Synthesis of Poly-Sodium-Acrylate (PSA)-Coated Magnetic Nanoparticles for Use in Forward Osmosis Draw Solutions

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
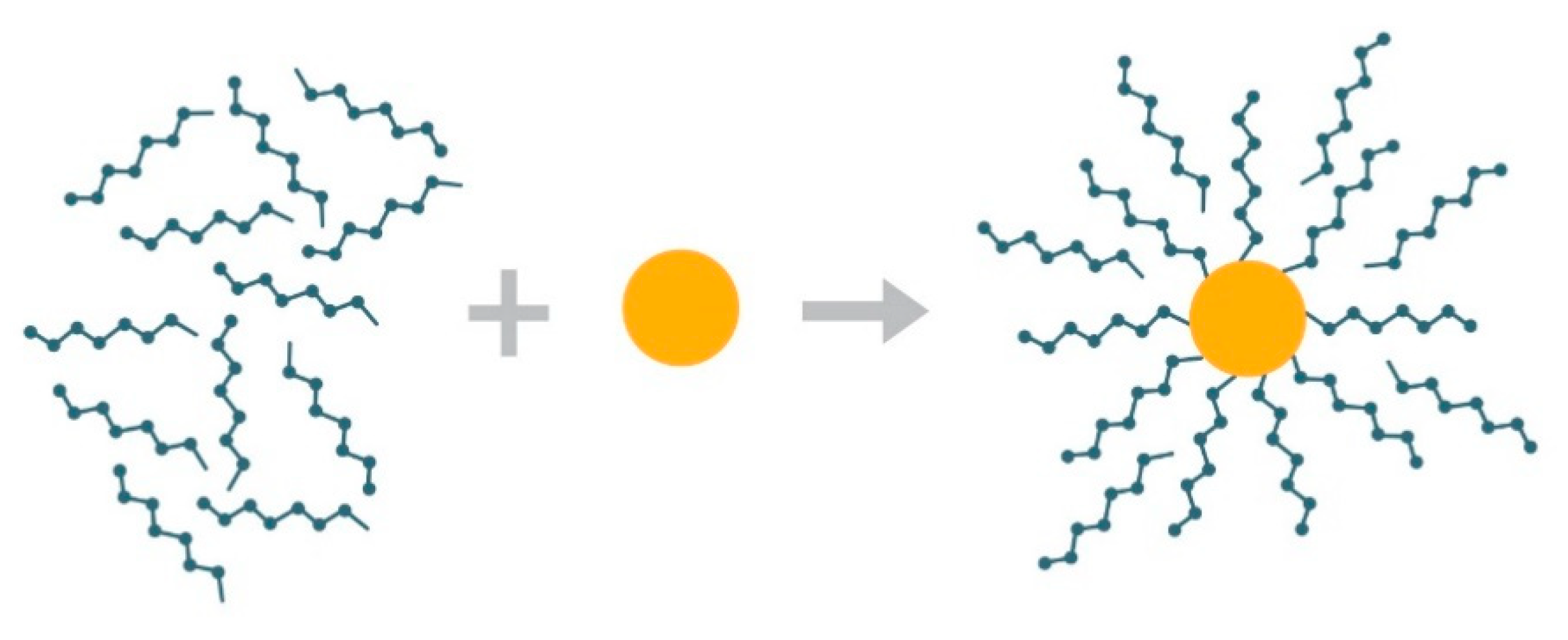
2.2. Preparation of PSA-MNPs Solutions
- Sample A: Stock solution PSA (20 mmol PSA in 10 mL DI water):Stock solution MNPs = 1:1,
- Sample B: Stock solution PSA (1 mmol PSA in 10 mL DI water):Stock solution MNPs = 1:2 and
- Sample C: Stock solution (0.66 mmol PSA in 10 mL DI water) PSA:Stock solution MNPs = 1:3.
2.3. Characterization of Nanoparticles
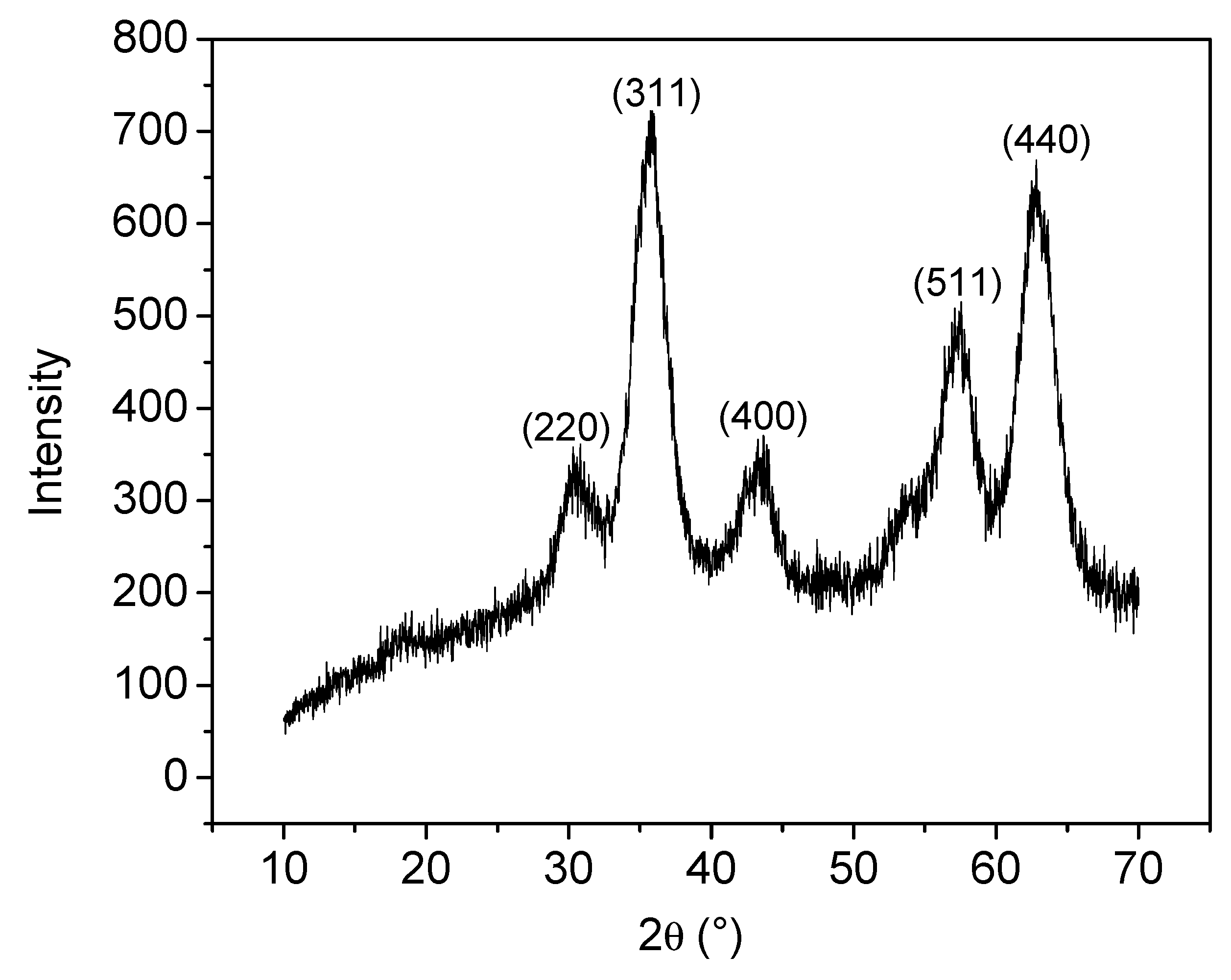
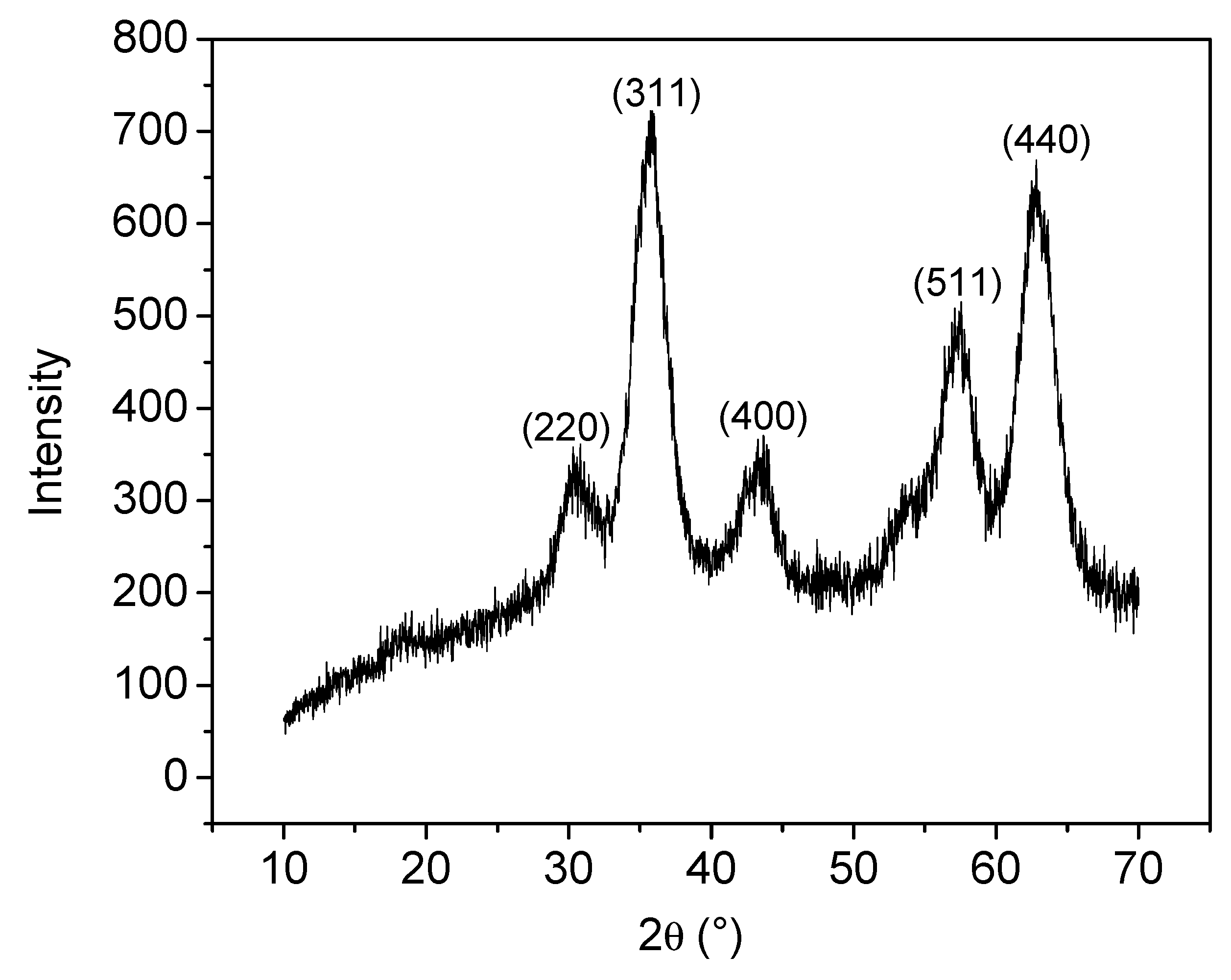
2.3.1. Structural X-Ray Powder Diffraction
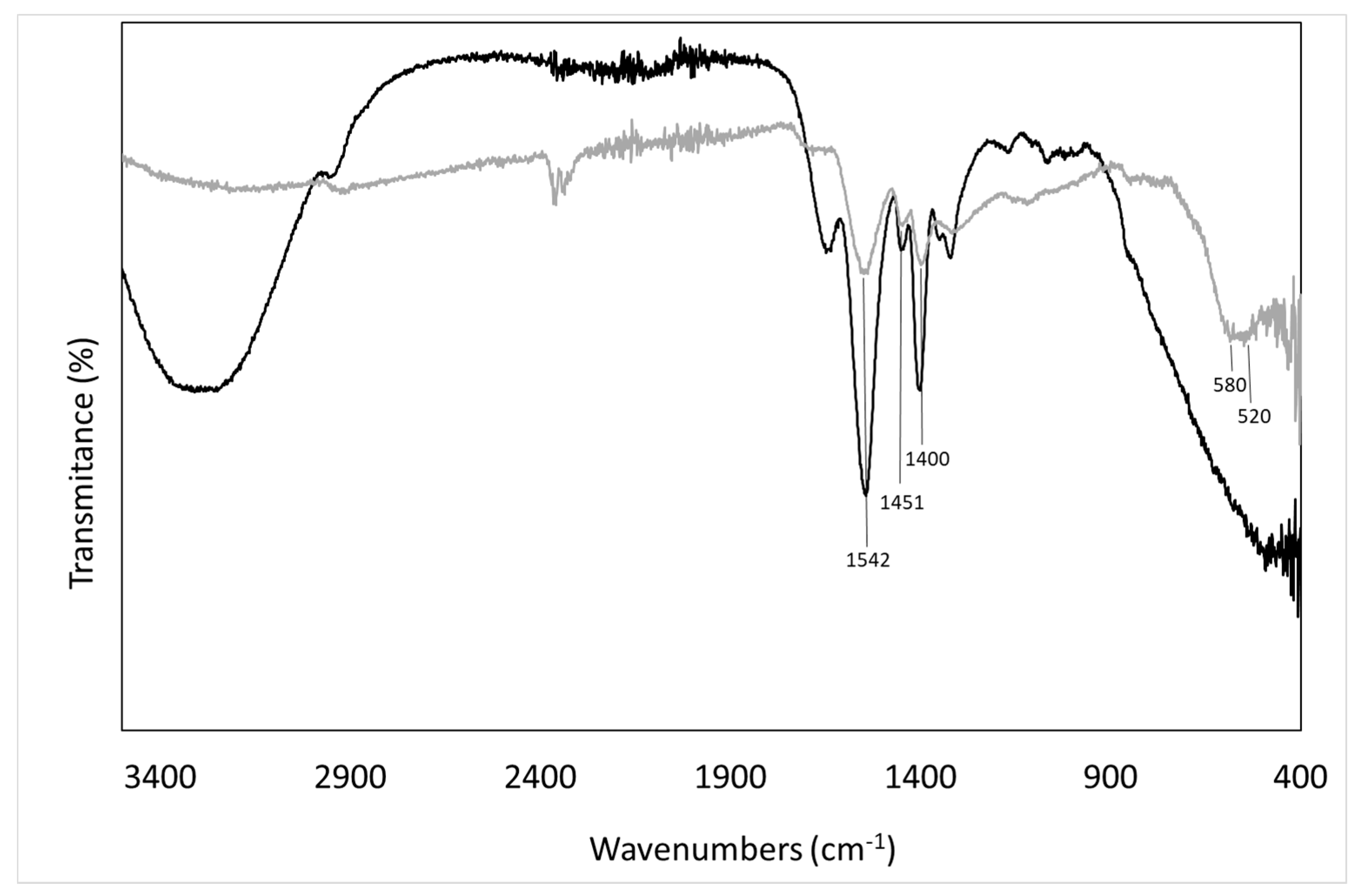
2.3.2. Fourier Transform Infrared Spectroscopy (FTIR)
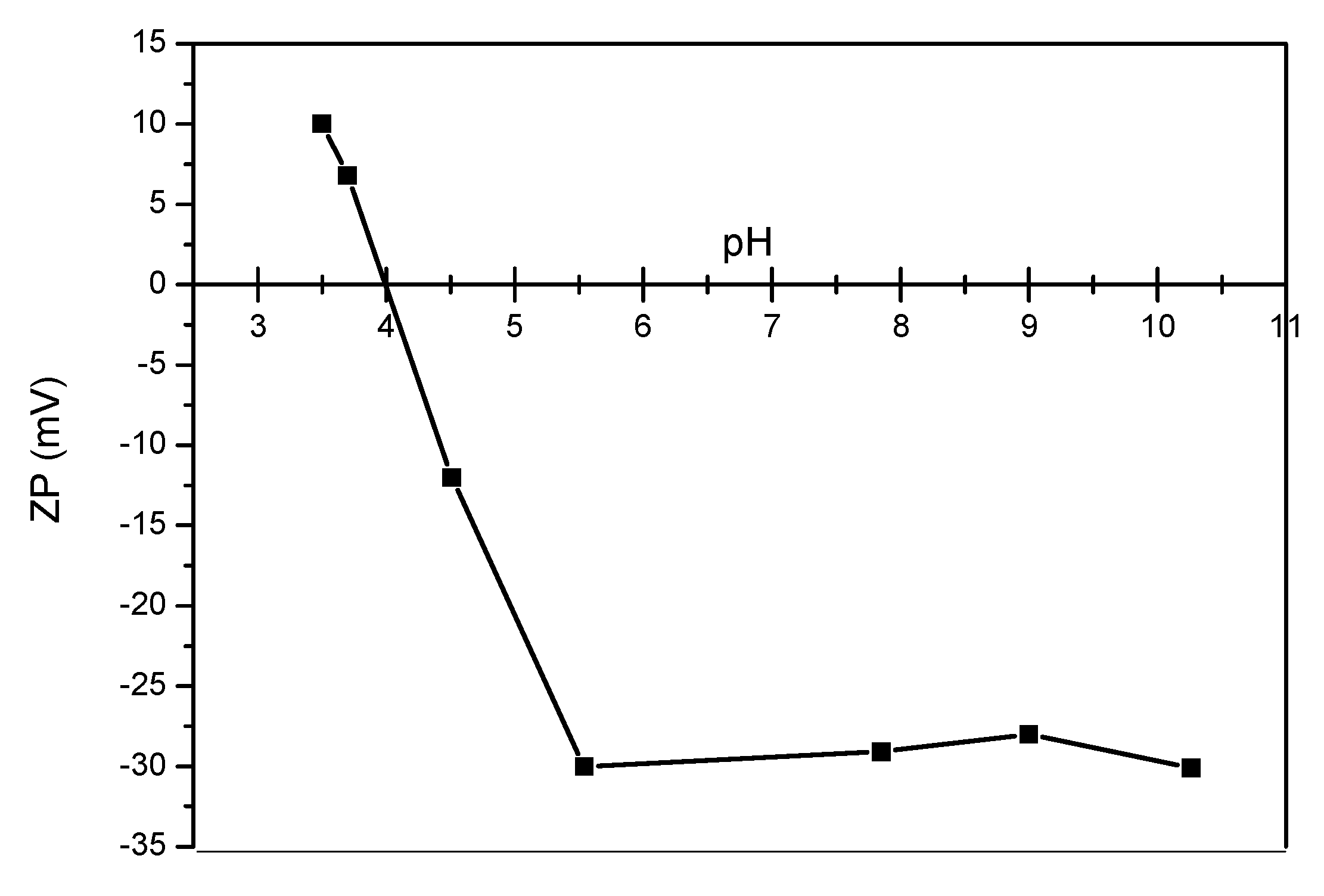
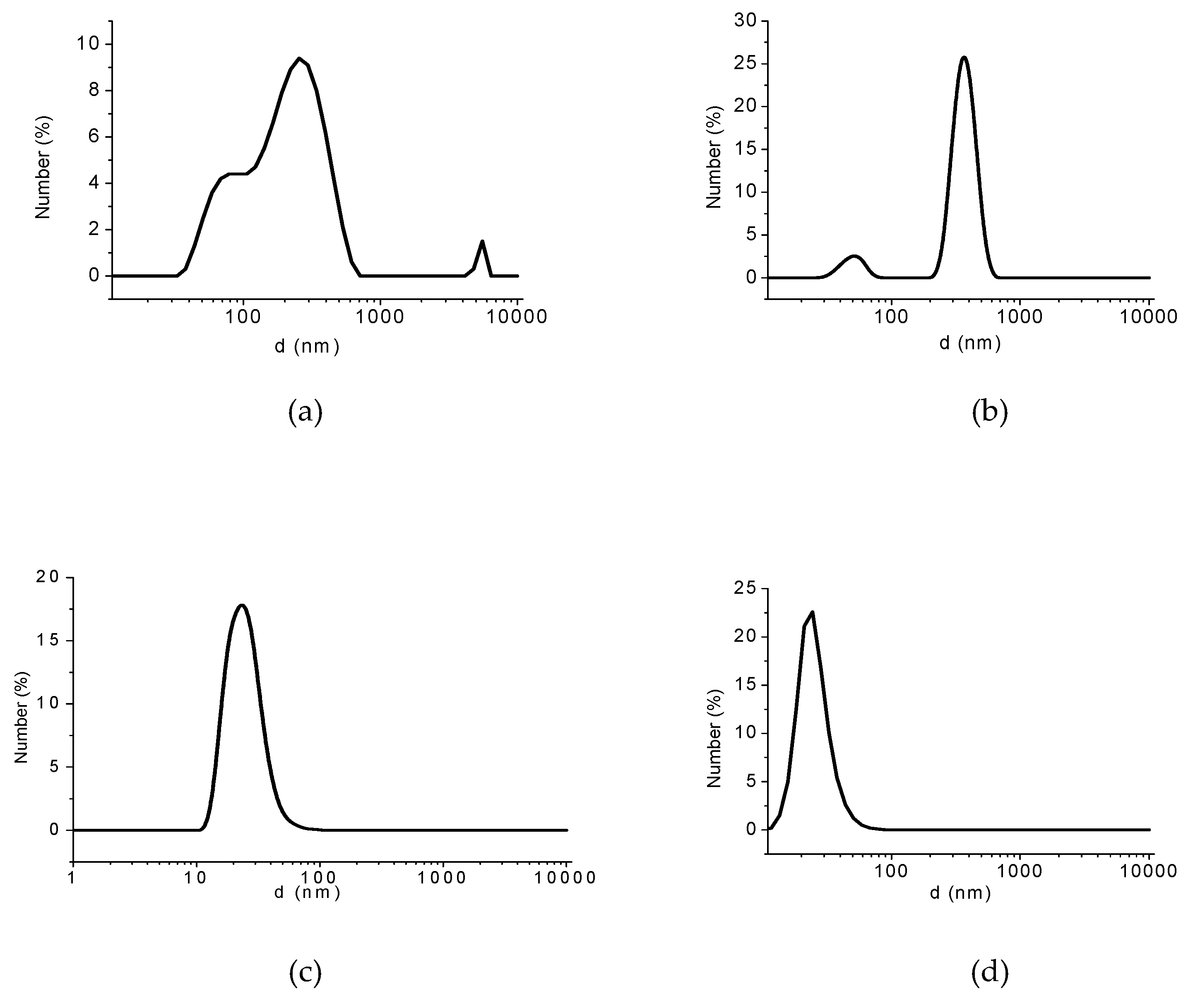
2.3.3. Surface Charge, Hydrodynamic Diameter, and Size Distribution Measurements
2.3.4. Particle Morphology Determined by Transmission Electron Microscopy (TEM)
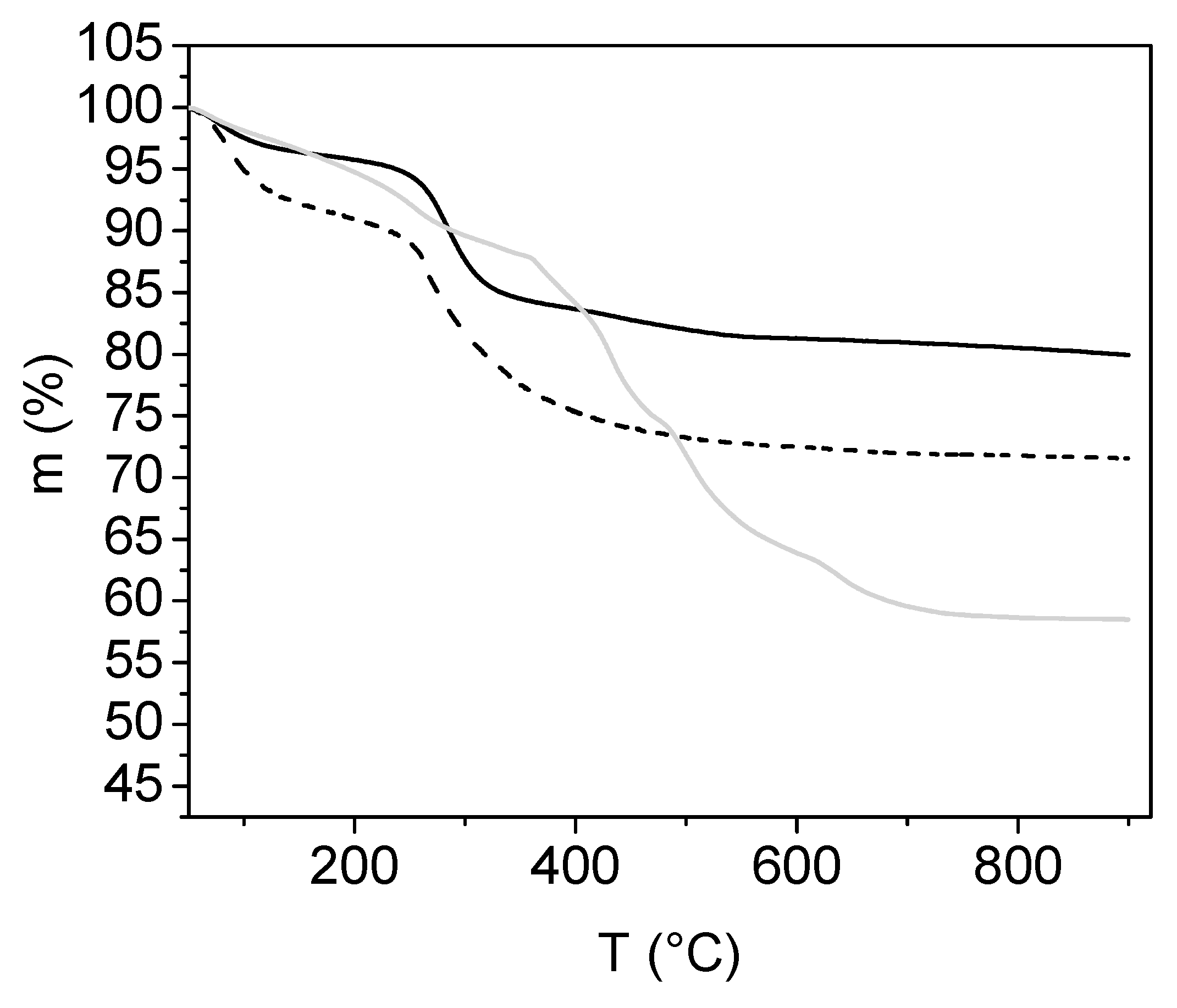
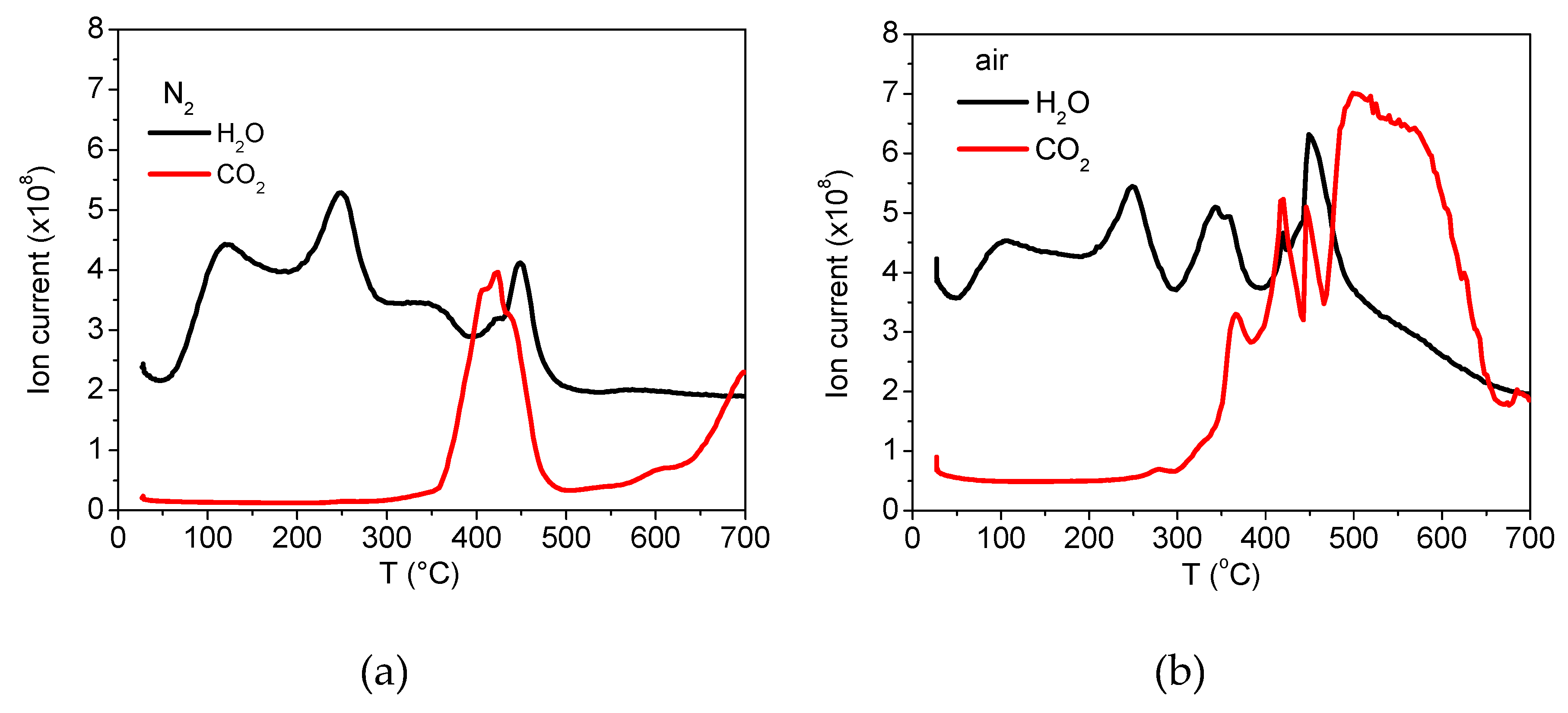
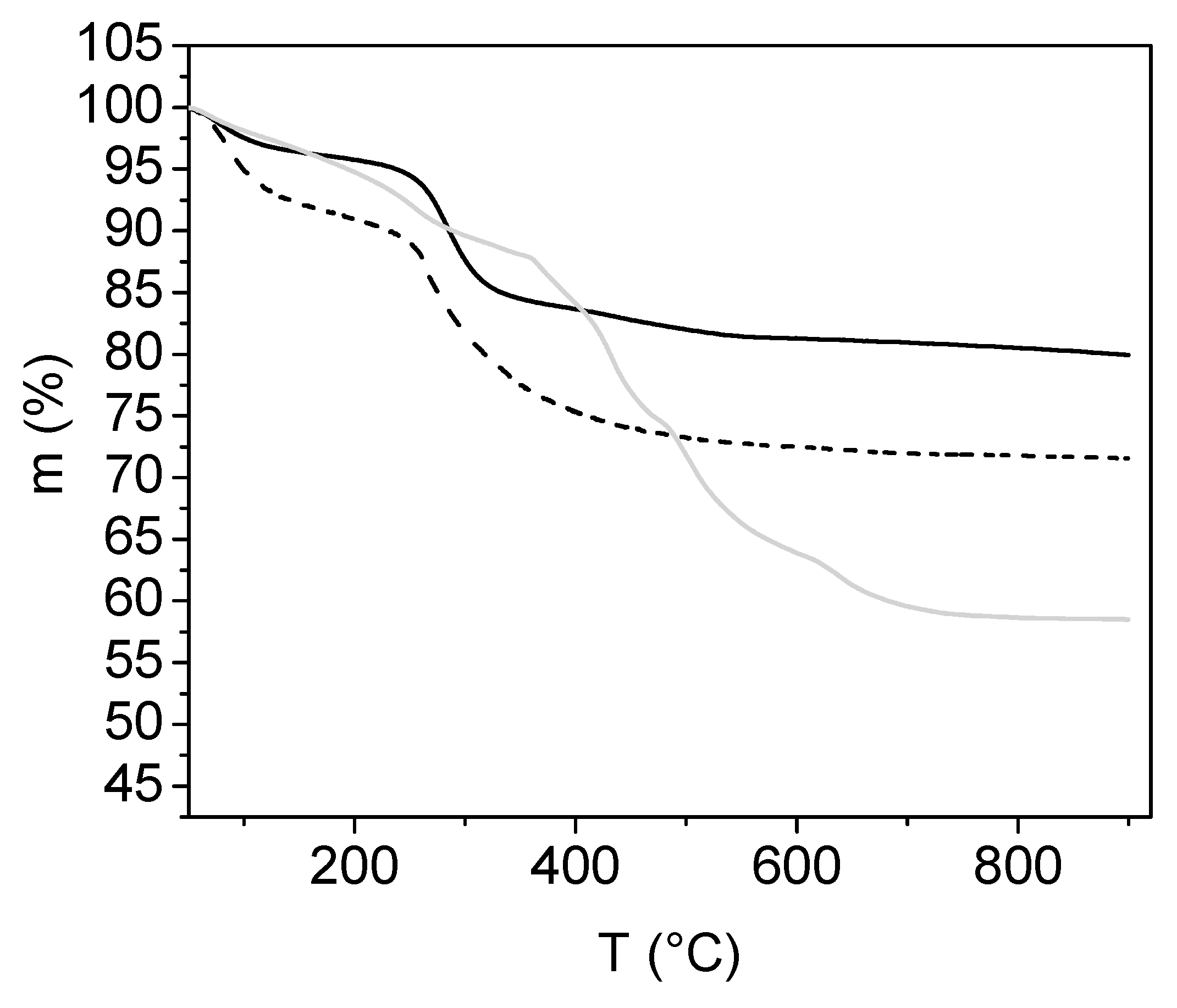
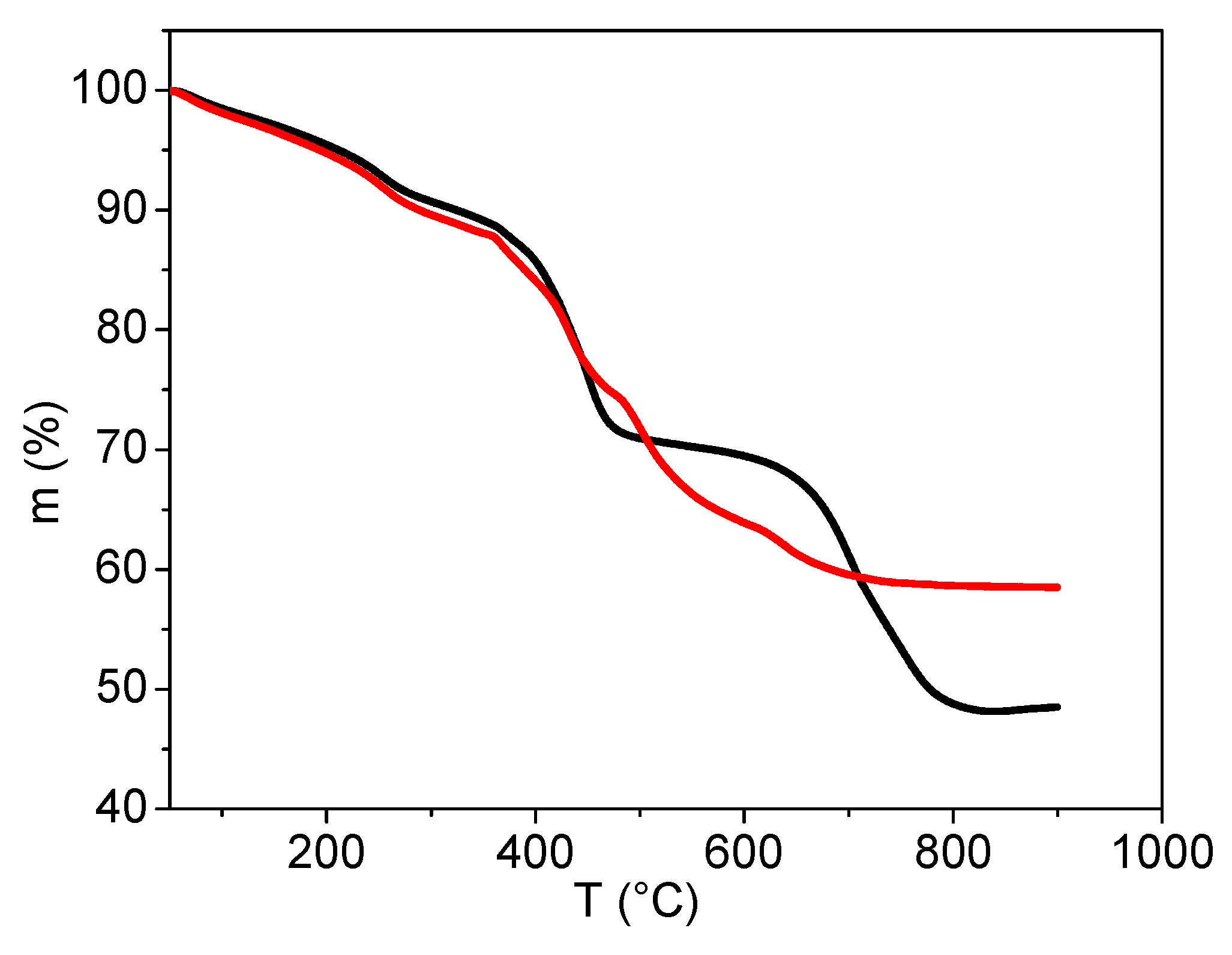
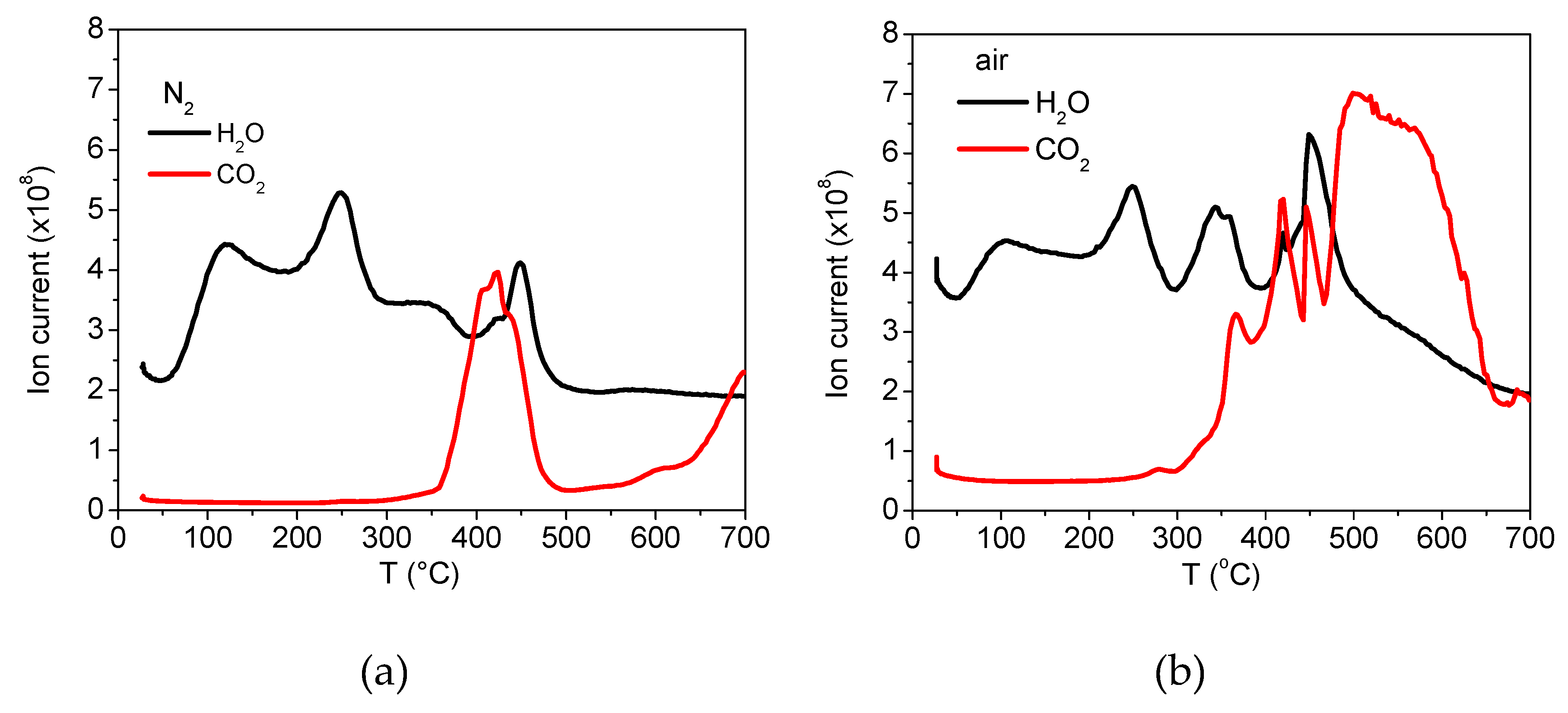
2.3.5. Characterisation by Thermogravimetric Analysis
2.3.6. Osmolality Measurements
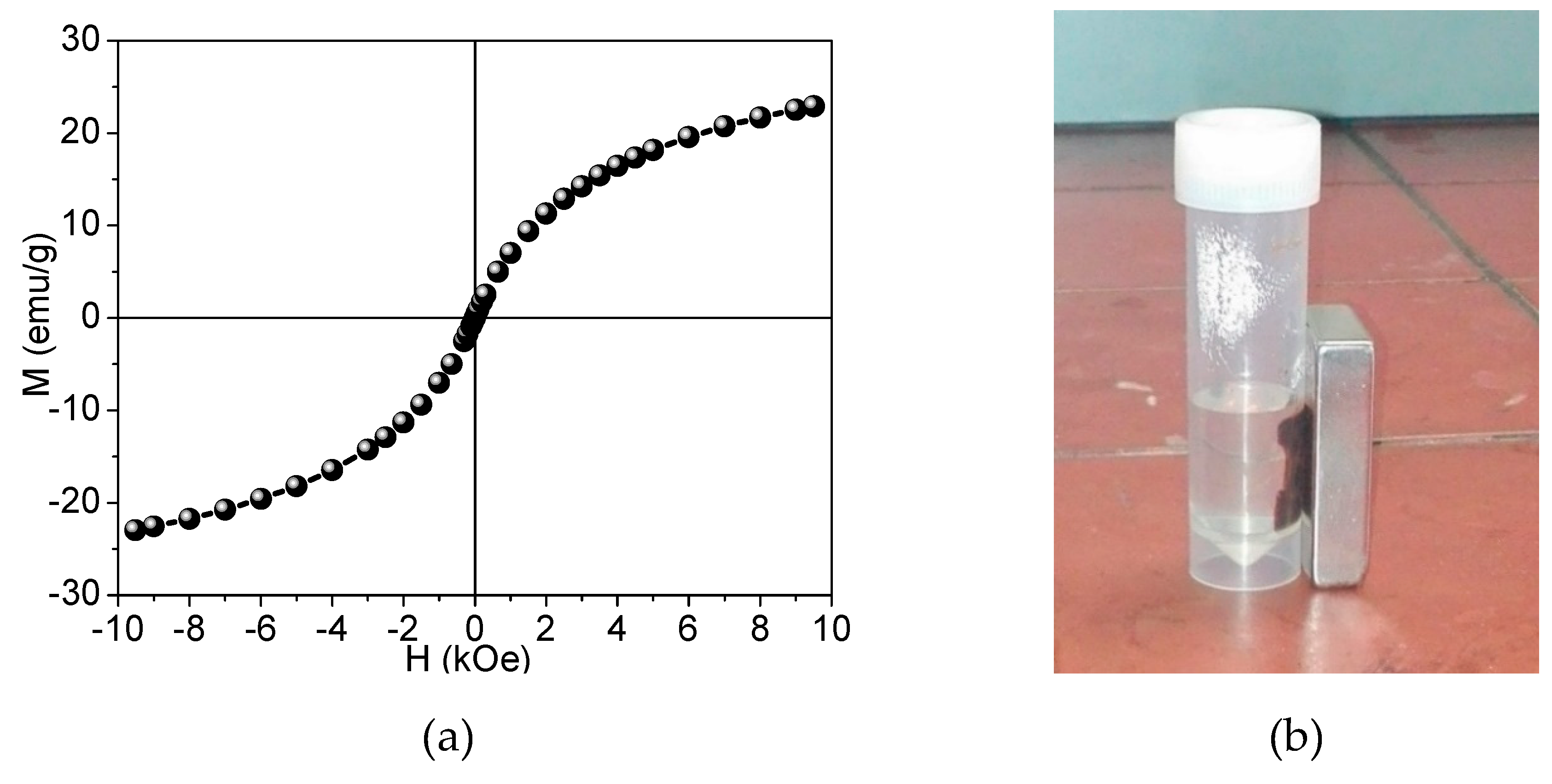
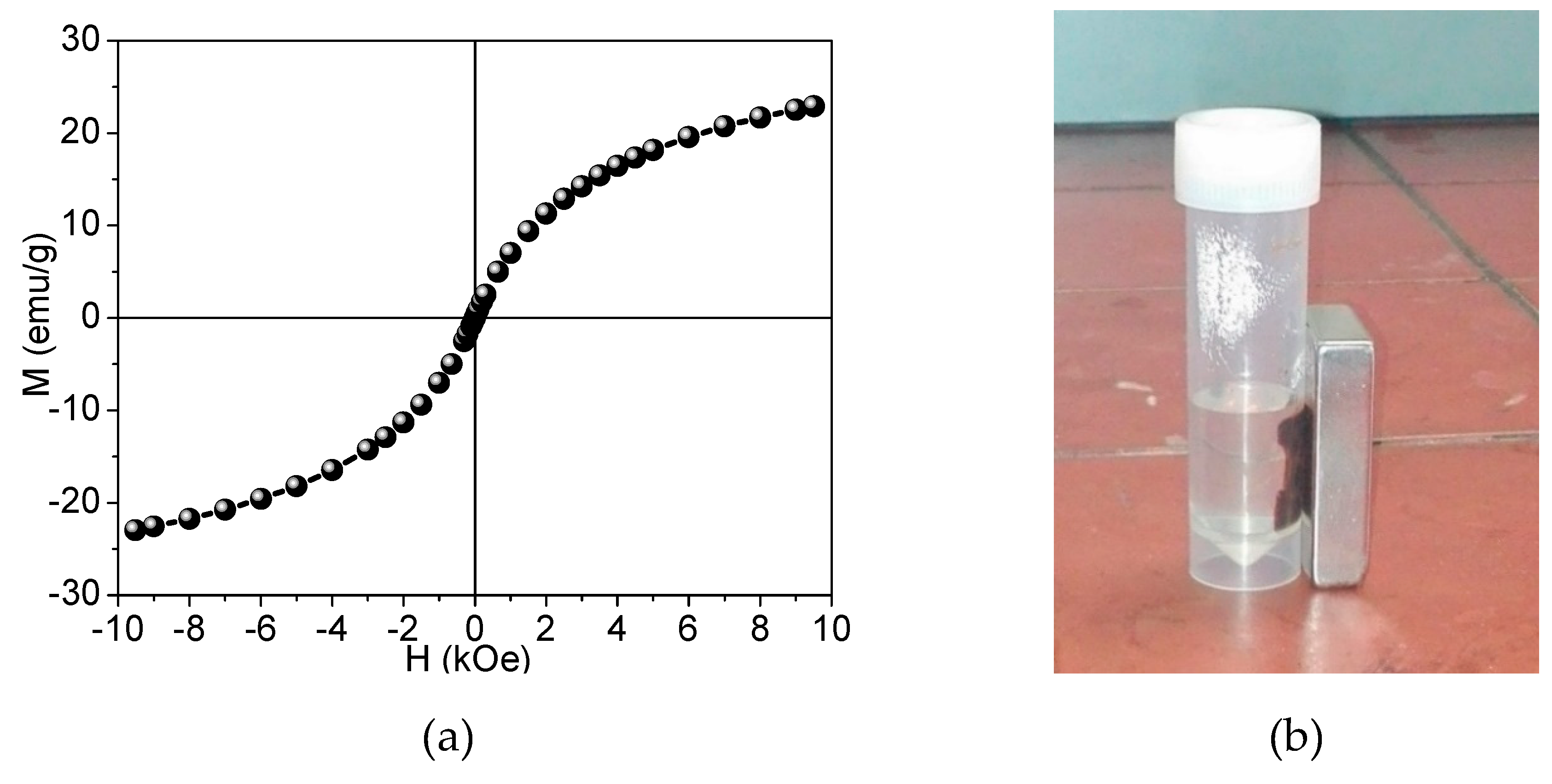
2.3.7. Magnetization Study by Vibrating Sample Magnetometry
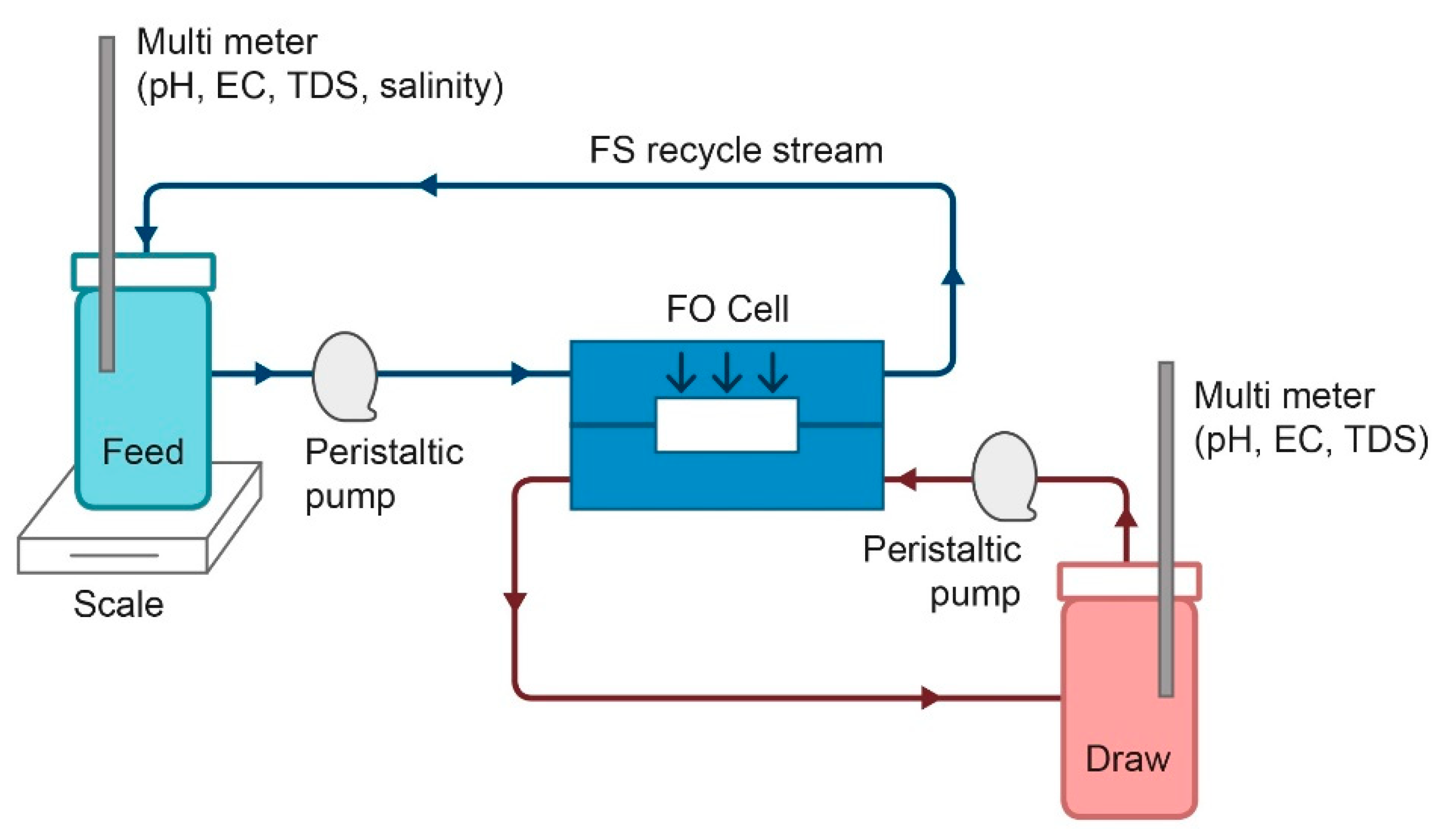
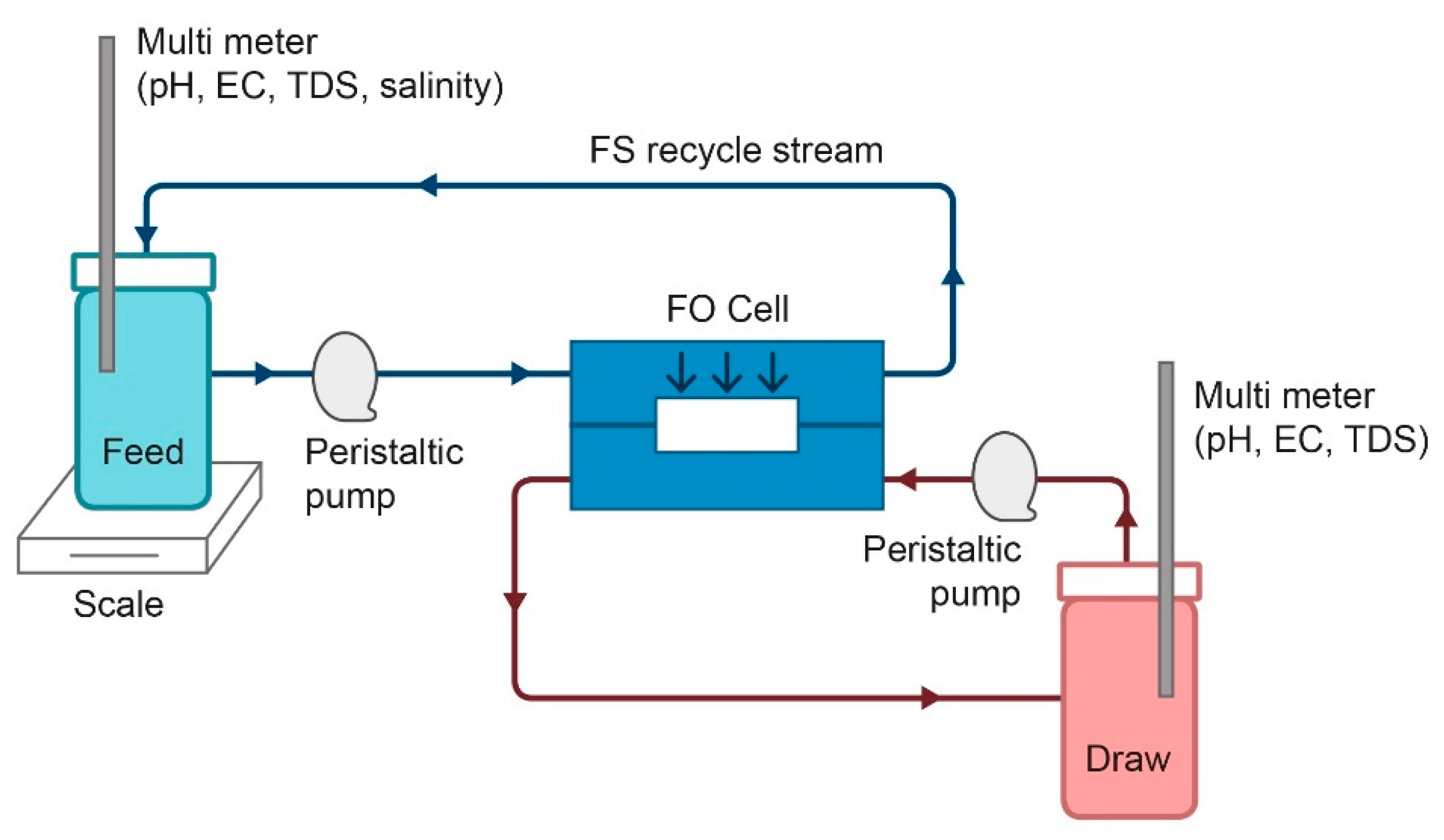
2.4. Forward Osmosis (FO) Experiments
3. Results and Discussion
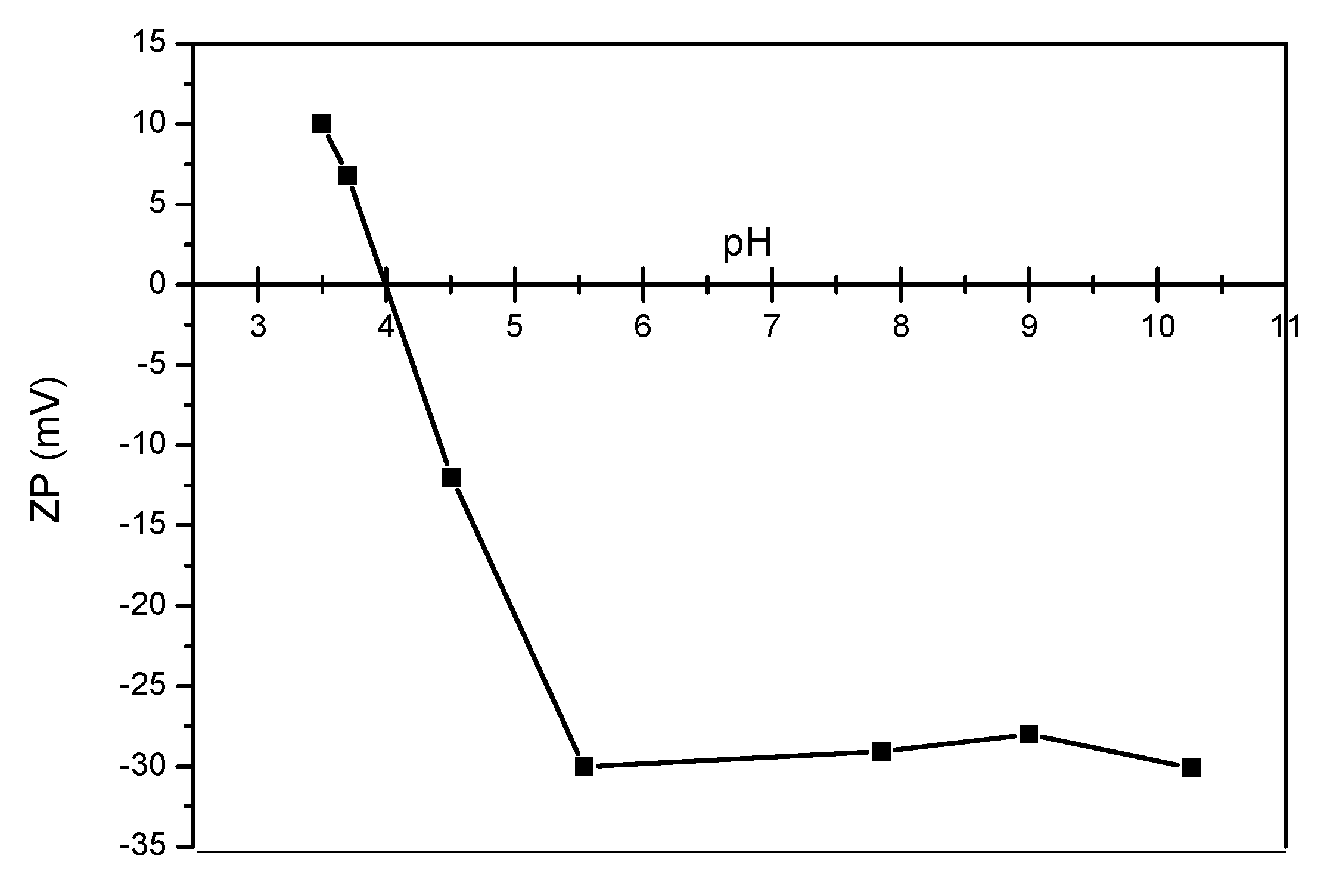
3.1. Analyses of Uncoated and Coated Samples A, B and C Using XRD, FTIR, Thermogravimetric Analysis (TGA) and Zeta Potential
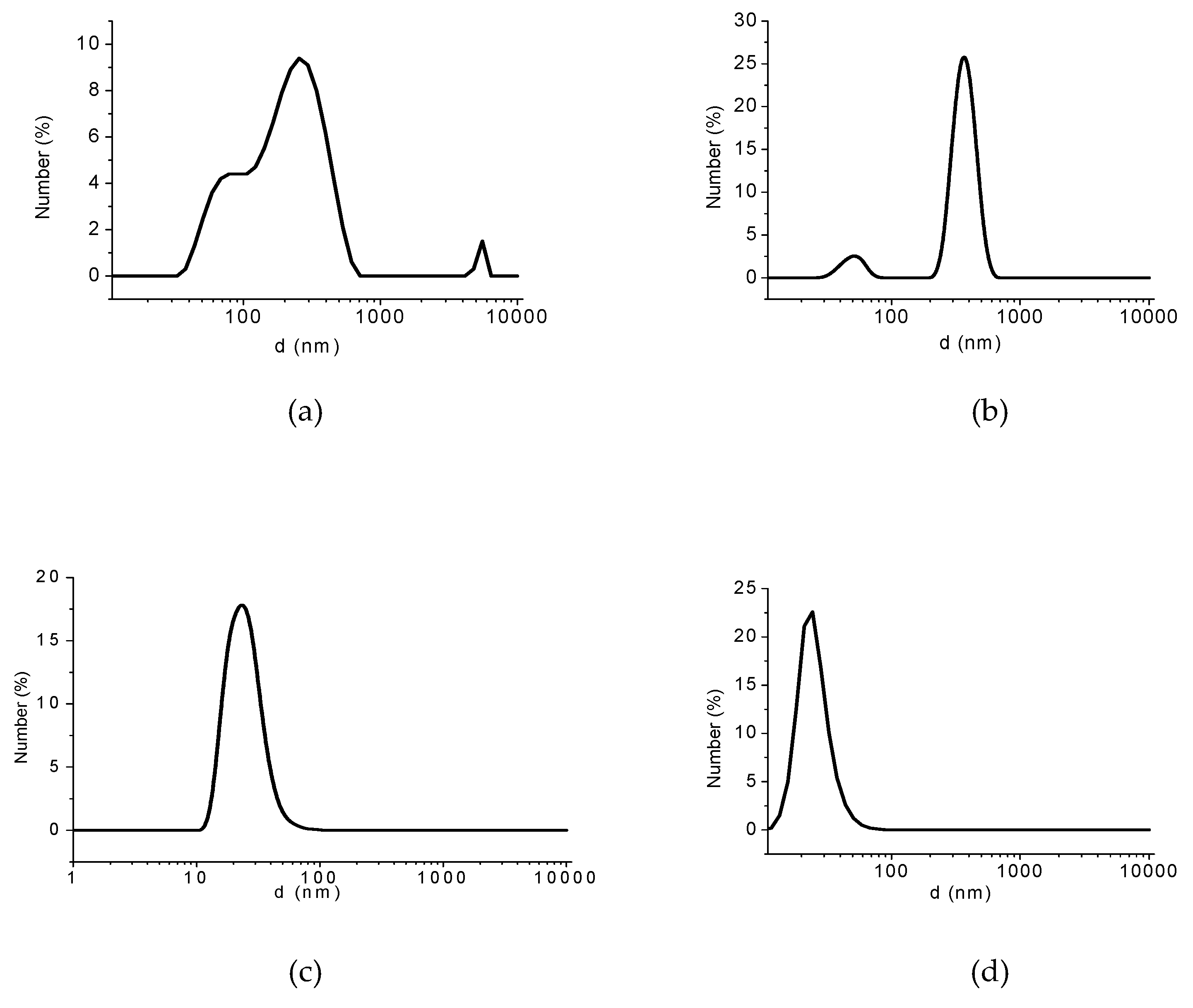
3.2. Analyses of Sample A
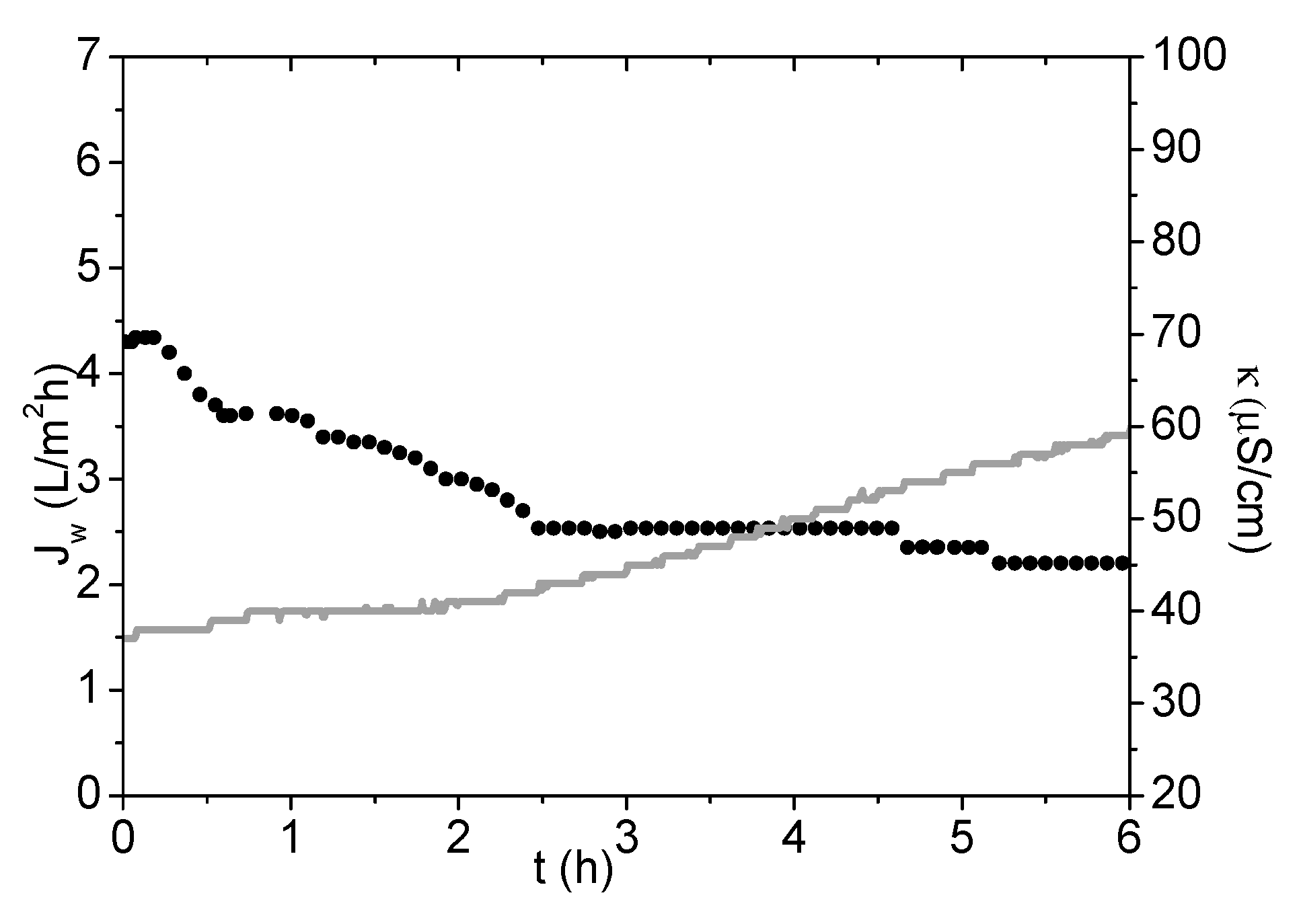
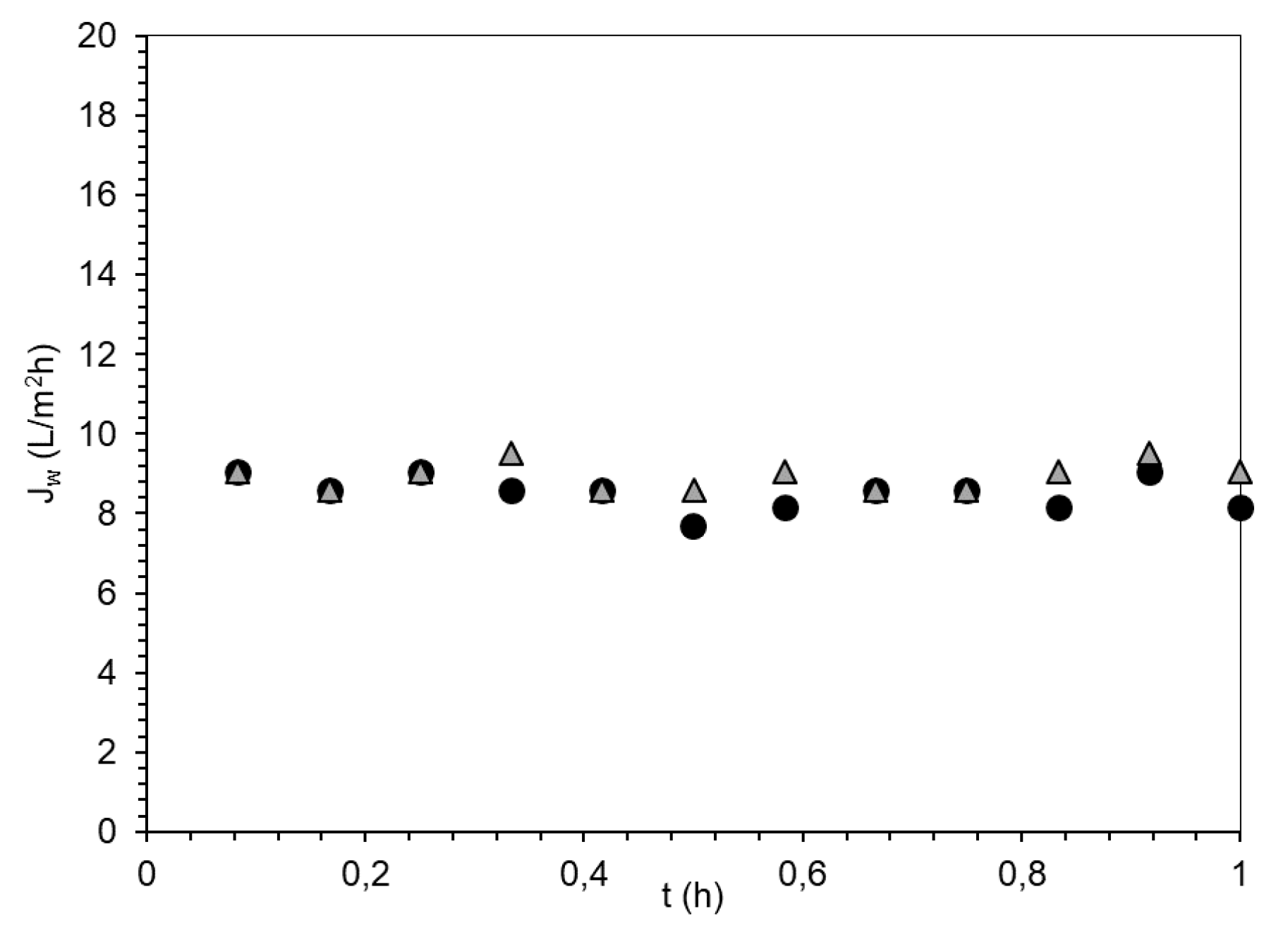
3.3. Evaluation of PSA-MNPs as Draw Solution (DS) in FO
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yang, H.M.; Woo Park, C.; Jin Han, M.; Seo, B.-K.; Moon, J.K.; Lee, K.W. Hyperbranched Polyglycerol Carboxylate-Coated Magnetic Nanoparticles as a Draw Solute in a Combined Forward Osmosis and Ultrafiltration Process. J. Nanosci. Nanotechnol. 2016, 16, 10858–10863. [Google Scholar] [CrossRef]
- Akther, N.; Sodiq, A.; Giwa, A.; Daer, S.; Arafat, H.A.; Hasan, S.W. Recent advancements in forward osmosis desalination: A review. Chem. Eng. J. 2015, 281, 502–522. [Google Scholar] [CrossRef]
- Ling, M.M.; Yu Wang, K.; Chung, T.S. Highly Water-Soluble Magnetic Nanoparticles as Novel Draw Solutes in Forward Osmosis for Water Reuse. Ind. Eng. Chem. Res. 2010, 49, 5869–5876. [Google Scholar] [CrossRef]
- Ge, Q.; Ling, M.; Chung, T.S. Draw solutions for forward osmosis processes: Developments, challenges, and prospects for the future. J. Membr. Sci. 2013, 442, 225–237. [Google Scholar] [CrossRef]
- Chekli, L.; Phuntsho, S.; Shon, H.-K.; Vigneswaran, S.; Kandasamy, J.; Chanan, A. A review of draw solutes in forward osmosis process and their use in modern applications. Desalin. Water Treat. 2012, 43, 167–184. [Google Scholar] [CrossRef]
- Ge, Q.; Su, J.; Chung, T.S.; Amy, G. Hydrophilic Superparamagnetic Nanoparticles: Synthesis, Characterization, and Performance in Forward Osmosis Processes. Ind. Eng. Chem. Res. 2011, 50, 382–388. [Google Scholar] [CrossRef]
- Zufía-Rivas, J.; Morales, P.; Veintemillas-Verdaguer, S. Effect of the Sodium Polyacrylate on the Magnetite Nanoparticles Produced by Green Chemistry Routes: Applicability in Forward Osmosis. Nanomaterials 2018, 8, 470. [Google Scholar] [CrossRef]
- Liu, J.-F.; Zhao, Z.-S.; Jiang, G.-B. Coating Fe3O4 Magnetic Nanoparticles with Humic Acid for High Efficient Removal of Heavy Metals in Water. Environ. Sci. Technol. 2008, 42, 6949–6954. [Google Scholar] [CrossRef]
- Dey, P.; Izake, E.L. Magnetic nanoparticles boosting the osmotic efficiency of a polymeric FO draw agent: Effect of polymer conformation. Desalination 2015, 373, 79–85. [Google Scholar] [CrossRef] [Green Version]
- Reddy, L.H.; Arias, J.L.; Nicolas, J.; Couvreur, P. Magnetic nanoparticles: Design and characterization, toxicity and biocompatibility, pharmaceutical and biomedical applications. Chem. Rev. 2012, 112, 5818–5878. [Google Scholar] [CrossRef]
- Alejo, T.; Arruebo, M.; Carcelen, V.; Monsalvo, V.M.; Sebastian, V. Advances in draw solutes for forward osmosis: Hybrid organic-inorganic nanoparticles and conventional solutes. Chem. Eng. J. 2017, 309, 738–752. [Google Scholar] [CrossRef]
- Li, D.; Zhang, X.; Yao, J.; Simon, G.P.; Wang, H. Stimuli-responsive polymer hydrogels as a new class of draw agent for forward osmosis desalination. Chem. Commun. 2011, 47, 1710–1712. [Google Scholar] [CrossRef] [PubMed]
- McGovern, R.K. On the potential of forward osmosis to energetically outperform reverse osmosis desalination. J. Membr. Sci. 2014, 469, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Shen, W.; Loo, S.L.; Krantz, W.B.; Wang, R.; Fane, A.G.; Hu, X. Towards temperature driven forward osmosis desalination using semi-IPN hydrogels as reversible draw agents. Water Res. 2013, 47, 3773–3781. [Google Scholar] [CrossRef] [PubMed]
- Achilli, A.; Cath, T.Y.; Childress, A.E. Selection of inorganic-based draw solutions for forward osmosis applications. J. Membr. Sci. 2010, 364, 233–241. [Google Scholar] [CrossRef]
- Ge, Q.; Su, J.; Amy, G.L.; Chung, T. Exploration of polyelectrolytes as draw solutes in forward osmosis processes. Water Res. 2012, 46, 1318–1326. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Wang, P.; Wan, C.; Chung, T. Polyelectrolyte-promoted forward osmosis membrane distillation (FO-MD) hybrid process for dye wastewater treatment. Environ. Sci. Technol. 2012, 46, 6236–6243. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Risbud, S.; Rabolt, J.F.; Stroeve, P. Synthesis and characterization of nanometer-size Fe3O4 and gamma-Fe2O3 particles. Chem. Mater. 1996, 8, 2209–2211. [Google Scholar] [CrossRef]
- Langford, J.I.; Wilson, A.J.; Seherrer, C. After Sixty Years. A Survey and Some New Results in the Determination of Crystallite Size. J. Appl. Crystallogr. 1978, 11, 102–103. [Google Scholar] [CrossRef]
- Kaszuba, M.; Corbett, J.; Watson, F.M.; Jones, A. High-concentration zeta potential measurements using light-scattering techniques. Philos. Trans. R. Soc. A 2010, 368, 4439–4451. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Ahn, H.W.; Chung, J.W.; Kwak, S.Y. Magnetic core-hydrophilic shell nanosphere as stability-enhanced draw solute for forward osmosis (FO) application. Desalination 2016, 397, 22–29. [Google Scholar] [CrossRef]
- Wilson, A.D.; Stewart, F.F. Deriving osmotic pressures of draw solutes used in osmotically driven membrane processes. J. Membr. Sci. 2013, 431, 205–211. [Google Scholar] [CrossRef]
- Chen, X.; Xu, J.; Lu, J.; Shan, B.; Gao, C. Enhanced performance of cellulose triacetate membranes using binary mixed additives for forward osmosis desalination. Desalination 2017, 405, 68–75. [Google Scholar] [CrossRef]
- Engelhardt, S.; Sadekb, A.; Duirk, S. Rejection of trace organic water contaminants by an Aquaporin-based biomimetic hollow fiber membrane. Sep. Purif. Technol. 2017, 197, 170–177. [Google Scholar] [CrossRef]
- Unterweger, H.; Subatzus, D.; Tietze, R.; Janko, C.; Poettler, M.; Stiegelschmitt, A.; Schuster, M.; Maake, C.; Boccaccini, A.; Alexiou, C. Hypericin-bearing magnetic iron oxide nanoparticles for selective drug delivery in photodynamic therapy. Int. J. Nanomed. 2015, 10, 6985–6996. [Google Scholar] [CrossRef] [Green Version]
- Dubinsky, S.; Grader, G.S.; Shter, G.E.; Silverstein, M.S. Thermal degradation of poly (acrylic acid) containing copper nitrate. Polym. Degrad. Stab. 2004, 86, 171–178. [Google Scholar] [CrossRef]
- Reith, D.; Müller, B.; Müller-Plathe, F.; Wiegand, S. How does the chain extension of poly (acrylic acid) scale in aqueous solution? A combined study with light scattering and computer simulation. J. Chem. Phys. 2002, 116, 9100–9106. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.; Zheng, Q.; An, L.; He, M.; Lin, J.; Tian, Q.; Yang, S. T1-Weight Magnetic Resonance Imaging Performances of Iron Oxide Nanoparticles Modified with a Natural Protein Macromolecule and an Artificial Macromolecule. Nanomaterials 2019, 9, 170. [Google Scholar] [CrossRef]
- Perry, M.; Madsen, S.; Jørgensen, T.; Braekevelt, S.; Lauritzen, K.; Hélix-Nielsen, C. Challenges in Commercializing Biomimetic Membranes. Membranes 2015, 5, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Li, H.; Pan, L.; Li, J.; Wang, X.; Jing, P.; Cheng, X.; Wang, W.; Wang, J.; Liu, Q. High saturation magnetization of γ-Fe2O3 nano-particles by a facile one-step synthesis approach. Sci. Rep. 2016, 6, 32360. [Google Scholar] [CrossRef]

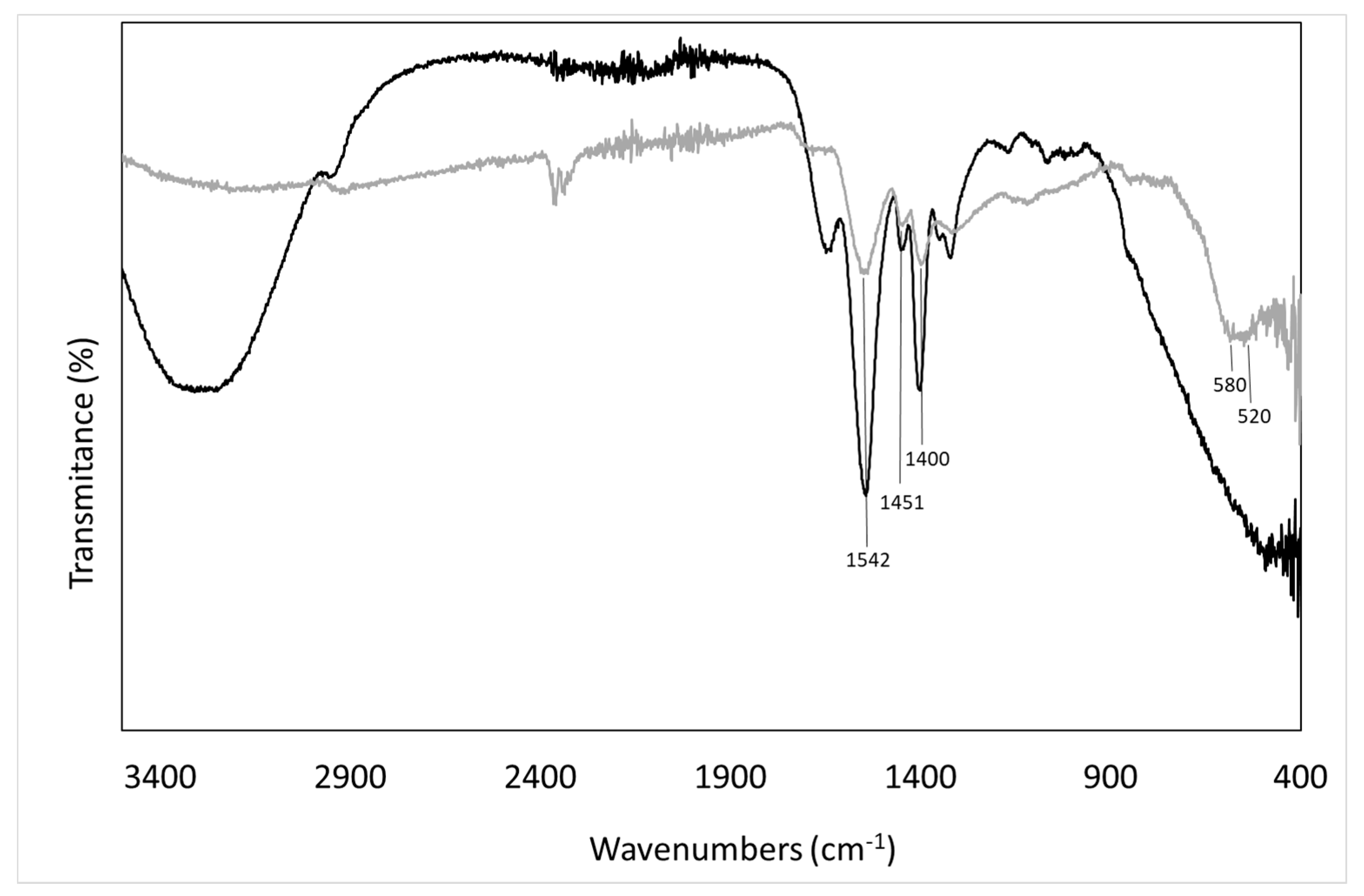



 ) and washed membrane (
) and washed membrane (  ).
) and washed membrane ( ).
).
) and washed membrane ( ).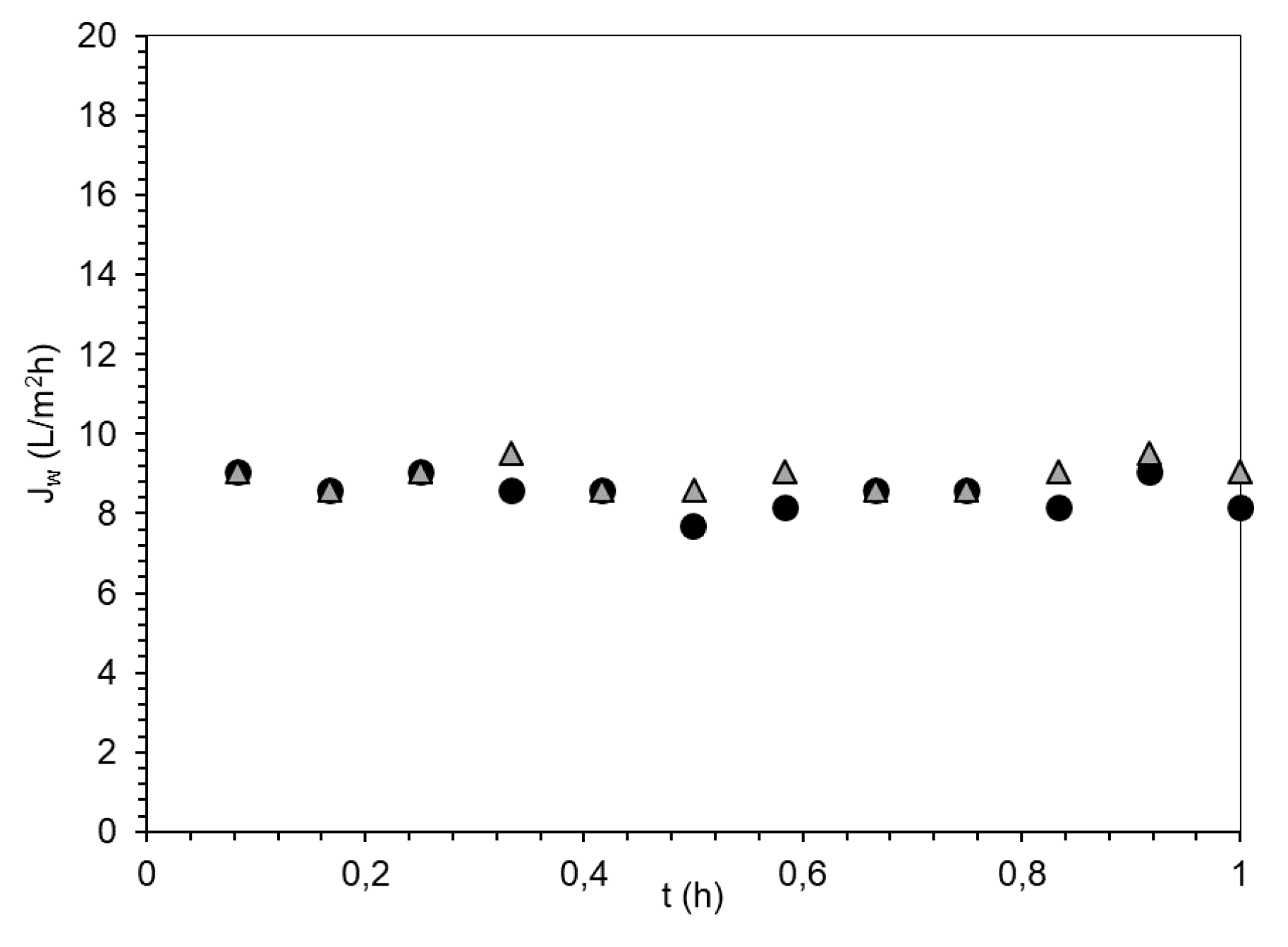

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molar Ratio PSA-MNPs. | pH | ζ-Potential (mV) | Osmotic Pressure (bar) | m (%) (air) | ns (molecules/nm2) |
|---|---|---|---|---|---|
| Sample A: 1:1 | 9.5 | −42.9 | 9 | 37 | 0.95 |
| Sample B: 1:2 | 8.3 | −35.5 | 5.5 | 22 | 0.46 |
| Sample C: 1:3 | 9.2 | −41.8 | 0.2 | 15 | 0.29 |
| Sample A. | Osmotic Pressure (bar) | Concentration (g/L) | d (nm) |
|---|---|---|---|
| As-prepared sample | 30 | 0.1 | 520 |
| Retentate from first UF purification | 9 | 0.1 | 290 |
| Retentate from second UF purification | 4 | 0.1 | 300 |
| Sonicated retentate after second UF purification | 6 | 0.1 | 50 |
| Retentate from third UF purification | 1.3 | 0.1 | 630 |
| Sonicated retentate after third UF purification | 2.3 | 0.1 | 35 |
| FS | DS | Initial Osmotic Pressure (bar) | Jw (L/m2h) | Js (g/m2·h) | Js/Jw (g/L) | Vpermeate in 1 h (mL) |
|---|---|---|---|---|---|---|
| DI water | PSA-MNP | 9 | 3.8 | 0.05 | 0.01 | 10.6 |
| Sample A | Conductivity (mS/cm) |
|---|---|
| Permeate from first UF purification | 15.9 |
| Permeate from second UF purification | 2.64 |
| Permeate from third UF purification | 1.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ban, I.; Markuš, S.; Gyergyek, S.; Drofenik, M.; Korenak, J.; Helix-Nielsen, C.; Petrinić, I. Synthesis of Poly-Sodium-Acrylate (PSA)-Coated Magnetic Nanoparticles for Use in Forward Osmosis Draw Solutions. Nanomaterials 2019, 9, 1238. https://doi.org/10.3390/nano9091238
Ban I, Markuš S, Gyergyek S, Drofenik M, Korenak J, Helix-Nielsen C, Petrinić I. Synthesis of Poly-Sodium-Acrylate (PSA)-Coated Magnetic Nanoparticles for Use in Forward Osmosis Draw Solutions. Nanomaterials. 2019; 9(9):1238. https://doi.org/10.3390/nano9091238
Chicago/Turabian StyleBan, Irena, Sabina Markuš, Sašo Gyergyek, Miha Drofenik, Jasmina Korenak, Claus Helix-Nielsen, and Irena Petrinić. 2019. "Synthesis of Poly-Sodium-Acrylate (PSA)-Coated Magnetic Nanoparticles for Use in Forward Osmosis Draw Solutions" Nanomaterials 9, no. 9: 1238. https://doi.org/10.3390/nano9091238