Electronic and Magnetic Properties of Stone–Wales Defected Graphene Decorated with the Half-Metallocene of M (M = Fe, Co, Ni): A First Principle Study

Abstract
:1. Introduction
2. Calculation Details
3. Results and Discussion
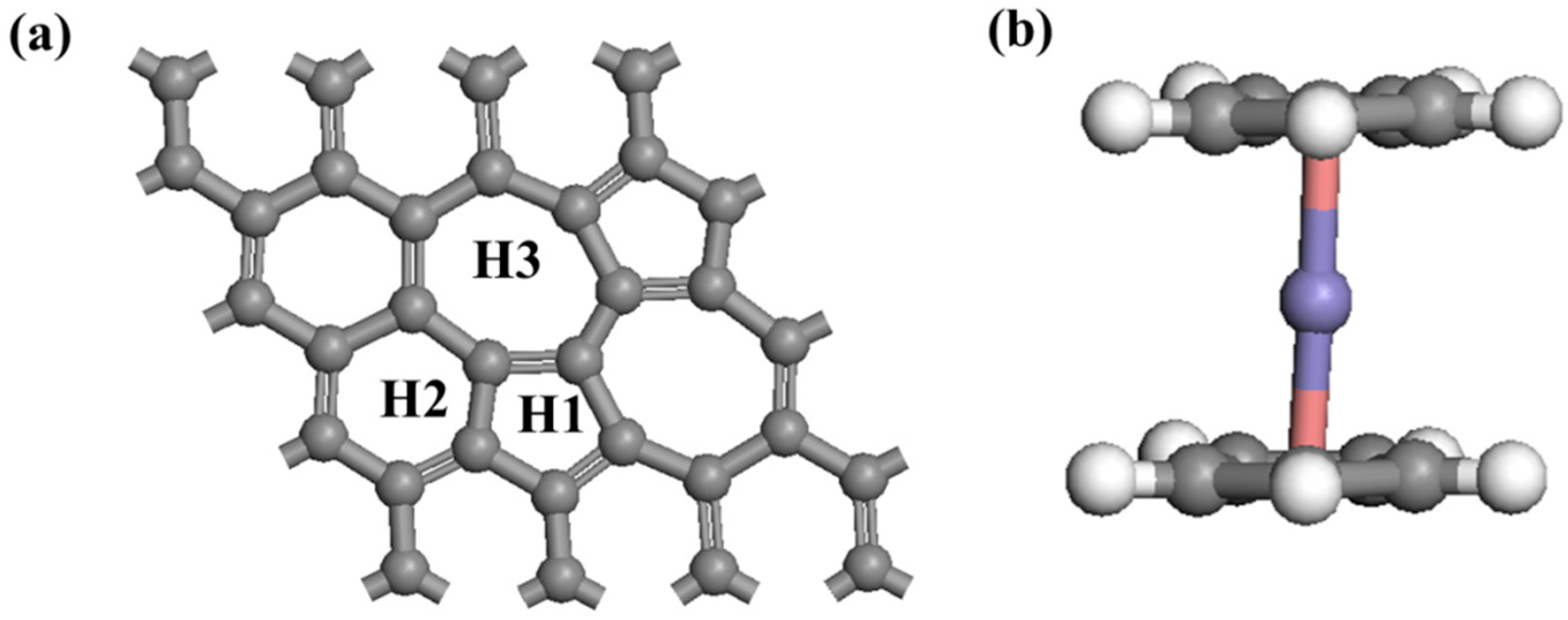
3.1. Adsorption Configurations and Energy
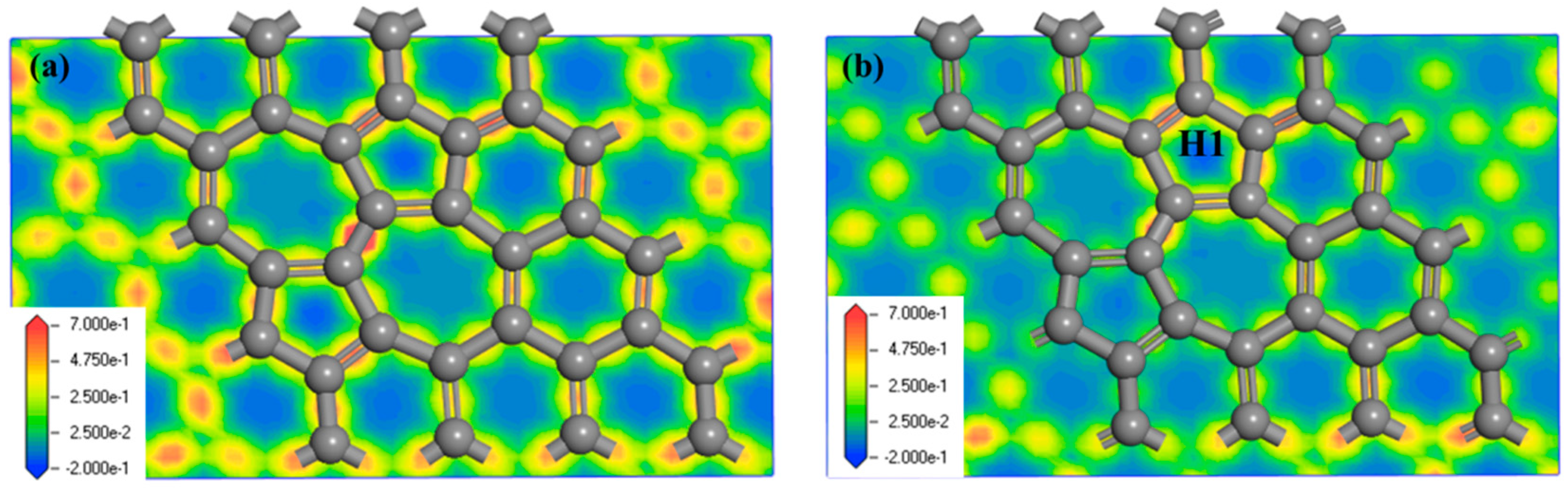
3.1.1. Adsorption of Cp in Stone-Wales Graphene (SWG)
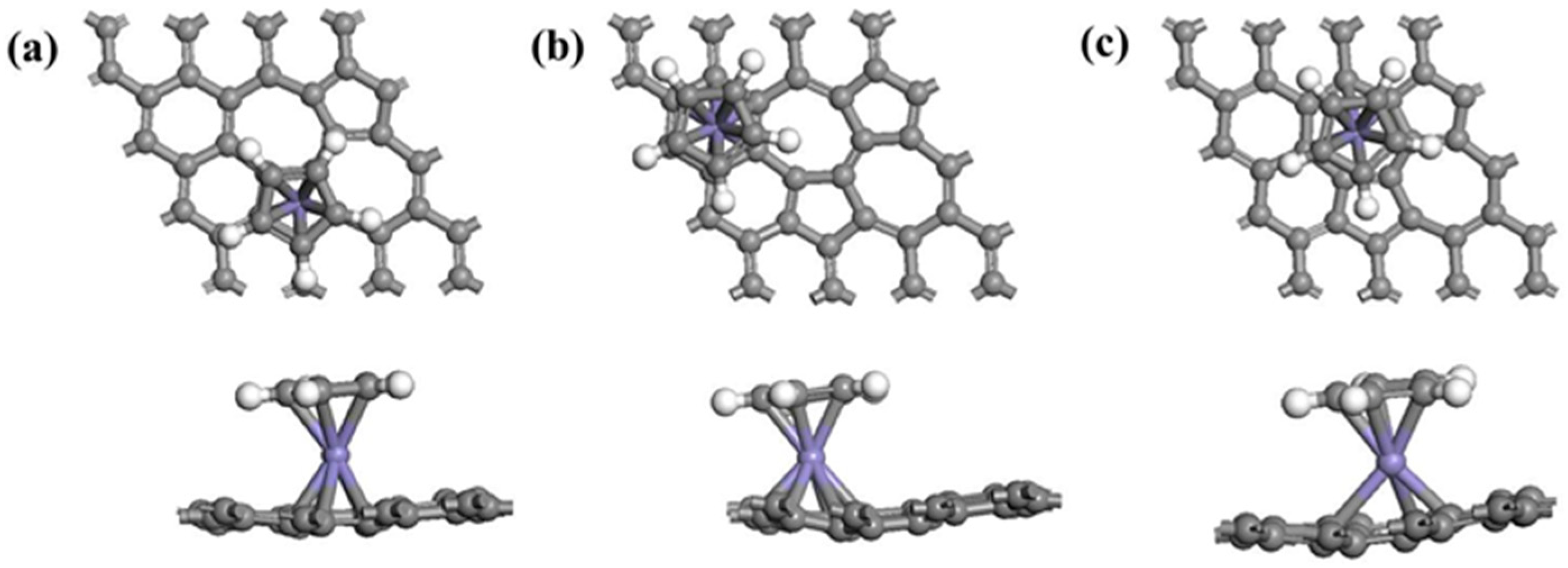
3.1.2. Adsorption of Half-Metallocene of M (M = Fe, Co, Ni) in SWG
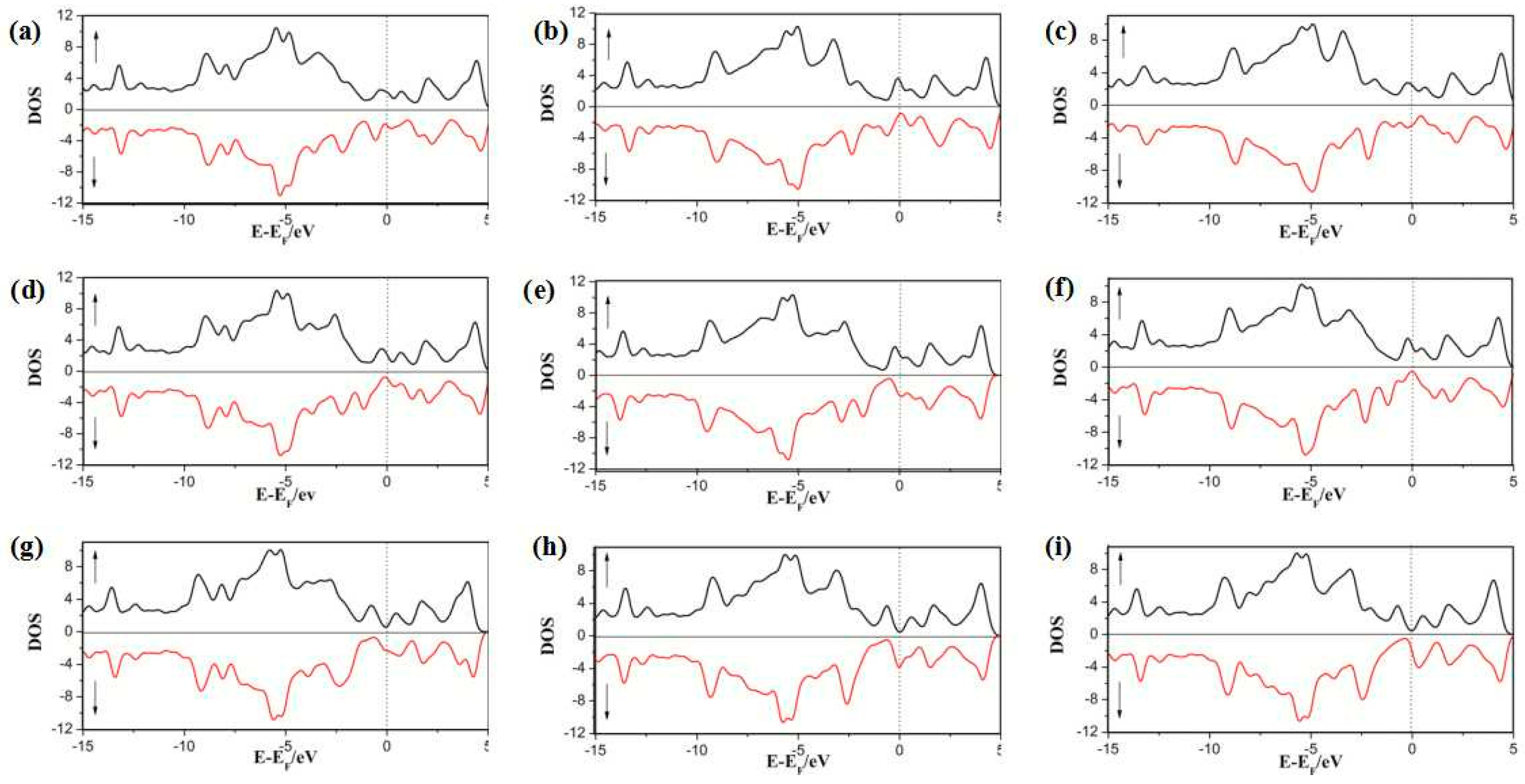
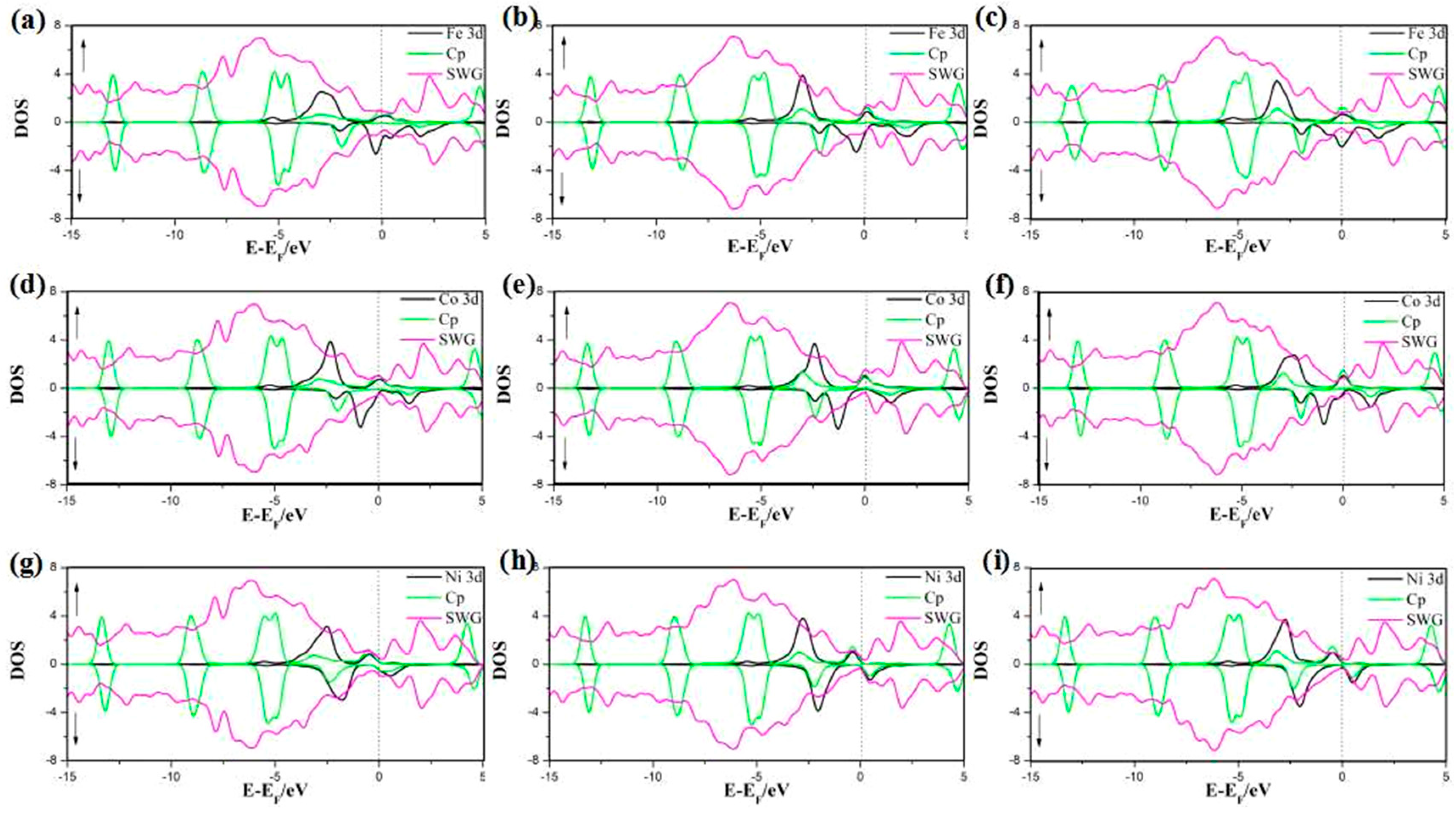
3.2. Density of States (DOS) of the SWG System
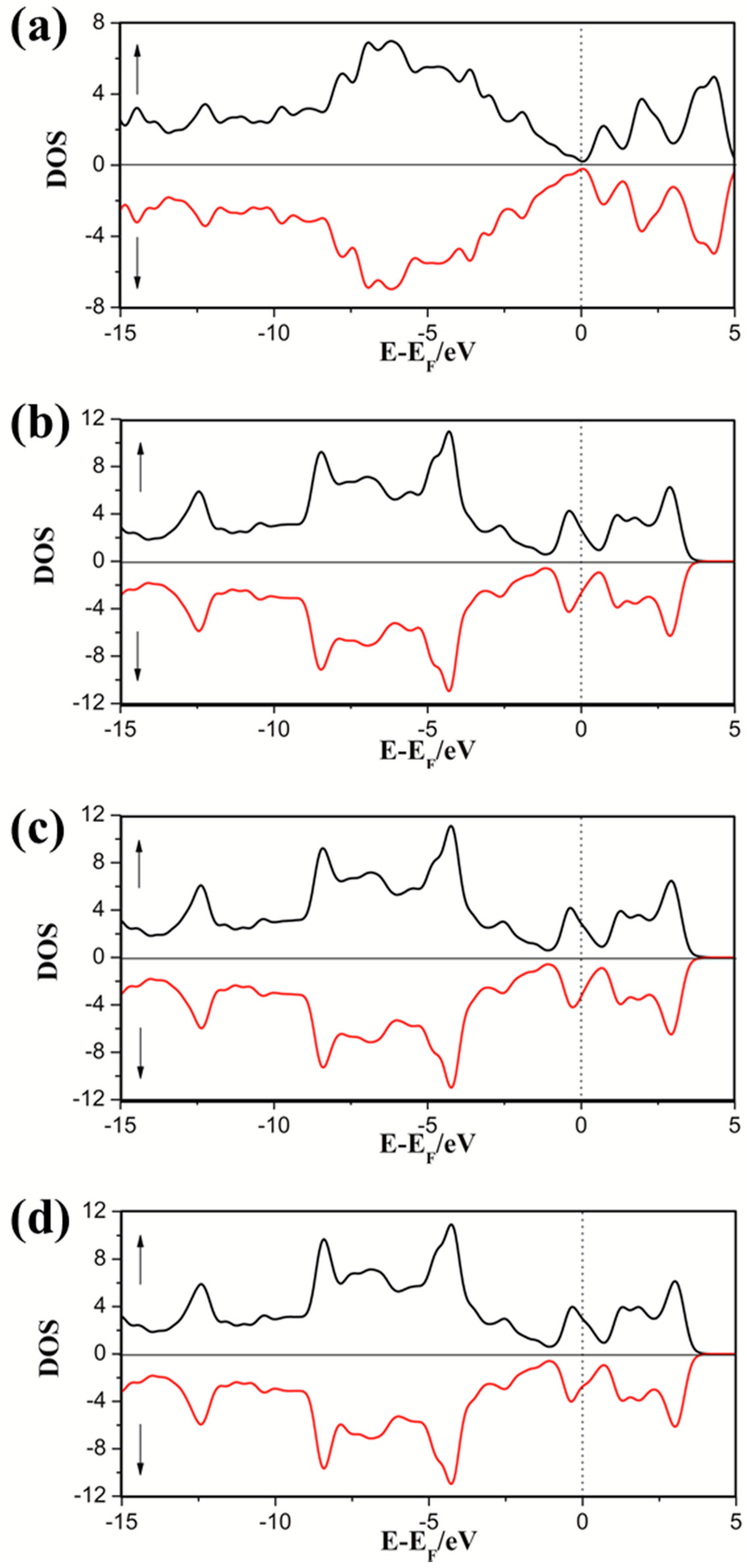
3.2.1. DOS of Cp in SWG
3.2.2. DOS of Half-Metallocene of M (M = Fe, Co, Ni) in SWG
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Khoo, K.H.; Wei, S.T.; John, T.L.Q.; Su, Y. Origin of contact resistance at ferromagnetic metal-graphene interfaces. ACS Nano 2016, 10, 11219–11227. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Wang, Z.; Wu, Y.; Liu, X.; He, Y.; Kim, J.K. Multilayer graphene enables higher efficiency in improving thermal conductivities of graphene/epoxy composites. Nano Lett. 2016, 16, 3585–3593. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Szpunar, J.A. Hydrogen storage performance in Pd/graphene nanocomposites. ACS Appl. Mater. Interface 2016, 8, 25933–25940. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Fu, Z.; Wang, C.; Tang, Y.; Zhang, H.; Yuan, L.; Yang, X. The electronic and magnetic properties of B-doping stone-wales defected graphene decorated with transition-metal atoms. Physica E 2015, 73, 257–261. [Google Scholar] [CrossRef]
- Mohammadi, A.; Haji-Nasiri, S. The electronic transport properties of defected bilayer sliding armchair graphene nanoribbons. Phys. Lett. A 2018, 382, 1040–1046. [Google Scholar] [CrossRef]
- Mombru, D.; Faccio, R.; Mombru, A.W. Possible causes for rippling in a multivacancy graphene system. Int. J. Quantum Chem. 2018, 118, e25529. [Google Scholar] [CrossRef]
- Tran, T.T.; Alotaibi, F.; Nine, M.J.; Silva, R.; Tran, D.N.H.; Janowska, I.; Losic, D. Engineering of highly conductive and ultra-thin nitrogen-doped graphene films by combined methods of microwave irradiation, ultrasonic spraying and thermal annealing. Chem. Eng. J. 2018, 338, 764–773. [Google Scholar]
- Wang, K.; Wang, J.; Wu, Y.; Zhao, S.; Wang, Z.; Wang, S. Nitrogen-doped graphene prepared by a millisecond photo-thermal process and its applications. Org. Electron. 2018, 56, 221–231. [Google Scholar] [CrossRef]
- Yokwana, K.; Ray, S.C.; Khenfouch, M.; Kuyarega, A.T.; Mamba, B.B.; Mhlanga, S.D.; Nxunnalo, E.N. Facile Synthesis of nitrogen doped graphene oxide from graphite flakes and powders: A comparison of their surface chemistry. J. Nanosci. Nanotechnol. 2018, 18, 5470–5484. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, F.M.D.; Angilella, G.G.N.; Pucci1, R. Effect of impurities in high-symmetry lattice positions on the local density of states and conductivity of grapheme. Phys. Rev. B 2009, 80, 094203. [Google Scholar] [CrossRef]
- Dang, J.S.; Wang, W.W.; Zheng, J.J.; Nagase, S.; Zhao, X. Formation of stone-wales edge: Multistep reconstruction and growth mechanisms of zigzag nanographene. J. Comp. Chem. 2017, 38, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, S.; Azizi, M. The effect of high concentrations and orientations of stone-wales defects on the thermal conductivity of graphene nanoribbons. Mol. Simulat. 2018, 44, 236–242. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, H.; Cheng, X.L.; Xu, Y. Coherent Resonance of quantum plasmons in stone-wales defected graphene-silver nanowire hybrid system. Front. Phys. 2017, 12, 99–107. [Google Scholar] [CrossRef]
- Zaminpayma, E.; Razavi, M.E.; Nayebi, P. Electronic properties of graphene with single vacancy and stone-wales defects. Appl. Surf. Sci. 2017, 414, 101–106. [Google Scholar] [CrossRef]
- Jaskolski, W.; Pelc, M.; Chico, L.; Ayuela, A. Existence of nontrivial topologically protected states at grain boundaries in bilayer graphene: Signatures and electrical switching. Nanoscale 2016, 8, 6079–6084. [Google Scholar] [CrossRef] [PubMed]
- King, R.B. Some Aspects of the symmetry and topology of possible carbon allotrope structures. J. Math. Chem. 1998, 23, 197–227. [Google Scholar] [CrossRef]
- Feng, P.X.L. Tuning in to a graphene oscillator. Nat. Nanotechnol. 2013, 8, 897–898. [Google Scholar] [CrossRef] [PubMed]
- Salary, M.M.; Inampudi, S.; Zhang, K.; Tadmor, E.B.; Mosallaei, H. Mechanical actuation of graphene sheets via optically induced forces. Phys. Rev. B 2016, 94, 235–403. [Google Scholar] [CrossRef]
- Majidi, R.; Tabrizi, K.G. Electronic properties of defect-free and defective bilayer graphene in an electric field. Nanotubes Carbon Nanostruct. 2011, 19, 532–539. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Hu, H.F.; Gu, L.; Wang, W.; Jia, J.F. Electronic and optical properties of zigzag graphene nanoribbon with stone-wales defect. Acta Phys. Sin. 2011, 60. [Google Scholar]
- Zhang, Y.H.; Zhou, K.G.; Xie, K.F.; Gou, X.C.; Zeng, J.; Zhang, H.L.; Peng, Y. Effects of stone-wales defect on the interactions between NH3, NO2 and graphene. J. Nanosci. Nanotechnol. 2010, 10, 7347–7350. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Lee, B.; Kim, J.; Cho, K. Ozone Adsorption on graphene: Ab initio study and experimental validation. J. Phys. Chem. C 2009, 113, 14225–14229. [Google Scholar] [CrossRef]
- Chen, F.; Tao, N.J. Electron transport in single molecules: From benzene to graphene. Accounts Chem. Res. 2009, 42, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.D.; Singh, M.R.; Gumbs, G.; Anton, M.A.; Carreno, F. Dipole-dipole interaction between a quantum dot and a graphene nanodisk. Phys. Rev. B 2012, 86, 7035. [Google Scholar] [CrossRef]
- Kim, S.S.; Kuang, Z.; Ngo, Y.H.; Farmer, B.L.; Naik, R.R. Biotic-abiotic interactions: Factors that influence peptide-graphene interactions. ACS Appl. Mater. Interface 2015, 7, 20447–20453. [Google Scholar] [CrossRef] [PubMed]
- Podeszwa, R. Interactions of graphene sheets deduced from properties of polycyclic aromatic hydrocarbons. J. Chem. Phys. 2010, 132, 044704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lu, W.C.; Zhang, H.X.; Ho, K.M.; Wang, C.Z. Hydrogen adatom interaction on graphene: A first principles study. Carbon 2018, 131, 137–141. [Google Scholar] [CrossRef]
- Enriquez, J.I.G.; Villagracia, A.R. Hydrogen adsorption on pristine, defected, and 3d-block transition metal-doped penta-graphene. Int. J. Hydrog. Energy 2016, 41, 15411. [Google Scholar] [CrossRef]
- Gao, J.; Yip, J.; Zhao, J.; Yakobson, B.I.; Ding, F. Graphene nucleation on transition metal surface: Structure transformation and role of the metal step edge. J. Am. Chem. Soc. 2012, 134, 9534. [Google Scholar] [CrossRef]
- Yu, X.; Qu, B.; Zhao, Y.; Li, C.; Chen, Y.; Sun, C.; Gao, P.; Zhu, C. Growth of hollow transition metal (Fe, Co., Ni) oxide nanoparticles on graphene sheets through kirkendall effect as anodes for high-performance lithium-ion batteries. Chem. Eur. J. 2016, 22, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Adsorption of H2O, NH3, CO, NO2, and NO on graphene: A first-principles study. Phys. Rev. B 2008, 77, 125416. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d | E | Qe | |
|---|---|---|---|
| SWG-Cp | |||
| 5 | 3.901 | −1.15 | 0.51 |
| 6 | 3.489 | −1.10 | 0.31 |
| 7 | 3.413 | −1.09 | 0.36 |
| SWG-Fe-Cp | |||
| 5 | 3.692 | −5.54 | 0.38 |
| 6 | 3.384 | −5.63 | 0.54 |
| 7 | 3.603 | −5.72 | 0.58 |
| SWG-Co-Cp | |||
| 5 | 3.653 | −6.12 | 0.38 |
| 6 | 3.383 | −6.21 | 0.44 |
| 7 | 3.456 | −6.67 | 0.48 |
| SWG-Ni-Cp | |||
| 5 | 3.751 | −3.82 | 0.32 |
| 6 | 3.608 | −3.83 | 0.36 |
| 7 | 3.507 | −3.85 | 0.38 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, K.; Jia, Q.; Zhang, X.; Fu, L.; Zhao, G. Electronic and Magnetic Properties of Stone–Wales Defected Graphene Decorated with the Half-Metallocene of M (M = Fe, Co, Ni): A First Principle Study. Nanomaterials 2018, 8, 552. https://doi.org/10.3390/nano8070552
Xie K, Jia Q, Zhang X, Fu L, Zhao G. Electronic and Magnetic Properties of Stone–Wales Defected Graphene Decorated with the Half-Metallocene of M (M = Fe, Co, Ni): A First Principle Study. Nanomaterials. 2018; 8(7):552. https://doi.org/10.3390/nano8070552
Chicago/Turabian StyleXie, Kefeng, Qiangqiang Jia, Xiangtai Zhang, Li Fu, and Guohu Zhao. 2018. "Electronic and Magnetic Properties of Stone–Wales Defected Graphene Decorated with the Half-Metallocene of M (M = Fe, Co, Ni): A First Principle Study" Nanomaterials 8, no. 7: 552. https://doi.org/10.3390/nano8070552