iversen, B.B.; Chutia, A.; Besenbacher, F. Optimised photocatalytic hydrogen production using core–shell aupd promoters with controlled shell thickness. Phys. Chem. Chem. Phys. 2014, 16, 26638–26644. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Marelli, M.; Psaro, R.; Gombac, V.; Montini, T.; Fornasiero, P.; Pievo, R.; Santo, V.D. Bimetallic au-pt/TiO2 photocatalysts active under uv-a and simulated sunlight for H2 production from ethanol. Green Chem. 2012, 14, 330–333. [Google Scholar] [CrossRef]
- Douthwaite, M.; Huang, X.; Iqbal, S.; Miedziak, P.J.; Brett, G.L.; Kondrat, S.A.; Edwards, J.K.; Sankar, M.; Knight, D.W.; Bethell, D. The controlled catalytic oxidation of furfural to furoic acid using aupd/mg (oh) 2. Catal. Sci. Technol. 2017, 7, 5284–5293. [Google Scholar] [CrossRef]
- Redina, E.; Greish, A.; Novikov, R.; Strelkova, A.; Kirichenko, O.; Tkachenko, O.; Kapustin, G.; Sinev, I.; Kustov, L. Au/pt/TiO2 catalysts prepared by redox method for the chemoselective 1,2-propanediol oxidation to lactic acid and an nmr spectroscopy approach for analyzing the product mixture. Appl. Catal. A Gen. 2015, 491, 170–183. [Google Scholar] [CrossRef]
- Tan, T.H.; Scott, J.A.; Ng, Y.H.; Taylor, R.A.; Aguey-Zinsou, K.-F.; Amal, R. Plasmon enhanced selective electronic pathways in TiO2 supported atomically ordered bimetallic au-cu alloys. J. Catal. 2017, 352, 638–648. [Google Scholar] [CrossRef]
- Durán-Álvarez, J.C.; Avella, E.; Ramírez-Zamora, R.M.; Zanella, R. Photocatalytic degradation of ciprofloxacin using mono-(au, ag and cu) and bi-(au–ag and au–cu) metallic nanoparticles supported on TiO2 under uv-c and simulated sunlight. Catal. Today 2016, 266, 175–187. [Google Scholar] [CrossRef]
- Villa, A.; Jouve, A.; Sanchez Trujillo, F.J.; Motta, D.; Prati, L.; Dimitratos, N. Exploring the effect of au/pt ratio on glycerol oxidation in presence and absence of a base. Catalysts 2018, 8, 54. [Google Scholar] [CrossRef]
- Dimitratos, N.; Porta, F.; Prati, L. Au, pd (mono and bimetallic) catalysts supported on graphite using the immobilisation method: Synthesis and catalytic testing for liquid phase oxidation of glycerol. Appl. Catal. A Gen. 2005, 291, 210–214. [Google Scholar] [CrossRef]
- Evans, C.D.; Kondrat, S.A.; Smith, P.J.; Manning, T.D.; Miedziak, P.J.; Brett, G.L.; Armstrong, R.D.; Bartley, J.K.; Taylor, S.H.; Rosseinsky, M.J. The preparation of large surface area lanthanum based perovskite supports for aupt nanoparticles: Tuning the glycerol oxidation reaction pathway by switching the perovskite b site. Faraday Discuss. 2016, 188, 427–450. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Anthonykutty, J.M.; Brett, G.; Carley, A.F.; Tiruvalam, R.C.; Herzing, A.A.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Oxidation of glycerol using gold–palladium alloy-supported nanocrystals. Phys. Chem. Chem. Phys. 2009, 11, 4952–4961. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.L.; Canton, P.; Dimitratos, N.; Porta, F.; Prati, L. Selective oxidation of glycerol with oxygen using mono and bimetallic catalysts based on au, pd and pt metals. Catal. Today 2005, 102, 203–212. [Google Scholar] [CrossRef]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Lennon, D.; Porta, F.; Prati, L.; Villa, A. Effect of particle size on monometallic and bimetallic (au, pd)/c on the liquid phase oxidation of glycerol. Catal. Lett. 2006, 108, 147–153. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Karamerou, E.E.; Kondarides, D.I. Kinetics and mechanism of glycerol photo-oxidation and photo-reforming reactions in aqueous TiO2 and pt/TiO2 suspensions. Catal. Today 2013, 209, 91–98. [Google Scholar] [CrossRef]
- Jedsukontorn, T.; Saito, N.; Hunsom, M. Photocatalytic behavior of metal-decorated TiO2 and their catalytic activity for transformation of glycerol to value added compounds. Mol. Catal. 2017, 432, 160–171. [Google Scholar] [CrossRef]
- Ning, X.; Li, Y.; Yu, H.; Peng, F.; Wang, H.; Yang, Y. Promoting role of bismuth and antimony on pt catalysts for the selective oxidation of glycerol to dihydroxyacetone. J. Catal. 2016, 335, 95–104. [Google Scholar] [CrossRef]
- Brandner, A.; Lehnert, K.; Bienholz, A.; Lucas, M.; Claus, P. Production of biomass-derived chemicals and energy: Chemocatalytic conversions of glycerol. Top. Catal. 2009, 52, 278–287. [Google Scholar] [CrossRef]
- Xue, W.; Wang, Z.; Liang, Y.; Xu, H.; Liu, L.; Dong, J. Promoting role of bismuth on hydrotalcite-supported platinum catalysts in aqueous phase oxidation of glycerol to dihydroxyacetone. Catalysts 2018, 8, 20. [Google Scholar] [CrossRef]
- Porta, F.; Prati, L. Selective oxidation of glycerol to sodium glycerate with gold-on-carbon catalyst: An insight into reaction selectivity. J. Catal. 2004, 224, 397–403. [Google Scholar] [CrossRef]
- Naldoni, A.; Riboni, F.; Marelli, M.; Bossola, F.; Ulisse, G.; Di Carlo, A.; Píš, I.; Nappini, S.; Malvestuto, M.; Dozzi, M.V. Influence of TiO2 electronic structure and strong metal–support interaction on plasmonic au photocatalytic oxidations. Catal.Sci. Technol. 2016, 6, 3220–3229. [Google Scholar] [CrossRef]
- Link, S.; El-Sayed, M.A. Size and temperature dependence of the plasmon absorption of colloidal gold nanoparticles. J. Phys. Chem. B 1999, 103, 4212–4217. [Google Scholar] [CrossRef]
- De, G.; Rao, C. Au–pt alloy nanocrystals incorporated in silica films. J. Mater. Chem. 2005, 15, 891–894. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in xps analysis of first row transition metals, oxides and hydroxides: Sc, ti, v, cu and zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, C.; Zhang, L.; Zhang, X.; Yao, H.; Shi, J. Pd-catalyzed instant hydrogenation of TiO2 with enhanced photocatalytic performance. Energy Environ. Sci. 2016, 9, 2410–2417. [Google Scholar] [CrossRef]
- Gallo, A.; Montini, T.; Marelli, M.; Minguzzi, A.; Gombac, V.; Psaro, R.; Fornasiero, P.; Dal Santo, V. H2 production by renewables photoreforming on pt–au/TiO2 catalysts activated by reduction. ChemSusChem 2012, 5, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Pang, G.; He, G.; Li, Y.; He, C.; Hao, Z. Layered sphere-shaped TiO2 capped with gold nanoparticles on structural defects and their catalysis of formaldehyde oxidation. J. Environ. Sci. 2016, 39, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Boyen, H.-G.; Kästle, G.; Weigl, F.; Koslowski, B.; Dietrich, C.; Ziemann, P.; Spatz, J.P.; Riethmüller, S.; Hartmann, C.; Möller, M. Oxidation-resistant gold-55 clusters. Science 2002, 297, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, X.; Zhu, Z.; Gao, Y.; Wang, Q.; Zhu, F.; Xie, Z. Enhanced visible light photocatalytic performance of cds sensitized TiO2 nanorod arrays decorated with au nanoparticles as electron sinks. Sci. Rep. 2017, 7, 973. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Liu, Y.; Zhang, M.-X.; Min, F.-F. Electronic structure and visible-light absorption of transition metals (tm= cr, mn, fe, co) and zn-codoped srtio3: A first-principles study. Chin. Phys. Lett. 2018, 35, 017101. [Google Scholar] [CrossRef]
- Minero, C.; Bedini, A.; Maurino, V. Glycerol as a probe molecule to uncover oxidation mechanism in photocatalysis. Appl. Catal. B Environ. 2012, 128, 135–143. [Google Scholar] [CrossRef]
- Jedsukontorn, T.; Meeyoo, V.; Saito, N.; Hunsom, M. Effect of electron acceptors H2O2 and O2 on the generated reactive oxygen species 1O2 and OH in TiO2-catalyzed photocatalytic oxidation of glycerol. Chin. J. Catal. 2016, 37, 1975–1981. [Google Scholar] [CrossRef]
- Devarajan, S.; Bera, P.; Sampath, S. Bimetallic nanoparticles: A single step synthesis, stabilization, and characterization of au–ag, au–pd, and au–pt in sol–gel derived silicates. J. Colloid Interface Sci. 2005, 290, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Liz-Marzan, L.M.; Philipse, A.P. Stable hydrosols of metallic and bimetallic nanoparticles immobilized on imogolite fibers. J. Phys. Chem. 1995, 99, 15120–15128. [Google Scholar] [CrossRef]
- Feng, N.; Liu, F.; Huang, M.; Zheng, A.; Wang, Q.; Chen, T.; Cao, G.; Xu, J.; Fan, J.; Deng, F. Unravelling the efficient photocatalytic activity of boron-induced Ti3+ species in the surface layer of TiO2. Sci. Rep. 2016, 6, 34765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pan, F.; Li, Y. A review on the effects of TiO2 surface point defects on CO2 photoreduction with H2O. J. Materiomics 2017, 3, 17–32. [Google Scholar] [CrossRef]
- Yi, C.-W.; Luo, K.; Wei, T.; Goodman, D. The composition and structure of pd−au surfaces. J. Phys. Chem. B 2005, 109, 18535–18540. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, X.; Sun, H.; Huo, M. Photocatalytic degradation of malathion in aqueous solution using an au–pd–TiO2 nanotube film. J. Hazard. Mater. 2010, 184, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Tripković, A.; Popović, K.D.; Stevanović, R.; Socha, R.; Kowal, A. Activity of a ptbi alloy in the electrochemical oxidation of formic acid. Electrochem. Commun. 2006, 8, 1492–1498. [Google Scholar] [CrossRef]
- Zhang, L.-M.; Wang, Z.-B.; Sui, X.-L.; Li, C.-Z.; Zhao, L.; Gu, D.-M. Nitrogen-doped carbon with mesoporous structure as high surface area catalyst support for methanol oxidation reaction. RSC Adv. 2016, 6, 39310–39316. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, B.; Dai, Y.; Qin, X.; Zhang, X.; Wang, Z.; Jiang, M. Visible-light photocatalytic activity of the metastable Bi20TiO32 synthesized by a high-temperature quenching method. J. Solid State Chem. 2009, 182, 2274–2278. [Google Scholar] [CrossRef]
- Wong, R.J.; Scott, J.; Kappen, P.; Low, G.K.-C.; Hart, J.N.; Amal, R. Enhancing bimetallic synergy with light: The effect of uv light pre-treatment on catalytic oxygen activation by bimetallic au–pt nanoparticles on a TiO2 support. Catal. Sci. Technol. 2017, 7, 4792–4805. [Google Scholar] [CrossRef]
Figure 1.
Representative X-ray diffraction (XRD) patterns of the (a) Aux/TiO2 and (b) Au3M3/TiO2 photocatalysts.
Figure 1.
Representative X-ray diffraction (XRD) patterns of the (a) Aux/TiO2 and (b) Au3M3/TiO2 photocatalysts.
Figure 2.
(Left) Representative SEM images of the different Aux/TiO2 photocatalysts and (Right) the EDS elemental analysis of the corresponding area shown in the rectangle on the SEM image.
Figure 2.
(Left) Representative SEM images of the different Aux/TiO2 photocatalysts and (Right) the EDS elemental analysis of the corresponding area shown in the rectangle on the SEM image.
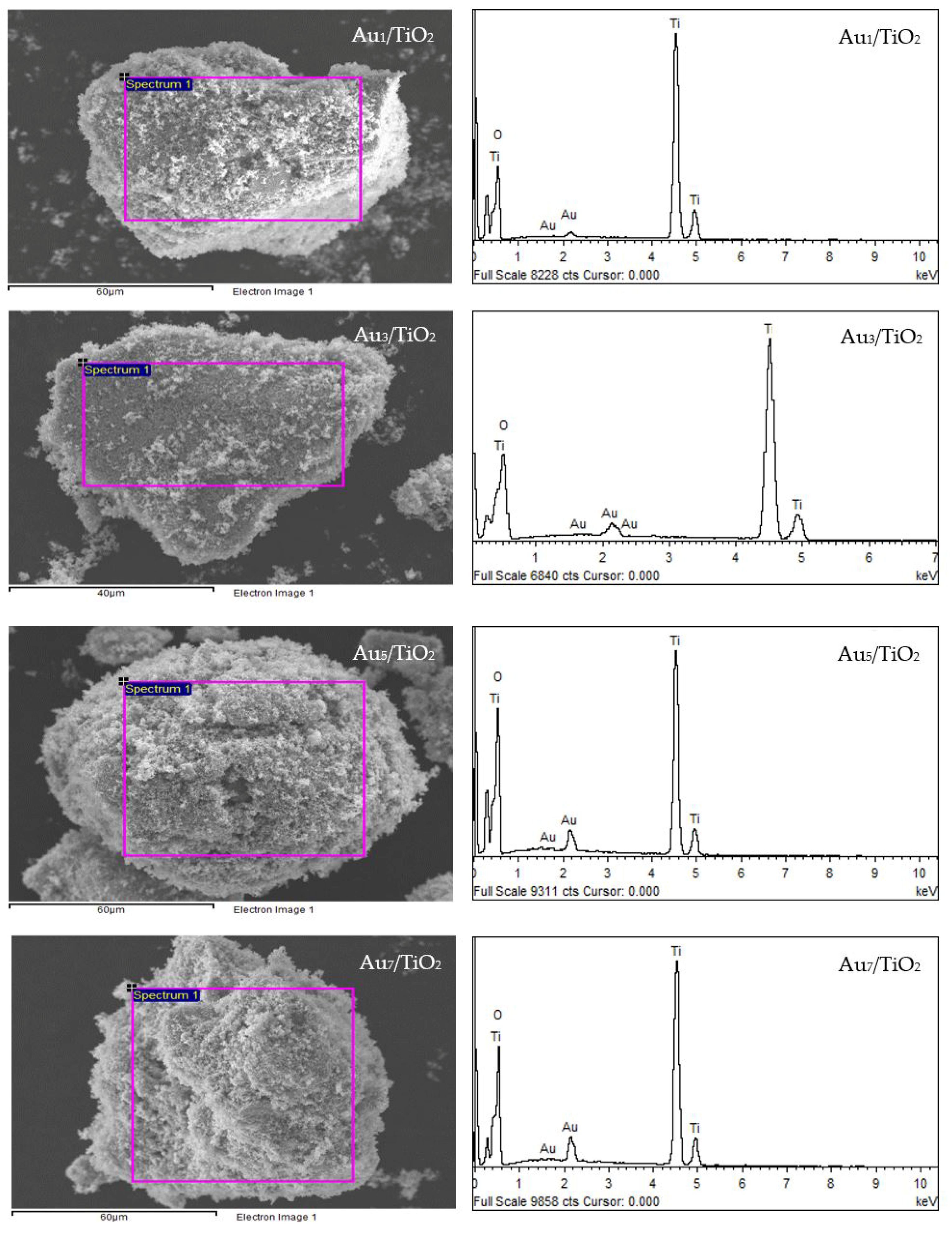
Figure 3.
(Left) Representative TEM images of the different Aux/TiO2 photocatalysts, and (Right) the derived Au nanoparticle (NP) size distribution on the surface of the TiO2.
Figure 3.
(Left) Representative TEM images of the different Aux/TiO2 photocatalysts, and (Right) the derived Au nanoparticle (NP) size distribution on the surface of the TiO2.
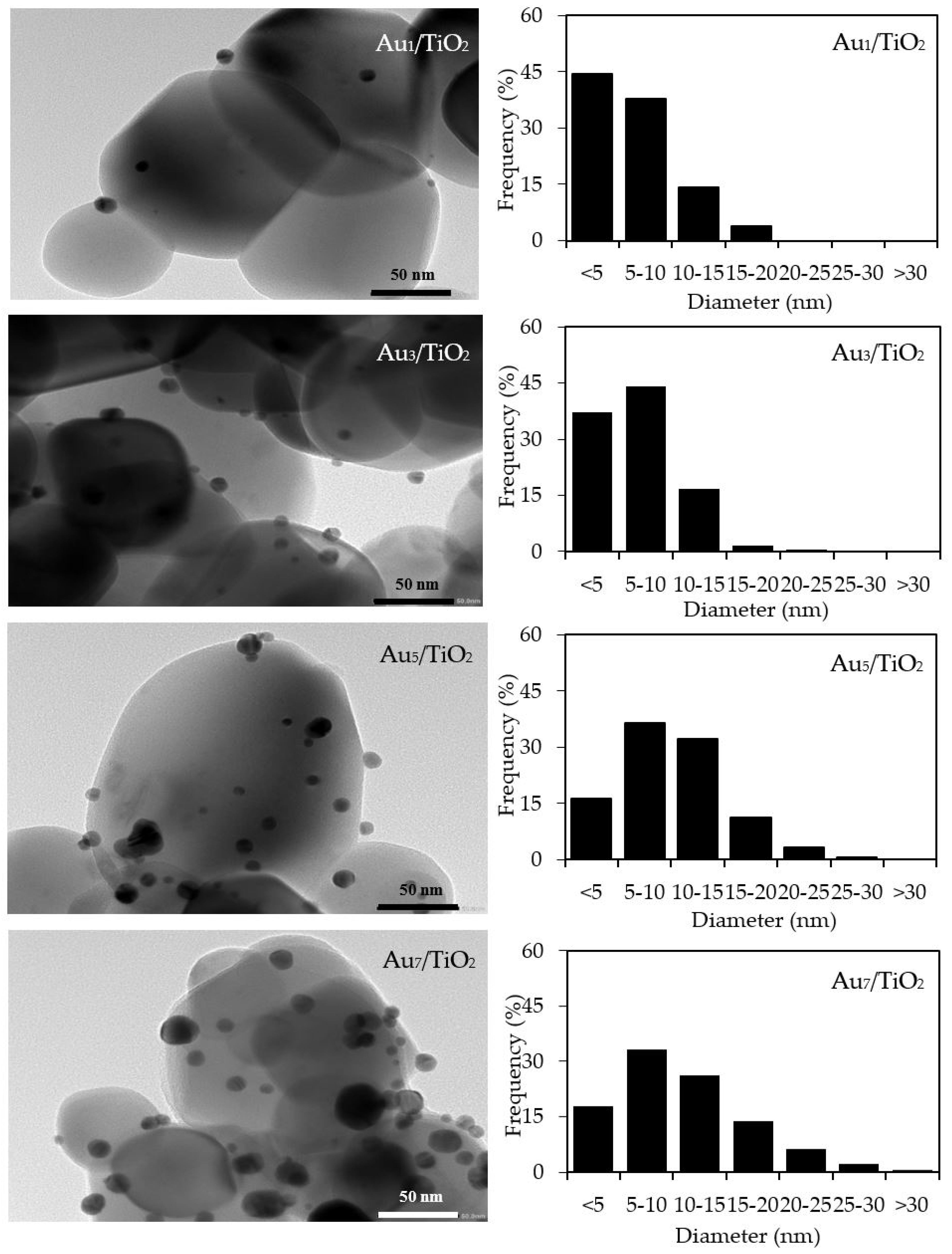
Figure 4.
Representative ultra violet-visible (UV-Vis) spectra of the TiO2 and different (a) Aux/TiO2 and (b) Au3M3/TiO2 photocatalysts with (insert) the respective Tauc’s plots.
Figure 4.
Representative ultra violet-visible (UV-Vis) spectra of the TiO2 and different (a) Aux/TiO2 and (b) Au3M3/TiO2 photocatalysts with (insert) the respective Tauc’s plots.
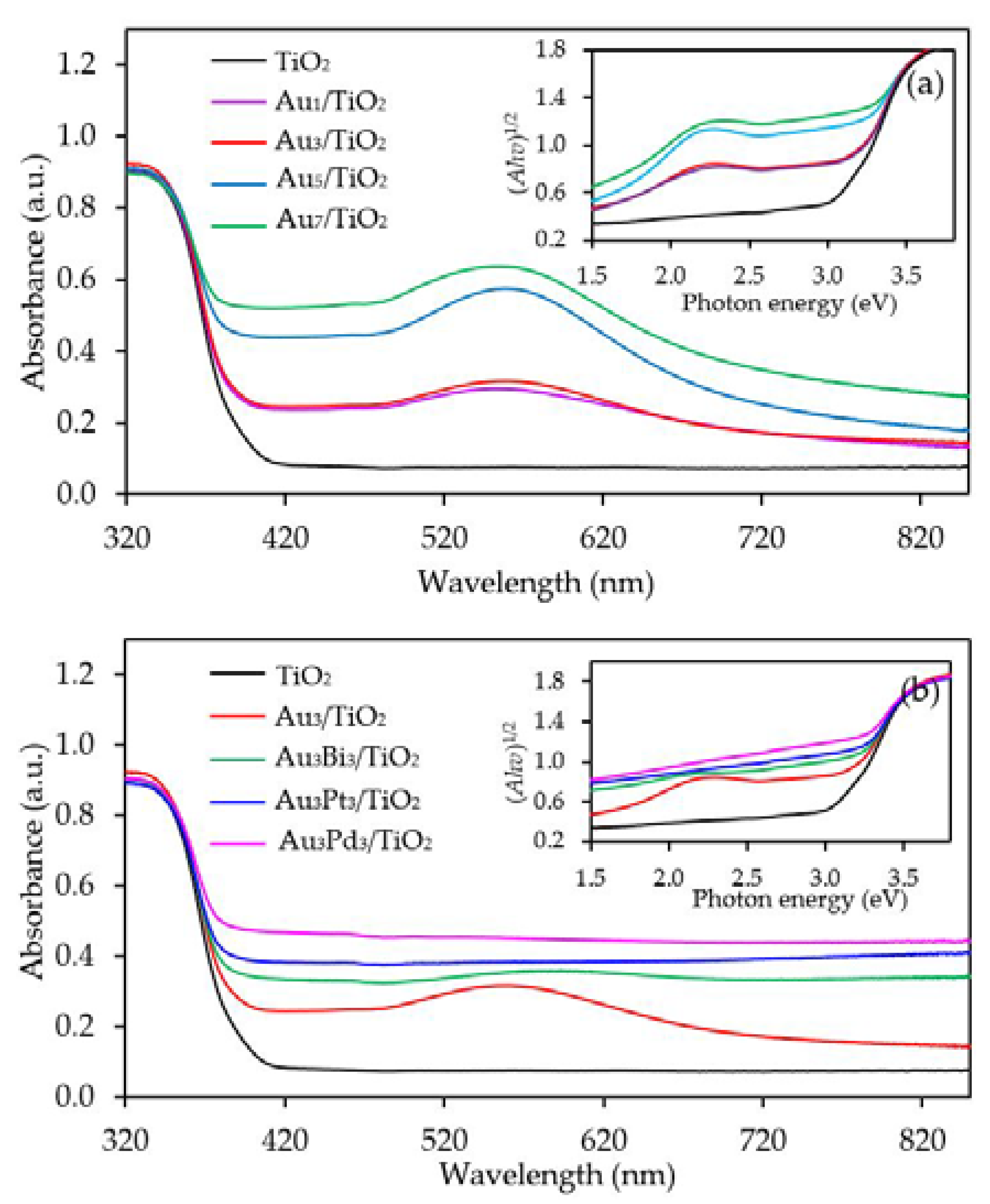
Figure 5.
Representative c spectra of the (a) Ti2p and (b) Au4f of the TiO2 and different Aux/TiO2 photocatalysts.
Figure 5.
Representative c spectra of the (a) Ti2p and (b) Au4f of the TiO2 and different Aux/TiO2 photocatalysts.

Figure 6.
Representative (a) glycerol conversion and (b) plot of ln (Ct/C0) as a function of time in the presence of the TiO2 or different Aux/TiO2 (x = 1–7) photocatalysts at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2 and using O2 as the electron acceptor.
Figure 6.
Representative (a) glycerol conversion and (b) plot of ln (Ct/C0) as a function of time in the presence of the TiO2 or different Aux/TiO2 (x = 1–7) photocatalysts at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2 and using O2 as the electron acceptor.
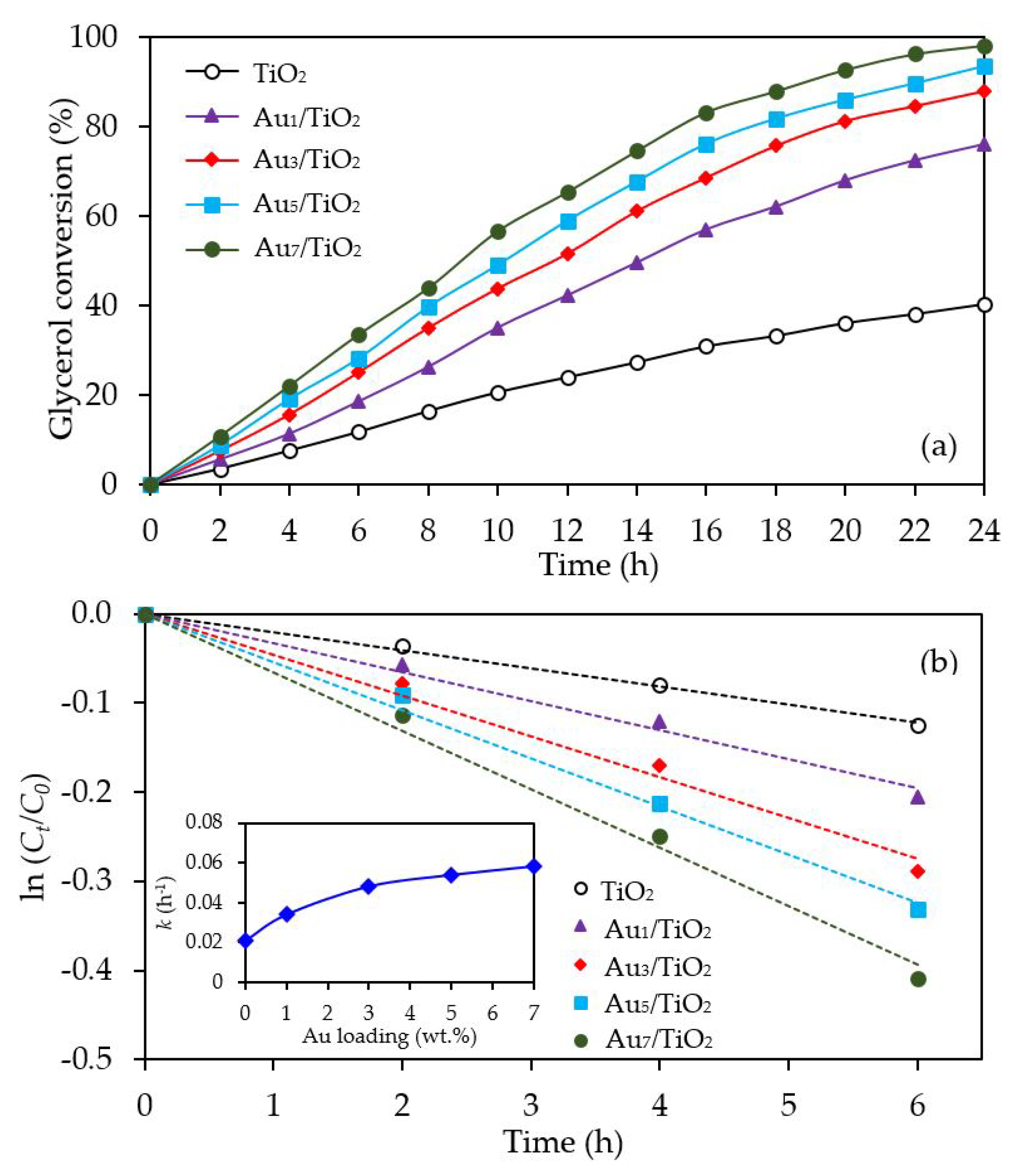
Figure 7.
Yield of selected products obtained from the glycerol photocatalytic conversion via the TiO2 and different Aux/TiO2 photocatalysts at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2 and using O2 as the electron acceptor.
Figure 7.
Yield of selected products obtained from the glycerol photocatalytic conversion via the TiO2 and different Aux/TiO2 photocatalysts at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2 and using O2 as the electron acceptor.
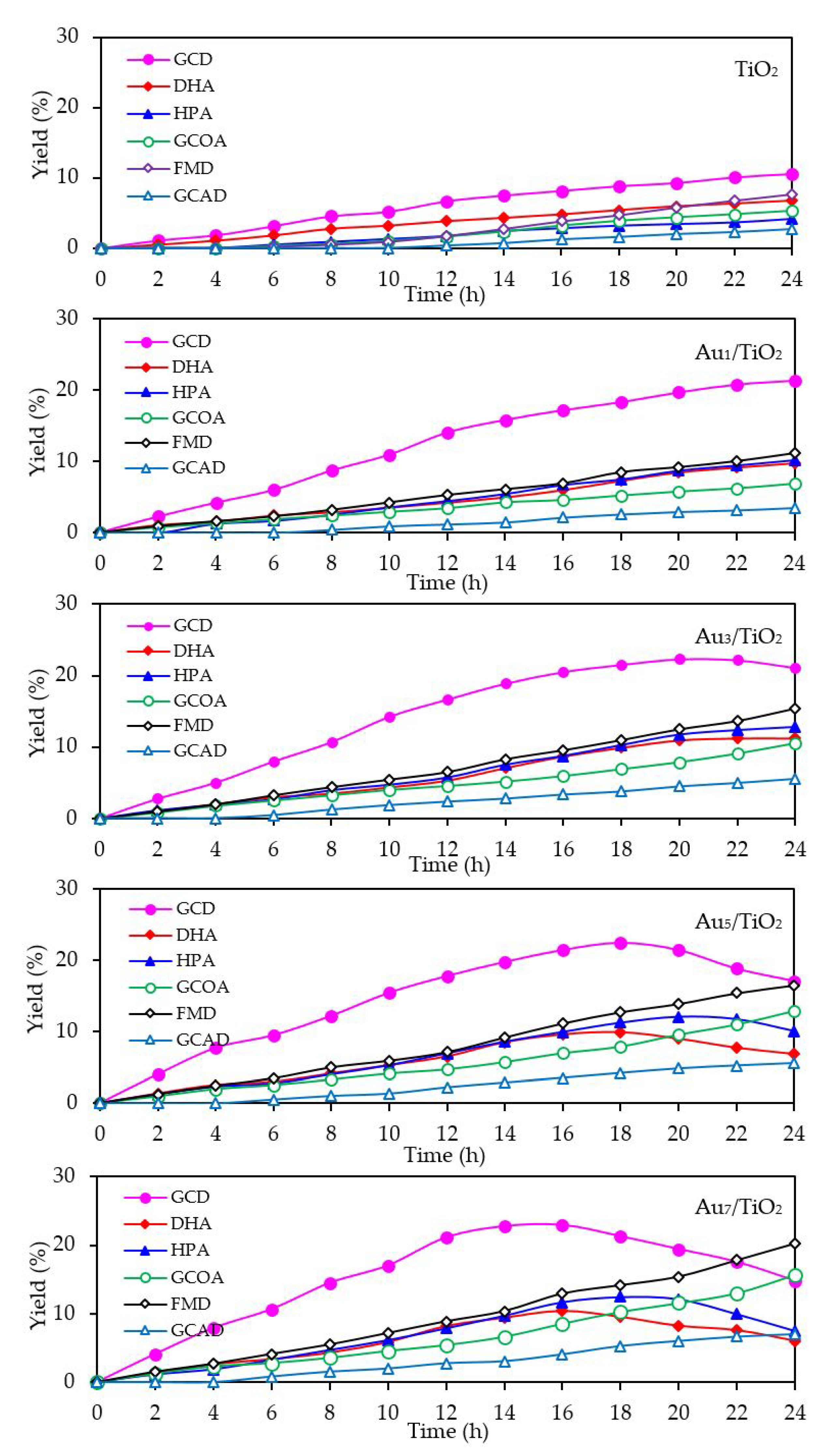
Figure 8.
(Left) Representative SEM images of the different Au3M3/TiO2 photocatalysts and (Right) EDS elemental analysis of the corresponding area shown in the rectangle on the SEM image.
Figure 8.
(Left) Representative SEM images of the different Au3M3/TiO2 photocatalysts and (Right) EDS elemental analysis of the corresponding area shown in the rectangle on the SEM image.
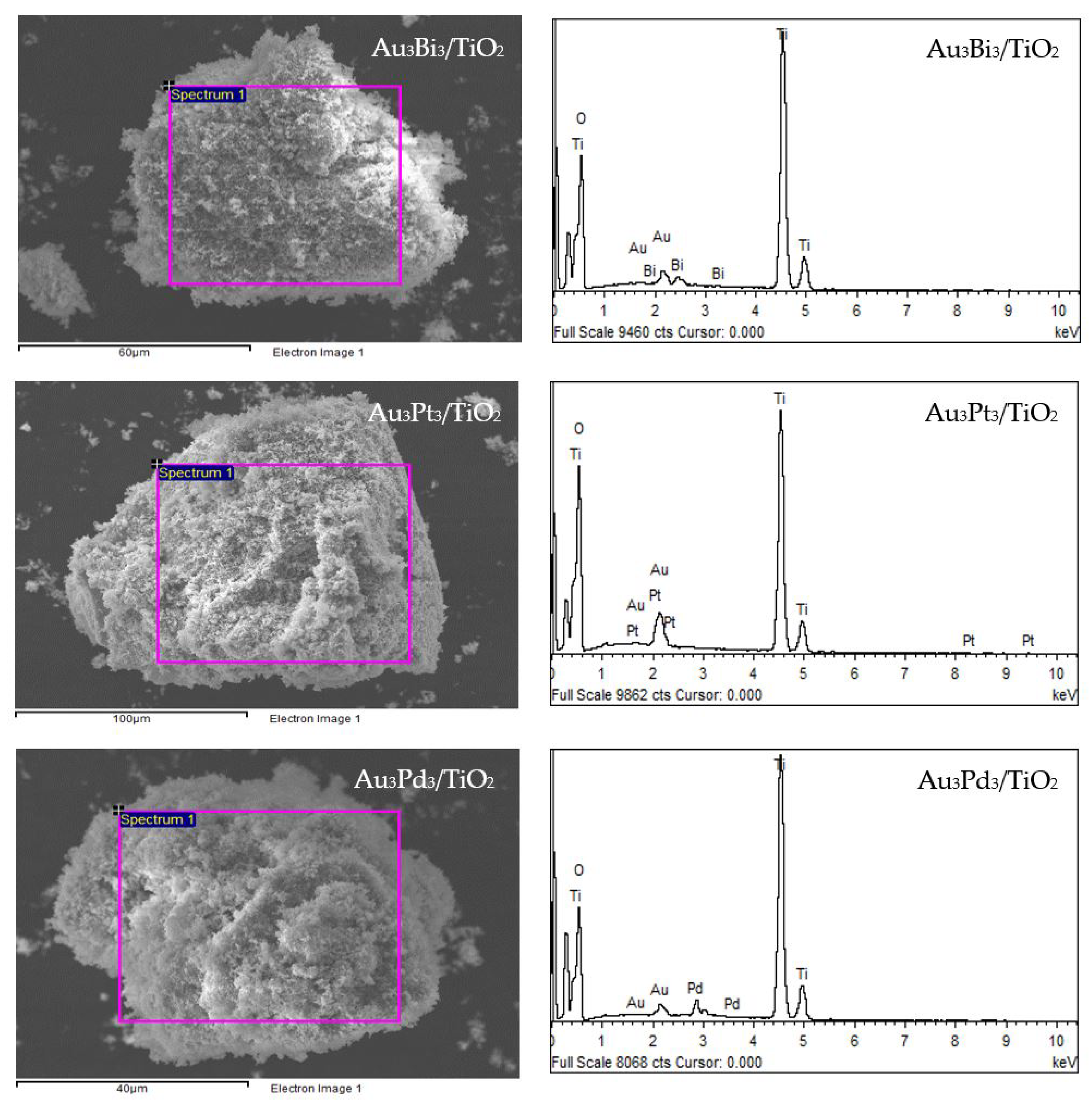
Figure 9.
(Left) Representative TEM images of the Au3/TiO2 and different Au3M3/TiO2 photocatalysts and (Right) the derived respective AuM NP size distribution on the surface of the TiO2.
Figure 9.
(Left) Representative TEM images of the Au3/TiO2 and different Au3M3/TiO2 photocatalysts and (Right) the derived respective AuM NP size distribution on the surface of the TiO2.
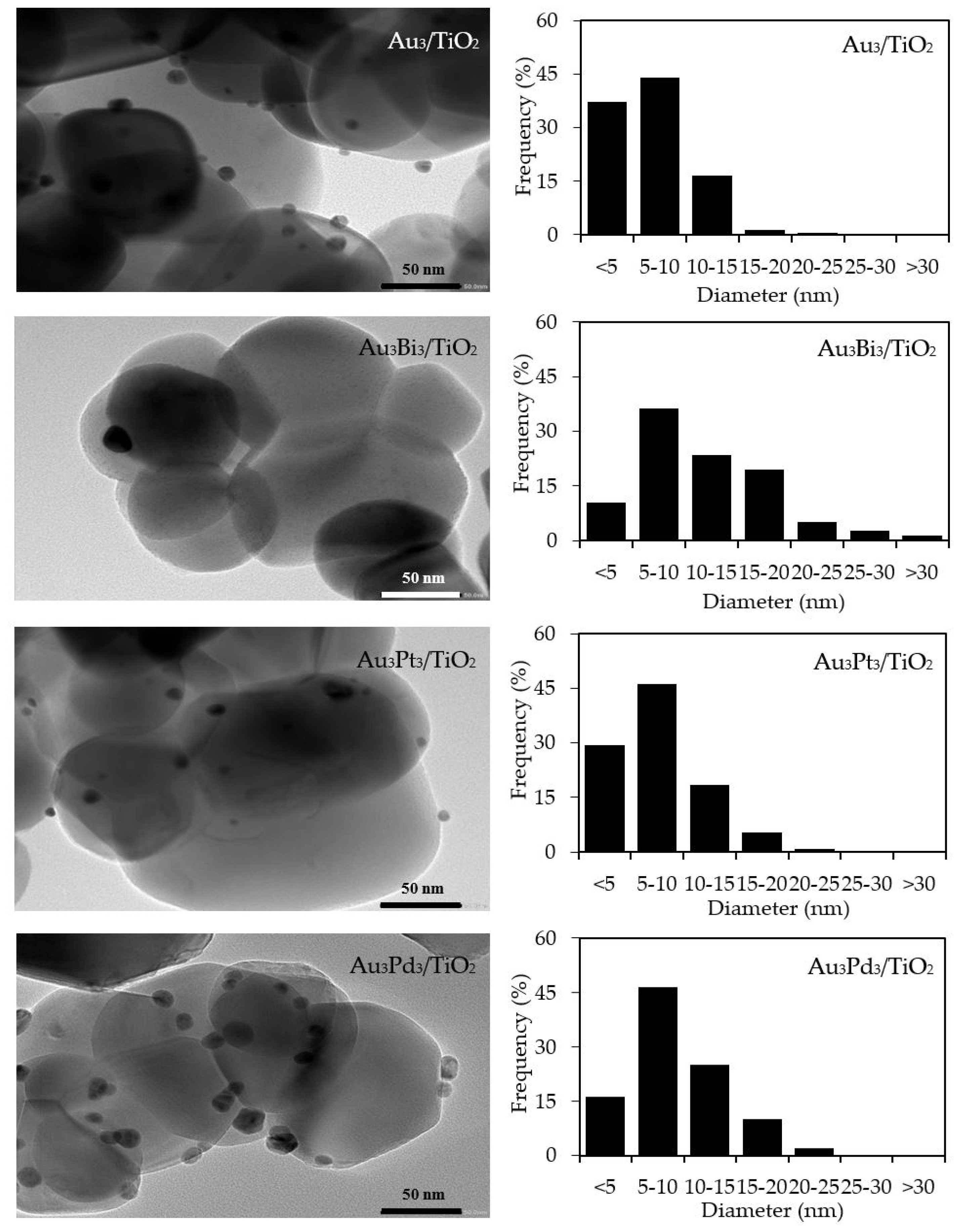
Figure 10.
Representative TEM–EDS mapping images and line-scan profile of the different of Au3M3/TiO2 photocatalysts.
Figure 10.
Representative TEM–EDS mapping images and line-scan profile of the different of Au3M3/TiO2 photocatalysts.
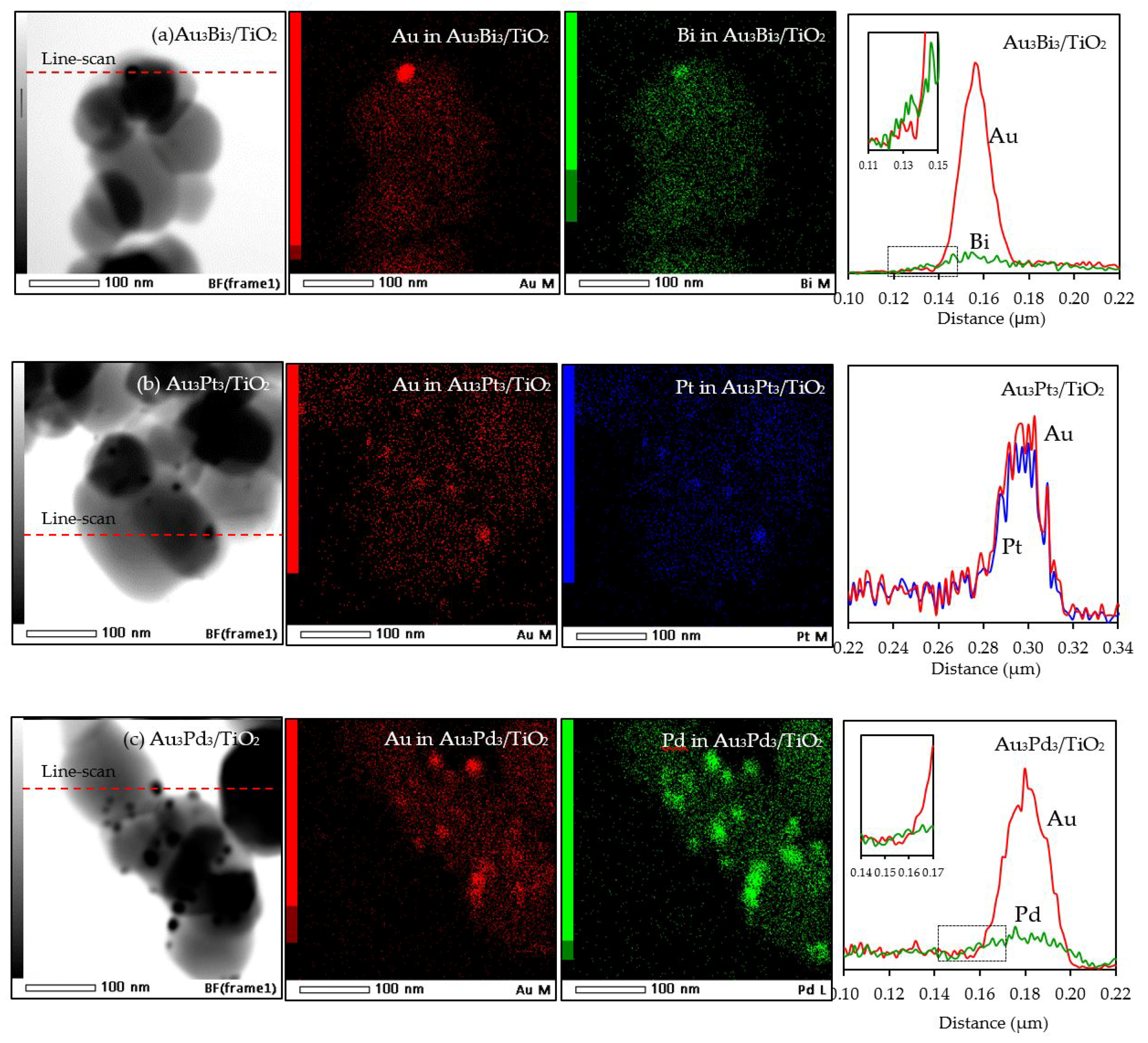
Figure 11.
Representative HR-XPS spectra of (a) Ti2p and (b) Au4f of the TiO2, Au3/TiO2 and the different Au3M3/TiO2 photocatalysts.
Figure 11.
Representative HR-XPS spectra of (a) Ti2p and (b) Au4f of the TiO2, Au3/TiO2 and the different Au3M3/TiO2 photocatalysts.
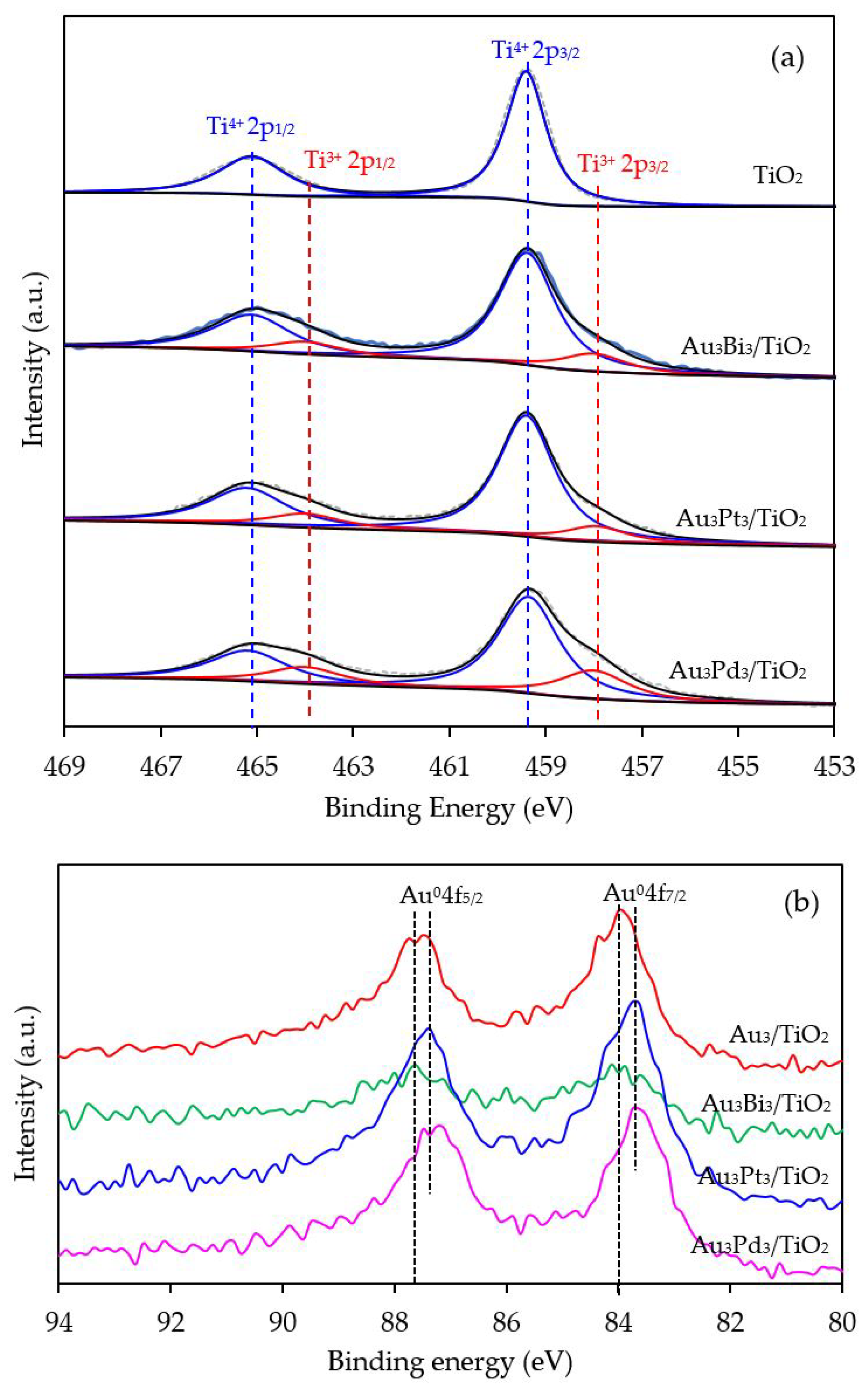
Figure 12.
Representative HR-XPS spectra of Bi 4f, Pt 4f and Pd 3d in the respective Au3M3/TiO2 photocatalysts.
Figure 12.
Representative HR-XPS spectra of Bi 4f, Pt 4f and Pd 3d in the respective Au3M3/TiO2 photocatalysts.
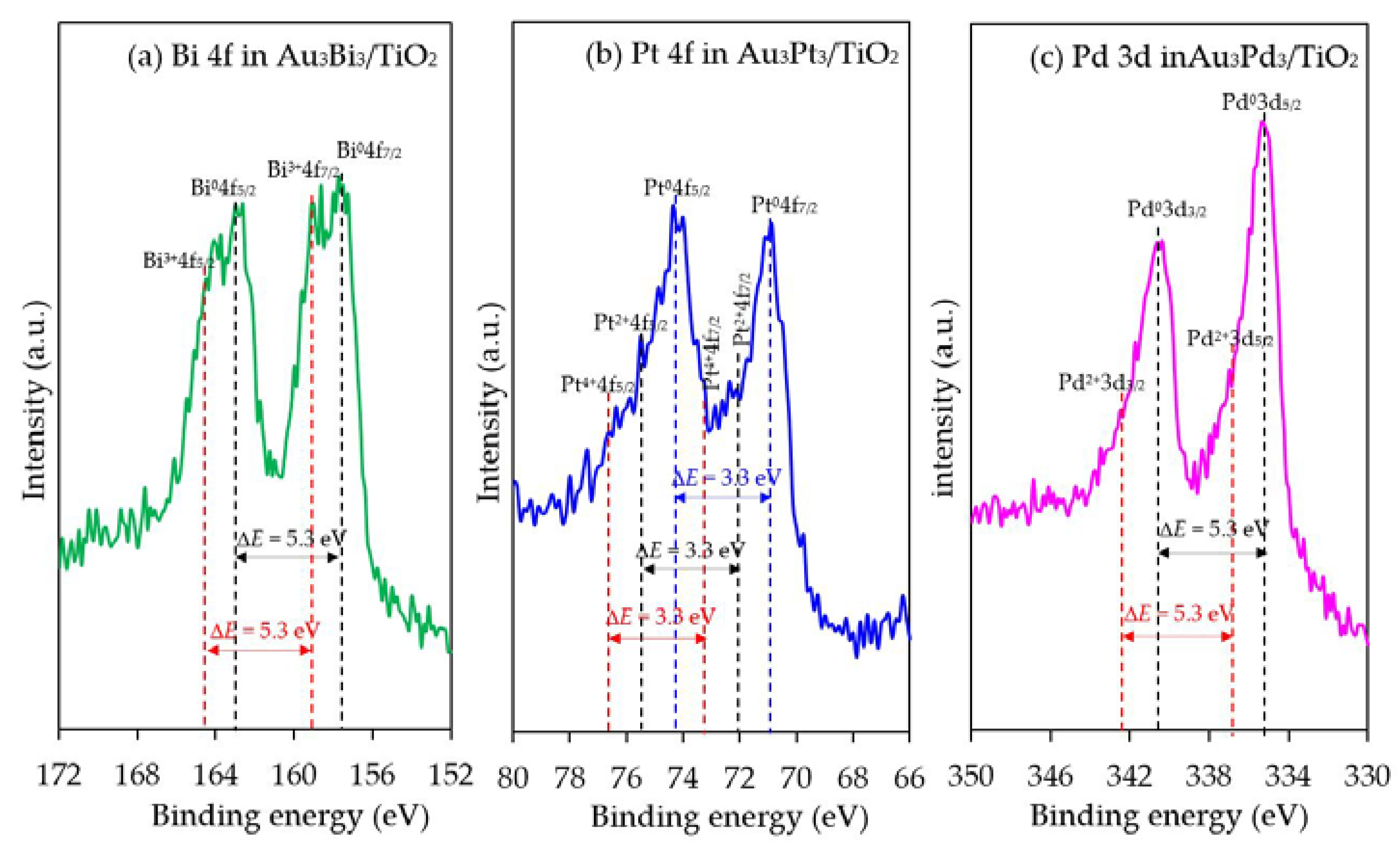
Figure 13.
Representative (a) glycerol conversion and (b) a plot of ln (Ct/C0) as a function of time in the presence of the respective Au3M3/TiO2 photocatalyst at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2 and using O2 as the electron acceptor.
Figure 13.
Representative (a) glycerol conversion and (b) a plot of ln (Ct/C0) as a function of time in the presence of the respective Au3M3/TiO2 photocatalyst at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2 and using O2 as the electron acceptor.
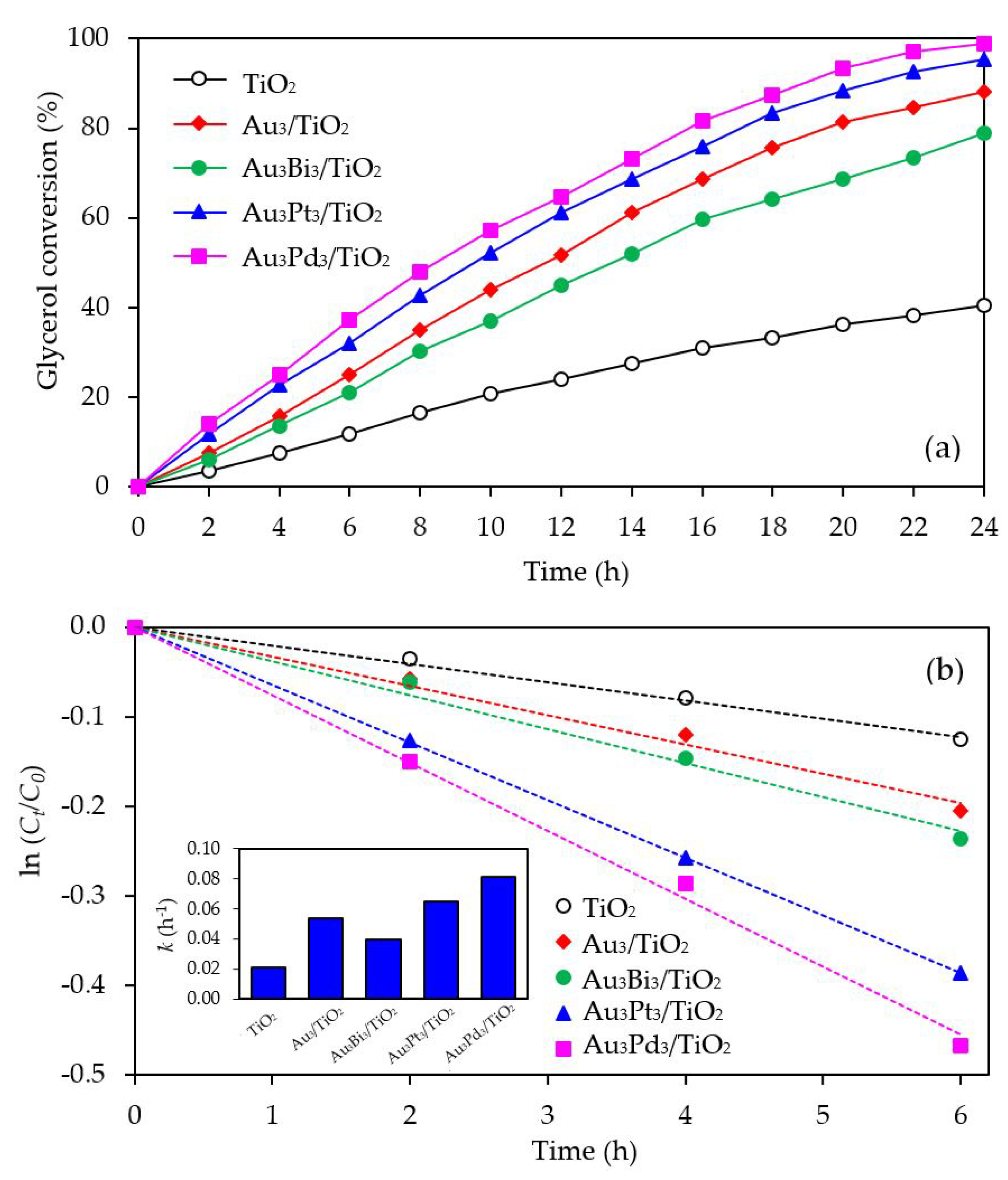
Figure 14.
Schematic profiles of the contact metal with work function in the bimetallic NPs system.
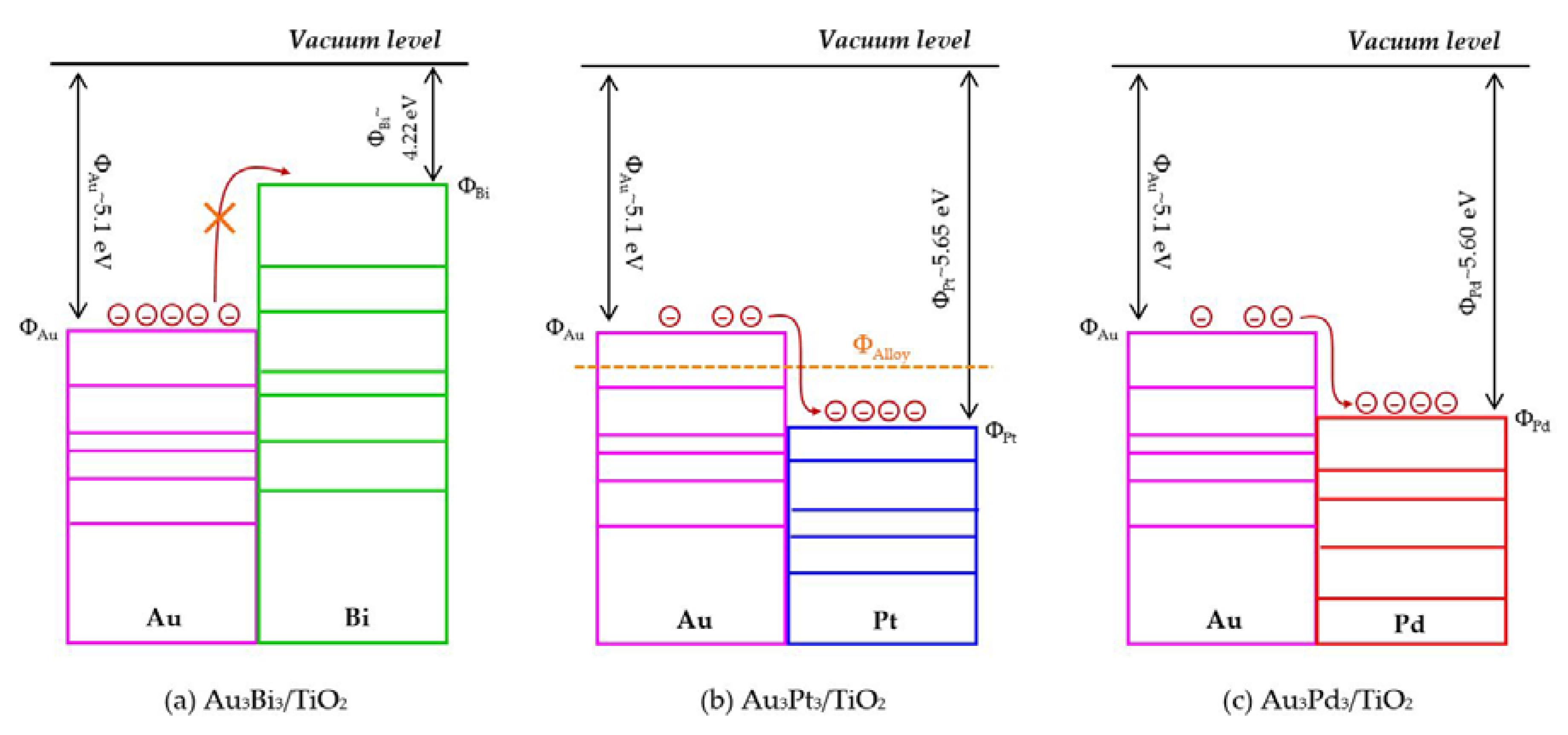
Figure 15.
Yield of selected products obtained from the glycerol photocatalytic conversion via the TiO2, Au3/TiO2 and respective Au3M3/TiO2 photocatalysts at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2, and using O2 as the electron acceptor.
Figure 15.
Yield of selected products obtained from the glycerol photocatalytic conversion via the TiO2, Au3/TiO2 and respective Au3M3/TiO2 photocatalysts at a loading of 3.0 g/L, light intensity of 4.7 mW/cm2, and using O2 as the electron acceptor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Properties of the as-synthesized Aux/TiO2 and Au3M3/TiO2 photocatalysts.
| Photocatalyst | Au Loading (wt. %) a | M Loading (wt. %) a | Decorated Metal Particle Size (nm) b | Bandgap Energy (eV) c | Ti3+/Ti4+ d |
|---|---|---|---|---|---|
| Au1/TiO2 | 0.98 ± 0.10 | n.d. | 6.36 ± 0.75 | 3.00 | 0.1220 |
| Au3/TiO2 | 3.02 ± 0.21 | n.d. | 6.69 ± 0.86 | 3.00 | 0.1478 |
| Au5/TiO2 | 4.96 ± 0.19 | n.d. | 10.04 ± 1.11 | 2.94 | 0.2198 |
| Au7/TiO2 | 7.08 ± 0.35 | n.d. | 10.84 ± 1.01 | 2.80 | 0.2412 |
| Au3Bi3/TiO2 | 2.96 ± 0.12 | 3.11 ± 0.28 | 11.84 ± 1.11 | 2.85 | 0.1960 |
| Au3Pt3/TiO2 | 3.06 ± 0.10 | 3.01 ± 0.20 | 7.61 ± 0.91 | 2.85 | 0.1941 |
| Au3Pd3/TiO2 | 3.09 ± 0.14 | 3.05 ± 0.22 | 9.66 ± 1.06 | 2.79 | 0.3561 |
a Estimated from the scanning electron microscopy–energy-dispersive X-ray spectrometry (SEM–EDS) analysis; b Average (± standard deviation (SD)) metal particle size obtained from the transmission electron microscopy (TEM) analysis; c Calculated from Tauc’s plot (plot of (αhν)1/n against (hν)); d Calculated from area under deconvoluted high-resolution X-ray photoelectron spectroscopy (HR-XPS) peak of Ti2p; n.d., not detected.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jedsukontorn, T.; Saito, N.; Hunsom, M. Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts. Nanomaterials 2018, 8, 269. https://doi.org/10.3390/nano8040269
AMA Style
Jedsukontorn T, Saito N, Hunsom M. Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts. Nanomaterials. 2018; 8(4):269. https://doi.org/10.3390/nano8040269
Chicago/Turabian StyleJedsukontorn, Trin, Nagahiro Saito, and Mali Hunsom. 2018. "Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts" Nanomaterials 8, no. 4: 269. https://doi.org/10.3390/nano8040269
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.